US20030045698A1 - Compounds, processes and intermediates for synthesis of mixed backbone oligomeric compounds - Google Patents
Compounds, processes and intermediates for synthesis of mixed backbone oligomeric compounds Download PDFInfo
- Publication number
- US20030045698A1 US20030045698A1 US10/117,267 US11726702A US2003045698A1 US 20030045698 A1 US20030045698 A1 US 20030045698A1 US 11726702 A US11726702 A US 11726702A US 2003045698 A1 US2003045698 A1 US 2003045698A1
- Authority
- US
- United States
- Prior art keywords
- phosphorothioate
- phosphoramidate
- phosphodiester
- independently
- oligomeric compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 117
- 238000000034 method Methods 0.000 title abstract description 78
- 230000008569 process Effects 0.000 title abstract description 9
- 239000000543 intermediate Substances 0.000 title abstract description 7
- 238000003786 synthesis reaction Methods 0.000 title description 43
- 230000015572 biosynthetic process Effects 0.000 title description 42
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 claims abstract description 70
- 150000004713 phosphodiesters Chemical class 0.000 claims abstract description 63
- PTMHPRAIXMAOOB-UHFFFAOYSA-L phosphoramidate Chemical compound NP([O-])([O-])=O PTMHPRAIXMAOOB-UHFFFAOYSA-L 0.000 claims abstract description 56
- 239000007787 solid Substances 0.000 claims description 53
- 108091034117 Oligonucleotide Proteins 0.000 claims description 44
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 35
- 239000002777 nucleoside Substances 0.000 claims description 32
- 150000003833 nucleoside derivatives Chemical class 0.000 claims description 12
- 125000000623 heterocyclic group Chemical group 0.000 claims description 9
- 239000002773 nucleotide Substances 0.000 claims description 6
- 125000003729 nucleotide group Chemical group 0.000 claims description 6
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 claims description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 3
- OJMIONKXNSYLSR-UHFFFAOYSA-N phosphorous acid Chemical group OP(O)O OJMIONKXNSYLSR-UHFFFAOYSA-N 0.000 claims 3
- 230000003647 oxidation Effects 0.000 abstract description 42
- 238000007254 oxidation reaction Methods 0.000 abstract description 42
- 230000015556 catabolic process Effects 0.000 abstract description 9
- 238000006731 degradation reaction Methods 0.000 abstract description 9
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 150
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 80
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 75
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 75
- 239000000243 solution Substances 0.000 description 41
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 40
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 34
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 29
- 230000001590 oxidative effect Effects 0.000 description 28
- QGJOPFRUJISHPQ-UHFFFAOYSA-N Carbon disulfide Chemical compound S=C=S QGJOPFRUJISHPQ-UHFFFAOYSA-N 0.000 description 27
- 239000007800 oxidant agent Substances 0.000 description 27
- 210000004027 cell Anatomy 0.000 description 25
- -1 N(R4)(R5) OR4 Chemical group 0.000 description 22
- 239000000178 monomer Substances 0.000 description 22
- 238000010532 solid phase synthesis reaction Methods 0.000 description 22
- 239000003153 chemical reaction reagent Substances 0.000 description 21
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 20
- 239000000203 mixture Substances 0.000 description 19
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 18
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 18
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 18
- 125000003835 nucleoside group Chemical group 0.000 description 18
- 229950005499 carbon tetrachloride Drugs 0.000 description 17
- 239000003960 organic solvent Substances 0.000 description 17
- 125000000824 D-ribofuranosyl group Chemical group [H]OC([H])([H])[C@@]1([H])OC([H])(*)[C@]([H])(O[H])[C@]1([H])O[H] 0.000 description 16
- 102000015271 Intercellular Adhesion Molecule-1 Human genes 0.000 description 16
- 108010064593 Intercellular Adhesion Molecule-1 Proteins 0.000 description 16
- 230000014509 gene expression Effects 0.000 description 16
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 15
- 238000005859 coupling reaction Methods 0.000 description 14
- 238000011160 research Methods 0.000 description 14
- 125000004432 carbon atom Chemical group C* 0.000 description 13
- 238000003776 cleavage reaction Methods 0.000 description 13
- 239000005289 controlled pore glass Substances 0.000 description 13
- 230000008878 coupling Effects 0.000 description 13
- 238000010168 coupling process Methods 0.000 description 13
- 230000007017 scission Effects 0.000 description 13
- 235000000346 sugar Nutrition 0.000 description 13
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- 125000000217 alkyl group Chemical group 0.000 description 12
- 238000012986 modification Methods 0.000 description 12
- 230000004048 modification Effects 0.000 description 12
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical group [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 11
- 0 [1*]C1C(C)OC(CCC2C(C[3H])OC(C)C2[1*])C1[3H][3H] Chemical compound [1*]C1C(C)OC(CCC2C(C[3H])OC(C)C2[1*])C1[3H][3H] 0.000 description 11
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 11
- 239000012043 crude product Substances 0.000 description 11
- 238000010511 deprotection reaction Methods 0.000 description 11
- 108090000623 proteins and genes Proteins 0.000 description 11
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 10
- 108020004414 DNA Proteins 0.000 description 10
- 125000006239 protecting group Chemical group 0.000 description 10
- 229940074993 carbon disulfide Drugs 0.000 description 9
- 125000005842 heteroatom Chemical group 0.000 description 9
- 238000002360 preparation method Methods 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- 102000004169 proteins and genes Human genes 0.000 description 9
- 238000003756 stirring Methods 0.000 description 9
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 9
- YIMATHOGWXZHFX-WCTZXXKLSA-N (2r,3r,4r,5r)-5-(hydroxymethyl)-3-(2-methoxyethoxy)oxolane-2,4-diol Chemical group COCCO[C@H]1[C@H](O)O[C@H](CO)[C@H]1O YIMATHOGWXZHFX-WCTZXXKLSA-N 0.000 description 8
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 8
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 8
- 125000003118 aryl group Chemical group 0.000 description 8
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 8
- 125000004122 cyclic group Chemical group 0.000 description 8
- 239000006260 foam Substances 0.000 description 8
- NCGWKCHAJOUDHQ-UHFFFAOYSA-N n,n-diethylethanamine;formic acid Chemical compound OC=O.OC=O.CCN(CC)CC NCGWKCHAJOUDHQ-UHFFFAOYSA-N 0.000 description 8
- 239000012074 organic phase Substances 0.000 description 8
- 229910052760 oxygen Inorganic materials 0.000 description 8
- 239000001301 oxygen Substances 0.000 description 8
- 229910052938 sodium sulfate Inorganic materials 0.000 description 8
- 235000011152 sodium sulphate Nutrition 0.000 description 8
- 239000002904 solvent Substances 0.000 description 8
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 7
- 102000001788 Proto-Oncogene Proteins c-raf Human genes 0.000 description 7
- 108010029869 Proto-Oncogene Proteins c-raf Proteins 0.000 description 7
- QGJOPFRUJISHPQ-NJFSPNSNSA-N carbon disulfide-14c Chemical compound S=[14C]=S QGJOPFRUJISHPQ-NJFSPNSNSA-N 0.000 description 7
- 201000010099 disease Diseases 0.000 description 7
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 7
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 7
- 108020004999 messenger RNA Proteins 0.000 description 7
- DILRJUIACXKSQE-UHFFFAOYSA-N n',n'-dimethylethane-1,2-diamine Chemical compound CN(C)CCN DILRJUIACXKSQE-UHFFFAOYSA-N 0.000 description 7
- 108020004707 nucleic acids Proteins 0.000 description 7
- 102000039446 nucleic acids Human genes 0.000 description 7
- 150000007523 nucleic acids Chemical class 0.000 description 7
- 239000007790 solid phase Substances 0.000 description 7
- 229910052717 sulfur Inorganic materials 0.000 description 7
- 239000011593 sulfur Substances 0.000 description 7
- 230000001225 therapeutic effect Effects 0.000 description 7
- 238000005406 washing Methods 0.000 description 7
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 description 6
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 6
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 6
- 125000005647 linker group Chemical group 0.000 description 6
- 125000000467 secondary amino group Chemical class [H]N([*:1])[*:2] 0.000 description 6
- JVSFQJZRHXAUGT-UHFFFAOYSA-N 2,2-dimethylpropanoyl chloride Chemical compound CC(C)(C)C(Cl)=O JVSFQJZRHXAUGT-UHFFFAOYSA-N 0.000 description 5
- NSPMIYGKQJPBQR-UHFFFAOYSA-N 4H-1,2,4-triazole Chemical compound C=1N=CNN=1 NSPMIYGKQJPBQR-UHFFFAOYSA-N 0.000 description 5
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 5
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 5
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 230000001413 cellular effect Effects 0.000 description 5
- 238000010348 incorporation Methods 0.000 description 5
- 229910052740 iodine Inorganic materials 0.000 description 5
- 239000011630 iodine Substances 0.000 description 5
- 229910052757 nitrogen Inorganic materials 0.000 description 5
- FAIAAWCVCHQXDN-UHFFFAOYSA-N phosphorus trichloride Chemical compound ClP(Cl)Cl FAIAAWCVCHQXDN-UHFFFAOYSA-N 0.000 description 5
- 230000035484 reaction time Effects 0.000 description 5
- 230000014616 translation Effects 0.000 description 5
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 description 4
- XCOBLONWWXQEBS-KPKJPENVSA-N N,O-bis(trimethylsilyl)trifluoroacetamide Chemical compound C[Si](C)(C)O\C(C(F)(F)F)=N\[Si](C)(C)C XCOBLONWWXQEBS-KPKJPENVSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 125000001931 aliphatic group Chemical group 0.000 description 4
- 238000013459 approach Methods 0.000 description 4
- 239000008346 aqueous phase Substances 0.000 description 4
- BYKCUMSOQIPHSR-UHFFFAOYSA-N boron;n-ethyl-n-propan-2-ylpropan-2-amine Chemical compound [B].CCN(C(C)C)C(C)C BYKCUMSOQIPHSR-UHFFFAOYSA-N 0.000 description 4
- 239000007853 buffer solution Substances 0.000 description 4
- WONCKQSITUBTLE-UHFFFAOYSA-N carbonic acid;1,2,3,4,5,7,8,9-octahydropyrido[1,2-b]diazepine Chemical compound OC(O)=O.C1CCCNN2CCCC=C21 WONCKQSITUBTLE-UHFFFAOYSA-N 0.000 description 4
- 230000000295 complement effect Effects 0.000 description 4
- 230000001419 dependent effect Effects 0.000 description 4
- 238000005516 engineering process Methods 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 125000001188 haloalkyl group Chemical group 0.000 description 4
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 4
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 4
- 150000003141 primary amines Chemical class 0.000 description 4
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 4
- 239000012264 purified product Substances 0.000 description 4
- 238000010898 silica gel chromatography Methods 0.000 description 4
- 238000006884 silylation reaction Methods 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 4
- 238000011282 treatment Methods 0.000 description 4
- 229940035893 uracil Drugs 0.000 description 4
- 238000004679 31P NMR spectroscopy Methods 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- 229930024421 Adenine Natural products 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- 101710163270 Nuclease Proteins 0.000 description 3
- 229960000643 adenine Drugs 0.000 description 3
- 125000003342 alkenyl group Chemical group 0.000 description 3
- 125000000304 alkynyl group Chemical group 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- 230000000692 anti-sense effect Effects 0.000 description 3
- 229940104302 cytosine Drugs 0.000 description 3
- NAGJZTKCGNOGPW-UHFFFAOYSA-K dioxido-sulfanylidene-sulfido-$l^{5}-phosphane Chemical compound [O-]P([O-])([S-])=S NAGJZTKCGNOGPW-UHFFFAOYSA-K 0.000 description 3
- 238000009510 drug design Methods 0.000 description 3
- 230000003993 interaction Effects 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 238000002515 oligonucleotide synthesis Methods 0.000 description 3
- BQJRUJTZSGYBEZ-YVQNUNKESA-N phorbol 12,13-dibutanoate Chemical compound C([C@]1(O)C(=O)C(C)=C[C@H]1[C@@]1(O)[C@H](C)[C@H]2OC(=O)CCC)C(CO)=C[C@H]1[C@H]1[C@]2(OC(=O)CCC)C1(C)C BQJRUJTZSGYBEZ-YVQNUNKESA-N 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 238000005987 sulfurization reaction Methods 0.000 description 3
- 238000013518 transcription Methods 0.000 description 3
- 230000035897 transcription Effects 0.000 description 3
- 238000013519 translation Methods 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- MYRTYDVEIRVNKP-UHFFFAOYSA-N 1,2-Divinylbenzene Chemical compound C=CC1=CC=CC=C1C=C MYRTYDVEIRVNKP-UHFFFAOYSA-N 0.000 description 2
- LDGWQMRUWMSZIU-LQDDAWAPSA-M 2,3-bis[(z)-octadec-9-enoxy]propyl-trimethylazanium;chloride Chemical compound [Cl-].CCCCCCCC\C=C/CCCCCCCCOCC(C[N+](C)(C)C)OCCCCCCCC\C=C/CCCCCCCC LDGWQMRUWMSZIU-LQDDAWAPSA-M 0.000 description 2
- FZWGECJQACGGTI-UHFFFAOYSA-N 2-amino-7-methyl-1,7-dihydro-6H-purin-6-one Chemical compound NC1=NC(O)=C2N(C)C=NC2=N1 FZWGECJQACGGTI-UHFFFAOYSA-N 0.000 description 2
- 125000004080 3-carboxypropanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C(O[H])=O 0.000 description 2
- LRFVTYWOQMYALW-UHFFFAOYSA-N 9H-xanthine Chemical compound O=C1NC(=O)NC2=C1NC=N2 LRFVTYWOQMYALW-UHFFFAOYSA-N 0.000 description 2
- 101150112014 Gapdh gene Proteins 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 2
- 239000012124 Opti-MEM Substances 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- 102000040945 Transcription factor Human genes 0.000 description 2
- 108091023040 Transcription factor Proteins 0.000 description 2
- 102000004142 Trypsin Human genes 0.000 description 2
- 108090000631 Trypsin Proteins 0.000 description 2
- DRTQHJPVMGBUCF-XVFCMESISA-N Uridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-XVFCMESISA-N 0.000 description 2
- HMNZFMSWFCAGGW-XPWSMXQVSA-N [3-[hydroxy(2-hydroxyethoxy)phosphoryl]oxy-2-[(e)-octadec-9-enoyl]oxypropyl] (e)-octadec-9-enoate Chemical compound CCCCCCCC\C=C\CCCCCCCC(=O)OCC(COP(O)(=O)OCCO)OC(=O)CCCCCCC\C=C\CCCCCCCC HMNZFMSWFCAGGW-XPWSMXQVSA-N 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 229940041181 antineoplastic drug Drugs 0.000 description 2
- 239000000010 aprotic solvent Substances 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- 238000002405 diagnostic procedure Methods 0.000 description 2
- JXTHNDFMNIQAHM-UHFFFAOYSA-N dichloroacetic acid Chemical compound OC(=O)C(Cl)Cl JXTHNDFMNIQAHM-UHFFFAOYSA-N 0.000 description 2
- NFORZJQPTUSMRL-UHFFFAOYSA-N dipropan-2-yl hydrogen phosphite Chemical compound CC(C)OP(O)OC(C)C NFORZJQPTUSMRL-UHFFFAOYSA-N 0.000 description 2
- AUZONCFQVSMFAP-UHFFFAOYSA-N disulfiram Chemical compound CCN(CC)C(=S)SSC(=S)N(CC)CC AUZONCFQVSMFAP-UHFFFAOYSA-N 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 125000001475 halogen functional group Chemical group 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 125000004366 heterocycloalkenyl group Chemical group 0.000 description 2
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- YACKEPLHDIMKIO-UHFFFAOYSA-N methylphosphonic acid Chemical compound CP(O)(O)=O YACKEPLHDIMKIO-UHFFFAOYSA-N 0.000 description 2
- 238000010369 molecular cloning Methods 0.000 description 2
- 210000003463 organelle Anatomy 0.000 description 2
- 229920000570 polyether Polymers 0.000 description 2
- 238000003752 polymerase chain reaction Methods 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 230000008685 targeting Effects 0.000 description 2
- CIHOLLKRGTVIJN-UHFFFAOYSA-N tert‐butyl hydroperoxide Chemical compound CC(C)(C)OO CIHOLLKRGTVIJN-UHFFFAOYSA-N 0.000 description 2
- HJUGFYREWKUQJT-UHFFFAOYSA-N tetrabromomethane Chemical compound BrC(Br)(Br)Br HJUGFYREWKUQJT-UHFFFAOYSA-N 0.000 description 2
- 229940113082 thymine Drugs 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 239000012588 trypsin Substances 0.000 description 2
- STGXGJRRAJKJRG-JDJSBBGDSA-N (3r,4r,5r)-5-(hydroxymethyl)-3-methoxyoxolane-2,4-diol Chemical compound CO[C@H]1C(O)O[C@H](CO)[C@H]1O STGXGJRRAJKJRG-JDJSBBGDSA-N 0.000 description 1
- JUDOLRSMWHVKGX-UHFFFAOYSA-N 1,1-dioxo-1$l^{6},2-benzodithiol-3-one Chemical compound C1=CC=C2C(=O)SS(=O)(=O)C2=C1 JUDOLRSMWHVKGX-UHFFFAOYSA-N 0.000 description 1
- SCZNXLWKYFICFV-UHFFFAOYSA-N 1,2,3,4,5,7,8,9-octahydropyrido[1,2-b]diazepine Chemical compound C1CCCNN2CCCC=C21 SCZNXLWKYFICFV-UHFFFAOYSA-N 0.000 description 1
- MFUZBRDKDBLLDJ-UHFFFAOYSA-N 1-chloro-4-[(4-chlorophenyl)sulfonyldisulfanyl]sulfonylbenzene Chemical compound C1=CC(Cl)=CC=C1S(=O)(=O)SSS(=O)(=O)C1=CC=C(Cl)C=C1 MFUZBRDKDBLLDJ-UHFFFAOYSA-N 0.000 description 1
- OZNXTQSXSHODFR-UHFFFAOYSA-N 1-chloroadamantane Chemical compound C1C(C2)CC3CC2CC1(Cl)C3 OZNXTQSXSHODFR-UHFFFAOYSA-N 0.000 description 1
- JAPYIBBSTJFDAK-UHFFFAOYSA-N 2,4,6-tri(propan-2-yl)benzenesulfonyl chloride Chemical compound CC(C)C1=CC(C(C)C)=C(S(Cl)(=O)=O)C(C(C)C)=C1 JAPYIBBSTJFDAK-UHFFFAOYSA-N 0.000 description 1
- VZMUCIBBVMLEKC-UHFFFAOYSA-N 2-chloro-5,5-dimethyl-1,3,2$l^{5}-dioxaphosphinane 2-oxide Chemical compound CC1(C)COP(Cl)(=O)OC1 VZMUCIBBVMLEKC-UHFFFAOYSA-N 0.000 description 1
- KZMAWJRXKGLWGS-UHFFFAOYSA-N 2-chloro-n-[4-(4-methoxyphenyl)-1,3-thiazol-2-yl]-n-(3-methoxypropyl)acetamide Chemical compound S1C(N(C(=O)CCl)CCCOC)=NC(C=2C=CC(OC)=CC=2)=C1 KZMAWJRXKGLWGS-UHFFFAOYSA-N 0.000 description 1
- KLDLRDSRCMJKGM-UHFFFAOYSA-N 3-[chloro-(2-oxo-1,3-oxazolidin-3-yl)phosphoryl]-1,3-oxazolidin-2-one Chemical compound C1COC(=O)N1P(=O)(Cl)N1CCOC1=O KLDLRDSRCMJKGM-UHFFFAOYSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- LOJNBPNACKZWAI-UHFFFAOYSA-N 3-nitro-1h-pyrrole Chemical group [O-][N+](=O)C=1C=CNC=1 LOJNBPNACKZWAI-UHFFFAOYSA-N 0.000 description 1
- OVONXEQGWXGFJD-UHFFFAOYSA-N 4-sulfanylidene-1h-pyrimidin-2-one Chemical compound SC=1C=CNC(=O)N=1 OVONXEQGWXGFJD-UHFFFAOYSA-N 0.000 description 1
- RZIJZXUDFRSADE-UHFFFAOYSA-N 5-ethoxy-1,2,4-dithiazol-3-one Chemical compound CCOC1=NC(=O)SS1 RZIJZXUDFRSADE-UHFFFAOYSA-N 0.000 description 1
- MSSXOMSJDRHRMC-UHFFFAOYSA-N 9H-purine-2,6-diamine Chemical compound NC1=NC(N)=C2NC=NC2=N1 MSSXOMSJDRHRMC-UHFFFAOYSA-N 0.000 description 1
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 1
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 1
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 241000195493 Cryptophyta Species 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- 230000006820 DNA synthesis Effects 0.000 description 1
- SHIBSTMRCDJXLN-UHFFFAOYSA-N Digoxigenin Natural products C1CC(C2C(C3(C)CCC(O)CC3CC2)CC2O)(O)C2(C)C1C1=CC(=O)OC1 SHIBSTMRCDJXLN-UHFFFAOYSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 241000206602 Eukaryota Species 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- 101710203526 Integrase Proteins 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 101100523539 Mus musculus Raf1 gene Proteins 0.000 description 1
- 108020005187 Oligonucleotide Probes Proteins 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 230000006819 RNA synthesis Effects 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 102100040247 Tumor necrosis factor Human genes 0.000 description 1
- BHIIGRBMZRSDRI-UHFFFAOYSA-N [chloro(phenoxy)phosphoryl]oxybenzene Chemical compound C=1C=CC=CC=1OP(=O)(Cl)OC1=CC=CC=C1 BHIIGRBMZRSDRI-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- PPQRONHOSHZGFQ-LMVFSUKVSA-N aldehydo-D-ribose 5-phosphate Chemical group OP(=O)(O)OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PPQRONHOSHZGFQ-LMVFSUKVSA-N 0.000 description 1
- 125000002723 alicyclic group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000002877 alkyl aryl group Chemical group 0.000 description 1
- 125000005600 alkyl phosphonate group Chemical group 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- DRTQHJPVMGBUCF-PSQAKQOGSA-N beta-L-uridine Natural products O[C@H]1[C@@H](O)[C@H](CO)O[C@@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-PSQAKQOGSA-N 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 239000013060 biological fluid Substances 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- IOVVFSGCNWQFQT-UHFFFAOYSA-N bis(2,3,4,5,6-pentafluorophenyl) carbonate Chemical compound FC1=C(F)C(F)=C(F)C(F)=C1OC(=O)OC1=C(F)C(F)=C(F)C(F)=C1F IOVVFSGCNWQFQT-UHFFFAOYSA-N 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 125000001589 carboacyl group Chemical group 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 125000005626 carbonium group Chemical group 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- ITVPBBDAZKBMRP-UHFFFAOYSA-N chloro-dioxido-oxo-$l^{5}-phosphane;hydron Chemical class OP(O)(Cl)=O ITVPBBDAZKBMRP-UHFFFAOYSA-N 0.000 description 1
- UOALEFQKAOQICC-UHFFFAOYSA-N chloroborane Chemical compound ClB UOALEFQKAOQICC-UHFFFAOYSA-N 0.000 description 1
- 210000003763 chloroplast Anatomy 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 238000009223 counseling Methods 0.000 description 1
- 150000003983 crown ethers Chemical class 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- 230000005595 deprotonation Effects 0.000 description 1
- 238000010537 deprotonation reaction Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 229960005215 dichloroacetic acid Drugs 0.000 description 1
- QONQRTHLHBTMGP-UHFFFAOYSA-N digitoxigenin Natural products CC12CCC(C3(CCC(O)CC3CC3)C)C3C11OC1CC2C1=CC(=O)OC1 QONQRTHLHBTMGP-UHFFFAOYSA-N 0.000 description 1
- SHIBSTMRCDJXLN-KCZCNTNESA-N digoxigenin Chemical compound C1([C@@H]2[C@@]3([C@@](CC2)(O)[C@H]2[C@@H]([C@@]4(C)CC[C@H](O)C[C@H]4CC2)C[C@H]3O)C)=CC(=O)OC1 SHIBSTMRCDJXLN-KCZCNTNESA-N 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 150000002019 disulfides Chemical class 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 1
- DWYMPOCYEZONEA-UHFFFAOYSA-L fluoridophosphate Chemical compound [O-]P([O-])(F)=O DWYMPOCYEZONEA-UHFFFAOYSA-L 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- 238000009396 hybridization Methods 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical compound [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 1
- GTTBQSNGUYHPNK-UHFFFAOYSA-N hydroxymethylphosphonic acid Chemical compound OCP(O)(O)=O GTTBQSNGUYHPNK-UHFFFAOYSA-N 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 201000005296 lung carcinoma Diseases 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- VUZPPFZMUPKLLV-UHFFFAOYSA-N methane;hydrate Chemical compound C.O VUZPPFZMUPKLLV-UHFFFAOYSA-N 0.000 description 1
- CAAULPUQFIIOTL-UHFFFAOYSA-N methyl dihydrogen phosphate Chemical compound COP(O)(O)=O CAAULPUQFIIOTL-UHFFFAOYSA-N 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- ZSMNRKGGHXLZEC-UHFFFAOYSA-N n,n-bis(trimethylsilyl)methanamine Chemical compound C[Si](C)(C)N(C)[Si](C)(C)C ZSMNRKGGHXLZEC-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 238000010606 normalization Methods 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 229940046166 oligodeoxynucleotide Drugs 0.000 description 1
- 239000002751 oligonucleotide probe Substances 0.000 description 1
- 125000003431 oxalo group Chemical group 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 150000004965 peroxy acids Chemical class 0.000 description 1
- 239000008194 pharmaceutical composition Substances 0.000 description 1
- 125000005561 phenanthryl group Chemical group 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 150000003003 phosphines Chemical class 0.000 description 1
- 150000004714 phosphonium salts Chemical class 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 238000001243 protein synthesis Methods 0.000 description 1
- 125000001725 pyrenyl group Chemical group 0.000 description 1
- 239000002719 pyrimidine nucleotide Substances 0.000 description 1
- 125000002112 pyrrolidino group Chemical group [*]N1C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 230000037425 regulation of transcription Effects 0.000 description 1
- 230000009711 regulatory function Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- IXGZXXBJSZISOO-UHFFFAOYSA-N s-(2-phenylacetyl)sulfanyl 2-phenylethanethioate Chemical compound C=1C=CC=CC=1CC(=O)SSC(=O)CC1=CC=CC=C1 IXGZXXBJSZISOO-UHFFFAOYSA-N 0.000 description 1
- XTDHBAVVPOKCKF-UHFFFAOYSA-N s-(benzoyltrisulfanyl) benzenecarbothioate Chemical compound C=1C=CC=CC=1C(=O)SSSSC(=O)C1=CC=CC=C1 XTDHBAVVPOKCKF-UHFFFAOYSA-N 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- JRPHGDYSKGJTKZ-UHFFFAOYSA-N selenophosphoric acid Chemical compound OP(O)([SeH])=O JRPHGDYSKGJTKZ-UHFFFAOYSA-N 0.000 description 1
- 238000002741 site-directed mutagenesis Methods 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 229940126585 therapeutic drug Drugs 0.000 description 1
- 125000004001 thioalkyl group Chemical group 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 230000014621 translational initiation Effects 0.000 description 1
- SIOVKLKJSOKLIF-HJWRWDBZSA-N trimethylsilyl (1z)-n-trimethylsilylethanimidate Chemical compound C[Si](C)(C)OC(/C)=N\[Si](C)(C)C SIOVKLKJSOKLIF-HJWRWDBZSA-N 0.000 description 1
- 210000003606 umbilical vein Anatomy 0.000 description 1
- DRTQHJPVMGBUCF-UHFFFAOYSA-N uracil arabinoside Natural products OC1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-UHFFFAOYSA-N 0.000 description 1
- 229940045145 uridine Drugs 0.000 description 1
- 125000005500 uronium group Chemical group 0.000 description 1
- 229940075420 xanthine Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/11—Compounds covalently bound to a solid support
Definitions
- the present invention is directed to methods for oxidizing an H-phosphonate internucleoside linkage in a compound which has at least one 2′-substituent group.
- This invention further relates to methods for the preparation of mixed backbone oligomeric compounds having internucleoside linkages including phosphodiester, phosphorothioate, phosphoramidate and boranophosphate linkages.
- the present invention also relates to synthetic intermediates useful in such methods.
- mixed backbone oligomeric compounds having phosphodiester and boranophosphate internucleoside linkages or phosphodiester and boranophosphate internucleoside linkages in combination with other internucleoside linkages such as phosphorothioate and phosphoramidate internucleoside linkages.
- the methods of the present invention include a novel oxidation step that allows oxidation of a region having one or more H-phosphonate internucleoside linkages to phosphodiester internucleoside linkages without degradation of adjacent phosphorothioate, phosphoramidate or boranophosphate internucleoside linkages.
- Oligonucleotides and their analogs have been developed and used in molecular biology in a variety of procedures as probes, primers, linkers, adapters, and gene fragments. Modifications to oligonucleotides used in these procedures include labeling with nonisotopic labels, e.g., fluorescein, biotin, digoxigenin, alkaline phosphatase or other reporter molecules. Other modifications have been made to the ribose phosphate backbone to increase the nuclease stability of the resulting analog. Examples of such modifications include incorporation of methyl phosphonate, phosphorothioate, or phosphorodithioate linkages, and 2′-O-methyl ribose sugar units. Further modifications include those made to modulate uptake and cellular distribution. With the success of these compounds for both diagnostic and therapeutic uses, there exists an ongoing demand for improved oligonucleotides, their analogs and synthetic processes for their preparation.
- nonisotopic labels e.g., fluor
- oligonucleotides especially oligonucleotides which are complementary to a specific target messenger RNA (MRNA) sequence.
- MRNA target messenger RNA
- oligonucleotides are currently undergoing clinical trials for such use.
- Phosphorothioate oligonucleotides are presently being used as such antisense agents in human clinical trials for various disease states, including use as antiviral agents.
- oligonucleotides and their analogs also have found use in diagnostic tests. Such diagnostic tests can be performed using biological fluids, tissues, intact cells or isolated cellular components. As with inhibition of gene expression, diagnostic applications utilize the ability of oligonucleotides and their analogs to hybridize with a complementary strand of nucleic acid. Hybridization is the sequence-specific hydrogen bonding of oligomeric compounds via Watson-Crick and/or Hoogsteen base pairs to RNA or DNA. The bases of such base pairs are said to be complementary to one another.
- Oligonucleotides and their analogs are also widely used as research reagents. They are useful for understanding the function of many other biological molecules as well as in the preparation of other biological molecules. For example, the use of oligonucleotides and their analogs as primers in PCR reactions has given rise to an expanding commercial industry. PCR has become a mainstay of commercial and research laboratories, and applications of PCR have multiplied. For example, PCR technology now finds use in the fields of forensics, paleontology, evolutionary studies and genetic counseling. Commercialization has led to the development of kits which assist non-molecular biology-trained personnel in applying PCR. Oligonucleotides and their analogs, both natural and synthetic, are employed as primers in such PCR technology.
- Oligonucleotides and their analogs are also used in other laboratory procedures. Several of these uses are described in common laboratory manuals such as Molecular Cloning, A Laboratory Manual , Second Ed., Sambrook et al, Eds., Cold Spring Harbor Laboratory Press, 1989; and Current Protocols In Molecular Biology , Ausubel et al., Eds., Current Publications, 1993. Such uses include as synthetic oligonucleotide probes, in screening expression libraries with antibodies and oligomeric compounds, DNA sequencing, in vitro amplification of DNA by the polymerase chain reaction, and in site-directed mutagenesis of cloned DNA. See Book 2 of Molecular Cloning, A Laboratory Manual , supra. See, also, “DNA-protein interactions and The Polymerase Chain Reaction” in Vol. 2 of Current Protocols In Molecular Biology , supra.
- Oligonucleotides and their analogs can be synthesized to have customized properties that can be tailored for desired uses. Thus a number of chemical modifications have been introduced into oligomeric compounds to increase their usefulness in diagnostics, as research reagents and as therapeutic entities.
- Such modifications include those designed to increase binding to a target strand (i.e., increase their melting temperatures, Tm), to assist in identification of the oligonucleotide or an oligonucleotide-target complex, to increase cell penetration, to stabilize against nucleases and other enzymes that degrade or interfere with the structure or activity of the oligonucleotides and their analogs, to provide a mode of disruption (terminating event) once sequence-specifically bound to a target, and to improve the pharmacokinetic properties of the oligonucleotide.
- Tm melting temperatures
- the chemical literature discloses numerous processes for coupling nucleosides through phosphorous-containing covalent linkages to produce oligonucleotides of defined sequence.
- One such method utilizes H-phosphonate monomers to prepare oliogmeric compounds.
- the standard H-phosphonate method has been used for the synthesis of uniformly modified oligomeric compounds containing phosphodiester, phosphorothioate or phosphoramidate internucleoside linkages.
- the method has not been effective for the synthesis of mixed backbone oligomers because during the oxidation step previously oxidized regions in an oligomer are degraded. Such degradation has been observed for the oxidation of oligomers having phosphorothioate and or phosphoramidate internucleoside linkages when further H-phosphonate internucleoside linkages are oxidized to phosphodiester linkages.
- H-phosphonate methods and techniques are disclosed in numerous publications. See, Mackie and Hogrefe, Glen Research, H-phosphonate Chemistry , Glen Research Corp, Va.; Wada et al, J. Am. Chem. Soc., 1997, 119, 12710-12721; and Sergueev et al., J. Am. Chem. Soc., 1998, 120, 9417-9427.
- Phosphoramidate oligodeoxynucleotides have been prepared using H-phosphonate chemistry. Dagle et al., Nucleic Acids Research, 1991, 19, 1805-1810.
- the present invention provides methods for oxidizing H-phosphonate internucleoside linkages in a compound which bears at least one 2′-substituent group. This oxidation is effected by an oxidizing solution comprising an oxidizing agent, an aprotic solvent, a base and water.
- the present invention also provides methods for the preparation of mixed backbone oligomeric compounds of formula:
- each Z is, independently, a phosphodiester, phosphorothioate, phosphoramidate or boranophosphate internucleoside linkage;
- each T 1 and T 2 is, independently, hydroxyl or a protected hydroxyl
- Bx is a heterocyclic base moiety
- each R 1 is, independently, H, hydroxyl, a protected hydroxyl, a 2′-substituent group or a protected 2′-substituent group;
- n is an integer greater than 1;
- At least one of said Z is a phosphodiester internucleoside linkage and at least another of said Z is a phosphorothioate, phosphoramidate or boranophosphate internucleoside linkage;
- Pg is an acid labile hydroxyl protecting group
- T 3 is a base labile hydroxyl protecting group or a covalent attachment to a solid support
- the mixed backbone oligomeric compound comprises contiguous regions of phosphodiester and phosphorothioate internucleoside linkages.
- the mixed backbone oligomeric compound comprises contiguous regions of phosphodiester and phosphoramidate internucleoside linkages.
- the oxidizing solution comprises an oxidizing agent, an aprotic organic solvent, a base and water.
- the oxidizing solution comprises from about 18% to 45% oxidizing agent. In a preferred embodiment the oxidizing solution comprises from about 26% to about 40% oxidizing agent. In a more preferred embodiment the oxidizing solution comprises about 33% oxidizing agent.
- T 4 is hydroxyl or a protected hydroxyl
- Bx is a heterocyclic base moiety
- R 2 has one of formula I or II:
- E is C 1 -C 10 alkyl, N(R 4 )(R 5 ) or N ⁇ C(R 4 )(R 5 );
- R 3 is OX, SX, or N(X) 2 ;
- each R 4 and R 5 is, independently, H, C 1 -C 10 alkyl, a nitrogen protecting group, or R 4 and R 5 , together, are a nitrogen protecting group, or R 4 and R 5 are joined in a ring structure that can include at least one additional heteroatom selected from a group consisting of nitrogen and oxygen;
- each X is, independently, H, C 1 -C 8 alkyl, C 1 -C 8 haloalkyl, C( ⁇ NH)N(H)Z, C( ⁇ O)N(H)Z or OC( ⁇ O)N(H)Z;
- Z is H or C 1 -C 8 alkyl
- L 1 , L 2 and L 3 comprise a ring system having from about 4 to about 7 carbon atoms or having from about 3 to about 6 carbon atoms and at least 1 heteroatom, said heteroatom being selected from a group consisting of oxygen, nitrogen and sulfur, said ring system being aliphatic, unsaturated aliphatic, aromatic, or saturated or unsaturated heterocyclic;
- Y is alkyl or haloalkyl having 1 to about 10 carbon atoms, alkenyl having 2 to about 10 carbon atoms, alkynyl having 2 to about 10 carbon atoms, aryl having 6 to about 14 carbon atoms, N(R 4 )(R 5 ) OR 4 , halo, SR 4 or CN;
- each q 1 is, independently, an integer from 2 to 10;
- each q 2 is, independently, 0 or 1;
- p is an integer from 1 to 10;
- r is an integer from 1 to 10
- R 2 is a 2′-substituent group.
- the 2′-substituent group is —O—CH 2 CH 2 —O—CH 3 , —O—CH 2 CH 2 —O—N(R 6 )(R 7 ) or —O—CH 2 CH 2 —O—CH 2 CH 2 —N(R 6 )(R 7 ), wherein each of R 6 and R 7 is, independently, H or C 1 -C 10 alkyl.
- each R 6 and R 7 is —CH 3 .
- the present invention also provides chimeric oligomeric compounds having the formula:
- each L 4 , L 5 , and L 6 is, independently, an internucleoside linkage selected from phosphodiester, phosphorothioate, phosphoramidate or boranophosphate, with the provision that L 4 and L 6 are different from L 5 and that one of L 4 , L 5 and L 6 are other than phosphodiester and phosphorothioate;
- each z 1 , z 2 , and z 3 is, independently, an integer greater than 1;
- each Nu is a nucleoside having the formula:
- Bx is a heterocyclic base moiety
- each R 1 is, independently, H, hydroxyl, a protected hydroxyl, a 2′-substituent group or a protected 2′-substituent group;
- said 5′- and said -3′ ends of said chimeric oligomeric compound are, independently, hydroxyl, a protected hydroxyl, a linkage to a solid support, an activated phosphate group, an activated phosphate group, a reactive group for forming an internucleotide linkage, a nucleotide, a nucleoside, or an oligonucleotide.
- each L 4 and L 6 is a phosphoramidate internucleotide linkage and each L 5 is a phosphorothioate internucleoside linkage.
- each L 4 and L 6 is a phosphoramidate internucleotide linkage and each L 5 is a phosphodiester internucleoside linkage.
- each L 4 and L 6 is a boranophsphate internucleotide linkage and each L 5 is a phosphodiester internucleoside linkage.
- each L 4 , L 5 and L 6 is phosphoramidate, phosphorothioate or phosphodiester.
- each R 1 be, independently, —O—CH 2 CH 2 —O—CH 3 , —O—CH 2 CH 2 —O—N(R 6 )(R 7 ) or —O—CH 2 CH 2 —O—CH 2 CH 2 —N(R 6 )(R 7 ). It is also preferred that each of R 6 and R 7 be, independently, H or C 1 -C 10 alkyl.
- each R 6 and R 7 be -CH 3 .
- the present invention provides compounds and methods for the preparation of mixed backbone oligomeric , or chimeric, compounds having phosphodiester internucleoside linkages in addition to phosphorothioate and/or phosphoramidate internucleoside linkages.
- the methods also include incorporation of boranophosphate internucleoside linkages into oligomeric compounds of the invention.
- the methods utilize H-phosphonate intermediates that are coupled together forming contiguous regions of one or more H-phosphonate internucleoside linkages. Each contiguous region is subsequently oxidized to phosphodiester, phosphorothioate, phosphoramidate or boranophosphate internucleoside linkages prior to further elongation.
- Oligomeric compounds of the invention are prepared in this manner by oxidizing adjacent regions with different reagents. Oligomeric compounds of the invention are prepared using novel oxidation steps that oxidize a region of one or more H-phosphonate internucleoside linkages without degrading existing linkages that have been previously oxidized.
- the H-phosphonate method of oligomer synthesis has been used for the preparation of oligonucleotides and their analogs having a variety of internucleotide linkages. These internucleotide linkages have included phosphorothioate, phosphoramidate, methyl phosphate, methylphosphonate, alkylphosphonate, S-aryl phosphorothioate, acylphosphonate, phosphorofluoridate, phosphorodithioate, selenophosphate, (hydroxymethyl)phosphonate and boranophosphate.
- chimeric oligomeric compound or “mixed backbone oligomeric compound” refers to oligomeric compounds comprising nucleoside monomer subunits and containing at least two different internucleoside linkages.
- the mixed backbone oligomeric compounds of the present invention may contain a plurality of nucleoside monomer subunits that are joined together by more than one type of internucleoside linkages. At least one internucleoside linkage is a phosphodiester linkage and at least one other linkage is a phosphorothioate, a phosphoramidate or a boranophosphate internucleoside linkage.
- oligomeric compound includes oligonucleosides, oligonucleotides, their analogs, and synthetic oligonucleotides.
- a first monomer attached to a solid support is elongated using H-phosphonate monomers.
- the methods of the present invention have been used with automated DNA synthesizers to synthesize mixed backbone oligomeric compounds of desired length and sequence. Some oligomeric compounds have been synthesized having only phosphodiester and phosphorothioate internucleoside linkages. Other oligomeric compounds have been synthesized having phosphodiester, phosphorothioate and phosphoramidate internucleoside linkages.
- Example 14 illustrates the synthesis of a boranophosphate mixed backbone oligomeric compound. Table I illustrates selected oligomeric compounds that were synthesized using methods and intermediates of the present invention.
- the methods of the present invention are employed for use in iterative solid phase oligonucleotide synthetic regimes.
- Representative solid phase techniques are those typically employed for DNA and RNA synthesis utilizing standard H-phosphonate chemistry.
- the H-phosphonate method of oligonucleotide synthesis is disclosed in U.S. Pat. No. 5,149,798, issued Sep. 22, 1992, and U.S. Pat. No. 5,548,076, issued Aug. 20, 1996. The entire disclosure of each is incorporated herein by reference.
- a preferred synthetic solid phase synthesis utilizes H-phosphonates as activated phosphate compounds.
- an H-phosphonate monomer is reacted with a free hydroxyl on the growing oligomer chain to produce an intermediate phosphite compound, which is subsequently oxidized to the P v state.
- This technique is commonly used for the synthesis of several types of linkages including phosphodiester, phosphorothioate, and phosphoramidate linkages. Oxidation of H-phosphonate linkages to phosphodiester linkages using standard H-phosphonate chemistry results in the degradation of phosphorothioate and phosphoramidate linkages when present in a growing oligomeric compound. This degradation of adjacent linkages within an oligomeric compound has been a limitation to the use of standard H-phosphonate chemistry with mixed backbone syntheses.
- the first step in such a process is attachment of a monomer or higher order subunit containing a protected 5′-hydroxyl to a solid support, usually through a linker, using standard methods and procedures known in the art. See, e.g., Oligonucleotides And Analogues A Practical Approach , Ekstein, F. Ed., IRL Press, N.Y., 1991, hereby incorporated by reference in its entirety.
- the support-bound monomer or higher order first synthon is then treated to remove the 5′-protecting group. Typically, this is accomplished by treatment with acid.
- the solid support bound monomer is then reacted with a second monomer or higher order synthon having a reactive group for forming an internucleoside linkage.
- the coupling reaction is performed under anhydrous conditions in the presence of a condensing reagent.
- a capping step is performed to cap unreacted hydroxyl groups. It is generally preferable to perform a capping step either prior to or after oxidation or sulfurization of the internucleoside linkage. Such a capping step is generally known to be beneficial by preventing shortened oligomer chains, by blocking chains that have not reacted in the coupling cycle.
- One representative reagent used for capping is acetic anhydride.
- Other suitable capping reagents and methodologies can be found in U.S. Pat. No. 4,816,571, issued Mar. 28, 1989, hereby incorporated by reference in its entirety.
- an “oxidizing solution” comprises a mixture of reagents effective to oxidize an H-phosphonate internucleoside linkage or silylated derivative thereof to a phosphodiester, phosphorothioate, phosphoramidate or boranophosphate internucleoside linkage. Such oxidation can be performed on a region of an oligomeric compound without causing degradation of other previously oxidized portions of the same oligomeric compound. Oxidation of each type of internucleoside linkage is illustrated in the examples below. Oxidation of the internucleoside linkage can be performed iteratively after each coupling step, or can be performed after a desired number or sequence of monomers have been coupled.
- gapmer-type oligomeric compounds having one or more regions of one type of linkage with one or more other regions having another type of linkage.
- a representative example of a gapmer is an oligomeric compound having a phosphorothioate region at the 3′ and 5′ ends with a phosphodiester region located internally.
- Treatment of the growing oligomeric compound with an acid removes the 5′-hydroxyl protecting group enabling another synthetic iteration.
- the growing oligomeric compound is extended with subsequent oxidation at selected stages with selected oxidation agents until an oligomeric compound of desired length is produced.
- the completed oligomer is then cleaved from the solid support.
- the cleavage step can precede or follow deprotection of protected functional groups.
- deblocking solution as used herein is meant to encompass solutions routinely used for the deblocking of oligomeric compounds prepared either by solution or solid phase techniques.
- a common solution used for deblocking of oligomers synthesized on solid support is aqueous ammonia.
- heterocyclic base moiety is intended to include nucleobases.
- Nucleobases useful in the compounds and methods described herein include but are not limited to adenine, guanine, cytosine, uridine, and thymine, as well as other non-naturally occurring and natural nucleobases such as xanthine, hypoxanthine, 2-aminoadenine, 6-methyl and other alkyl derivatives of adenine and guanine, 2-propyl and other alkyl derivatives of adenine and guanine, 5-halo uracil and cytosine, 6-azo uracil, cytosine and thymine, 5-uracil (pseudo uracil), 4-thiouracil, 8-halo, oxa, amino, thiol, thioalkyl, hydroxyl and other 8-substituted adenines and guanines, 5-trifluor
- nucleobases include those disclosed in U.S. Pat. No. 3,687,808 (Merigan et al.); in Sanghvi, in Antisense Research and Application , Chapter 15, S. T. Crooke and B. Lebleu, Eds., CRC Press, 1993; in Englisch et al., Angewandte Chemie , International Edition, 1991, 30, 613-722 (particularly, pages 622 and 623); in the Concise Encyclopedia of Polymer Science and Engineering , J. I.
- nucleosidic base moiety is further intended to include heterocyclic compounds that can serve as nucleosidic bases, including certain ‘universal bases’ that are not nucleosidic bases in the most classical sense but serve as nucleosidic bases. Especially mentioned as a universal base is 3-nitropyrrole.
- 2′-substituent group includes 2′-sugar modifications.
- 2 ⁇ 0-Sugar modifications of the present invention include fluoro, O-alkyl, O-alkylamino, O-alkylalkoxy, protected O-alkylamino, O-alkylaminoalkyl, O-alkyl imidazole, and polyethers of the formula (O-alkyl) m , where m is 1 to about 10.
- polyethers linear and cyclic polyethylene glycols (PEGs), and PEG-containing groups, such as crown ethers and those which are disclosed by Ouchi et al., Drug Design and Discovery 1992, 9, 93; Ravasio et al., J. Org. Chem. 1991, 56, 4329; and Delgardo et. al., Critical Reviews in Therapeutic Drug Carrier Systems 1992, 9, 249, each of which is hereby incorporated by reference in its entirety. Further sugar modifications are disclosed in Cook, Anti-Cancer Drug Design, 1991, 6, 585-607.
- Additional 2′-sugar modifications of use in the present invention include 2′-SR and 2′-NR 2 groups, where each R is, independently, hydrogen, a protecting group or substituted or unsubstituted alkyl, alkenyl, or alkynyl.
- 2′-SR nucleosides are disclosed in U.S. Pat. No. 5,670,633, issued Sep. 23, 1997, hereby incorporated by reference in its entirety. Incorporation of 2′-SR monomer synthons is disclosed by Hamm et al., J. Org. Chem., 1997, 62, 3415-3420.
- 2′-NR 2 nucleosides are disclosed by Goettingen, J. Org. Chem., 1996, 61, 6273-6281; and Polushin et al., Tetrahedron Lett., 1996, 37, 3227-3230.
- E is C 1 -C 10 alkyl, N(R 4 )(R 5 ) or N ⁇ C(R 4 )(R 5 );
- each R 4 and R 5 is, independently, H, C 1 -C 10 alkyl, a nitrogen protecting group, or R 4 and R 5 , together, are a nitrogen protecting group, or R 4 and R 5 are joined in a ring structure that can include at least one additional heteroatom selected from a group consisting of nitrogen and oxygen;
- R 3 is OX, SX, or N(X) 2 ;
- each X is, independently, H, C 1 -C 8 alkyl, C 1 -C 8 haloalkyl, C( ⁇ NH)N(H)Z, C( ⁇ O)N(H)Z or OC( ⁇ O)N(H)Z;
- Z is H or C 1 -C 8 alkyl
- L 1 , L 2 and L 3 comprise a ring system having from about 4 to about 7 carbon atoms, or having from about 3 to about 6 carbon atoms and at least one heteroatom, said heteroatom being selected from a group consisting of oxygen, nitrogen and sulfur, said ring system being aliphatic, unsaturated aliphatic, aromatic, or saturated or unsaturated heterocyclic;
- Y is alkyl or haloalkyl having 1 to about 10 carbon atoms, alkenyl having 2 to about 10 carbon atoms, alkynyl having 2 to about 10 carbon atoms, aryl having 6 to about 14 carbon atoms, N(R 4 )(R 5 ) OR 4 , halo, SR 4 or CN;
- each q 1 is, independently, from 2 to 10;
- each q 2 is, 0 or 1;
- p is from 1 to 10;
- r is from 1 to 10, provided that when p is 0, r is greater than 1.
- alkyl includes, but is not limited to, straight chain, branched chain, and alicyclic hydrocarbon groups. Alkyl groups of the present invention may be substituted. Representative alkyl substituents are disclosed in U.S. Pat. No. 5,212,295, at column 12, lines 41-50, hereby incorporated by reference in its entirety. As used herein, the term “lower alkyl” is intended to mean alkyl having 6 or fewer carbons.
- aralkyl denotes alkyl groups which bear aryl groups, for example, benzyl groups.
- alkaryl denotes aryl groups which bear alkyl groups, for example, methylphenyl groups.
- aryl denotes aromatic cyclic groups including but not limited to phenyl, naphthyl, anthracyl, phenanthryl, and pyrenyl.
- alkanoyl has its accustomed meaning as a group of formula —C( ⁇ O)—alkyl.
- a preferred alkanoyl group is the acetyl group.
- hetero denotes an atom other than carbon, preferably but not exclusively nitrogen, oxygen or sulfur.
- heterocycloalkyl denotes an alkyl ring system having one or more heteroatoms, i.e., non-carbon atoms.
- Preferred heterocycloalkyl groups include, for example, morpholino groups.
- heterocycloalkenyl denotes a ring system having one or more double bonds and one or more heteroatoms.
- Preferred heterocycloalkenyl groups include, for example, pyrrolidino groups.
- oligomer synthesis is performed on an automated synthesizer utilizing a solid support.
- Solid supports are substrates which are capable of serving as the solid phase in solid phase synthetic methodologies, such as those described in U.S. Pat. Nos. 4,415,732; 4,458,066; 4,500,707; 4,668,777; 4,973,679; 5,132,418; 4,725,677 and Re. 34,069.
- Linkers are known in the art as short molecules which serve to connect a solid support to functional groups, e.g., hydroxyl groups, of initial synthon molecules in solid phase synthetic techniques. Suitable linkers are disclosed in, for example, Oligonucleotides And Analogues A Practical Approach , Ecstein, F., Ed., IRL Press, N.Y., 1991, Chapter 1, pages 1-23.
- Solid supports according to the invention include those generally known in the art to be suitable for use in solid phase methodologies including, for example, controlled pore glass (CPG), oxalyl-controlled pore glass (see, e.g., Alul et al., Nucleic Acids Research 1991, 19, 1527, hereby incorporated by reference in its entirety), TentaGel Support; an aminopolyethyleneglycol derivatized support (see, e.g., Wright et al., Tetrahedron Letters 1993, 34, 3373, hereby incorporated by reference in its entirety) and Poros; a copolymer of polystyrene/divinylbenzene.
- CPG controlled pore glass
- oxalyl-controlled pore glass see, e.g., Alul et al., Nucleic Acids Research 1991, 19, 1527, hereby incorporated by reference in its entirety
- TentaGel Support an aminopolyethyleneglycol derivatized support (see,
- each of T 1 and T 2 is, independently, a protected hydroxyl group
- T 3 is a hydroxyl protecting group.
- hydroxyl protecting groups can be employed in the methods of the invention.
- the protecting group is stable under either acidic or basic conditions. This enables removal of a base-stable protecting group under acidic conditions, and removal of an acid-stable protecting group under basic conditions.
- protecting groups render chemical functionalities inert to specific reaction conditions, and can be appended to and removed from such functionalities in a molecule without substantially damaging the remainder of the molecule.
- hydroxyl protecting groups are disclosed by Beaucage et al., Tetrahedron 1992, 48, 2223-2311, and also in Greene and Wuts, Protective Groups in Organic Synthesis , Chapter 2, 2d ed, John Wiley & Sons, N.Y., 1991, each of which is hereby incorporated by reference in its entirety.
- Preferred protecting groups include dimethoxytrityl (DMT), monomethoxytrityl (MMT), 9-phenylxanthen-9-yl (Pixyl) and 9-(p-methoxyphenyl)xanthen-9-yl (Mox).
- amino groups are appended to alkyl or other groups such as, for example, 2′-alkoxy groups. Such amino groups are also commonly present in naturally-occurring and non-naturally-occurring nucleobases. It is generally preferred that these amino groups be in protected form during the synthesis of oligomeric compounds of the invention. Representative amino protecting groups suitable for these purposes are discussed in Greene and Wuts, Protective Groups in Organic Synthesis , Chapter 7, 2d ed, John Wiley & Sons, N.Y., 1991. Generally, as used herein, the term “protected,” when used in connection with a molecular moiety such as “nucleobase,” indicates that the molecular moiety contains one or more functionalities protected by protecting groups.
- the condensing reagent is selected from a group consisting of pivaloyl chloride, adamantyl chloride, 2,4,6 -triisopropyl-benzenesulfonyl chloride, 2-chloro-5,5-dimethyl-2 -oxo-1,3,2-dioxaphosphinane, diphenyl phosphorochloridate, bis(2-oxo-3 -oxazolidinyl)phosphinic chloride, bis(pentafluorophenyl)carbonate, 2-(1H-benzotriazole- 1 -yl)-1,1,3,3-tetramethyluronium hexafluorophosphate, O-(azabenzotriazol- 1 -yl)- 1,1,3,3-tetramethyl uronium hexafluorophosphate, 6-(trifluoromethyl)benzotriazol-1 -yl-oxy-tris-pyrrolidino-
- Sulfurizing agents used during oxidation to form phosphorothioate and phosphorodithioate linkages include Beaucage reagent (Iyer et. al., J. Chem. Soc., 1990, 112, 1253-1254; and Iyer et. al., J. Org. Chem., 1990, 55, 4693-4699); tetraethylthiuram disulfide (Vu and Hirschbein, Tetrahedron Lett., 1991, 32, 3005-3008); dibenzoyl tetrasulfide (Rao, et.
- Useful oxidizing agents used to form the phosphodiester or phosphorothioate linkages, when they are the only linkages present, include iodine/tetrahydrofuran/water/pyridine, hydrogen peroxide/water, tert-butyl hydro peroxide or any peracid like m-chloroperbenzoic acid.
- oxidation using a sulfur species the reaction is performed under anhydrous conditions with the exclusion of air, in particular oxygen, whereas in the case of oxidation the reaction can be performed under aqueous conditions.
- the oxidation of H-phosphonate internucleoside linkages, without simultaneous degradation of existing, previously oxidized linkages, is performed using a novel oxidizing solution.
- the oxidizing solution comprises particular compounds/reagents dependent upon what oxidized linkages are being prepared from the H-phosphonate linkages.
- the oxidation solution comprises an oxidizing agent, an aprotic organic solvent, and a base.
- Bases are present in the oxidizing solution to assist in the oxidation process.
- Selected bases have a pka ranging from about 9 to about 12, which is believed to be the optimum range for deprotonation of H-phosphonate or water without causing cleavage of the backbone of the oligomeric compound. While not wanting to be bound by theory, it is believed that bases within this pKa range, and present in the oxidation solution in a concentration of from about 0.01 to about 0.8 M, are necessary for oxidation of H-phosphonate linkages without simultaneous degradation of existing oxidized linkages. A more preferred range for the concentration of the base is from about 0.05 to about 0.2 M in the oxidation solution.
- Representative bases that are useful in the present invention include 4-(dimethylamino)pyridine (DMAP), triethylamine (TEA) and N,N-diisopropylethylamine (DIEA).
- aprotic organic solvents known to those skilled in the art are amenable to the present invention.
- Some representative aprotic organic solvents include pyridine, acetonitrile and dimethylformamide.
- oxidizing agent is dependent on the particular internucleoside linkage that is desired for the region of H-phosphonate internucleoside linkages that is being oxidized.
- Representative oxidizing agents useful for the preparation of phosphodiester internucleoside linkages include carbon tetrachloride, carbon tetrabromide, N-chlorosuccinimide and N-bromosuccinimide.
- water is a necessary component of the oxidizing solution for preparing phosphodiester linkages.
- Phosphorothioate internucleoside linkages are prepared using an oxidizing agent that oxidizes phosphorus by addition of sulfur rather than oxygen. Elemental sulfur has been used to sulfrize H-phosphonate to form phosphorothioate internucleoside linkages as illustrated in the examples below. The use of elemental sulfur in the oxidizing solution affords the desired phosphorothioate internucleoside linkages while leaving unaffected any preexisting oxidized linkages in the same oligomeric compound.
- the selective oxidation of H-phosphonate internucleoside linkages to phosphodiester, phosphorothioate, phosphoramidate and boranophosphate internucleoside linkages in a mixed backbone oligomeric compound is dependent on the oxidizing solution.
- the oxidizing solution must not affect preexisting linkages in the oligomeric compound that is undergoing oxidation.
- the oxidizing solution is a mixture of an oxidizing agent, an aprotic solvent and a base.
- water is also a component of the oxidizing solution.
- concentration of reagents in the oxidizing solution for a given oxidation can vary. Optimal concentration of reagents can be discerned by the art-skilled without undue experimentation.
- composition and concentration of reagents in the oxidizing solution used to oxidize an H-phosphonate linkages in a mixed backbone oligomeric compound is variable dependent on whether the desired oxidized linkage is phosphodiester, phosphorothioate or phosphoramidate.
- a silylation step is also involved.
- the two main components are the base and the oxidizing agent.
- an aprotic organic solvent is also used for phosphodiester and phosphorothioate internucleoside linkages. Water is also used in the oxidizing solution when phosphodiester linkages are prepared.
- the oxidizing solution used for preparing phosphodiester internucleoside linkages in a mixed backbone oligomeric compound of the invention generally has from about 18% to about 45% oxidizing agent, from about 2% to about 15% water, from about 40% to about 80% aprotic organic solvent, and from about 0.01 M to about 0.8 M base dissolved in the aprotic organic solvent.
- a more preferred range of concentrations is from about 26% to about 40% oxidizing agent, from about 4% to about 10% water, from about 50% to about 70% aprotic organic solvent, and from about 0.04 M to about 0.4 M base dissolved in the aprotic organic solvent.
- Even more preferred is about 33% oxidizing agent, about 7% water, about 60% aprotic organic solvent, and from about 0.05 M to about 0.2 M base dissolved in the aprotic organic solvent.
- the oxidizing solution used for preparing phosphorothioate internucleoside linkages in a mixed backbone oligomeric compound of the invention generally has from about 1% to about 15% oxidizing agent, about 40% to about 60% solvent that will solubilize the oxidizing agent, from about 40% to about 60% aprotic organic solvent, and from about 0.01 M to about 0.8 M base dissolved in the aprotic organic solvent.
- a more preferred range of concentrations is from about 1% to about 10% oxidizing agent, about 40% to about 60% solvent that will solubilize the oxidizing agent, from about 40% to about 60% aprotic organic solvent, and from about 0.02 M to about 0.5 M base dissolved in the aprotic organic solvent.
- Even more preferred is about 3% to about 8% oxidizing agent, about 40% to about 60% solvent that will solubilize the oxidizing agent, from about 40% to about 60% aprotic organic solvent, and from about 0.04 M to about 0.1 M base dissolved in the aprotic organic solvent.
- the oxidizing solution used for preparing phosphoramidate internucleoside linkages in a mixed backbone oligomeric compound of the invention generally has a primary or secondary amine in combination with an oxidizing agent.
- An oxidizing agent that acts as a solvent for the primary or secondary amine is preferable.
- a preferred oxidizing agent is carbon tetrachloride.
- the oxidizing solution has from about 1% to about 15% of the primary or secondary amine, and from about 85% to about 99% oxidizing agent.
- a preferred concentration is about 1% to about 10% of the primary or secondary amine, and from about 90% to about 99% oxidizing agent.
- a more preferred concentration of primary or secondary amine is about 2% to about 5% by volume in the oxidizing solution.
- the silylation step is followed by oxidation with borane-N,N-diisopropylethylamine (BH 3 ⁇ DIEA, 0.1 mmol to 1 mmol), borane-2-chloro-pyridine or borane-aniline.
- BH 3 ⁇ DIEA borane-N,N-diisopropylethylamine
- the compounds of the invention are used to modulate RNA or DNA encoding a protein whose formation or activity is to be modulated.
- the targeting portion of the composition to be employed is, thus, selected to be complementary to the preselected portion of DNA or RNA. That is, the targeting portion of the composition is hybridizable to the preselected portion of DNA.
- the methods of the present invention can be used to prepare oligomeric compounds that are used in diagnostics, therapeutics, and as research reagents and kits. They can be used in pharmaceutical compositions by including a suitable pharmaceutically acceptable diluent or carrier. They further can be used for treating organisms having a disease characterized by the undesired production of a protein. The organism should be contacted with an oligonucleotide having a sequence that is capable of specifically hybridizing with a strand of nucleic acid encoding the undesirable protein. Treatments of this type can be practiced on a variety of organisms ranging from unicellular prokaryotic and eukaryotic organisms to multicellular eukaryotic organisms.
- Any organism that utilizes DNA-RNA transcription or RNA-protein translation as a fundamental part of its hereditary, metabolic or cellular control is susceptible to therapeutic and/or prophylactic treatment in accordance with the invention. Seemingly diverse organisms such as bacteria, yeast, protozoa, algae, all plants and all higher animal forms, including warm-blooded animals, can be treated. Further, each cell of multicellular eukaryotes can be treated, as they include both DNA-RNA transcription and RNA-protein translation as integral parts of their cellular activity. Furthermore, many of the organelles (e.g., mitochondria and chloroplasts) of eukaryotic cells also include transcription and translation mechanisms. Thus, single cells, cellular populations or organelles can also be included within the definition of organisms that can be treated with therapeutic or diagnostic oligonucleotides.
- organelles e.g., mitochondria and chloroplasts
- Solid phase synthesis was carried out using an Applied Biosystems (Perkin Elmer Corporation) DNA/RNA synthesizer 380B and controlled pore glass (5′-O-DMT-2′-MOE mononucleoside-3′-succinate: R. I. Chemical, Costa Mesa, Calif.) or primer support (5′-O-DMT-2′-MOE mononucleoside-3′-succinate: Pharmacia) as the solid supports.
- the H-phosphonate method of oligonucleotide synthesis is disclosed in U.S. Pat. No. 5,149,798, issued Sep. 22, 1992, and U.S. Pat. No. 5,548,076, issued Aug. 20, 1996. The entire disclosure of each is incorporated herein by reference.
- Phosphorothioate linkages were prepared via oxidation of H-phosphonate linkages using 5% elemental sulfur in carbon disulfide/pyridine/triethylamine (10:10:1). Prior to sulfurization, the solid support was thoroughly washed with acetonitrile and dried under vacuum. The reaction time was 60 to 120 minutes.
- oligomeric compounds containing phosphorothioate and phosphoramidate linkages the oxidation is performed using 5% elemental sulfur and DMAP (0.1 M) or triethylamine (0.1 M) in carbon disulfide/pyridine(1:1). Prior to sulfurization, the solid support is thoroughly washed with acetonitrile and dried under vacuum. The reaction time is 60-120 min.
- Oligomeric compounds containing phosphoramidate linkages were prepared via oxidation of the corresponding H-phosphonate oligomer blocks on the solid support using a mixture of a primary or secondary amine in carbon tetrachloride (10%:90%, v/v). Prior to oxidation, the solid support was thoroughly washed with acetonitrile and dried under vacuum. The reaction time was 60 to 120 minutes.
- Oligomeric compounds containing boranophosphate linkages are prepared by treatment of H-phosphonate internucleoside linkages with N,O-bis(trimethylsilyl)trifluoroacetamide (0.3 M) in dry tetrahydrofuran for 20 to 30 minutes, washing with tetrahydrofuran and then treating with borane-N,N-diisopropyl-ethylamine (BH 3 :DIEA, 0.1 mmol:1 mmol), borane-2-chloropyridine or borane-aniline. Prior to oxidation, the solid support is thoroughly washed with acetonitrile and dried under vacuum. The reaction time is 60 to 240 minutes.
- the crude product was purified by silica gel column chromatography using a step gradient of 0.5% triethylamine in dichloromethane to 0.5% triethylamine in dichloromethane/methanol (9:1). The combined product fractions were evaporated to a foam and evaporated twice with acetonitrile to remove excess triethylamine. Subsequently the purified product was dissolved in dichloromethane (300 mL), washed twice each with 0.2 M diazabicyclo[5.4.0]undec-7-ene bicarbonate buffer (350 mL, pH 8.7), dried over sodium sulfate and evaporated to a foam. Prior to its use for solid phase synthesis the product was dried under high vacuum to give 4.62 g (85%) of the title compound.
- the crude material was purified by silica gel column chromatography using a step gradient of 0.5% triethylamine in dichloromethane to 0.5% triethylamine in dichloromethane/ methanol (9:1).
- the combined product fractions were evaporated to a foam and coevaporated twice with acetonitrile to remove trace triethylamine.
- the purified product was dissolved in dichloromethane (350 mL), washed twice with diazabicyclo[5.4.0]undec-7-ene bicarbonate buffer (0.2 M, 350 mL, pH 8.7), dried over sodium sulfate, and evaporated to a foam. Prior to its use for solid phase synthesis the product was dried under high vacuum to give 5.5 g (90%) of the title compound.
- the crude product was purified by silica gel column chromatography using a step gradient of 3% triethylamine in dichloromethane to 3% triethylamine in dichloromethane/methanol (9:1). The combined product fractions were evaporated to a foam and evaporated twice with acetonitrile to remove trace triethylamine. Subsequently the purified product was dissolved in dichloromethane (300 mL), washed twice with diazabicyclo[5.4.0]undec-7-ene bicarbonate buffer (300 mL, 0.2 M, pH 8.7), dried over sodium sulfate, and evaporated to a foam. Prior to its use for solid phase synthesis the product was dried under high vacuum to give 3.9 g (63%) of the title compound. 31 P-NMR (CDCl 3 , using 85% H 3 PO 4 as an external standard): 3.76 ppm.
- the crude product was purified by silica gel column chromatography using a step gradient of 3% triethylamine in dichloromethane to 3% triethylamine in dichloromethane/methanol (9:1). The combined product fractions were evaporated to a foam and coevaporated twice with acetonitrile to remove trace triethylamine. Subsequently the purified product was dissolved in dichloromethane (50 mL), washed twice with diazabicyclo[5.4.0]undec-7-ene bicarbonate buffer (50 mL, 0.2 M, pH 8.7), dried over sodium sulfate, and evaporated to a foam. Prior to its use for solid phase synthesis the product was dried under high vacuum to give 0.8 g (12%) of the title compound. 31 P-NMR (CDCl 3 , external standard: 85% H 3 PO 4 ): 3.68 ppm.
- the oligomer was oxidized using iodine (0.1 M) in pyridine/water (98:2) and washed with acetonitrile/pyridine (1:1) and acetonitrile. Deprotection and cleavage from the solid support was performed with aqueous ammonia (25-28%), as described in the General Procedures. Analysis of the crude product by electrospray mass spectrometry gave a relative molecular mass of 8023.5 (calculated mass: 8022.3).
- the oligomer was oxidized using iodine (0.1 M) in pyridine/water (98:2) and washed with acetonitrile/pyridine (1:1) and acetonitrile. Deprotection and cleavage from the solid support was performed with aqueous ammonia (25-28%), as described in the General Procedures. Analysis of the crude product by electrospray mass spectrometry gave a relative molecular mass of 6435.5 (calculated mass: 6440.7).
- the oligomer was oxidized using elemental sulfur (5%) in carbon disulfide/pyridine/triethylamine (10:10:1) and washed with carbon disulfide, acetonitrile/pyridine (1:1) and acetonitrile. Deprotection and cleavage from the solid support was carried out with aqueous ammonia (25-28%), as described in the General Procedures. Analysis of the crude product by electrospray mass spectrometry gave a relative molecular mass of 6873.4 (calculated mass: 6874.8).
- the solid support Prior to synthesis of the next segment, the solid support was thoroughly washed with carbondisulfide, acetonitrile/pyridine (1:1) and acetonitrile. After completion of solid phase synthesis, the support-bound oligomer was finally treated with DMAP (0.1 M) in pyridine/carbontetrachloride/water (9:5:1) and washed with acetonitrile/pyridine (1:1) and acetonitrile. Deprotection and cleavage from the solid support was performed with aqueous ammonia (25-28%), as described in the General Procedures. Analysis of the crude product by electrospray mass spectrometry gave a relative molecular mass of 7425.9 (calculated mass: 7426.0).
- the synthesis cycle is described in the General Procedures. After the first 2 couplings the oligomer was oxidized using 2 -dimethylaminoethylamine (10%) in carbontetrachloride. Prior to synthesis of the next segment, the solid support was thoroughly washed with acetonitrile. After incorporation of fourteen additional H-phosphonate monomers (to give a 17-mer) the oligomer was oxidized using elemental sulfur (5%) in carbondisulfide/pyridine/ triethylamine (10:10:1), as described in the General Procedures. Prior to synthesis of the next segment, the solid support was thoroughly washed with carbondisulfide, acetonitrile/pyridine (1:1) and acetonitrile.
- the solid support Prior to synthesis of the next segment, the solid support was thoroughly washed with carbondisulfide, acetonitrile/pyridine (1:1) and acetonitrile. After introduction of three additional H-phosphonate monomers (to give an 18-mer) the oligomer was oxidized using triethylamine (0.1 M) in pyridine/carbontetrachloride/water (9:5:1) and washed thoroughly with acetonitrile/pyridine (1:1) and then acetonitrile. After completion of solid phase synthesis, the support-bound oligomer was finally treated with 2 -dimethyl-aminoethylamine (10%) in carbontetrachloride.
- the oligomer was oxidized using elemental sulfur (5%) in carbondisulfide/pyridine/ triethylamine (10:10:1). Prior to synthesis of the next segment, the solid support was thoroughly washed with carbondisulfide, acetonitrile/pyridine (1:1), and acetonitrile. After introduction of three additional H-phosphonate monomers (to give an 18-mer) the oligomer was oxidized using DMAP (0.1 M) in pyridine/carbontetrachloride/water (9:5:1) and thoroughly washed with acetonitrile/pyridine (1:1) and acetonitrile.
- DMAP 0.1 M
- Table I below shows the oligomeric compounds prepared according to the procedures described in the examples above.
- TABLE 1 SEQ ID No. Sequence 5′-3′ Mr calc Mr found 1 T*T*T* T*T*T* T*T*T* T*T*T* T*T*T* 7429.5 7424.8 T*T*T* T*T 2 T*T*T* T*GA CAC GGC AT*T* T*T* 6440.7 6435.5 3 T* N T* N T* N G S A S C S A S C S C S A S 6886.0 n.a.
- the solid support Prior to further elongation the solid support is washed with acetonitrile. After coupling of a desired number of further nucleosides oxidation to for phosphorothioate internucleoside linkages is performed using elemental sulfur (5%) and 4-(dimethylamino)pyridine (0.1 M) in carbon disulfide/pyridine (1:1). The solid support is again washed with carbon disulfide, acetonitrile/pyridine (1:1) and acetonitrile. After addition of a desired number of further nucleosides, the support-bound oligomer is treated with 10% 2-dimethylaminoethylamine in carbon tetrachloride. Deprotection and cleavage from the solid support is performed with aqueous ammonia (25-28%) to give the desired oligomeric compound.
- the resulting oligomeric compound is treated with N,O-bis(trimethylsilyl)trifluoroacetamide (0.3 M) in dry tetrahydrofuran for 30 minutes and washed with tetrahydrofuran. Subsequently, the support-bound oligomer is treated with borane-N,N-diisopropylethylamine (0.5 M) complex in dry tetrahydrofuran for 2 hours at room temperature. Prior to synthesis of the next segment, the solid support is washed with acetonitrile.
- the H-phosphonate internucleoside linkages are oxidized to phosphorothioate internucleoside linkages using elemental sulfur (5%) and 4-(dimethylamino)pyridine (0.1 M) in carbon disulfide/pyridine (1:1).
- elemental sulfur 5%)
- 4-(dimethylamino)pyridine 0.1 M
- carbon disulfide/pyridine 1:1
- acetonitrile/pyridine acetonitrile
- the H-phosphonate internucleoside linkages are treated at room temperature with N,O-bis(trimethylsilyl)trifluoroacetamide (0.3 M) in dry tetrahydrofuran for 30 minutes and washed with tetrahydrofuran. Subsequently, the support-bound oligomer is treated with 0.5 M borane-N,N-diisopropylethylamine complex in dry tetrahydrofuran for 2 hours at room temperature. Deprotection and cleavage from the solid support is performed with aqueous ammonia (25-28%).
- Targeted oligomeric compounds were prepared and assayed using HUVEC cells to determine their ability to inhibit the expression of ICAM- 1.
- HUVEC cells were washed three times with Opti-MEM (Life Technologies, Inc.) that was prewarmed to 37° C.
- Oligomeric compounds were premixed with Lipofectin (10 ⁇ g/mL, Life Technologies, Inc.) in Opti-MEM, serially diluted to the desired concentrations, and applied to washed cells.
- Basal and untreated (no oligomeric compounds added) control cells were also treated with Lipofectin.
- Cells were incubated for 4 hours at 37° C., at which time the medium was removed and replaced with standard growth medium with or without TNF-a (5 mg/mL, R&D Systems). Incubation at 37° C. was continued for different times as indicated below.
- a fluorescence-activated cell sorter was used to quantitate ICAM- 1 protein expression.
- the cells were removed from the plate surface by brief trypsinization with trypsin (0.25%) in PBS. Trypsin activity was quenched with a solution of bovine serum albumin (2%) and sodium azide (0.2%) in PBS (+Mg/Ca). Cells were pelleted by centrifugation (1000 rpm, Beckman GPR centrifuge), resuspended in PBS, and stained with ICAM-1 specific antibody (3 ⁇ L/105 cells), CD54-PE (Pharmingin). The antibodies were incubated with the cells for 30 minutes at 4° C. in the dark, under gentle agitation. Cells were washed by centrifugation procedures and then resuspended in FacsFlow buffer (0.3 mL, Becton Dickinson) with formaldehyde (0.5%, Polysciences).
- ICAM-1 2′-O-(2-methoxy)ethyl-modified anti-intercellular adhesion molecule 1 (ICAM-1) oligomeric compounds were shown to selectively increase the ICAM- 1 MRNA level and inhibit formation of the ICAM-1 translation initiation complex in human umbilical vein endothelial cells (Baker et al., The Journal of Biological Chemistry, 1997, 272, 11994-12000).
- ICAM-1 2′-O-(2-methoxy)ethyl-modified anti-intercellular adhesion molecule 1
- the oligomers are presumably working by a direct binding RNase H independent mechanism. Both compounds synthesized from different chemistries display dose response in inhibiting ICAM-1 expression between 3 and 100 nM range.
- Gapmers designed in this study are based on the parent sequence of ATG CAT TCT GCC CCC AAG GA (SEQ ID No. 9) which is a uniform deoxy phosphorothioate. This sequence is an antisense oligomeric compound targeted to mouse c-raf.
- c-raf m-RNA expression assay was carried out according to a reported procedure (Monia et al., Nature Medicinek, 1996, 2, 668).
- Human A549 lung carcinoma cells were obtained from the American Type Tissue Collection. These were grown in Dulbecco's modified Eagle's medium containing 1 g of glucose/liter (DMEM) and FCS (10%) and routinely passaged when 90-95% confluent.
- A549 cells were plated in 6-well plates (Falcon Labware, Lincoln Park, N.J.) and 24-48 hours later (when 80-90% confluent) treated with 1 ⁇ M phorbol 12,13-dibutyrate (PDBu) for 18 hours. This procedure removes greater than 75% of immunoreactive c-raf protein from the cells. Cells were then washed three times with DMEM (3 mL) (to remove PDBu), and DMEM (1 mL) containing DOTMA/DOPE solution (20 ⁇ g/mL, LipofectinR, Bethesda Research Laboratories) was added.
- Oligonucleoside gapmers were then added to the required concentration (for our initial screen, 1 ⁇ M) from a 10 ⁇ M stock solution, and the two solutions were mixed by swirling of the dish.
- the cells were incubated at 37° C. for 4 hours, washed once with DMEM+FCS (10%) to remove the DOTMA/DOPE solution, and then additional DMEM (3 mL) +10% FCS was added and the cells were allowed to recover for another 10 hours.
- Total RNA is prepared 24 hours after the experiment and analyzed for c-raf and G3PDH mRNA levels after normalization to G3PDH MRNA.
Abstract
Synthetic processes are provided wherein mixed backbone oligomeric compounds are prepared having at least one phosphodiester internucleoside linkage in addition to one or more phosphorothioate, phosphoramidate and boranophosphate internucleoside linkages. Novel H-phosphonate intermediates are also disclosed that are useful in the synthetic processes. The synthetic processes use a novel oxidation step to oxidize H-phosphonate internucleoside linkages to phosphodiester internucleoside linkages without degradation of adjacent phosphorothioate, phosphoramidate and boranophosphate internucleoside linkages. Also provided are synthetic intermediates useful in such processes.
Description
- This application is a continuation of patent application Ser. No. 09/726,096, filed Nov. 29, 2000, which is a divisional of patent application Ser. No. 09/250,075, filed Feb. 12, 1999, each of which is incorporated herein by reference in its entirety.
- The present invention is directed to methods for oxidizing an H-phosphonate internucleoside linkage in a compound which has at least one 2′-substituent group. This invention further relates to methods for the preparation of mixed backbone oligomeric compounds having internucleoside linkages including phosphodiester, phosphorothioate, phosphoramidate and boranophosphate linkages. The present invention also relates to synthetic intermediates useful in such methods. Also included are mixed backbone oligomeric compounds having phosphodiester and boranophosphate internucleoside linkages or phosphodiester and boranophosphate internucleoside linkages in combination with other internucleoside linkages such as phosphorothioate and phosphoramidate internucleoside linkages. The methods of the present invention include a novel oxidation step that allows oxidation of a region having one or more H-phosphonate internucleoside linkages to phosphodiester internucleoside linkages without degradation of adjacent phosphorothioate, phosphoramidate or boranophosphate internucleoside linkages.
- Oligonucleotides and their analogs have been developed and used in molecular biology in a variety of procedures as probes, primers, linkers, adapters, and gene fragments. Modifications to oligonucleotides used in these procedures include labeling with nonisotopic labels, e.g., fluorescein, biotin, digoxigenin, alkaline phosphatase or other reporter molecules. Other modifications have been made to the ribose phosphate backbone to increase the nuclease stability of the resulting analog. Examples of such modifications include incorporation of methyl phosphonate, phosphorothioate, or phosphorodithioate linkages, and 2′-O-methyl ribose sugar units. Further modifications include those made to modulate uptake and cellular distribution. With the success of these compounds for both diagnostic and therapeutic uses, there exists an ongoing demand for improved oligonucleotides, their analogs and synthetic processes for their preparation.
- It is well known that most of the bodily states in multicellular organisms, including most disease states, are effected by proteins. Such proteins, either acting directly or through their enzymatic or other functions, contribute in major proportion to many diseases and regulatory functions in animals and man. For disease states, classical therapeutics has generally focused upon interactions with such proteins in efforts to moderate their disease-causing or disease-potentiating functions. In newer therapeutic approaches, modulation of the actual production of such proteins is desired. By interfering with the production of proteins, the maximum therapeutic effect may be obtained with minimal side effects. It is therefore a general object of such therapeutic approaches to interfere with or otherwise modulate gene expression, which would lead to undesired protein formation.
- One method for inhibiting specific gene expression is via the use of oligonucleotides, especially oligonucleotides which are complementary to a specific target messenger RNA (MRNA) sequence. Several oligonucleotides are currently undergoing clinical trials for such use. Phosphorothioate oligonucleotides are presently being used as such antisense agents in human clinical trials for various disease states, including use as antiviral agents.
- Transcription factors interact with double-stranded DNA during regulation of transcription. Oligonucleotides can serve as competitive inhibitors of transcription factors to modulate their action. Several recent reports describe such interactions. Bielinska, et al., Science, 1990, 250, 997-1000; and Wu, et al., Gene, 1990, 89, 203-209.
- In addition to their use as indirect and direct regulators of proteins, oligonucleotides and their analogs also have found use in diagnostic tests. Such diagnostic tests can be performed using biological fluids, tissues, intact cells or isolated cellular components. As with inhibition of gene expression, diagnostic applications utilize the ability of oligonucleotides and their analogs to hybridize with a complementary strand of nucleic acid. Hybridization is the sequence-specific hydrogen bonding of oligomeric compounds via Watson-Crick and/or Hoogsteen base pairs to RNA or DNA. The bases of such base pairs are said to be complementary to one another.
- Oligonucleotides and their analogs are also widely used as research reagents. They are useful for understanding the function of many other biological molecules as well as in the preparation of other biological molecules. For example, the use of oligonucleotides and their analogs as primers in PCR reactions has given rise to an expanding commercial industry. PCR has become a mainstay of commercial and research laboratories, and applications of PCR have multiplied. For example, PCR technology now finds use in the fields of forensics, paleontology, evolutionary studies and genetic counseling. Commercialization has led to the development of kits which assist non-molecular biology-trained personnel in applying PCR. Oligonucleotides and their analogs, both natural and synthetic, are employed as primers in such PCR technology.
- Oligonucleotides and their analogs are also used in other laboratory procedures. Several of these uses are described in common laboratory manuals such as Molecular Cloning, A Laboratory Manual, Second Ed., Sambrook et al, Eds., Cold Spring Harbor Laboratory Press, 1989; and Current Protocols In Molecular Biology, Ausubel et al., Eds., Current Publications, 1993. Such uses include as synthetic oligonucleotide probes, in screening expression libraries with antibodies and oligomeric compounds, DNA sequencing, in vitro amplification of DNA by the polymerase chain reaction, and in site-directed mutagenesis of cloned DNA. See Book 2 of Molecular Cloning, A Laboratory Manual, supra. See, also, “DNA-protein interactions and The Polymerase Chain Reaction” in Vol. 2 of Current Protocols In Molecular Biology, supra.
- Oligonucleotides and their analogs can be synthesized to have customized properties that can be tailored for desired uses. Thus a number of chemical modifications have been introduced into oligomeric compounds to increase their usefulness in diagnostics, as research reagents and as therapeutic entities. Such modifications include those designed to increase binding to a target strand (i.e., increase their melting temperatures, Tm), to assist in identification of the oligonucleotide or an oligonucleotide-target complex, to increase cell penetration, to stabilize against nucleases and other enzymes that degrade or interfere with the structure or activity of the oligonucleotides and their analogs, to provide a mode of disruption (terminating event) once sequence-specifically bound to a target, and to improve the pharmacokinetic properties of the oligonucleotide.
- The chemical literature discloses numerous processes for coupling nucleosides through phosphorous-containing covalent linkages to produce oligonucleotides of defined sequence. One such method utilizes H-phosphonate monomers to prepare oliogmeric compounds. The standard H-phosphonate method has been used for the synthesis of uniformly modified oligomeric compounds containing phosphodiester, phosphorothioate or phosphoramidate internucleoside linkages. The method has not been effective for the synthesis of mixed backbone oligomers because during the oxidation step previously oxidized regions in an oligomer are degraded. Such degradation has been observed for the oxidation of oligomers having phosphorothioate and or phosphoramidate internucleoside linkages when further H-phosphonate internucleoside linkages are oxidized to phosphodiester linkages.
- H-phosphonate methods and techniques are disclosed in numerous publications. See, Mackie and Hogrefe, Glen Research, H-phosphonate Chemistry, Glen Research Corp, Va.; Wada et al, J. Am. Chem. Soc., 1997, 119, 12710-12721; and Sergueev et al., J. Am. Chem. Soc., 1998, 120, 9417-9427.
- Phosphoramidate oligodeoxynucleotides have been prepared using H-phosphonate chemistry. Dagle et al., Nucleic Acids Research, 1991, 19, 1805-1810.
- 2′-Methoxymethyl oligonucleotides have been prepared using H-phosphonate monomers. Bizdena et al., Collect. Czech. Chem. Commun., 1996, 61, 283-286.
- The present invention provides methods for oxidizing H-phosphonate internucleoside linkages in a compound which bears at least one 2′-substituent group. This oxidation is effected by an oxidizing solution comprising an oxidizing agent, an aprotic solvent, a base and water.
- The present invention also provides methods for the preparation of mixed backbone oligomeric compounds of formula:

- wherein:
- each Z is, independently, a phosphodiester, phosphorothioate, phosphoramidate or boranophosphate internucleoside linkage;
- each T 1 and T2 is, independently, hydroxyl or a protected hydroxyl;
- Bx is a heterocyclic base moiety;
- each R 1 is, independently, H, hydroxyl, a protected hydroxyl, a 2′-substituent group or a protected 2′-substituent group; and
- n is an integer greater than 1;
- provided that at least one of said Z is a phosphodiester internucleoside linkage and at least another of said Z is a phosphorothioate, phosphoramidate or boranophosphate internucleoside linkage;
- comprising the steps of:
- (a) providing a compound of formula:

- wherein:
- Pg is an acid labile hydroxyl protecting group; and
- T 3 is a base labile hydroxyl protecting group or a covalent attachment to a solid support;
- (b) deblocking said acid labile hydroxyl protecting group to form a deblocked hydroxyl group;
- (c) treating said deblocked hydroxyl group with a further compound having the formula:
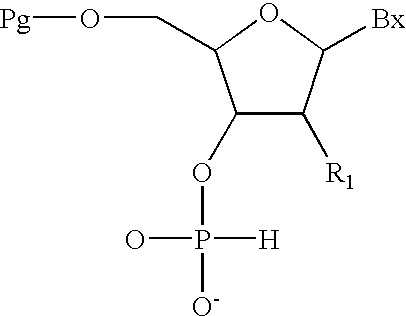
- and a condensing reagent in a solvent under conditions of time, temperature and pressure effective to form an extended compound having an added H-phosphonate internucleoside linkage;
- (d) optionally treating said extended compound with a capping agent to form a capped compound;
- (e) optionally treating said capped compound with a silylating agent to give a silylated compound;
- (f) optionally treating said extended compound, said capped compound, or said silylated compound with an oxidizing solution inert to phosphodiester, phosphorothioate, phosphoramidate or boranophosphate internucleoside linkages, thereby oxidizing said H-phosphonate internucleoside linkages to phosphodiester, phosphorothioate, phosphoramidate or boranophosphate internucleoside linkages;
- (g) optionally repeating steps (b), (c), (d), (e) and (f) to form a protected oligomeric compound; and
- (h) treating said protected oligomeric compound with a deblocking solution to form said mixed backbone oligomeric compound.
- In a preferred embodiment, the mixed backbone oligomeric compound comprises contiguous regions of phosphodiester and phosphorothioate internucleoside linkages.
- In another preferred embodiment, the mixed backbone oligomeric compound comprises contiguous regions of phosphodiester and phosphoramidate internucleoside linkages.
- In a further preferred embodiment, the oxidizing solution comprises an oxidizing agent, an aprotic organic solvent, a base and water.
- In one embodiment of the present invention the oxidizing solution comprises from about 18% to 45% oxidizing agent. In a preferred embodiment the oxidizing solution comprises from about 26% to about 40% oxidizing agent. In a more preferred embodiment the oxidizing solution comprises about 33% oxidizing agent.
- The present invention also provides compounds of formula:

- wherein:
- T 4 is hydroxyl or a protected hydroxyl;
- Bx is a heterocyclic base moiety; and
- R 2 has one of formula I or II:

- wherein:
- E is C 1-C10 alkyl, N(R4)(R5) or N═C(R4)(R5);
- R 3is OX, SX, or N(X)2;
- each R 4 and R5 is, independently, H, C1-C10 alkyl, a nitrogen protecting group, or R4 and R5, together, are a nitrogen protecting group, or R4 and R5 are joined in a ring structure that can include at least one additional heteroatom selected from a group consisting of nitrogen and oxygen;
- each X is, independently, H, C 1-C8 alkyl, C1-C8 haloalkyl, C(═NH)N(H)Z, C(═O)N(H)Z or OC(═O)N(H)Z;
- Z is H or C 1-C8 alkyl;
- L 1, L2 and L3 comprise a ring system having from about 4 to about 7 carbon atoms or having from about 3 to about 6 carbon atoms and at least 1 heteroatom, said heteroatom being selected from a group consisting of oxygen, nitrogen and sulfur, said ring system being aliphatic, unsaturated aliphatic, aromatic, or saturated or unsaturated heterocyclic;
- Y is alkyl or haloalkyl having 1 to about 10 carbon atoms, alkenyl having 2 to about 10 carbon atoms, alkynyl having 2 to about 10 carbon atoms, aryl having 6 to about 14 carbon atoms, N(R 4)(R5) OR4, halo, SR4 or CN;
- each q 1 is, independently, an integer from 2 to 10;
- each q 2 is, independently, 0 or 1;
- p is an integer from 1 to 10; and
- r is an integer from 1 to 10,
- provided that when p is 0, r is an integer greater than 1.
- In a preferred embodiment R 2 is a 2′-substituent group. In another preferred embodiment the 2′-substituent group is —O—CH2CH2—O—CH3, —O—CH2CH2—O—N(R6)(R7) or —O—CH2CH2—O—CH2CH2—N(R6)(R7), wherein each of R6 and R7 is, independently, H or C1-C10 alkyl. In a further preferred embodiment each R6 and R7 is —CH3.
- The present invention also provides chimeric oligomeric compounds having the formula:
- 5′—Nu—(L4—Nu)z1—(L5—Nu)z2—(L6—Nu)z3—3′
- wherein:
- each L 4, L5, and L6 is, independently, an internucleoside linkage selected from phosphodiester, phosphorothioate, phosphoramidate or boranophosphate, with the provision that L4 and L6 are different from L5 and that one of L4, L5 and L6 are other than phosphodiester and phosphorothioate;
- each z 1, z2, and z3 is, independently, an integer greater than 1;
- each Nu is a nucleoside having the formula:

- wherein:
- Bx is a heterocyclic base moiety;
- each R 1 is, independently, H, hydroxyl, a protected hydroxyl, a 2′-substituent group or a protected 2′-substituent group; and
- said 5′- and said -3′ ends of said chimeric oligomeric compound are, independently, hydroxyl, a protected hydroxyl, a linkage to a solid support, an activated phosphate group, an activated phosphate group, a reactive group for forming an internucleotide linkage, a nucleotide, a nucleoside, or an oligonucleotide.
- In one embodiment, each L 4 and L6 is a phosphoramidate internucleotide linkage and each L5 is a phosphorothioate internucleoside linkage.
- In another embodiment, each L 4 and L6 is a phosphoramidate internucleotide linkage and each L5 is a phosphodiester internucleoside linkage.
- In yet another embodiment, each L 4 and L6 is a boranophsphate internucleotide linkage and each L5 is a phosphodiester internucleoside linkage.
- In a further embodiment, each L 4, L5 and L6 is phosphoramidate, phosphorothioate or phosphodiester.
- It is preferred that each R 1 be, independently, —O—CH2CH2—O—CH3, —O—CH2CH2—O—N(R6)(R7) or —O—CH2CH2—O—CH2CH2—N(R6)(R7). It is also preferred that each of R6and R7 be, independently, H or C1-C10 alkyl.
- It is further preferred that each R 6 and R7 be -CH3.
- The present invention provides compounds and methods for the preparation of mixed backbone oligomeric , or chimeric, compounds having phosphodiester internucleoside linkages in addition to phosphorothioate and/or phosphoramidate internucleoside linkages. The methods also include incorporation of boranophosphate internucleoside linkages into oligomeric compounds of the invention. The methods utilize H-phosphonate intermediates that are coupled together forming contiguous regions of one or more H-phosphonate internucleoside linkages. Each contiguous region is subsequently oxidized to phosphodiester, phosphorothioate, phosphoramidate or boranophosphate internucleoside linkages prior to further elongation. Mixed backbone oligomeric compounds are prepared in this manner by oxidizing adjacent regions with different reagents. Oligomeric compounds of the invention are prepared using novel oxidation steps that oxidize a region of one or more H-phosphonate internucleoside linkages without degrading existing linkages that have been previously oxidized.
- The H-phosphonate method of oligomer synthesis has been used for the preparation of oligonucleotides and their analogs having a variety of internucleotide linkages. These internucleotide linkages have included phosphorothioate, phosphoramidate, methyl phosphate, methylphosphonate, alkylphosphonate, S-aryl phosphorothioate, acylphosphonate, phosphorofluoridate, phosphorodithioate, selenophosphate, (hydroxymethyl)phosphonate and boranophosphate.
- As used herein, the term “chimeric oligomeric compound” or “mixed backbone oligomeric compound” refers to oligomeric compounds comprising nucleoside monomer subunits and containing at least two different internucleoside linkages. The mixed backbone oligomeric compounds of the present invention may contain a plurality of nucleoside monomer subunits that are joined together by more than one type of internucleoside linkages. At least one internucleoside linkage is a phosphodiester linkage and at least one other linkage is a phosphorothioate, a phosphoramidate or a boranophosphate internucleoside linkage. Thus the term “oligomeric compound” includes oligonucleosides, oligonucleotides, their analogs, and synthetic oligonucleotides.
- In some preferred embodiments, a first monomer attached to a solid support is elongated using H-phosphonate monomers. The methods of the present invention have been used with automated DNA synthesizers to synthesize mixed backbone oligomeric compounds of desired length and sequence. Some oligomeric compounds have been synthesized having only phosphodiester and phosphorothioate internucleoside linkages. Other oligomeric compounds have been synthesized having phosphodiester, phosphorothioate and phosphoramidate internucleoside linkages. Example 14 illustrates the synthesis of a boranophosphate mixed backbone oligomeric compound. Table I illustrates selected oligomeric compounds that were synthesized using methods and intermediates of the present invention.
- In preferred embodiments, the methods of the present invention are employed for use in iterative solid phase oligonucleotide synthetic regimes. Representative solid phase techniques are those typically employed for DNA and RNA synthesis utilizing standard H-phosphonate chemistry. The H-phosphonate method of oligonucleotide synthesis is disclosed in U.S. Pat. No. 5,149,798, issued Sep. 22, 1992, and U.S. Pat. No. 5,548,076, issued Aug. 20, 1996. The entire disclosure of each is incorporated herein by reference. A preferred synthetic solid phase synthesis utilizes H-phosphonates as activated phosphate compounds. In this technique, an H-phosphonate monomer is reacted with a free hydroxyl on the growing oligomer chain to produce an intermediate phosphite compound, which is subsequently oxidized to the P v state. This technique is commonly used for the synthesis of several types of linkages including phosphodiester, phosphorothioate, and phosphoramidate linkages. Oxidation of H-phosphonate linkages to phosphodiester linkages using standard H-phosphonate chemistry results in the degradation of phosphorothioate and phosphoramidate linkages when present in a growing oligomeric compound. This degradation of adjacent linkages within an oligomeric compound has been a limitation to the use of standard H-phosphonate chemistry with mixed backbone syntheses.
- Typically, the first step in such a process is attachment of a monomer or higher order subunit containing a protected 5′-hydroxyl to a solid support, usually through a linker, using standard methods and procedures known in the art. See, e.g., Oligonucleotides And Analogues A Practical Approach, Ekstein, F. Ed., IRL Press, N.Y., 1991, hereby incorporated by reference in its entirety. The support-bound monomer or higher order first synthon is then treated to remove the 5′-protecting group. Typically, this is accomplished by treatment with acid. The solid support bound monomer is then reacted with a second monomer or higher order synthon having a reactive group for forming an internucleoside linkage. In preferred embodiments the coupling reaction is performed under anhydrous conditions in the presence of a condensing reagent.
- After the addition of each monomer or higher order synthon, a capping step is performed to cap unreacted hydroxyl groups. It is generally preferable to perform a capping step either prior to or after oxidation or sulfurization of the internucleoside linkage. Such a capping step is generally known to be beneficial by preventing shortened oligomer chains, by blocking chains that have not reacted in the coupling cycle. One representative reagent used for capping is acetic anhydride. Other suitable capping reagents and methodologies can be found in U.S. Pat. No. 4,816,571, issued Mar. 28, 1989, hereby incorporated by reference in its entirety.
- As used herein an “oxidizing solution” comprises a mixture of reagents effective to oxidize an H-phosphonate internucleoside linkage or silylated derivative thereof to a phosphodiester, phosphorothioate, phosphoramidate or boranophosphate internucleoside linkage. Such oxidation can be performed on a region of an oligomeric compound without causing degradation of other previously oxidized portions of the same oligomeric compound. Oxidation of each type of internucleoside linkage is illustrated in the examples below. Oxidation of the internucleoside linkage can be performed iteratively after each coupling step, or can be performed after a desired number or sequence of monomers have been coupled. This allows for the preparation of gapmer-type oligomeric compounds having one or more regions of one type of linkage with one or more other regions having another type of linkage. A representative example of a gapmer is an oligomeric compound having a phosphorothioate region at the 3′ and 5′ ends with a phosphodiester region located internally.
- Treatment of the growing oligomeric compound with an acid removes the 5′-hydroxyl protecting group enabling another synthetic iteration. The growing oligomeric compound is extended with subsequent oxidation at selected stages with selected oxidation agents until an oligomeric compound of desired length is produced.
- The completed oligomer is then cleaved from the solid support. The cleavage step can precede or follow deprotection of protected functional groups. The term “deblocking solution” as used herein is meant to encompass solutions routinely used for the deblocking of oligomeric compounds prepared either by solution or solid phase techniques. A common solution used for deblocking of oligomers synthesized on solid support is aqueous ammonia.
- As used herein “heterocyclic base moiety” is intended to include nucleobases. Nucleobases useful in the compounds and methods described herein include but are not limited to adenine, guanine, cytosine, uridine, and thymine, as well as other non-naturally occurring and natural nucleobases such as xanthine, hypoxanthine, 2-aminoadenine, 6-methyl and other alkyl derivatives of adenine and guanine, 2-propyl and other alkyl derivatives of adenine and guanine, 5-halo uracil and cytosine, 6-azo uracil, cytosine and thymine, 5-uracil (pseudo uracil), 4-thiouracil, 8-halo, oxa, amino, thiol, thioalkyl, hydroxyl and other 8-substituted adenines and guanines, 5-trifluoromethyl and other 5-substituted uracils and cytosines, 7-methylguanine. Further naturally- and non-naturally-occurring nucleobases include those disclosed in U.S. Pat. No. 3,687,808 (Merigan et al.); in Sanghvi, in Antisense Research and Application, Chapter 15, S. T. Crooke and B. Lebleu, Eds., CRC Press, 1993; in Englisch et al., Angewandte Chemie, International Edition, 1991, 30, 613-722 (particularly, pages 622 and 623); in the Concise Encyclopedia of Polymer Science and Engineering, J. I. Kroschwitz, Ed., John Wiley & Sons, 1990, pages 858-859; and in Cook, Anti-Cancer Drug Design, 1991, 6, 585-607, each of which is hereby incorporated by reference in its entirety. The term “nucleosidic base moiety” is further intended to include heterocyclic compounds that can serve as nucleosidic bases, including certain ‘universal bases’ that are not nucleosidic bases in the most classical sense but serve as nucleosidic bases. Especially mentioned as a universal base is 3-nitropyrrole.
- As used herein, the term “2′-substituent group” includes 2′-sugar modifications. 2∝0-Sugar modifications of the present invention include fluoro, O-alkyl, O-alkylamino, O-alkylalkoxy, protected O-alkylamino, O-alkylaminoalkyl, O-alkyl imidazole, and polyethers of the formula (O-alkyl) m, where m is 1 to about 10. Preferred among these polyethers are linear and cyclic polyethylene glycols (PEGs), and PEG-containing groups, such as crown ethers and those which are disclosed by Ouchi et al., Drug Design and Discovery 1992, 9, 93; Ravasio et al., J. Org. Chem. 1991, 56, 4329; and Delgardo et. al., Critical Reviews in Therapeutic Drug Carrier Systems 1992, 9, 249, each of which is hereby incorporated by reference in its entirety. Further sugar modifications are disclosed in Cook, Anti-Cancer Drug Design, 1991, 6, 585-607. Fluoro, O-alkyl, O-alkylamino, O-alkyl imidazole, O-alkylaminoalkyl, and alkyl amino substitutions are described in U.S. patent application Ser. No. 08/398,901, filed Mar. 6, 1995, entitled “Oligomeric Compounds having Pyrimidine Nucleotide(s) with 2′ and 5′ Substitutions,” hereby incorporated by reference in its entirety.
- Additional 2′-sugar modifications of use in the present invention include 2′-SR and 2′-NR 2 groups, where each R is, independently, hydrogen, a protecting group or substituted or unsubstituted alkyl, alkenyl, or alkynyl. 2′-SR nucleosides are disclosed in U.S. Pat. No. 5,670,633, issued Sep. 23, 1997, hereby incorporated by reference in its entirety. Incorporation of 2′-SR monomer synthons is disclosed by Hamm et al., J. Org. Chem., 1997, 62, 3415-3420. 2′-NR2 nucleosides are disclosed by Goettingen, J. Org. Chem., 1996, 61, 6273-6281; and Polushin et al., Tetrahedron Lett., 1996, 37, 3227-3230.
- Some representative 2′-sugar modifications amenable to the present invention include those having one of formula I or II:

- wherein:
- E is C 1-C10 alkyl, N(R4)(R5) or N═C(R4)(R5);
- each R 4 and R5 is, independently, H, C1-C10 alkyl, a nitrogen protecting group, or R4 and R5, together, are a nitrogen protecting group, or R4 and R5 are joined in a ring structure that can include at least one additional heteroatom selected from a group consisting of nitrogen and oxygen;
- R 3 is OX, SX, or N(X)2;
- each X is, independently, H, C 1-C8 alkyl, C1-C8 haloalkyl, C(═NH)N(H)Z, C(═O)N(H)Z or OC(═O)N(H)Z;
- Z is H or C 1-C8 alkyl;
- L 1, L2 and L3 comprise a ring system having from about 4 to about 7 carbon atoms, or having from about 3 to about 6 carbon atoms and at least one heteroatom, said heteroatom being selected from a group consisting of oxygen, nitrogen and sulfur, said ring system being aliphatic, unsaturated aliphatic, aromatic, or saturated or unsaturated heterocyclic;
- Y is alkyl or haloalkyl having 1 to about 10 carbon atoms, alkenyl having 2 to about 10 carbon atoms, alkynyl having 2 to about 10 carbon atoms, aryl having 6 to about 14 carbon atoms, N(R 4)(R5) OR4, halo, SR4 or CN;
- each q 1 is, independently, from 2 to 10;
- each q 2 is, 0 or 1;
- p is from 1 to 10; and
- r is from 1 to 10, provided that when p is 0, r is greater than 1.
- Representative 2′-sugar substituents of formula I are disclosed in U.S. patent application Ser. No. 09/130,973, filed Aug. 7, 1998, entitled “Capped 2′-Oxyethoxy Oligonucleotides,” hereby incorporated by reference in its entirety.
- Representative cyclic 2′-sugar substituents of formula II are disclosed in U.S. patent application Ser. No. 09/123,108, filed Jul. 27, 1998, entitled “RNA Targeted 2′-Modified Oligonucleotides that are Conformationally Preorganized,” hereby incorporated by reference in its entirety.
- Sugars wherein the ring oxygen atom is replaced by other atoms are also amenable to the present invention. Representative substitutions for the ring oxygen include S, CH 2, CHF, and CF2. See, e.g., Secrist et al., Abstract 21, Program & Abstracts, Tenth International Roundtable, Nucleosides, Nucleotides and their Biological Applications, Park City, Utah, Sep. 16-20, 1992, hereby incorporated by reference in its entirety.
- As used herein, the term “alkyl” includes, but is not limited to, straight chain, branched chain, and alicyclic hydrocarbon groups. Alkyl groups of the present invention may be substituted. Representative alkyl substituents are disclosed in U.S. Pat. No. 5,212,295, at column 12, lines 41-50, hereby incorporated by reference in its entirety. As used herein, the term “lower alkyl” is intended to mean alkyl having 6 or fewer carbons.
- As used herein, the term “aralkyl” denotes alkyl groups which bear aryl groups, for example, benzyl groups. The term “alkaryl” denotes aryl groups which bear alkyl groups, for example, methylphenyl groups. As used herein the term “aryl” denotes aromatic cyclic groups including but not limited to phenyl, naphthyl, anthracyl, phenanthryl, and pyrenyl.
- As used herein, the term “alkanoyl” has its accustomed meaning as a group of formula —C(═O)—alkyl. A preferred alkanoyl group is the acetyl group.
- In general, the term “hetero” denotes an atom other than carbon, preferably but not exclusively nitrogen, oxygen or sulfur. Accordingly, the term “heterocycloalkyl” denotes an alkyl ring system having one or more heteroatoms, i.e., non-carbon atoms. Preferred heterocycloalkyl groups include, for example, morpholino groups. As used herein, the term “heterocycloalkenyl” denotes a ring system having one or more double bonds and one or more heteroatoms. Preferred heterocycloalkenyl groups include, for example, pyrrolidino groups.
- In a preferred embodiment of the present invention, oligomer synthesis is performed on an automated synthesizer utilizing a solid support. Solid supports are substrates which are capable of serving as the solid phase in solid phase synthetic methodologies, such as those described in U.S. Pat. Nos. 4,415,732; 4,458,066; 4,500,707; 4,668,777; 4,973,679; 5,132,418; 4,725,677 and Re. 34,069. Linkers are known in the art as short molecules which serve to connect a solid support to functional groups, e.g., hydroxyl groups, of initial synthon molecules in solid phase synthetic techniques. Suitable linkers are disclosed in, for example, Oligonucleotides And Analogues A Practical Approach, Ecstein, F., Ed., IRL Press, N.Y., 1991, Chapter 1, pages 1-23.
- Solid supports according to the invention include those generally known in the art to be suitable for use in solid phase methodologies including, for example, controlled pore glass (CPG), oxalyl-controlled pore glass (see, e.g., Alul et al., Nucleic Acids Research 1991, 19, 1527, hereby incorporated by reference in its entirety), TentaGel Support; an aminopolyethyleneglycol derivatized support (see, e.g., Wright et al., Tetrahedron Letters 1993, 34, 3373, hereby incorporated by reference in its entirety) and Poros; a copolymer of polystyrene/divinylbenzene.
- In some embodiments of the invention, each of T 1 and T2 is, independently, a protected hydroxyl group, and T3 is a hydroxyl protecting group. A wide variety of hydroxyl protecting groups can be employed in the methods of the invention. Preferably, the protecting group is stable under either acidic or basic conditions. This enables removal of a base-stable protecting group under acidic conditions, and removal of an acid-stable protecting group under basic conditions. In general, protecting groups render chemical functionalities inert to specific reaction conditions, and can be appended to and removed from such functionalities in a molecule without substantially damaging the remainder of the molecule. Representative hydroxyl protecting groups are disclosed by Beaucage et al., Tetrahedron 1992, 48, 2223-2311, and also in Greene and Wuts, Protective Groups in Organic Synthesis, Chapter 2, 2d ed, John Wiley & Sons, N.Y., 1991, each of which is hereby incorporated by reference in its entirety. Preferred protecting groups include dimethoxytrityl (DMT), monomethoxytrityl (MMT), 9-phenylxanthen-9-yl (Pixyl) and 9-(p-methoxyphenyl)xanthen-9-yl (Mox).
- In some preferred embodiments of the invention amino groups are appended to alkyl or other groups such as, for example, 2′-alkoxy groups. Such amino groups are also commonly present in naturally-occurring and non-naturally-occurring nucleobases. It is generally preferred that these amino groups be in protected form during the synthesis of oligomeric compounds of the invention. Representative amino protecting groups suitable for these purposes are discussed in Greene and Wuts, Protective Groups in Organic Synthesis, Chapter 7, 2d ed, John Wiley & Sons, N.Y., 1991. Generally, as used herein, the term “protected,” when used in connection with a molecular moiety such as “nucleobase,” indicates that the molecular moiety contains one or more functionalities protected by protecting groups.
- There are currently many useful condensing reagents known to the art skilled that are amenable to the H-phosphonate method of oligonucleotide synthesis. Wada et al., J. Am. Chem. Soc., 1997, 119, 12710-12721. Useful condensing reagents include, but are not limited to, acid chlorides, chlorophosphates, carbonates, carbonium type compounds and phosphonium type compounds. In a preferred embodiment the condensing reagent is selected from a group consisting of pivaloyl chloride, adamantyl chloride, 2,4,6 -triisopropyl-benzenesulfonyl chloride, 2-chloro-5,5-dimethyl-2 -oxo-1,3,2-dioxaphosphinane, diphenyl phosphorochloridate, bis(2-oxo-3 -oxazolidinyl)phosphinic chloride, bis(pentafluorophenyl)carbonate, 2-(1H-benzotriazole- 1 -yl)-1,1,3,3-tetramethyluronium hexafluorophosphate, O-(azabenzotriazol- 1 -yl)- 1,1,3,3-tetramethyl uronium hexafluorophosphate, 6-(trifluoromethyl)benzotriazol-1 -yl-oxy-tris-pyrrolidino-phosphonium hexafluorophosphate, bromo-tris-pyrrolidino-phosphonium hexafluorophosphate, benzotriazole-1-yl-oxy-tris-pyrrolidino-phosphonium hexafluorophosphate and 2-(benzotriazol-1 -yloxy)- 1,3-dimethyl-2 -pyrrolidin- 1 -yl- 1,3,2-diazaphospholidinium hexafluorophosphate.
- Sulfurizing agents used during oxidation to form phosphorothioate and phosphorodithioate linkages, when they are the only linkages present, include Beaucage reagent (Iyer et. al., J. Chem. Soc., 1990, 112, 1253-1254; and Iyer et. al., J. Org. Chem., 1990, 55, 4693-4699); tetraethylthiuram disulfide (Vu and Hirschbein, Tetrahedron Lett., 1991, 32, 3005-3008); dibenzoyl tetrasulfide (Rao, et. al., Tetrahedron Lett., 1992, 33, 4839-4842); di(phenylacetyl)disulfide (Kamer, Tetrahedron Lett., 1989, 30, 6757-6760); Bis(O,O-diisopropoxy phosphinothioyl)disulfides (Stec et al, Tetrahedron Lett., 1993, 34, 5317-5320); 3-ethoxy- 1,2,4-dithiazoline-5-one (Nucleic Acids Research, 1996 24, 1602-1607; and Nucleic Acids Research, 1996 24, 3643-3644); bis(p-chlorobenzenesulfonyl)-disulfide (Nucleic Acids Research, 1995 23, 4029-4033); sulfur, and sulfur in combination with ligands such as triaryl, trialkyl, triaralkyl, or trialkaryl phosphines. Each of the foregoing references is hereby incorporated by reference in its entirety.
- Useful oxidizing agents used to form the phosphodiester or phosphorothioate linkages, when they are the only linkages present, include iodine/tetrahydrofuran/water/pyridine, hydrogen peroxide/water, tert-butyl hydro peroxide or any peracid like m-chloroperbenzoic acid. In the case of oxidation using a sulfur species, the reaction is performed under anhydrous conditions with the exclusion of air, in particular oxygen, whereas in the case of oxidation the reaction can be performed under aqueous conditions.
- The oxidation of H-phosphonate internucleoside linkages, without simultaneous degradation of existing, previously oxidized linkages, is performed using a novel oxidizing solution. The oxidizing solution comprises particular compounds/reagents dependent upon what oxidized linkages are being prepared from the H-phosphonate linkages. In general, the oxidation solution comprises an oxidizing agent, an aprotic organic solvent, and a base.
- Bases are present in the oxidizing solution to assist in the oxidation process. Selected bases have a pka ranging from about 9 to about 12, which is believed to be the optimum range for deprotonation of H-phosphonate or water without causing cleavage of the backbone of the oligomeric compound. While not wanting to be bound by theory, it is believed that bases within this pKa range, and present in the oxidation solution in a concentration of from about 0.01 to about 0.8 M, are necessary for oxidation of H-phosphonate linkages without simultaneous degradation of existing oxidized linkages. A more preferred range for the concentration of the base is from about 0.05 to about 0.2 M in the oxidation solution. Representative bases that are useful in the present invention include 4-(dimethylamino)pyridine (DMAP), triethylamine (TEA) and N,N-diisopropylethylamine (DIEA).
- Many aprotic organic solvents known to those skilled in the art are amenable to the present invention. Some representative aprotic organic solvents include pyridine, acetonitrile and dimethylformamide.
- The choice of oxidizing agent is dependent on the particular internucleoside linkage that is desired for the region of H-phosphonate internucleoside linkages that is being oxidized. Representative oxidizing agents useful for the preparation of phosphodiester internucleoside linkages include carbon tetrachloride, carbon tetrabromide, N-chlorosuccinimide and N-bromosuccinimide. In addition to an oxidizing agent, water is a necessary component of the oxidizing solution for preparing phosphodiester linkages.
- Phosphorothioate internucleoside linkages are prepared using an oxidizing agent that oxidizes phosphorus by addition of sulfur rather than oxygen. Elemental sulfur has been used to sulfrize H-phosphonate to form phosphorothioate internucleoside linkages as illustrated in the examples below. The use of elemental sulfur in the oxidizing solution affords the desired phosphorothioate internucleoside linkages while leaving unaffected any preexisting oxidized linkages in the same oligomeric compound.
- The selective oxidation of H-phosphonate internucleoside linkages to phosphodiester, phosphorothioate, phosphoramidate and boranophosphate internucleoside linkages in a mixed backbone oligomeric compound is dependent on the oxidizing solution. The oxidizing solution must not affect preexisting linkages in the oligomeric compound that is undergoing oxidation. The oxidizing solution is a mixture of an oxidizing agent, an aprotic solvent and a base. For phosphodiester linkages water is also a component of the oxidizing solution. The concentration of reagents in the oxidizing solution for a given oxidation can vary. Optimal concentration of reagents can be discerned by the art-skilled without undue experimentation.
- The composition and concentration of reagents in the oxidizing solution used to oxidize an H-phosphonate linkages in a mixed backbone oligomeric compound is variable dependent on whether the desired oxidized linkage is phosphodiester, phosphorothioate or phosphoramidate. In the case of boranophosphate internucleoside linkages, a silylation step is also involved. The two main components are the base and the oxidizing agent. For phosphodiester and phosphorothioate internucleoside linkages, an aprotic organic solvent is also used. Water is also used in the oxidizing solution when phosphodiester linkages are prepared. The oxidizing solution used for preparing phosphodiester internucleoside linkages in a mixed backbone oligomeric compound of the invention generally has from about 18% to about 45% oxidizing agent, from about 2% to about 15% water, from about 40% to about 80% aprotic organic solvent, and from about 0.01 M to about 0.8 M base dissolved in the aprotic organic solvent. A more preferred range of concentrations is from about 26% to about 40% oxidizing agent, from about 4% to about 10% water, from about 50% to about 70% aprotic organic solvent, and from about 0.04 M to about 0.4 M base dissolved in the aprotic organic solvent. Even more preferred is about 33% oxidizing agent, about 7% water, about 60% aprotic organic solvent, and from about 0.05 M to about 0.2 M base dissolved in the aprotic organic solvent.
- The oxidizing solution used for preparing phosphorothioate internucleoside linkages in a mixed backbone oligomeric compound of the invention generally has from about 1% to about 15% oxidizing agent, about 40% to about 60% solvent that will solubilize the oxidizing agent, from about 40% to about 60% aprotic organic solvent, and from about 0.01 M to about 0.8 M base dissolved in the aprotic organic solvent. A more preferred range of concentrations is from about 1% to about 10% oxidizing agent, about 40% to about 60% solvent that will solubilize the oxidizing agent, from about 40% to about 60% aprotic organic solvent, and from about 0.02 M to about 0.5 M base dissolved in the aprotic organic solvent. Even more preferred is about 3% to about 8% oxidizing agent, about 40% to about 60% solvent that will solubilize the oxidizing agent, from about 40% to about 60% aprotic organic solvent, and from about 0.04 M to about 0.1 M base dissolved in the aprotic organic solvent.
- The oxidizing solution used for preparing phosphoramidate internucleoside linkages in a mixed backbone oligomeric compound of the invention generally has a primary or secondary amine in combination with an oxidizing agent. An oxidizing agent that acts as a solvent for the primary or secondary amine is preferable. A preferred oxidizing agent is carbon tetrachloride. The oxidizing solution has from about 1% to about 15% of the primary or secondary amine, and from about 85% to about 99% oxidizing agent. A preferred concentration is about 1% to about 10% of the primary or secondary amine, and from about 90% to about 99% oxidizing agent. A more preferred concentration of primary or secondary amine is about 2% to about 5% by volume in the oxidizing solution.
- The conversion of H-phosphonate internucleoside linkages to the boranophosphate (O═P—BH 3) internucleoside linkages requires an additional silylation step prior to the oxidation step. Sergueev et al., J. Am. Chem. Soc., 1998, 120, 9417-9427. A number of silylating agents have been shown to be effective in the silylation step including chlorotrimethyl-silane, N, O-bis-(trimethylsilyl)acetamide, heptamethyldisilazane and N, O-bis-(trimethylsilyl)trifluoroacetamide. The silylation step is followed by oxidation with borane-N,N-diisopropylethylamine (BH3×DIEA, 0.1 mmol to 1 mmol), borane-2-chloro-pyridine or borane-aniline.
- In one aspect of the present invention, the compounds of the invention are used to modulate RNA or DNA encoding a protein whose formation or activity is to be modulated. The targeting portion of the composition to be employed is, thus, selected to be complementary to the preselected portion of DNA or RNA. That is, the targeting portion of the composition is hybridizable to the preselected portion of DNA.
- The methods of the present invention can be used to prepare oligomeric compounds that are used in diagnostics, therapeutics, and as research reagents and kits. They can be used in pharmaceutical compositions by including a suitable pharmaceutically acceptable diluent or carrier. They further can be used for treating organisms having a disease characterized by the undesired production of a protein. The organism should be contacted with an oligonucleotide having a sequence that is capable of specifically hybridizing with a strand of nucleic acid encoding the undesirable protein. Treatments of this type can be practiced on a variety of organisms ranging from unicellular prokaryotic and eukaryotic organisms to multicellular eukaryotic organisms. Any organism that utilizes DNA-RNA transcription or RNA-protein translation as a fundamental part of its hereditary, metabolic or cellular control is susceptible to therapeutic and/or prophylactic treatment in accordance with the invention. Seemingly diverse organisms such as bacteria, yeast, protozoa, algae, all plants and all higher animal forms, including warm-blooded animals, can be treated. Further, each cell of multicellular eukaryotes can be treated, as they include both DNA-RNA transcription and RNA-protein translation as integral parts of their cellular activity. Furthermore, many of the organelles (e.g., mitochondria and chloroplasts) of eukaryotic cells also include transcription and translation mechanisms. Thus, single cells, cellular populations or organelles can also be included within the definition of organisms that can be treated with therapeutic or diagnostic oligonucleotides.
- As will be recognized, the steps of the methods of the present invention need not be performed any particular number of times or in any particular sequence. Additional objects, advantages, and novel features of this invention will become apparent to those skilled in the art upon examination of the following examples thereof, which are intended to be illustrative and not intended to be limiting.
- Reagents
- Selected solvents and reagents were purchased from Aldrich Chemical Company and J. T. Baker Company. Phosphorus trichloride, pivaloyl chloride, N-methylmorpholine, triethylamine, diazabicyclo[5.4.0]undec-7-ene and 1,2,4-triazole were purchased from Fluka. Pivaloyl chloride was distilled prior to its use for solid phase synthesis. The 5′-O-DMT-2′-O-methoxyethyl mononucleosides were prepared as per known literature methods. Martin, Helv. Chim. Acta, 1995, 78, 486-504; Altmann et al., Chimia, 1996, 50, 168-176; Altmann et al., Biochem. Soc. Trans., 1996, 24, 630-637; and Altmann et al., Nucleosides Nucleotides, 1997, 16, 917-926. Unmodified H-phosphonate mononucleotides, and the capping reagent isopropylphosphite were purchased from Glen Research.
- Solid Phase Synthesis
- Solid phase synthesis was carried out using an Applied Biosystems (Perkin Elmer Corporation) DNA/RNA synthesizer 380B and controlled pore glass (5′-O-DMT-2′-MOE mononucleoside-3′-succinate: R. I. Chemical, Costa Mesa, Calif.) or primer support (5′-O-DMT-2′-MOE mononucleoside-3′-succinate: Pharmacia) as the solid supports. The H-phosphonate method of oligonucleotide synthesis is disclosed in U.S. Pat. No. 5,149,798, issued Sep. 22, 1992, and U.S. Pat. No. 5,548,076, issued Aug. 20, 1996. The entire disclosure of each is incorporated herein by reference.
- The synthesis cycle was based on the H-phosphonate method and included three reaction steps and several washing steps as follows:
- (i) Deblocking (DMT cleavage) with dichloroacetic acid (3%) in dichloromethane;
- Washing with acetonitrile; and
- Washing with acetonitrile/pyridine (1: 1).
- (ii) Coupling of the 5′-O-DMT-H-phosphonate mononucleotide with the deblocked nucleotide using 10 equivalents of mononucleotide and 40 equivalents of pivaloylchloride (0.2 M) in acetonitrile/pyridine (1:1); and
- Washing with acetonitrile/pyridine (1:1).
- (iii) Capping (optional) of unreacted 5′-hydroxy functions using 16 equivalents of isopropylphosphite and 64 equivalents of pivaloylchloride (0.2 M) in acetonitrile/ pyridine (1:1);
- Washing with acetonitrile/pyridine (1:1); and
- Washing with acetonitrile.
- Cleavage from the solid support and deprotection of the oligonucleotides was carried out using aqueous ammonia (25-28%) for 1.5 hours on a column followed by heating at 55° C. for 6 hours.
- Phosphodiester Internucleoside Linkages in Mixed Backbone Oligomeric Compounds
- Mixed backbone oligomeric compounds were prepared having phosphodiester linkages as well as phosphorothioate and/or phosphoramidate linkages. The oxidation of H-phosphonate internucleoside linkages to phosphodiester linkages was performed with reagents inert to existing phosphorothioate and phosphoramidate linkages. One oxidation solution that was effective to convert H-phosphonate to phosphodiester and that was inert to existing phosphorothioate and phosphoramidate internucleoside linkages was triethylamine (0.1 M) or 4-(dimethylamino)pyridine (DMAP, 0.1 M) in pyridine/carbon tetrachloride/water (9:5:1). The reaction time for this oxidation step was 60 to 120 minutes.
- The standard oxidation procedures used for H-phosphonate synthesis are based on various formulations containing iodine as the oxidation reagent. However, when preparing mixed backbone oligomeric compounds having in addition to phosphodiester one or both of phosphorothioate and phosphoramidate internucleoside linkages present, the use of iodine significantly affects product homogeneity leading to partial degradation of phosphorothioate linkages or replacement of phosphoramidate linkages with phosphodiester linkages.
- The mild oxidation procedures shown in the reaction scheme below illustrate the facile preparation of mixed backbone oligomeric compounds. The oxidation of one or more consecutive internucleoside linkages from H-phosphonate to phosphodiester is performed under reaction conditions which are inert to phosphorothioate or phosphoramidate internucleoside linkages.

- Phosphorothioate Internucleoside Linkages
- Phosphorothioate linkages were prepared via oxidation of H-phosphonate linkages using 5% elemental sulfur in carbon disulfide/pyridine/triethylamine (10:10:1). Prior to sulfurization, the solid support was thoroughly washed with acetonitrile and dried under vacuum. The reaction time was 60 to 120 minutes.
- For oligomeric compounds containing phosphorothioate and phosphoramidate linkages the oxidation is performed using 5% elemental sulfur and DMAP (0.1 M) or triethylamine (0.1 M) in carbon disulfide/pyridine(1:1). Prior to sulfurization, the solid support is thoroughly washed with acetonitrile and dried under vacuum. The reaction time is 60-120 min.
- Phosphoramidate Internucleoside Linkages
- Oligomeric compounds containing phosphoramidate linkages were prepared via oxidation of the corresponding H-phosphonate oligomer blocks on the solid support using a mixture of a primary or secondary amine in carbon tetrachloride (10%:90%, v/v). Prior to oxidation, the solid support was thoroughly washed with acetonitrile and dried under vacuum. The reaction time was 60 to 120 minutes.
- Boranophosphate Internucleoside Linkages
- Oligomeric compounds containing boranophosphate linkages are prepared by treatment of H-phosphonate internucleoside linkages with N,O-bis(trimethylsilyl)trifluoroacetamide (0.3 M) in dry tetrahydrofuran for 20 to 30 minutes, washing with tetrahydrofuran and then treating with borane-N,N-diisopropyl-ethylamine (BH 3:DIEA, 0.1 mmol:1 mmol), borane-2-chloropyridine or borane-aniline. Prior to oxidation, the solid support is thoroughly washed with acetonitrile and dried under vacuum. The reaction time is 60 to 240 minutes.
- 5′-O-DMT-2′-MOE-Thymidine-3′-H-Phosphonate
- To a stirred solution of 1,2,4-triazole (7.75 g, 0.112 mole) and N-methylmorpholine (33.4 g, 0.33 mole) in dichloromethane (330 mL) was added phosphorus trichloride (4.53 g, 0.033 mole). After stirring for 30 minutes at room temperature the mixture was cooled down to ° C. Subsequently 5′-O-DMT-2′-MOE-thymidine (4 g, 6.6 mmole) in dichloromethane (100 mL) was added dropwise over a period of 20 minutes. After stirring for another 20 min, the mixture was allowed to warm up to room temperature and poured into aqueous triethylammonium bicarbonate buffer (350 mL, pH 8.5) with stirring. The mixture was transferred to a separatory funnel and separated. The aqueous phase was extracted once with dichloromethane (300 mL) and the combined organic phases were washed with another triethylammonium bicarbonate buffer (350 mL). After drying the organic phase over sodium sulfate, the solvent was evaporated and the crude product was dried under vacuum. The crude product was purified by silica gel column chromatography using a step gradient of 0.5% triethylamine in dichloromethane to 0.5% triethylamine in dichloromethane/methanol (9:1). The combined product fractions were evaporated to a foam and evaporated twice with acetonitrile to remove excess triethylamine. Subsequently the purified product was dissolved in dichloromethane (300 mL), washed twice each with 0.2 M diazabicyclo[5.4.0]undec-7-ene bicarbonate buffer (350 mL, pH 8.7), dried over sodium sulfate and evaporated to a foam. Prior to its use for solid phase synthesis the product was dried under high vacuum to give 4.62 g (85%) of the title compound.
- 31P-NMR (CDCl3, using 2% H3PO4 as an external standard) 4.22 ppm.
- 5′-O-DMT-2′-MOE-N-6-Benzoyladenosine-3′-H-Phosphonate
- To a stirred solution of 8.03 g (0.116 mole) 1,2,4-triazole and 34.6 g (0.342 mole) N-methylmorpholine in 340 mL dichloromethane 4.7 g (0.0342 mole) of phosphorus trichloride were added. The mixture was stirred for 30 min at room temperature and cooled down to 0° C. Subsequently, 5′-O-DMT-2′-MOE-N-6-benzoyladenosine (5 g, 6.84 mmole) in dichloromethane (100 mL) was added dropwise over a period of 20 minutes. After stirring for another 20 minutes, the mixture was allowed to warm up to room temperature and poured into aqueous triethylammonium bicarbonate buffer (350 mL, pH 8.5) with stirring. The mixture was transferred to a separatory funnel and separated. The aqueous phase was extracted once with dichloromethane (300 mL) and the combined organic phases were washed with triethylammonium bicarbonate buffer (350 mL, pH 8.5). The organic phase was dried over sodium sulfate, filtered, concentrated under vacuum, and the residue dried under vacuum. The crude material was purified by silica gel column chromatography using a step gradient of 0.5% triethylamine in dichloromethane to 0.5% triethylamine in dichloromethane/ methanol (9:1). The combined product fractions were evaporated to a foam and coevaporated twice with acetonitrile to remove trace triethylamine. Subsequently, the purified product was dissolved in dichloromethane (350 mL), washed twice with diazabicyclo[5.4.0]undec-7-ene bicarbonate buffer (0.2 M, 350 mL, pH 8.7), dried over sodium sulfate, and evaporated to a foam. Prior to its use for solid phase synthesis the product was dried under high vacuum to give 5.5 g (90%) of the title compound. 31pNMR (CDCl3, external standard: 85% H3PO4): 3.77 ppm.
- 5∝0-O-DMT-2′-MOE-N-4-benzoyl-5-methyl-cytidine-3′-H-phosphonate
- To a stirred solution of 1,2,4-triazole (8.29 g, 0.12 mole) and N-methylmorpholine (35.7 g, 0.353 mole) in dichloromethane (350 mL) phosphorus trichloride (4.85g, 35.3 mmole) was added. The mixture was stirred for 30 minutes at room temperature and cooled down to 0° C. Subsequently 5′-O-DMT-2′-MOE-N-4-benzoylcytidine (5 g, 7.06 mmole) in dichloromethane (100 mL) was added dropwise over a period of 20 minutes. After stirring for another 20 minutes the mixture was allowed to warm up to room temperature and poured into aqueous triethylammonium bicarbonate buffer (350 mL, pH 8.5) with stirring. The mixture was transferred to a separatory funnel and separated. The aqueous phase was extracted once with dichloromethane (300 mL) and the combined organic phases were washed with aqueous triethylammonium bicarbonate buffer (350 mL, pH 8.5). The organic phase was dried over sodium sulfate, filtered, evaporated under vacuum and the resulting residue dried under high vacuum. The crude product was purified by silica gel column chromatography using a step gradient of 3% triethylamine in dichloromethane to 3% triethylamine in dichloromethane/methanol (9:1). The combined product fractions were evaporated to a foam and evaporated twice with acetonitrile to remove trace triethylamine. Subsequently the purified product was dissolved in dichloromethane (300 mL), washed twice with diazabicyclo[5.4.0]undec-7-ene bicarbonate buffer (300 mL, 0.2 M, pH 8.7), dried over sodium sulfate, and evaporated to a foam. Prior to its use for solid phase synthesis the product was dried under high vacuum to give 3.9 g (63%) of the title compound. 31P-NMR (CDCl3, using 85% H3PO4 as an external standard): 3.76 ppm.
- 5′-O-DMT-2′-MOE-N-2-Isobutyrylguanosine-3′-H-phosphonate
- To a stirred solution of 1,2,4-triazole (11.84 g, 0.171 mole) and N-methylmorpholine (50.98 g, 0.504 mole) in dichloromethane (350 mL), phosphorus trichloride (6.92 g, 0.050 mole) was added. The mixture was stirred for 30 minutes at room temperature and cooled down to −20° C. 5′-O-DMT-2′-MOE-isobutyrylguanosine (6 g, 8.4 mmole) in dichloromethane (100 mL) was added dropwise over a period of 20 minutes. After stirring for another 20 minutes, the mixture was allowed to warm to room temperature and poured into aqueous triethylammonium bicarbonate buffer (350 mL, pH 8.5) with stirring. The mixture was transferred to a separatory funnel and separated. The aqueous phase was extracted once with dichloromethane (300 mL) and the combined organic phases were washed with aqueous triethylammonium bicarbonate buffer (350 mL, pH 8.5). After drying the organic phase over sodium sulfate, the solvent was evaporated and the crude product was dried under vacuum. The crude product was purified by silica gel column chromatography using a step gradient of 3% triethylamine in dichloromethane to 3% triethylamine in dichloromethane/methanol (9:1). The combined product fractions were evaporated to a foam and coevaporated twice with acetonitrile to remove trace triethylamine. Subsequently the purified product was dissolved in dichloromethane (50 mL), washed twice with diazabicyclo[5.4.0]undec-7-ene bicarbonate buffer (50 mL, 0.2 M, pH 8.7), dried over sodium sulfate, and evaporated to a foam. Prior to its use for solid phase synthesis the product was dried under high vacuum to give 0.8 g (12%) of the title compound. 31P-NMR (CDCl3, external standard: 85% H3PO4): 3.68 ppm.
- Synthesis of Uniform 2′-MOE-Modified Phosphodiester Oligonucleotides
- The solid phase synthesis of a uniform phosphodiester oligonucleotide (SEQ ID No: 8, 20-mer) containing T, C, A and G nucleobases and having a 2′-methoxyethoxy (MOE) on each ribosyl sugar moiety, was performed on a 1 μmole scale using 5′-O-DMT-2′-MOE-5-methylcytidine-3′-succinyl CPG as the solid support. The synthesis cycle is described in the General Procedures above. After completion of synthesis the oligomer was oxidized using iodine (0.1 M) in pyridine/water (98:2) and washed with acetonitrile/pyridine (1:1) and acetonitrile. Deprotection and cleavage from the solid support was performed with aqueous ammonia (25-28%), as described in the General Procedures. Analysis of the crude product by electrospray mass spectrometry gave a relative molecular mass of 8023.5 (calculated mass: 8022.3).
- Synthesis of 2′-MOE-Modified Gapmer Phosphodiester Oligonucleotides
- The solid phase synthesis of a uniform phosphodiester oligonucleotide (SEQ ID No.: 2, 18-mer) containing T, C, A and G nucleobases and having a 2′-MOE group on 4 of the ribosyl sugar moieties at the 5′ end of the oligonucleotide and on 5 ribosyl sugar moieties at the 3′ end of the oligonucleotide was performed in a 1 μmole scale using 5′-O-DMT-2′-MOE-thymidine-3′-succinyl CPG as the solid support. The synthesis cycle is described in the General Procedures of Example 1. After synthesis the oligomer was oxidized using iodine (0.1 M) in pyridine/water (98:2) and washed with acetonitrile/pyridine (1:1) and acetonitrile. Deprotection and cleavage from the solid support was performed with aqueous ammonia (25-28%), as described in the General Procedures. Analysis of the crude product by electrospray mass spectrometry gave a relative molecular mass of 6435.5 (calculated mass: 6440.7).
- Synthesis of 2′-MOE-Modified Gapmer Phosphorothioate Oligonucleosides
- The solid phase synthesis of a uniform phosphorothioate oligonucleotide (SEQ ID No: 6, 19-mer) containing T, C, A and G nucleobases and having a 2′-MOE group on 5 of the ribosyl sugar moieties at the 5′ end of the oligonucleotide and on 6 ribosyl sugar moieties at the 3′ end of the oligonucleotide was performed in a 1 μmole scale using 5∝0-O-DMT-2′-MOE-thymidine-3′-succinyl CPG as the solid support. The synthesis cycle is described in the General Procedures. After completion of synthesis the oligomer was oxidized using elemental sulfur (5%) in carbon disulfide/pyridine/triethylamine (10:10:1) and washed with carbon disulfide, acetonitrile/pyridine (1:1) and acetonitrile. Deprotection and cleavage from the solid support was carried out with aqueous ammonia (25-28%), as described in the General Procedures. Analysis of the crude product by electrospray mass spectrometry gave a relative molecular mass of 6873.4 (calculated mass: 6874.8).
- Synthesis of a 2′-MOE-Modified Gapmer Oligomeric Compounds With a PO-PS-PO Mixed Backbone
- The solid phase synthesis of a gapped oligomer (SEQ ID No: 9, 20-mer) having phosphodiester internucleoside linkages on the 3′ and the 5′ ends with phosphorothioate internucleoside linkages between nucleosides 6-15 and having a 2′-MOE group on 6 of the ribosyl sugar moieties at each of the 5′ and 3′ ends of the gapped oligomer with internal ribosyl moieties being deoxyribo moieties, was performed on a 1 μmole scale using 5′-O-DMT-2′-MOE-adenosine-3′-succinyl CPG as the solid support. The synthesis cycle is described in the General Procedures of Example 1. After the first 5 couplings the oligomer was oxidized using 0.1 M DMAP in pyridine/carbontetrachloride/water (9:5: 1). Prior to synthesis of the next segment, the solid support was thoroughly washed with acetonitrile/pyridine (1:1) and acetonitrile. After incorporation of nine additional H-phosphonate monomers (to give a 15-mer) the oligomer was oxidized using elemental sulfur (5%) in carbondisulfide/ pyridine/triethylamine (10:10:1), as described in the General Procedures. Prior to synthesis of the next segment, the solid support was thoroughly washed with carbondisulfide, acetonitrile/pyridine (1:1) and acetonitrile. After completion of solid phase synthesis, the support-bound oligomer was finally treated with DMAP (0.1 M) in pyridine/carbontetrachloride/water (9:5:1) and washed with acetonitrile/pyridine (1:1) and acetonitrile. Deprotection and cleavage from the solid support was performed with aqueous ammonia (25-28%), as described in the General Procedures. Analysis of the crude product by electrospray mass spectrometry gave a relative molecular mass of 7425.9 (calculated mass: 7426.0).
- Synthesis of a 2′-MOE-Modified Gapmer Oligomeric Compounds With PN-PS-PN Mixed Backbone
- The solid phase synthesis of a gapmer oligomer (SEQ ID No: 4, 19-mer) having two dimethylaminoethyl (DMAE) phosphoramidate internucleoside linkages on the 3′ and the 5′ ends with phosphorothioate internucleoside linkages between nucleosides 4-16 and having a 2′-MOE group on 3 of the ribosyl sugar moieties at the 5′end and on 4 of the ribosyl sugar moieties at the 5′ end of the oligomeric compound with internal ribosyl moieties being deoxyribo moieties was performed in a 1 μmole scale using 5′-O-DMT-2′-MOE-thymidine-3′-succinyl CPG as the solid support. The synthesis cycle is described in the General Procedures. After the first 2 couplings the oligomer was oxidized using 2 -dimethylaminoethylamine (10%) in carbontetrachloride. Prior to synthesis of the next segment, the solid support was thoroughly washed with acetonitrile. After incorporation of fourteen additional H-phosphonate monomers (to give a 17-mer) the oligomer was oxidized using elemental sulfur (5%) in carbondisulfide/pyridine/ triethylamine (10:10:1), as described in the General Procedures. Prior to synthesis of the next segment, the solid support was thoroughly washed with carbondisulfide, acetonitrile/pyridine (1:1) and acetonitrile. After completion of solid phase synthesis, the support-bound oligomer was finally treated with 2-dimethylaminoethylamine (10%) in carbontetrachloride. Deprotection and cleavage from the solid support was performed with aqueous ammonia (25-28%), as described in the General Procedures.
- Synthesis of a 2′-MOE-Modified Gapmer Oligomeric Compounds With PN-PO-PS-PO Mixed Backbone
- The solid phase synthesis of a gapmer oligomer (SEQ ID No: 10, 20-mer) having DMAE phosphoramidate internucleoside linkages between nucleosides 1-3 on the 5′ end, phosphodiester internucleoside linkages between nucleosides 3 to 6 and 15 to 20, phosphorothioate internucleoside linkages between nucleosides 6 to 15, 2′-MOE group on 6 of the ribosyl sugar moieties at the 5′ and the 3′ end of the oligomeric compound, and with remaining ribosyl units being 2′ -deoxy ribosyls, was performed in a 1 μmole scale using 5′-O-DMT-2′-MOE-adenosine-3′-succinyl CPG as the solid support. The synthesis cycle is described in the General Procedures of Example 1. After the first 5 couplings the oligomer was oxidized using triethylamine (0.1 M) in pyridine/carbontetrachloride/water (9:5:1). Prior to synthesis of the next segment, the solid support was thoroughly washed with acetonitrile/pyridine (1:1) and acetonitrile. After introduction of nine additional H-phosphonate monomers (to give an 15-mer) the oligomer was oxidized using elemental sulfur (5%) in carbondisulfide/ pyridine/triethylamine (10:10:1), as described in the General Procedures. Prior to synthesis of the next segment, the solid support was thoroughly washed with carbondisulfide, acetonitrile/pyridine (1:1) and acetonitrile. After introduction of three additional H-phosphonate monomers (to give an 18-mer) the oligomer was oxidized using triethylamine (0.1 M) in pyridine/carbontetrachloride/water (9:5:1) and washed thoroughly with acetonitrile/pyridine (1:1) and then acetonitrile. After completion of solid phase synthesis, the support-bound oligomer was finally treated with 2 -dimethyl-aminoethylamine (10%) in carbontetrachloride. Deprotection and cleavage from the solid support was performed with aqueous ammonia (25-28%), as described in the General Procedures. Analysis of the crude product by electrospray mass spectrometry gave a relative molecular mass of 7566.3 (calculated mass: 7566.3).
- Synthesis of a 2′-MOE-Modified Gapmer Oligorieric Compounds PN-PO-PS-PO-PN Mixed Backbone
- The solid phase synthesis of a gapmer oligomer (SEQ ID No: 11, 20-mer) having DMAE phosphoramidate internucleoside linkages between nucleosides 1-3 on the 5′ end and 18-20 on the 3′ end, phosphodiester internucleoside linkages between nucleosides 3 to 6 and 15 to 18, phosphorothioate internucleoside linkages between nucleosides 6 to 15, 2′-MOE group on 6 of the ribosyl sugar moieties at the 5′ and the 3′ end of the oligonucleotide, and with remaining ribosyl units being 2′-deoxy ribosyls was performed in a 1 μmole scale using 5′-O-DMT-2′-MOE-adenosine-3′-succinyl CPG as the solid support. The synthesis cycle is described in the General Procedures of Example 1. After the first 2 couplings the oligomer was oxidized, using 2-dimethylaminoethylamine (10%) in carbon-tetrachloride. Prior to synthesis of the next segment, the solid support was thoroughly washed with acetonitrile. After introduction of three additional H-phosphonate monomers (to give a 6-mer) the oligomer was oxidized using DMAP (0.1 M) in pyridine/carbontetrachloride/water (9:5:1) and thoroughly washed with acetonitrile/pyridine (1:1) and then acetonitrile. After introduction of nine additional H-phosphonate monomers (to give a 15-mer) the oligomer was oxidized using elemental sulfur (5%) in carbondisulfide/pyridine/ triethylamine (10:10:1). Prior to synthesis of the next segment, the solid support was thoroughly washed with carbondisulfide, acetonitrile/pyridine (1:1), and acetonitrile. After introduction of three additional H-phosphonate monomers (to give an 18-mer) the oligomer was oxidized using DMAP (0.1 M) in pyridine/carbontetrachloride/water (9:5:1) and thoroughly washed with acetonitrile/pyridine (1:1) and acetonitrile. After completion of solid phase synthesis, the support-bound oligomer was treated with 2-dimethylaminoethylamine (10%) in carbontetrachloride. Deprotection and cleavage from the solid support was performed with aqueous ammonia (25-28%). Analysis of the crude product by electrospray mass spectrometry gave a relative molecular mass of 7704.6 (calculated mass: 7706.6).
- Table I below shows the oligomeric compounds prepared according to the procedures described in the examples above.
TABLE 1 SEQ ID No. Sequence 5′-3′ Mrcalc Mrfound 1 T*T*T* T*T*T* T*T*T* T*T*T* T*T*T* 7429.5 7424.8 T*T*T* T*T 2 T*T*T* T*GA CAC GGC AT*T* T*T*T* 6440.7 6435.5 3 T*NT*NT*N GSASCS ASCSGS GSCSAS CSCSAS 6886.0 n.a. T*NT*NT*N T* 4 T*NT*NT*S GSASCS ASCSGS GSCSAS CSCSAS 6777.8 n.a. T*ST*NT*N T* 5 T*ST*ST*S T*ST*ST*S T*ST*ST*S T*ST*ST*S 6625.8 6626.0 T*ST*ST*S T*ST*S 6 T*SA*SA*S T*ST*SCS GSASAS CSGSGS CST*SA*S 6874.8 6873.4 T*SA*SA*ST*S 7 T*SA*SA*S T*ST*SCS GSASAS CSGSGS CSA*SA*S 6892.9 6891.0 A*SA*SA*ST*S 8 T*C*T* G*A*G* T*A*G* C*A*G* A*G*G* 8022.3 8023.5 A*G*C* T*C* 9 A*T*G* C*A*T*S TSCSTS GSCSCS CSCSC* 7426.0 7425.9 A*A*G* G*A* 10 A*NT*NG* C*A*T*S TSCSTS GSCSCS CSCSC* 7566.3 7566.3 A*A*G* G*A* 11 A*NT*NG* C*A*T*S TSCSTS GSCSCS CSCSC* 7706.6 7704.6 A*A*G*N G*NA* - General Procedure for the Synthesis of 2′-MOE-Modified Gapmers With PN-PS-PN Mixed Backbone
- The solid phase synthesis of a 2′-MOE gapped oligomer having phosphoramidate internucleoside linkages at the 5′ and 3′ ends and internal deoxyribo moieties having phosphorothioate linkages is performed on a 1 mmole scale using a 5′-O-DMT-2′-nucleoside attached via a succinyl linking group to CPG as the solid support. The synthesis cycle is as described above in the general procedures of Example 1. After coupling a desired number of further nucleosides to the solid support bound nucleoside the resulting oligomer is oxidized using 2-dimethylaminoethylamine (10%) in carbon tetrachloride. Prior to further elongation the solid support is washed with acetonitrile. After coupling of a desired number of further nucleosides oxidation to for phosphorothioate internucleoside linkages is performed using elemental sulfur (5%) and 4-(dimethylamino)pyridine (0.1 M) in carbon disulfide/pyridine (1:1). The solid support is again washed with carbon disulfide, acetonitrile/pyridine (1:1) and acetonitrile. After addition of a desired number of further nucleosides, the support-bound oligomer is treated with 10% 2-dimethylaminoethylamine in carbon tetrachloride. Deprotection and cleavage from the solid support is performed with aqueous ammonia (25-28%) to give the desired oligomeric compound.
- General Procedure for the Synthesis of 2′-MOE-Modified Gapmers With PBH 3-PS-PBH3 Mixed Backbone
- The solid phase synthesis of a 2′-MOE gapped oligomer having boranophosphate internucleoside linkages at the 5′ and 3′ ends and internal deoxyribo moieties having phosphorothioate linkages is performed on a 1 mmole scale using a 5′-O-DMT-2′-nucleoside attached via a succinyl linking group to CPG as the solid support. The synthesis cycle is as described above in the general procedures of Example 1. After coupling a desired number of further nucleosides to the solid support bound nucleoside the resulting oligomeric compound is treated with N,O-bis(trimethylsilyl)trifluoroacetamide (0.3 M) in dry tetrahydrofuran for 30 minutes and washed with tetrahydrofuran. Subsequently, the support-bound oligomer is treated with borane-N,N-diisopropylethylamine (0.5 M) complex in dry tetrahydrofuran for 2 hours at room temperature. Prior to synthesis of the next segment, the solid support is washed with acetonitrile. After coupling of a desired number of further nucleosides, the H-phosphonate internucleoside linkages are oxidized to phosphorothioate internucleoside linkages using elemental sulfur (5%) and 4-(dimethylamino)pyridine (0.1 M) in carbon disulfide/pyridine (1:1). Prior to elongation of the oligomer the solid support is washed with carbon disulfide, acetonitrile/pyridine (1:1) and acetonitrile. After coupling of a desired number of further nucleosides the H-phosphonate internucleoside linkages are treated at room temperature with N,O-bis(trimethylsilyl)trifluoroacetamide (0.3 M) in dry tetrahydrofuran for 30 minutes and washed with tetrahydrofuran. Subsequently, the support-bound oligomer is treated with 0.5 M borane-N,N-diisopropylethylamine complex in dry tetrahydrofuran for 2 hours at room temperature. Deprotection and cleavage from the solid support is performed with aqueous ammonia (25-28%).
- ICAM-1 Expression
- Targeted oligomeric compounds were prepared and assayed using HUVEC cells to determine their ability to inhibit the expression of ICAM- 1. HUVEC cells were washed three times with Opti-MEM (Life Technologies, Inc.) that was prewarmed to 37° C. Oligomeric compounds were premixed with Lipofectin (10μg/mL, Life Technologies, Inc.) in Opti-MEM, serially diluted to the desired concentrations, and applied to washed cells. Basal and untreated (no oligomeric compounds added) control cells were also treated with Lipofectin. Cells were incubated for 4 hours at 37° C., at which time the medium was removed and replaced with standard growth medium with or without TNF-a (5 mg/mL, R&D Systems). Incubation at 37° C. was continued for different times as indicated below.
- A fluorescence-activated cell sorter was used to quantitate ICAM- 1 protein expression. The cells were removed from the plate surface by brief trypsinization with trypsin (0.25%) in PBS. Trypsin activity was quenched with a solution of bovine serum albumin (2%) and sodium azide (0.2%) in PBS (+Mg/Ca). Cells were pelleted by centrifugation (1000 rpm, Beckman GPR centrifuge), resuspended in PBS, and stained with ICAM-1 specific antibody (3 μL/105 cells), CD54-PE (Pharmingin). The antibodies were incubated with the cells for 30 minutes at 4° C. in the dark, under gentle agitation. Cells were washed by centrifugation procedures and then resuspended in FacsFlow buffer (0.3 mL, Becton Dickinson) with formaldehyde (0.5%, Polysciences).
- Expression of cell surface ICAM-1 was determined by flow cytometry using a Becton Dickinson FACScan. The percentage of ICAM-1 expression in the control was calculated as follows: [(oligomeric compound-treated ICAM-1 value)-(basal ICAM-1 value)/(non-treated ICAM- 1 value) - (basal ICAM- 1 value))]. In one study, 2′-O-(2-methoxy)ethyl-modified anti-intercellular adhesion molecule 1 (ICAM-1) oligomeric compounds were shown to selectively increase the ICAM- 1 MRNA level and inhibit formation of the ICAM-1 translation initiation complex in human umbilical vein endothelial cells (Baker et al., The Journal of Biological Chemistry, 1997, 272, 11994-12000).
- ICAM-1 expression data reveal that the uniform phosphorothioate MOE oligomer T*C*T* G*A*G* T*A*G* C*A*G* (*=2′-methoxyethoxy (MOE)) sequence synthesized by amidite chemistry and the same compound synthesized by H-phosphonate chemistry are efficacious in HUVEC cells in controlling ICAM-1 expression. The oligomers are presumably working by a direct binding RNase H independent mechanism. Both compounds synthesized from different chemistries display dose response in inhibiting ICAM-1 expression between 3 and 100 nM range.
- c-raf MRNA Expression in A549 Cells
- Gapmers designed in this study are based on the parent sequence of ATG CAT TCT GCC CCC AAG GA (SEQ ID No. 9) which is a uniform deoxy phosphorothioate. This sequence is an antisense oligomeric compound targeted to mouse c-raf. c-raf m-RNA expression assay was carried out according to a reported procedure (Monia et al., Nature Medicinek, 1996, 2, 668). Human A549 lung carcinoma cells were obtained from the American Type Tissue Collection. These were grown in Dulbecco's modified Eagle's medium containing 1 g of glucose/liter (DMEM) and FCS (10%) and routinely passaged when 90-95% confluent.
- Assay for Oligonucleoside Inhibition of c-raf Protein Synthesis
- A549 cells were plated in 6-well plates (Falcon Labware, Lincoln Park, N.J.) and 24-48 hours later (when 80-90% confluent) treated with 1 μM phorbol 12,13-dibutyrate (PDBu) for 18 hours. This procedure removes greater than 75% of immunoreactive c-raf protein from the cells. Cells were then washed three times with DMEM (3 mL) (to remove PDBu), and DMEM (1 mL) containing DOTMA/DOPE solution (20 μg/mL, LipofectinR, Bethesda Research Laboratories) was added. Oligonucleoside gapmers were then added to the required concentration (for our initial screen, 1 μM) from a 10 μM stock solution, and the two solutions were mixed by swirling of the dish. The cells were incubated at 37° C. for 4 hours, washed once with DMEM+FCS (10%) to remove the DOTMA/DOPE solution, and then additional DMEM (3 mL) +10% FCS was added and the cells were allowed to recover for another 10 hours. Total RNA is prepared 24 hours after the experiment and analyzed for c-raf and G3PDH mRNA levels after normalization to G3PDH MRNA.
- Results
- Gapped oligomeric compounds (SEQ ID NO.: 9, 10 and 11, Table 1) inhibit c-raf MRNA expression in A549 cells in a dose dependent manner between 50-400 nM range. The duration of the effect in reducing c-raf expression is much longer for P=N containing Gapmers as expected from the improved nuclease resistance.
- It is intended that each of the patents, applications, printed publications, and other published documents mentioned or referred to in this specification be herein incorporated by reference in their entirety.
- Those skilled in the art will appreciate that numerous changes and modifications may be made to the preferred embodiments of the invention and that such changes and modifications may be made without departing from the spirit of the invention. It is therefore intended that the appended claims cover all such equivalent variations as fall within the true spirit and scope of the invention.
Claims (13)
1. A chimeric oligomeric compound having the formula:
5′—Nu—(L4—Nu)z1—(L5—Nu)z2—(L6 -Nu)z3—3′
wherein:

each L4, L5, and L6 is, independently, an internucleoside linkage selected from phosphodiester, phosphorothioate, secondary phosphoramidate, tertiary phosphoramidate or boranophosphate, with the provision that L4 and L6 are different from L5 and that one of L4, L5 and L6 are other than phosphodiester and phosphorothioate;
each z1, z2, and z3 is, independently, an integer greater than 1;
each Nu is a nucleoside having the formula:
wherein:
Bx is a heterocyclic base moiety;
each R1 is, independently, H, hydroxyl, a protected hydroxyl, a 2′-substituent group or a protected 2′-substituent group; and
said 5′- and said -3′ ends of said chimeric oligomeric compound are, independently, hydroxyl, a protected hydroxyl, a linkage to a solid support, an activated phosphate group, an activated phosphite group, a reactive group for forming an internucleotide linkage, a nucleotide, a nucleoside, or an oligonucleotide
with the proviso that when one or both of said L4 and L6 is independently a phosphoramidate then at least one R1 is other than H.
2. The chimeric oligomeric compound of claim 1 wherein each L4 and L6 is a phosphoramidate internucleotide linkage and each L5 is a phosphorothioate internucleoside linkage.
3. The chimeric oligomeric compound of claim 1 wherein each L4 and L6 is a phosphoramidate internucleotide linkage and each L5 is a phosphodiester internucleoside linkage.
4. The chimeric oligomeric compound of claim 1 wherein each L4 and L6 is a boranophosphate internucleotide linkage and each L5 is a phosphodiester internucleoside linkage.
5. The chimeric oligomeric compound of claim 1 wherein each L4, L5 and L6 is a phosphoramidate, phosphorothioate or phosphodiester internucleoside linkage.
6. The chimeric oligomeric compound of claim 1 wherein each L4 and L6 is a phosphodiester internucleotide linkage and each L5 is a phosphorothioate internucleoside linkage.
7. The chimeric oligomeric compound of claim 1 wherein each L4 and L6 is a boranophosphate internucleotide linkage and each L5 is a phosphorothioate internucleoside linkage.
8. The chimeric oligomeric compound of claim 1 wherein each R1 is independently —O—CH2CH2—O—CH3, —O—CH2CH2—O—N(R6)(R7) or —O—CH2CH2 —O—CH2CH2—N(R6)(R7), wherein each of R6and R7 is, independently, H or C1-C10alkyl.
9. The chimeric oligomeric compound of claim 8 wherein each R6 and R7 is —CH3.
10. A chimeric oligomeric compound having the formula:
5′—Nu—(L4-Nu)z1—(L5—Nu)z2—(L6 —NU)z3—(L7—Nu)z4—3′
wherein:

each L4, L5, L6, and L7 is, independently, an internucleoside linkage selected from phosphodiester, phosphorothioate, secondary phosphoramidate, tertiary phosphoramidate or boranophosphate, with the provision that L4 and L6 are different from L5 and that one of L4, L5 and L6 are other than phosphodiester and phosphorothioate;
each z1, z2, z3, and z4 is, independently, an integer greater than 1;
each Nu is a nucleoside having the formula:
wherein:
Bx is a heterocyclic base moiety;
each R1 is, independently, H, hydroxyl, a protected hydroxyl, a 2′-substituent group or a protected 2′-substituent group; and
said 5′- and said -3′ ends of said chimeric oligomeric compound are, independently, hydroxyl, a protected hydroxyl, a linkage to a solid support, an activated phosphate group, an activated phosphite group, a reactive group for forming an internucleotide linkage, a nucleotide, a nucleoside, or an oligonucleotide with the proviso that when one or both of said L4 and L6 is independently a phosphoramidate then at least one R1 is other than H.
11. The chimeric oligomeric compound of claim 10 wherein L4 is a phosphoramidate internucleotide linkage, L5 and L7 are, independently, a phosphodiester internucleotide linkage and L6 is a phosphorothioate internucleotide linkage
12. A chimeric oligomeric compound having the formula:
5′—Nu—(L4—Nu)z1—(L5—Nu)z2—(L6—Nu)z3—(L7—Nu)z4(L8—Nu)z5—3′
wherein:

each L4, L5, L6, L7, and L8 is, independently, an internucleoside linkage selected from phosphodiester, phosphorothioate, secondary phosphoramidate, tertiary phosphoramidate or boranophosphate, with the provision that L4 and L6 are different from L5 and that one of L4, L5 and L6 are other than phosphodiester and phosphorothioate;
each z1, z2, z3, z4 and Z5 is, independently, an integer greater than 1;
each Nu is a nucleoside having the formula:
wherein:
Bx is a heterocyclic base moiety;
each R1 is, independently, H, hydroxyl, a protected hydroxyl, a 2′-substituent group or a protected 2′-substituent group; and
said 5′- and said -3′ ends of said chimeric oligomeric compound are, independently, hydroxyl, a protected hydroxyl, a linkage to a solid support, an activated phosphate group, an activated phosphite group, a reactive group for forming an internucleotide linkage, a nucleotide, a nucleoside, or an oligonucleotide
with the proviso that when one or both of said L4 and L6 is independently a phosphoramidate then at least one R1 is other than H.
13. The chimeric oligomeric compound of claim 12 wherein L4 is a phosphoramidate internucleotide linkage, L5 and L7 are, independently, a phosphodiester internucleotide linkage, L6 is a phosphorothioate internucleotide linkage and L8 is a phosphoramidate internucleotide linkage.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/117,267 US20030045698A1 (en) | 1999-02-12 | 2002-04-05 | Compounds, processes and intermediates for synthesis of mixed backbone oligomeric compounds |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US09/250,075 US6207819B1 (en) | 1999-02-12 | 1999-02-12 | Compounds, processes and intermediates for synthesis of mixed backbone oligomeric compounds |
| US09/726,096 US6462184B2 (en) | 1999-02-12 | 2000-11-29 | Compounds, processes and intermediates for synthesis of mixed backbone oligomeric compounds |
| US10/117,267 US20030045698A1 (en) | 1999-02-12 | 2002-04-05 | Compounds, processes and intermediates for synthesis of mixed backbone oligomeric compounds |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/726,096 Continuation US6462184B2 (en) | 1999-02-12 | 2000-11-29 | Compounds, processes and intermediates for synthesis of mixed backbone oligomeric compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20030045698A1 true US20030045698A1 (en) | 2003-03-06 |
Family
ID=22946217
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/250,075 Expired - Fee Related US6207819B1 (en) | 1999-02-12 | 1999-02-12 | Compounds, processes and intermediates for synthesis of mixed backbone oligomeric compounds |
| US09/726,096 Expired - Lifetime US6462184B2 (en) | 1999-02-12 | 2000-11-29 | Compounds, processes and intermediates for synthesis of mixed backbone oligomeric compounds |
| US10/117,267 Abandoned US20030045698A1 (en) | 1999-02-12 | 2002-04-05 | Compounds, processes and intermediates for synthesis of mixed backbone oligomeric compounds |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/250,075 Expired - Fee Related US6207819B1 (en) | 1999-02-12 | 1999-02-12 | Compounds, processes and intermediates for synthesis of mixed backbone oligomeric compounds |
| US09/726,096 Expired - Lifetime US6462184B2 (en) | 1999-02-12 | 2000-11-29 | Compounds, processes and intermediates for synthesis of mixed backbone oligomeric compounds |
Country Status (5)
| Country | Link |
|---|---|
| US (3) | US6207819B1 (en) |
| EP (1) | EP1159282A4 (en) |
| JP (1) | JP2002536453A (en) |
| AU (1) | AU2990900A (en) |
| WO (1) | WO2000047593A1 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040266707A1 (en) * | 2003-04-02 | 2004-12-30 | Devin Leake | Stabilized polynucleotides for use in RNA interference |
| US20050223427A1 (en) * | 2004-04-01 | 2005-10-06 | Dharmacon, Inc. | Modified polynucleotides for reducing off-target effects in RNA interference |
| US20060110766A1 (en) * | 2004-11-22 | 2006-05-25 | Barbara Robertson | Method of determining a cellular response to a biological agent |
| US20060115461A1 (en) * | 2004-11-22 | 2006-06-01 | Barbara Robertson | Apparatus and system having dry gene silencing compositions |
| US20060223777A1 (en) * | 2005-03-29 | 2006-10-05 | Dharmacon, Inc. | Highly functional short hairpin RNA |
| US20070269889A1 (en) * | 2004-02-06 | 2007-11-22 | Dharmacon, Inc. | Stabilized siRNAs as transfection controls and silencing reagents |
| US20080085869A1 (en) * | 2006-09-22 | 2008-04-10 | Dharmacon, Inc. | Duplex oligonucleotide complexes and methods for gene silencing by rna interference |
| US20090280567A1 (en) * | 2004-02-06 | 2009-11-12 | Dharmacon, Inc. | Stabilized sirnas as transfection controls and silencing reagents |
| US7923207B2 (en) | 2004-11-22 | 2011-04-12 | Dharmacon, Inc. | Apparatus and system having dry gene silencing pools |
| US8188060B2 (en) | 2008-02-11 | 2012-05-29 | Dharmacon, Inc. | Duplex oligonucleotides with enhanced functionality in gene regulation |
| WO2014028739A1 (en) * | 2012-08-15 | 2014-02-20 | Isis Pharmaceuticals, Inc. | Method of preparing oligomeric compounds using modified capping protocols |
Families Citing this family (48)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6639061B1 (en) * | 1999-07-07 | 2003-10-28 | Isis Pharmaceuticals, Inc. | C3′-methylene hydrogen phosphonate oligomers and related compounds |
| US20030026782A1 (en) * | 1995-02-07 | 2003-02-06 | Arthur M. Krieg | Immunomodulatory oligonucleotides |
| US6207646B1 (en) | 1994-07-15 | 2001-03-27 | University Of Iowa Research Foundation | Immunostimulatory nucleic acid molecules |
| US6693086B1 (en) * | 1998-06-25 | 2004-02-17 | National Jewish Medical And Research Center | Systemic immune activation method using nucleic acid-lipid complexes |
| US20030022854A1 (en) | 1998-06-25 | 2003-01-30 | Dow Steven W. | Vaccines using nucleic acid-lipid complexes |
| US6949520B1 (en) * | 1999-09-27 | 2005-09-27 | Coley Pharmaceutical Group, Inc. | Methods related to immunostimulatory nucleic acid-induced interferon |
| US7135565B2 (en) * | 2000-07-28 | 2006-11-14 | Agilent Technologies, Inc. | Synthesis of polynucleotides using combined oxidation/deprotection chemistry |
| KR100917101B1 (en) * | 2000-08-04 | 2009-09-15 | 도요 보세키 가부시키가이샤 | Flexible metal laminate and production method thereof |
| EP1350262B8 (en) * | 2000-12-08 | 2008-08-13 | Coley Pharmaceuticals GmbH | Cpg-like nucleic acids and methods of use thereof |
| WO2003002587A2 (en) * | 2001-06-29 | 2003-01-09 | Micrologix Biotech Inc. | Phosphoramide containing nucleic acid-based compounds and libraries as antivirals |
| AU2002339175A1 (en) * | 2001-11-21 | 2003-06-10 | Avecia Biotechnology Inc. | Coupling reagent for h-phosphonate chemistry |
| WO2003068102A1 (en) * | 2002-01-30 | 2003-08-21 | Duret Francois | Electro-optical device for the photo-polymerization of composite material |
| AR040996A1 (en) * | 2002-08-19 | 2005-04-27 | Coley Pharm Group Inc | IMMUNE STIMULATING NUCLEIC ACIDS |
| EP2241325B1 (en) | 2002-10-29 | 2012-02-08 | Coley Pharmaceutical Group, Inc. | Use of CPG oligonucleotides in the treatment of hepatitis C virus infection |
| US20050013812A1 (en) * | 2003-07-14 | 2005-01-20 | Dow Steven W. | Vaccines using pattern recognition receptor-ligand:lipid complexes |
| JP4989225B2 (en) * | 2003-09-25 | 2012-08-01 | コーリー ファーマシューティカル グループ,インコーポレイテッド | Nucleic acid lipophilic conjugate |
| SG123799A1 (en) * | 2003-10-30 | 2006-07-26 | Coley Pharm Gmbh | C-class oligonucleotide analogs with enchanced immunostimulatory potency |
| EP2484374A1 (en) | 2004-07-18 | 2012-08-08 | CSL Limited | Immuno stimulating complex and oligonucleotide formulations for inducing enhanced interferon-gamma responses |
| MY159370A (en) * | 2004-10-20 | 2016-12-30 | Coley Pharm Group Inc | Semi-soft-class immunostimulatory oligonucleotides |
| US20080009455A9 (en) * | 2005-02-24 | 2008-01-10 | Coley Pharmaceutical Group, Inc. | Immunostimulatory oligonucleotides |
| AU2006241149A1 (en) * | 2005-04-26 | 2006-11-02 | Coley Pharmaceutical Gmbh | Modified oligoribonucleotide analogs with enhanced immunostimulatory activity |
| JP2006311067A (en) * | 2005-04-27 | 2006-11-09 | Toshiba Corp | Electronic camera apparatus and history file creating method |
| US20080045473A1 (en) | 2006-02-15 | 2008-02-21 | Coley Pharmaceutical Gmbh | Compositions and methods for oligonucleotide formulations |
| KR101251707B1 (en) | 2006-09-27 | 2013-04-11 | 콜리 파마슈티칼 게엠베하 | CpG oligonucleotide analogs containing hydrophobic T analogs with enhanced immunostimulatory activity |
| EP2591787A1 (en) | 2007-08-13 | 2013-05-15 | Pfizer Inc | Combination motif immune stimulatory oligonucleotides with improved activity |
| CN101820908A (en) * | 2007-10-09 | 2010-09-01 | 科利制药公司 | The immune stimulatory oligonucleotide analogs that comprises modified sugar moieties |
| US10131904B2 (en) | 2008-02-11 | 2018-11-20 | Rxi Pharmaceuticals Corporation | Modified RNAi polynucleotides and uses thereof |
| WO2010008582A2 (en) | 2008-07-18 | 2010-01-21 | Rxi Pharmaceuticals Corporation | Phagocytic cell drug delivery system |
| WO2010033248A2 (en) | 2008-09-22 | 2010-03-25 | Rxi Pharmaceuticals Corporation | Neutral nanotransporters |
| US9074211B2 (en) | 2008-11-19 | 2015-07-07 | Rxi Pharmaceuticals Corporation | Inhibition of MAP4K4 through RNAI |
| WO2010078536A1 (en) | 2009-01-05 | 2010-07-08 | Rxi Pharmaceuticals Corporation | Inhibition of pcsk9 through rnai |
| WO2010090762A1 (en) | 2009-02-04 | 2010-08-12 | Rxi Pharmaceuticals Corporation | Rna duplexes with single stranded phosphorothioate nucleotide regions for additional functionality |
| EP4089169A1 (en) | 2009-10-12 | 2022-11-16 | Larry J. Smith | Methods and compositions for modulating gene expression using oligonucleotide based drugs administered in vivo or in vitro |
| JP6060071B2 (en) | 2010-03-24 | 2017-01-11 | アールエックスアイ ファーマシューティカルズ コーポレーション | RNA interference in skin and fibrosis applications |
| WO2011119871A1 (en) | 2010-03-24 | 2011-09-29 | Rxi Phrmaceuticals Corporation | Rna interference in ocular indications |
| EP2550000A4 (en) | 2010-03-24 | 2014-03-26 | Advirna Inc | Reduced size self-delivering rnai compounds |
| CN113151180A (en) | 2013-12-02 | 2021-07-23 | 菲奥医药公司 | Immunotherapy of cancer |
| CA2947270A1 (en) | 2014-04-28 | 2015-11-05 | Rxi Pharmaceuticals Corporation | Methods for treating cancer using nucleic acids targeting mdm2 or mycn |
| KR20230037676A (en) | 2014-09-05 | 2023-03-16 | 피오 파마슈티칼스 코프. | Methods for treating aging and skin disorders using nucleic acids targeting tyr or mmp1 |
| EP3240801B1 (en) | 2014-12-31 | 2021-01-20 | Checkmate Pharmaceuticals, Inc. | Combination tumor immunotherapy |
| KR20180026739A (en) | 2015-07-06 | 2018-03-13 | 알엑스아이 파마슈티칼스 코포레이션 | A nucleic acid molecule targeting superoxide dismutase 1 (SOD1) |
| US10808247B2 (en) | 2015-07-06 | 2020-10-20 | Phio Pharmaceuticals Corp. | Methods for treating neurological disorders using a synergistic small molecule and nucleic acids therapeutic approach |
| US11021707B2 (en) | 2015-10-19 | 2021-06-01 | Phio Pharmaceuticals Corp. | Reduced size self-delivering nucleic acid compounds targeting long non-coding RNA |
| EP3548005A4 (en) | 2016-11-29 | 2020-06-17 | Puretech Health LLC | Exosomes for delivery of therapeutic agents |
| US11597744B2 (en) | 2017-06-30 | 2023-03-07 | Sirius Therapeutics, Inc. | Chiral phosphoramidite auxiliaries and methods of their use |
| GB201714403D0 (en) * | 2017-09-07 | 2017-10-25 | Univ Oxford Innovation Ltd | Process for oligonucleotide synthesis |
| US10927140B2 (en) * | 2018-04-16 | 2021-02-23 | The University Of Toledo | Compositions and methods for reverse automated nucleic acid synthesis |
| CN113993876A (en) * | 2019-06-11 | 2022-01-28 | 豪夫迈·罗氏有限公司 | Method for preparing oligonucleotides using improved oxidation protocols |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5256775A (en) * | 1989-06-05 | 1993-10-26 | Gilead Sciences, Inc. | Exonuclease-resistant oligonucleotides |
| US5898031A (en) * | 1996-06-06 | 1999-04-27 | Isis Pharmaceuticals, Inc. | Oligoribonucleotides for cleaving RNA |
Family Cites Families (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3687808A (en) | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| US5132418A (en) | 1980-02-29 | 1992-07-21 | University Patents, Inc. | Process for preparing polynucleotides |
| US4458066A (en) | 1980-02-29 | 1984-07-03 | University Patents, Inc. | Process for preparing polynucleotides |
| US4500707A (en) | 1980-02-29 | 1985-02-19 | University Patents, Inc. | Nucleosides useful in the preparation of polynucleotides |
| US4668777A (en) | 1981-03-27 | 1987-05-26 | University Patents, Inc. | Phosphoramidite nucleoside compounds |
| US4415732A (en) | 1981-03-27 | 1983-11-15 | University Patents, Inc. | Phosphoramidite compounds and processes |
| US4973679A (en) | 1981-03-27 | 1990-11-27 | University Patents, Inc. | Process for oligonucleo tide synthesis using phosphormidite intermediates |
| USRE34069E (en) | 1983-08-18 | 1992-09-15 | Biosyntech Gmbh | Process for the preparation of oligonucleotides |
| DE3329892A1 (en) | 1983-08-18 | 1985-03-07 | Köster, Hubert, Prof. Dr., 2000 Hamburg | METHOD FOR PRODUCING OLIGONUCLEOTIDES |
| US4959463A (en) | 1985-10-15 | 1990-09-25 | Genentech, Inc. | Intermediates |
| US4816571A (en) | 1987-06-04 | 1989-03-28 | Applied Biosystems, Inc. | Chemical capping by phosphitylation during oligonucleotide synthesis |
| JP2976436B2 (en) * | 1988-04-27 | 1999-11-10 | 味の素株式会社 | Novel oligoribonucleotide derivatives and use as antiviral agents |
| US5149798A (en) | 1989-04-06 | 1992-09-22 | Worcester Foundation For Experimental Biology | Process for synthesizing oligonucleotides and their analogs adaptable to large scale syntheses |
| WO1990015065A1 (en) * | 1989-06-05 | 1990-12-13 | Gilead Sciences, Inc. | Exonuclease-resistant oligonucleotides and methods for preparing the same |
| US5670633A (en) | 1990-01-11 | 1997-09-23 | Isis Pharmaceuticals, Inc. | Sugar modified oligonucleotides that detect and modulate gene expression |
| US5212295A (en) | 1990-01-11 | 1993-05-18 | Isis Pharmaceuticals | Monomers for preparation of oligonucleotides having chiral phosphorus linkages |
| US5541307A (en) * | 1990-07-27 | 1996-07-30 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs and solid phase synthesis thereof |
| WO1994015946A1 (en) * | 1993-01-08 | 1994-07-21 | Hybridon, Inc. | Synthesis of dimmer blocks and their use in assembling oligonucleotides |
| US6060456A (en) * | 1993-11-16 | 2000-05-09 | Genta Incorporated | Chimeric oligonucleoside compounds |
| US6166197A (en) * | 1995-03-06 | 2000-12-26 | Isis Pharmaceuticals, Inc. | Oligomeric compounds having pyrimidine nucleotide (S) with 2'and 5 substitutions |
| US6140482A (en) * | 1995-06-01 | 2000-10-31 | Hybridon, Inc. | Primary phosphoramidate internucleoside linkages and oligonucleotides containing the same |
| US5955600A (en) * | 1996-12-27 | 1999-09-21 | Isis Pharmaceuticals, Inc. | Method for the synthesis of nucleotide or oligonucleotide phosphoramidites |
| US6172209B1 (en) * | 1997-02-14 | 2001-01-09 | Isis Pharmaceuticals Inc. | Aminooxy-modified oligonucleotides and methods for making same |
| US6127533A (en) * | 1997-02-14 | 2000-10-03 | Isis Pharmaceuticals, Inc. | 2'-O-aminooxy-modified oligonucleotides |
-
1999
- 1999-02-12 US US09/250,075 patent/US6207819B1/en not_active Expired - Fee Related
-
2000
- 2000-02-11 AU AU29909/00A patent/AU2990900A/en not_active Abandoned
- 2000-02-11 JP JP2000598512A patent/JP2002536453A/en active Pending
- 2000-02-11 EP EP00908597A patent/EP1159282A4/en not_active Withdrawn
- 2000-02-11 WO PCT/US2000/003543 patent/WO2000047593A1/en active Application Filing
- 2000-11-29 US US09/726,096 patent/US6462184B2/en not_active Expired - Lifetime
-
2002
- 2002-04-05 US US10/117,267 patent/US20030045698A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5256775A (en) * | 1989-06-05 | 1993-10-26 | Gilead Sciences, Inc. | Exonuclease-resistant oligonucleotides |
| US5898031A (en) * | 1996-06-06 | 1999-04-27 | Isis Pharmaceuticals, Inc. | Oligoribonucleotides for cleaving RNA |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20080242851A1 (en) * | 2003-04-02 | 2008-10-02 | Dharmacon, Inc. | Modified polynucleotides for reducing off-target effects in rna interference |
| US20040266707A1 (en) * | 2003-04-02 | 2004-12-30 | Devin Leake | Stabilized polynucleotides for use in RNA interference |
| US20070173476A1 (en) * | 2003-04-02 | 2007-07-26 | Dharmacon Inc. | Modified polynucleotides for use in rna interference |
| US7834171B2 (en) | 2003-04-02 | 2010-11-16 | Dharmacon, Inc. | Modified polynucleotides for reducing off-target effects in RNA interference |
| US20070269889A1 (en) * | 2004-02-06 | 2007-11-22 | Dharmacon, Inc. | Stabilized siRNAs as transfection controls and silencing reagents |
| US20090280567A1 (en) * | 2004-02-06 | 2009-11-12 | Dharmacon, Inc. | Stabilized sirnas as transfection controls and silencing reagents |
| US20050223427A1 (en) * | 2004-04-01 | 2005-10-06 | Dharmacon, Inc. | Modified polynucleotides for reducing off-target effects in RNA interference |
| US20060110766A1 (en) * | 2004-11-22 | 2006-05-25 | Barbara Robertson | Method of determining a cellular response to a biological agent |
| US20060115461A1 (en) * | 2004-11-22 | 2006-06-01 | Barbara Robertson | Apparatus and system having dry gene silencing compositions |
| US20060166234A1 (en) * | 2004-11-22 | 2006-07-27 | Barbara Robertson | Apparatus and system having dry control gene silencing compositions |
| US7935811B2 (en) | 2004-11-22 | 2011-05-03 | Dharmacon, Inc. | Apparatus and system having dry gene silencing compositions |
| US7923206B2 (en) | 2004-11-22 | 2011-04-12 | Dharmacon, Inc. | Method of determining a cellular response to a biological agent |
| US7923207B2 (en) | 2004-11-22 | 2011-04-12 | Dharmacon, Inc. | Apparatus and system having dry gene silencing pools |
| US20060223777A1 (en) * | 2005-03-29 | 2006-10-05 | Dharmacon, Inc. | Highly functional short hairpin RNA |
| US20080085869A1 (en) * | 2006-09-22 | 2008-04-10 | Dharmacon, Inc. | Duplex oligonucleotide complexes and methods for gene silencing by rna interference |
| US8252755B2 (en) | 2006-09-22 | 2012-08-28 | Dharmacon, Inc. | Duplex oligonucleotide complexes and methods for gene silencing by RNA interference |
| US8188060B2 (en) | 2008-02-11 | 2012-05-29 | Dharmacon, Inc. | Duplex oligonucleotides with enhanced functionality in gene regulation |
| WO2014028739A1 (en) * | 2012-08-15 | 2014-02-20 | Isis Pharmaceuticals, Inc. | Method of preparing oligomeric compounds using modified capping protocols |
| CN104736551A (en) * | 2012-08-15 | 2015-06-24 | Isis制药公司 | Method of preparing oligomeric compounds using modified capping protocols |
| US9403865B2 (en) | 2012-08-15 | 2016-08-02 | Ionis Pharmaceuticals, Inc. | Method of preparing oligomeric compounds using modified capping protocols |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2990900A (en) | 2000-08-29 |
| US6462184B2 (en) | 2002-10-08 |
| EP1159282A1 (en) | 2001-12-05 |
| US6207819B1 (en) | 2001-03-27 |
| JP2002536453A (en) | 2002-10-29 |
| EP1159282A4 (en) | 2002-08-28 |
| US20010016652A1 (en) | 2001-08-23 |
| WO2000047593A1 (en) | 2000-08-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6462184B2 (en) | Compounds, processes and intermediates for synthesis of mixed backbone oligomeric compounds | |
| US6169177B1 (en) | Processes for the synthesis of oligomeric compounds | |
| US6531590B1 (en) | Processes for the synthesis of oligonucleotide compounds | |
| US6020475A (en) | Process for the synthesis of oligomeric compounds | |
| US6326478B1 (en) | Process for the synthesis of oligomeric compounds | |
| US5705621A (en) | Oligomeric phosphite, phosphodiester, Phosphorothioate and phosphorodithioate compounds and intermediates for preparing same | |
| US6649750B1 (en) | Process for the preparation of oligonucleotide compounds | |
| US20060003952A1 (en) | Oligomeric compounds having modified phosphate groups | |
| AU721750B2 (en) | Method of synthesizing phosphorothioate oligonucleotides | |
| US6610842B1 (en) | Processes for the synthesis of oligomers using phosphoramidite compositions | |
| US6919437B1 (en) | Synthetic methods and intermediates for triester oligonucleotides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: ISIS PHARMACEUTICALS, INC., CALIFORNIA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:MANOHARAN, MUTHIAH;MAIER, MARTIN A.;REEL/FRAME:012781/0723;SIGNING DATES FROM 20001118 TO 20001128 |
|
| STCB | Information on status: application discontinuation |
Free format text: ABANDONED -- FAILURE TO RESPOND TO AN OFFICE ACTION |