CN1257912C - 用于治疗乙型肝炎的β-L-2′-脱氧-核苷 - Google Patents
用于治疗乙型肝炎的β-L-2′-脱氧-核苷 Download PDFInfo
- Publication number
- CN1257912C CN1257912C CNB998095532A CN99809553A CN1257912C CN 1257912 C CN1257912 C CN 1257912C CN B998095532 A CNB998095532 A CN B998095532A CN 99809553 A CN99809553 A CN 99809553A CN 1257912 C CN1257912 C CN 1257912C
- Authority
- CN
- China
- Prior art keywords
- purposes
- administration
- pharmaceutically acceptable
- compound
- applicable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Images


Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
- A61K31/7072—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid having two oxo groups directly attached to the pyrimidine ring, e.g. uridine, uridylic acid, thymidine, zidovudine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
- A61K31/708—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid having oxo groups directly attached to the purine ring system, e.g. guanosine, guanylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
Abstract
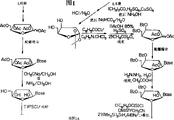
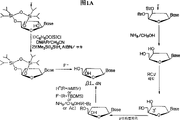
Description














| 抑制剂 | WHV DNAIC50 | DNAαKi b(μM) | DNA βKi b(μM) | DNA γKi b(μM) |
| β-L-dT-TP | 0.34 | >100 | >100 | >100 |
| β-L-dA-TP | 2.3 | >100 | >100 | >100 |
| β-L-dC-TP | 2.0 | >100 | >100 | >100 |
| β-L-dU-TP | 8 | >100 | >100 | >100 |
| 化合物 | HBV病毒颗粒aEC50(μM) | HBV RibEC50(μM) | 细胞毒性1C50(μM) | 选择性指数1C50/EC50 |
| β-L-dT | 0.05 | 0.05 | >200 | >4000 |
| β-L-dC | 0.05 | 0.05 | >200 | >4000 |
| β-L-dA | 0.05 | 0.05 | >200 | >2000 |
| β-L-dI | 1.0 | 1.0 | >200 | >200 |
| β-L-dU | 5.0 | 5.0 | >200 | >40 |
| 组合物 | 比例 | EC50 |
| L-dC+L-dT | 1∶3 | 0.023 |
| L-dC+L-dT | 1∶1 | 0.053 |
| L-dC+L-dT | 3∶1 | 0.039 |
| L-dC+L-dA | 1∶30 | 0.022 |
| L-dC+L-dA | 1∶10 | 0.041 |
| L-dC+L-dA | 1∶3 | 0.075 |
| L-dT+L-dA | 1∶30 | 0.054 |
| L-dT+L-dA | 1∶10 | 0.077 |
| L-dT+L-dA | 1∶3 | 0.035 |
| aβ-L-2′-脱氧腺苷(μM) | bβ-L-2′-脱氧胞苷(μM) | 抑制率% | CC.I. |
| 0.5 | 90 | ||
| 0.05 | 24 | ||
| 0.005 | 1 | ||
| 0.5 | 95 | ||
| 0.05 | 40 | ||
| 0.005 | 10 | ||
| 0.05 | 0.05 | 80 | 0.34 |
| 0.05 | 0.005 | 56 | 0.20 |
| 0.05 | 0.0005 | 50 | 0.56 |
| 0.005 | 0.05 | 72 | 0.35 |
| 0.005 | 0.005 | 54 | 0.35 |
| 0.005 | 0.0005 | 30 | 0.16 |
| 0.0005 | 0.05 | 50 | 0.83 |
| 0.0005 | 0.005 | 15 | 0.28 |
| 0.0005 | 0.0005 | 0 | N.A. |
| 天 | 对照 | LdA | LdT | LdC |
| ng/ml WHV-DNA/ml血清1,2 | ||||
| 013714 | 381398412446392 | 43636914010274 | 423451461 | 426123624620 |
| 化合物 | CFU-GM(μM) | BFU-E(μM) |
| β-L-dA | >10 | >10 |
| β-L-dC | >10 | >10 |
| β-L-dT | >10 | >10 |
| β-L-dU | >10 | >10 |
| 拉米夫定 | >10 | >10 |
Claims (31)

Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US9611098P | 1998-08-10 | 1998-08-10 | |
| US60/096,110 | 1998-08-10 | ||
| US13135299P | 1999-04-28 | 1999-04-28 | |
| US60/131,352 | 1999-04-28 |
Related Child Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB021265739A Division CN100482236C (zh) | 1998-08-10 | 1999-08-10 | 用于治疗乙型肝炎的β-L-2’-脱氧-核苷 |
| CN2006101007520A Division CN101120947B (zh) | 1998-08-10 | 1999-08-10 | 用于治疗乙型肝炎的β-L-2′-脱氧-核苷 |
| CN2006100997025A Division CN1911237B (zh) | 1998-08-10 | 1999-08-10 | 用于治疗乙型肝炎的β-L-2'-脱氧-核苷 |
| CNB2004100028639A Division CN100387237C (zh) | 1998-08-10 | 1999-08-10 | 用于治疗乙型肝炎的β-L-2'-脱氧-核苷 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1320128A CN1320128A (zh) | 2001-10-31 |
| CN1257912C true CN1257912C (zh) | 2006-05-31 |
Family
ID=26791121
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB2004100028639A Expired - Lifetime CN100387237C (zh) | 1998-08-10 | 1999-08-10 | 用于治疗乙型肝炎的β-L-2'-脱氧-核苷 |
| CNB021265739A Expired - Lifetime CN100482236C (zh) | 1998-08-10 | 1999-08-10 | 用于治疗乙型肝炎的β-L-2’-脱氧-核苷 |
| CNB998095532A Expired - Lifetime CN1257912C (zh) | 1998-08-10 | 1999-08-10 | 用于治疗乙型肝炎的β-L-2′-脱氧-核苷 |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB2004100028639A Expired - Lifetime CN100387237C (zh) | 1998-08-10 | 1999-08-10 | 用于治疗乙型肝炎的β-L-2'-脱氧-核苷 |
| CNB021265739A Expired - Lifetime CN100482236C (zh) | 1998-08-10 | 1999-08-10 | 用于治疗乙型肝炎的β-L-2’-脱氧-核苷 |
Country Status (22)
| Country | Link |
|---|---|
| US (5) | US6395716B1 (zh) |
| EP (2) | EP1104436B1 (zh) |
| JP (2) | JP4294870B2 (zh) |
| KR (4) | KR100568035B1 (zh) |
| CN (3) | CN100387237C (zh) |
| AT (1) | ATE313550T1 (zh) |
| AU (1) | AU5475799A (zh) |
| BR (1) | BRPI9912896B8 (zh) |
| CA (1) | CA2340156C (zh) |
| CY (2) | CY2007017I2 (zh) |
| DE (2) | DE122007000062I1 (zh) |
| DK (3) | DK1104436T3 (zh) |
| ES (3) | ES2255295T3 (zh) |
| FR (1) | FR07C0046I2 (zh) |
| HK (3) | HK1034083A1 (zh) |
| LU (1) | LU91348I2 (zh) |
| MX (1) | MXPA01001507A (zh) |
| NL (1) | NL300286I2 (zh) |
| PT (2) | PT2415776T (zh) |
| RU (1) | RU2424016C2 (zh) |
| SG (1) | SG132498A1 (zh) |
| WO (1) | WO2000009531A2 (zh) |
Families Citing this family (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1655033A1 (en) * | 1995-06-07 | 2006-05-10 | Emory University | Nucleosides with anti-hepatitis B virus activity |
| US6444652B1 (en) * | 1998-08-10 | 2002-09-03 | Novirio Pharmaceuticals Limited | β-L-2'-deoxy-nucleosides for the treatment of hepatitis B |
| CN100387237C (zh) * | 1998-08-10 | 2008-05-14 | 艾丹尼克斯(开曼)有限公司 | 用于治疗乙型肝炎的β-L-2'-脱氧-核苷 |
| TR200601784T2 (tr) * | 1999-11-12 | 2007-01-22 | Pharmasset Ltd. | 2'-deoksi-L-nükleositlerin sentezi |
| EP1600452A3 (en) | 1999-11-12 | 2008-09-10 | Pharmasset, Inc. | Synthesis of 2'-deoxy-L-nucleosides |
| HU230698B1 (hu) * | 2000-02-29 | 2017-09-28 | Bristol-Myers Squibb Holdings Ireland | Alacsony dózisú entecavir formuláció és alkalmazása |
| US20020056123A1 (en) * | 2000-03-09 | 2002-05-09 | Gad Liwerant | Sharing a streaming video |
| US6822089B1 (en) * | 2000-03-29 | 2004-11-23 | Isis Pharmaceuticals, Inc. | Preparation of deoxynucleosides |
| EP1284741B1 (en) * | 2000-04-13 | 2008-11-19 | Pharmasset, Inc. | 3'-or 2'-hydroxymethyl substituted nucleoside derivatives for treatment of viral infections |
| MY164523A (en) | 2000-05-23 | 2017-12-29 | Univ Degli Studi Cagliari | Methods and compositions for treating hepatitis c virus |
| CN101099745A (zh) * | 2000-05-26 | 2008-01-09 | 艾登尼科斯(开曼)有限公司 | 处理黄病毒和瘟病毒的方法和组合物 |
| US6787526B1 (en) * | 2000-05-26 | 2004-09-07 | Idenix Pharmaceuticals, Inc. | Methods of treating hepatitis delta virus infection with β-L-2′-deoxy-nucleosides |
| TR200402565T4 (tr) * | 2000-05-26 | 2004-12-21 | Idenix (Cayman) Limited | Hepatit delta virüsü enfeksiyonunun tedavi edilmesinde ß-L-2'-deoksi-nükleosidlerin kullanımı |
| US6875751B2 (en) | 2000-06-15 | 2005-04-05 | Idenix Pharmaceuticals, Inc. | 3′-prodrugs of 2′-deoxy-β-L-nucleosides |
| MY141594A (en) * | 2000-06-15 | 2010-05-14 | Novirio Pharmaceuticals Ltd | 3'-PRODRUGS OF 2'-DEOXY-ß-L-NUCLEOSIDES |
| DE60144573D1 (de) | 2000-11-29 | 2011-06-16 | Mitsui Chemicals Inc | Verfahren und Zwischenprodukte bei der Synthese von L-Thymidin |
| CN101250208B (zh) * | 2000-11-29 | 2012-06-20 | 三井化学株式会社 | L-核酸衍生物及其合成方法 |
| AU2002360697B2 (en) | 2001-12-20 | 2009-04-23 | Beth Israel Deaconess Medical Center | Treatment of EBV and KHSV infection and associated abnormal cellular proliferation |
| WO2003055896A2 (en) * | 2001-12-21 | 2003-07-10 | Micrologix Biotech Inc. | Anti-viral 7-deaza l-nucleosides |
| CA2477741A1 (en) * | 2002-02-28 | 2003-09-04 | Biota, Inc. | Nucleotide mimics and their prodrugs |
| JP2005533870A (ja) * | 2002-06-27 | 2005-11-10 | メディヴィル・アクチボラグ | アバカビールおよびアロブジンの相乗的相互作用 |
| PL374831A1 (en) | 2002-06-28 | 2005-10-31 | Idenix (Cayman) Limited | Modified 2' and 3' -nucleoside prodrugs for treating flaviridae infections |
| US7608600B2 (en) * | 2002-06-28 | 2009-10-27 | Idenix Pharmaceuticals, Inc. | Modified 2′ and 3′-nucleoside prodrugs for treating Flaviviridae infections |
| CN101172993A (zh) * | 2002-06-28 | 2008-05-07 | 埃迪尼克斯(开曼)有限公司 | 用于治疗黄病毒感染的2′-c-甲基-3′-o-l-缬氨酸酯核糖呋喃基胞苷 |
| RS113904A (en) * | 2002-06-28 | 2007-02-05 | Idenix (Cayman) Limited | 2'-c-methyl-3'-o-l-valine ester ribofuranosyl cytidine for treatment of flaviviridae infections |
| JP5087211B2 (ja) | 2002-06-28 | 2012-12-05 | イデニクス(ケイマン)リミテツド | フラビウィルス感染治療のための2′および3′−ヌクレオシドプロドラッグ |
| TWI244393B (en) | 2002-08-06 | 2005-12-01 | Idenix Pharmaceuticals Inc | Crystalline and amorphous forms of beta-L-2'-deoxythymidine |
| US7186700B2 (en) * | 2002-09-13 | 2007-03-06 | Idenix Pharmaceuticals, Inc. | β-L-2′-deoxynucleosides for the treatment of resistant HBV strains and combination therapies |
| BR0315795A (pt) | 2002-10-31 | 2005-09-13 | Metabasis Therapeutics Inc | Pró-medicamentos de monofosfato de citarabina |
| HUE033832T2 (en) | 2002-11-15 | 2018-01-29 | Idenix Pharmaceuticals Llc | 2'-methyl nucleosides in combination with interferon and Flaviviridae mutation |
| US7598373B2 (en) * | 2002-12-12 | 2009-10-06 | Idenix Pharmaceuticals, Inc. | Process for the production of 2-C-methyl-D-ribonolactone |
| WO2004084453A2 (en) * | 2003-03-20 | 2004-09-30 | Microbiologica Quimica E Farmaceutica Ltd. | METHODS OF MANUFACTURE OF 2'-DEOXY-β-L-NUCLEOSIDES |
| US7595390B2 (en) | 2003-04-28 | 2009-09-29 | Novartis Ag | Industrially scalable nucleoside synthesis |
| WO2004096197A2 (en) * | 2003-05-02 | 2004-11-11 | Universita Degli Studi Di Cagliari | 5-aza-7-deazapurine nucleosides for treating flaviviridae |
| KR100883703B1 (ko) | 2003-05-30 | 2009-02-12 | 파마셋 인코포레이티드 | 변형 불소화 뉴클레오시드 유사체 |
| AU2004254620A1 (en) * | 2003-06-30 | 2005-01-13 | Idenix (Cayman) Limited | Synthesis of beta-L-2-deoxy nucleosides |
| CN1315863C (zh) * | 2003-12-12 | 2007-05-16 | 河南省科学院质量检验与分析测试研究中心 | β-L-2'-脱氧-核苷衍生物、其合成方法及其药物用途 |
| SI3109244T1 (sl) | 2004-09-14 | 2019-06-28 | Gilead Pharmasset Llc | Priprava 2'fluoro-2'-alkil-substituiranih ali drugih neobvezno substituiranih ribofuranozil pirimidinov in purinov in njihovih derivatov |
| JP4516863B2 (ja) * | 2005-03-11 | 2010-08-04 | 株式会社ケンウッド | 音声合成装置、音声合成方法及びプログラム |
| US8076303B2 (en) | 2005-12-13 | 2011-12-13 | Spring Bank Pharmaceuticals, Inc. | Nucleotide and oligonucleotide prodrugs |
| CA2634749C (en) * | 2005-12-23 | 2014-08-19 | Idenix Pharmaceuticals, Inc. | Process for preparing a synthetic intermediate for preparation of branched nucleosides |
| ES2543840T3 (es) | 2006-04-11 | 2015-08-24 | Novartis Ag | Inhibidores espirocíclicos del VHC/VIH y sus usos |
| US7964580B2 (en) | 2007-03-30 | 2011-06-21 | Pharmasset, Inc. | Nucleoside phosphoramidate prodrugs |
| US20090082306A1 (en) * | 2007-09-26 | 2009-03-26 | Protia, Llc | Deuterium-enriched telbivudine |
| TW200946541A (en) | 2008-03-27 | 2009-11-16 | Idenix Pharmaceuticals Inc | Solid forms of an anti-HIV phosphoindole compound |
| NZ617066A (en) | 2008-12-23 | 2015-02-27 | Gilead Pharmasset Llc | Nucleoside analogs |
| CA2748057C (en) | 2008-12-23 | 2018-07-03 | Pharmasset, Inc. | Nucleoside phosphoramidates |
| EP2671888A1 (en) | 2008-12-23 | 2013-12-11 | Gilead Pharmasset LLC | 3',5'-cyclic nucleoside phosphate analogues |
| US8512690B2 (en) | 2009-04-10 | 2013-08-20 | Novartis Ag | Derivatised proline containing peptide compounds as protease inhibitors |
| US20110182850A1 (en) | 2009-04-10 | 2011-07-28 | Trixi Brandl | Organic compounds and their uses |
| US8618076B2 (en) | 2009-05-20 | 2013-12-31 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| TWI576352B (zh) | 2009-05-20 | 2017-04-01 | 基利法瑪席特有限責任公司 | 核苷磷醯胺 |
| KR101840479B1 (ko) * | 2009-11-16 | 2018-03-20 | 유니버시티 오브 조지아 리서치 파운데이션, 인코포레이티드 | 바이러스 감염 치료를 위한 2'―플루오로―6'―메틸렌 카보사이클릭 뉴클레오사이드 및 방법 |
| CL2011000716A1 (es) | 2010-03-31 | 2012-04-20 | Gilead Pharmasset Llc | Formas cristalinas 1 6 de (s)-isopropil-2-(((s)-(((2r.3r.4r.5r)-5-(2,4-dioxo-3,4-dihidropirimidin-1(2h)-il)-4-fluoro-3-hidroxi-4-metil tetrahidrofuran-2-il)metoxi)(fenoxi)fosforil)amino)propanoato; composicion y combinacion farmaceutica; y su uso para tratar una infeccion por el virus de la hepatitis c. |
| EP2624826B1 (en) | 2010-10-08 | 2018-07-18 | Novartis AG | Vitamin e formulations of sulfamide ns3 inhibitors |
| AU2011336632B2 (en) | 2010-11-30 | 2015-09-03 | Gilead Pharmasset Llc | Compounds |
| CN102649788B (zh) * | 2011-02-28 | 2015-03-25 | 四川大学 | β-L-2’-脱氧-胸腺嘧啶核苷衍生物及其制备方法和用途 |
| HUE036588T2 (hu) | 2011-09-16 | 2018-07-30 | Gilead Pharmasset Llc | Eljárások HCV kezelésére |
| US8889159B2 (en) | 2011-11-29 | 2014-11-18 | Gilead Pharmasset Llc | Compositions and methods for treating hepatitis C virus |
| MY172166A (en) | 2013-01-31 | 2019-11-15 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| EP4005560A1 (en) | 2013-08-27 | 2022-06-01 | Gilead Pharmasset LLC | Combination formulation of two antiviral compounds |
| CN114404427A (zh) | 2014-02-13 | 2022-04-29 | 配体药物公司 | 前药化合物及其用途 |
| CN106687118A (zh) | 2014-07-02 | 2017-05-17 | 配体药物公司 | 前药化合物及其用途 |
| CN110680806A (zh) * | 2018-07-04 | 2020-01-14 | 郑州泰丰制药有限公司 | 一种替比夫定颗粒的制备方法 |
| CN108570078A (zh) * | 2018-07-18 | 2018-09-25 | 荆门医药工业技术研究院 | 一种制备1-O-乙酰基-2,3,5-三-O-苯甲酰基-β-D-呋喃核糖的方法 |
| CN114133398B (zh) * | 2021-12-31 | 2022-11-22 | 中南民族大学 | 氨基酸取代的阿昔洛韦类三环核苷衍生物及其合成方法和应用 |
Family Cites Families (51)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB875971A (en) | 1959-01-14 | 1961-08-30 | Hoffmann La Roche | A process for the manufacture of uridine and thymidine derivatives and novel compounds concerned therein |
| US4522811A (en) | 1982-07-08 | 1985-06-11 | Syntex (U.S.A.) Inc. | Serial injection of muramyldipeptides and liposomes enhances the anti-infective activity of muramyldipeptides |
| US5223263A (en) | 1988-07-07 | 1993-06-29 | Vical, Inc. | Liponucleotide-containing liposomes |
| US4916122A (en) | 1987-01-28 | 1990-04-10 | University Of Georgia Research Foundation, Inc. | 3'-Azido-2',3'-dideoxyuridine anti-retroviral composition |
| US5190926A (en) | 1987-01-28 | 1993-03-02 | University Of Georgia Research Foundation, Inc. | 3'-azido-2',3'-dideoxypyrimidines and related compounds as antiviral agents |
| GB8719367D0 (en) | 1987-08-15 | 1987-09-23 | Wellcome Found | Therapeutic compounds |
| AU2526188A (en) | 1987-09-22 | 1989-04-18 | Regents Of The University Of California, The | Liposomal nucleoside analogues for treating aids |
| US6020322A (en) | 1993-11-09 | 2000-02-01 | Pro-Neuron, Inc. | Acyl deoxyribonucleoside derivatives and uses thereof |
| ATE142221T1 (de) | 1987-10-28 | 1996-09-15 | Pro Neuron Inc | Acyl deoxyribonukleosid-derivate und deren verwendungen |
| JP2675864B2 (ja) | 1988-07-05 | 1997-11-12 | キヤノン株式会社 | 被記録材及びこれを用いたインクジェット記録方法 |
| SE8802687D0 (sv) | 1988-07-20 | 1988-07-20 | Astra Ab | Nucleoside derivatives |
| US5411947A (en) | 1989-06-28 | 1995-05-02 | Vestar, Inc. | Method of converting a drug to an orally available form by covalently bonding a lipid to the drug |
| US5194654A (en) | 1989-11-22 | 1993-03-16 | Vical, Inc. | Lipid derivatives of phosphonoacids for liposomal incorporation and method of use |
| US5463092A (en) | 1989-11-22 | 1995-10-31 | Vestar, Inc. | Lipid derivatives of phosphonacids for liposomal incorporation and method of use |
| US5204466A (en) | 1990-02-01 | 1993-04-20 | Emory University | Method and compositions for the synthesis of bch-189 and related compounds |
| WO1991016920A1 (en) | 1990-05-07 | 1991-11-14 | Vical, Inc. | Lipid prodrugs of salicylate and nonsteroidal anti-inflammatory drugs |
| EP0531452A4 (en) | 1990-05-29 | 1993-06-09 | Vical, Inc. | Synthesis of glycerol di- and triphosphate derivatives |
| JP3347723B2 (ja) | 1990-06-13 | 2002-11-20 | グラツィエル,アーノルド | 含リンプロドラッグ |
| EP0543806B1 (en) | 1990-07-12 | 1996-01-17 | G-Drill Ab | Hydraulic down-the-hole rock drill |
| DE10399025I2 (de) * | 1990-09-14 | 2007-11-08 | Acad Of Science Czech Republic | Wirkstoffvorläufer von Phosphonaten |
| US5206244A (en) * | 1990-10-18 | 1993-04-27 | E. R. Squibb & Sons, Inc. | Hydroxymethyl (methylenecyclopentyl) purines and pyrimidines |
| US5543390A (en) | 1990-11-01 | 1996-08-06 | State Of Oregon, Acting By And Through The Oregon State Board Of Higher Education, Acting For And On Behalf Of The Oregon Health Sciences University | Covalent microparticle-drug conjugates for biological targeting |
| US5543389A (en) | 1990-11-01 | 1996-08-06 | State Of Oregon, Acting By And Through The Oregon State Board Of Higher Education On Behalf Of The Oregon Health Sciences University, A Non Profit Organization | Covalent polar lipid-peptide conjugates for use in salves |
| US5256641A (en) | 1990-11-01 | 1993-10-26 | State Of Oregon | Covalent polar lipid-peptide conjugates for immunological targeting |
| US5149794A (en) | 1990-11-01 | 1992-09-22 | State Of Oregon | Covalent lipid-drug conjugates for drug targeting |
| IT1246983B (it) | 1990-11-13 | 1994-12-12 | Consiglio Nazionale Ricerche | L-2'-desossiuridine e composizioni farmaceutiche che le contengono. |
| IL100502A (en) | 1991-01-03 | 1995-12-08 | Iaf Biochem Int | PHARMACEUTICAL PREPARATIONS CONTAINING CIS-4-AMINO-1-) 2-HYDROXIMETHIL-1,3-OXETYOLEN-5-IL (- |
| HUT64844A (en) | 1991-03-06 | 1994-03-28 | Wellcome Found | Application of 5-fluorine-2'-desoxi-3'-thia-cytidine for treatment of hepatitis b |
| US5220003A (en) | 1991-03-29 | 1993-06-15 | The Regents Of The University Of California | Process for the synthesis of 2',3'-dideoxynucleosides |
| WO1992018517A1 (en) | 1991-04-17 | 1992-10-29 | Yale University | Method of treating or preventing hepatitis b virus |
| JPH07500573A (ja) | 1991-07-12 | 1995-01-19 | ネクススター・ファーマシューティカルズ・インコーポレイテッド | 抗ウィルス性リポヌクレオシド:b型肝炎の治療 |
| US5554728A (en) | 1991-07-23 | 1996-09-10 | Nexstar Pharmaceuticals, Inc. | Lipid conjugates of therapeutic peptides and protease inhibitors |
| EP0691981A1 (en) * | 1993-03-10 | 1996-01-17 | The Wellcome Foundation Limited | Tumor targeting with l-enantiomeric oligonucleotide conjugates of immunoreagents and of chelated radionuclides |
| JP3693357B2 (ja) | 1993-04-09 | 2005-09-07 | 峯郎 実吉 | 逆転写酵素阻害剤 |
| CA2162574A1 (en) | 1993-05-12 | 1994-11-24 | Karl Y. Hostetler | Acyclovir derivatives for topical use |
| US5627160A (en) * | 1993-05-25 | 1997-05-06 | Yale University | L-2',3'-dideoxy nucleoside analogs as anti-hepatitis B (HBV) and anti-HIV agents |
| EP0717628A4 (en) | 1993-09-10 | 1999-05-26 | Univ Emory | NUCLEOSIDES ACTIVATED AGAINST HEPATITIS B VIRUS |
| US5587362A (en) | 1994-01-28 | 1996-12-24 | Univ. Of Ga Research Foundation | L-nucleosides |
| WO1996011204A1 (de) * | 1994-10-07 | 1996-04-18 | Max-Delbrück-Centrum für Molekulare Medizin | NEUE β-L-NUCLEOSIDE UND IHRE VERWENDUNG |
| US5559101A (en) * | 1994-10-24 | 1996-09-24 | Genencor International, Inc. | L-ribofuranosyl nucleosides |
| US5696277A (en) | 1994-11-15 | 1997-12-09 | Karl Y. Hostetler | Antiviral prodrugs |
| EP1655033A1 (en) * | 1995-06-07 | 2006-05-10 | Emory University | Nucleosides with anti-hepatitis B virus activity |
| US6025335A (en) | 1995-09-21 | 2000-02-15 | Lipitek International, Inc. | L-Nucleoside Dimer Compounds and therapeutic uses |
| EP1069903A1 (en) * | 1998-03-11 | 2001-01-24 | Lipitek International, Inc. | Novel nucleoside analogs and uses in treating disease |
| US6194391B1 (en) | 1998-06-24 | 2001-02-27 | Emory University | 3′-azido-2′,3′-dideoxyuridine administration to treat HIV and related test protocol |
| US6444652B1 (en) * | 1998-08-10 | 2002-09-03 | Novirio Pharmaceuticals Limited | β-L-2'-deoxy-nucleosides for the treatment of hepatitis B |
| CN100387237C (zh) * | 1998-08-10 | 2008-05-14 | 艾丹尼克斯(开曼)有限公司 | 用于治疗乙型肝炎的β-L-2'-脱氧-核苷 |
| MY164523A (en) * | 2000-05-23 | 2017-12-29 | Univ Degli Studi Cagliari | Methods and compositions for treating hepatitis c virus |
| CN101099745A (zh) * | 2000-05-26 | 2008-01-09 | 艾登尼科斯(开曼)有限公司 | 处理黄病毒和瘟病毒的方法和组合物 |
| TR200402565T4 (tr) * | 2000-05-26 | 2004-12-21 | Idenix (Cayman) Limited | Hepatit delta virüsü enfeksiyonunun tedavi edilmesinde ß-L-2'-deoksi-nükleosidlerin kullanımı |
| US6787526B1 (en) * | 2000-05-26 | 2004-09-07 | Idenix Pharmaceuticals, Inc. | Methods of treating hepatitis delta virus infection with β-L-2′-deoxy-nucleosides |
-
1999
- 1999-08-10 CN CNB2004100028639A patent/CN100387237C/zh not_active Expired - Lifetime
- 1999-08-10 ES ES99941027T patent/ES2255295T3/es not_active Expired - Lifetime
- 1999-08-10 AT AT99941027T patent/ATE313550T1/de active
- 1999-08-10 EP EP99941027A patent/EP1104436B1/en not_active Expired - Lifetime
- 1999-08-10 CN CNB021265739A patent/CN100482236C/zh not_active Expired - Lifetime
- 1999-08-10 CN CNB998095532A patent/CN1257912C/zh not_active Expired - Lifetime
- 1999-08-10 KR KR1020057009515A patent/KR100568035B1/ko not_active IP Right Cessation
- 1999-08-10 DK DK99941027T patent/DK1104436T3/da active
- 1999-08-10 ES ES04075926.8T patent/ES2531928T3/es not_active Expired - Lifetime
- 1999-08-10 WO PCT/US1999/018149 patent/WO2000009531A2/en active IP Right Grant
- 1999-08-10 KR KR1020067007304A patent/KR100702230B1/ko not_active IP Right Cessation
- 1999-08-10 JP JP2000564981A patent/JP4294870B2/ja not_active Expired - Lifetime
- 1999-08-10 PT PT101846574T patent/PT2415776T/pt unknown
- 1999-08-10 CA CA002340156A patent/CA2340156C/en not_active Expired - Lifetime
- 1999-08-10 PT PT4075926T patent/PT1431304E/pt unknown
- 1999-08-10 DK DK04075926T patent/DK1431304T3/en active
- 1999-08-10 MX MXPA01001507A patent/MXPA01001507A/es active IP Right Grant
- 1999-08-10 DK DK10184657.4T patent/DK2415776T3/en active
- 1999-08-10 DE DE200712000062 patent/DE122007000062I1/de active Pending
- 1999-08-10 DE DE69929060T patent/DE69929060T2/de not_active Expired - Lifetime
- 1999-08-10 US US09/371,747 patent/US6395716B1/en not_active Expired - Lifetime
- 1999-08-10 EP EP10184657.4A patent/EP2415776B1/en not_active Expired - Lifetime
- 1999-08-10 SG SG200302078-1A patent/SG132498A1/en unknown
- 1999-08-10 AU AU54757/99A patent/AU5475799A/en not_active Abandoned
- 1999-08-10 KR KR1020017001758A patent/KR100634342B1/ko active IP Right Grant
- 1999-08-10 ES ES10184657.4T patent/ES2579903T3/es not_active Expired - Lifetime
- 1999-08-10 RU RU2006147216/04A patent/RU2424016C2/ru active Protection Beyond IP Right Term
- 1999-08-10 BR BRPI9912896A patent/BRPI9912896B8/pt not_active IP Right Cessation
- 1999-08-10 KR KR1020067007301A patent/KR100691737B1/ko not_active IP Right Cessation
-
2001
- 2001-06-26 HK HK01104418A patent/HK1034083A1/xx not_active IP Right Cessation
- 2001-12-14 US US10/022,276 patent/US6569837B1/en not_active Expired - Lifetime
-
2003
- 2003-05-13 US US10/438,167 patent/US20030225028A1/en not_active Abandoned
-
2005
- 2005-09-22 US US11/232,818 patent/US7304043B2/en not_active Expired - Lifetime
-
2007
- 2007-05-07 JP JP2007122896A patent/JP2007269798A/ja active Pending
- 2007-05-21 HK HK07105364.0A patent/HK1097776A1/xx not_active IP Right Cessation
- 2007-07-18 LU LU91348C patent/LU91348I2/fr unknown
- 2007-08-06 CY CY200700017C patent/CY2007017I2/el unknown
- 2007-08-09 NL NL300286C patent/NL300286I2/nl unknown
- 2007-09-21 FR FR07C0046C patent/FR07C0046I2/fr active Active
- 2007-10-30 US US11/929,807 patent/US20080064655A1/en not_active Abandoned
-
2008
- 2008-06-24 HK HK08107009.6A patent/HK1111913A1/xx not_active IP Right Cessation
-
2015
- 2015-03-02 CY CY20151100218T patent/CY1116988T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1257912C (zh) | 用于治疗乙型肝炎的β-L-2′-脱氧-核苷 | |
| CA2410488C (en) | Methods of treating hepatitis delta virus infection with .beta.-l-2' deoxy-nucleosides | |
| US6444652B1 (en) | β-L-2'-deoxy-nucleosides for the treatment of hepatitis B | |
| AU2001263484A1 (en) | Methods of treating hepatitis delta virus infection with beta-l-2'- deoxy-nucleosides | |
| CN1911237B (zh) | 用于治疗乙型肝炎的β-L-2'-脱氧-核苷 | |
| AU2007216721B2 (en) | Beta-L-2'-Deoxy Nucleosides for the Treatment of Hepatitis B | |
| EP1431304B1 (en) | Beta - L-2'-Deoxy-Nucleosides for the treatment of Hepatitis B |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| ASS | Succession or assignment of patent right |
Owner name: AIDAN KNICKS (CAYMAN) CO., LTD. Free format text: FORMER OWNER: IDENIX PHARMACEUTICAL CO., LTD. |
|
| C41 | Transfer of patent application or patent right or utility model | ||
| TA01 | Transfer of patent application right |
Effective date of registration: 20040611 Address after: Cayman Islands Applicant after: Idenix Cayman Ltd. Co-applicant after: Centre National De La Recherche Scientifique Address before: Cayman Islands, Grand Cayman Applicant before: Aidan Nicks pharmaceuticals Co-applicant before: Centre National De La Recherche Scientifique |
|
| ASS | Succession or assignment of patent right |
Owner name: FRANCE SCIENTIFIC RESEARCH NATIONAL CENTER MONTPE Free format text: FORMER OWNER: FRANCE SCIENTIFIC RESEARCH NATIONAL CENTER Effective date: 20040924 |
|
| C41 | Transfer of patent application or patent right or utility model | ||
| TA01 | Transfer of patent application right |
Effective date of registration: 20040924 Address after: Cayman Islands Applicant after: Idenix Cayman Ltd. Co-applicant after: Centre National De La Recherche Scientifique Co-applicant after: Univ. Montpellier II Address before: Cayman Islands Applicant before: Idenix Cayman Ltd. Co-applicant before: Centre National De La Recherche Scientifique |
|
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| ASS | Succession or assignment of patent right |
Owner name: IDENIX PHARMACEUTICALS, INC. Free format text: FORMER OWNER: IDENIX (CAYMAN) LIMITED Effective date: 20120710 |
|
| C41 | Transfer of patent application or patent right or utility model | ||
| TR01 | Transfer of patent right |
Effective date of registration: 20120710 Address after: Massachusetts, USA Co-patentee after: Centre National De La Recherche Scientifique Patentee after: Aidan Nicks pharmaceuticals Ltd Co-patentee after: Univ. Montpellier II Address before: Cayman Islands Co-patentee before: Centre National De La Recherche Scientifique Patentee before: Idenix Pharmaceuticals Inc. Co-patentee before: Univ. Montpellier II |
|
| ASS | Succession or assignment of patent right |
Owner name: NOVARTIS CO., LTD. Free format text: FORMER OWNER: IDENIX PHARMACEUTICALS, INC. Effective date: 20130503 |
|
| C41 | Transfer of patent application or patent right or utility model | ||
| TR01 | Transfer of patent right |
Effective date of registration: 20130503 Address after: Basel Patentee after: Novartis Ag Patentee after: Centre National De La Recherche Scientifique Patentee after: Univ. Montpellier II Address before: Massachusetts, USA Patentee before: Aidan Nicks pharmaceuticals Ltd Patentee before: Centre National De La Recherche Scientifique Patentee before: Univ. Montpellier II |
|
| CX01 | Expiry of patent term | ||
| CX01 | Expiry of patent term |
Granted publication date: 20060531 |