US20100274052A1 - Preparation of radiolabelled haloaromatics via polymer-bound intermediates - Google Patents
Preparation of radiolabelled haloaromatics via polymer-bound intermediates Download PDFInfo
- Publication number
- US20100274052A1 US20100274052A1 US12/702,009 US70200910A US2010274052A1 US 20100274052 A1 US20100274052 A1 US 20100274052A1 US 70200910 A US70200910 A US 70200910A US 2010274052 A1 US2010274052 A1 US 2010274052A1
- Authority
- US
- United States
- Prior art keywords
- polymer
- mibg
- group
- preparation
- radiolabelled
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- 0 [2*]C1=C([2*])C([2*])=C(C)C([2*])=C1[2*] Chemical compound [2*]C1=C([2*])C([2*])=C(C)C([2*])=C1[2*] 0.000 description 9
- FFFKWVWHOZJITG-UHFFFAOYSA-M CCCC(CC)C1=CC=CC=C1.CCCC[Sn](Cl)(CCC)CCCC Chemical compound CCCC(CC)C1=CC=CC=C1.CCCC[Sn](Cl)(CCC)CCCC FFFKWVWHOZJITG-UHFFFAOYSA-M 0.000 description 3
- KUEHTDCKELPWMZ-UHFFFAOYSA-N CCCCC(CCCC)C1=CC(CNC(=N)N)=CC=C1 Chemical compound CCCCC(CCCC)C1=CC(CNC(=N)N)=CC=C1 KUEHTDCKELPWMZ-UHFFFAOYSA-N 0.000 description 3
- OQOKKBRALJLSHJ-UHFFFAOYSA-O CCCCC(CCCC)C1=CC(C[NH3+])=CC=C1 Chemical compound CCCCC(CCCC)C1=CC(C[NH3+])=CC=C1 OQOKKBRALJLSHJ-UHFFFAOYSA-O 0.000 description 3
- DLLSDZVFKPSBOG-UHFFFAOYSA-N C[Si]1(C)CC[Si](C)(C)N1CC1=CC=CC(Br)=C1 Chemical compound C[Si]1(C)CC[Si](C)(C)N1CC1=CC=CC(Br)=C1 DLLSDZVFKPSBOG-UHFFFAOYSA-N 0.000 description 2
- PDWUPXJEEYOOTR-UHFFFAOYSA-N [H]N(CC1=CC=CC(I)=C1)C(=N)N Chemical compound [H]N(CC1=CC=CC(I)=C1)C(=N)N PDWUPXJEEYOOTR-UHFFFAOYSA-N 0.000 description 2
- XCFZGSVZONFZQP-UHFFFAOYSA-O C#N.CCCC(CC)C1=CC=CC=C1.CCCC(CC)C1=CC=CC=C1.CCCC[Sn](CCC)(CCCC)C1=CC=CC(C[NH3+])=C1.N.[H]N(CC1=CC([Sn](CCC)(CCCC)CCCC)=CC=C1)C(=N)N Chemical compound C#N.CCCC(CC)C1=CC=CC=C1.CCCC(CC)C1=CC=CC=C1.CCCC[Sn](CCC)(CCCC)C1=CC=CC(C[NH3+])=C1.N.[H]N(CC1=CC([Sn](CCC)(CCCC)CCCC)=CC=C1)C(=N)N XCFZGSVZONFZQP-UHFFFAOYSA-O 0.000 description 1
- CAAOVGPITXEFMJ-UHFFFAOYSA-M C=CC.C=CC1=CC=CC=C1.CCCC(CC)C1=CC=CC=C1.CCCC[Sn](Cl)(CCC)CCCC Chemical compound C=CC.C=CC1=CC=CC=C1.CCCC(CC)C1=CC=CC=C1.CCCC[Sn](Cl)(CCC)CCCC CAAOVGPITXEFMJ-UHFFFAOYSA-M 0.000 description 1
- URCWVAGLEYNTHN-UHFFFAOYSA-O CC1=CC(CN)=CC=C1.CCCC(CC)C1=CC=CC=C1.CCCC[Sn](CCC)(CCCC)C1=CC=CC(C[NH3+])=C1.C[Si](C)(Cl)CC[Si](C)(C)Cl.C[Si]1(C)CC[Si](C)(C)N1CC1=CC=CC(Br)=C1 Chemical compound CC1=CC(CN)=CC=C1.CCCC(CC)C1=CC=CC=C1.CCCC[Sn](CCC)(CCCC)C1=CC=CC(C[NH3+])=C1.C[Si](C)(Cl)CC[Si](C)(C)Cl.C[Si]1(C)CC[Si](C)(C)N1CC1=CC=CC(Br)=C1 URCWVAGLEYNTHN-UHFFFAOYSA-O 0.000 description 1
- OSZHESZVXRFOFL-UHFFFAOYSA-O CCCC(CC)C1=CC=CC=C1.CCCC[Sn](CCC)(CCCC)C1=CC=CC(C[NH3+])=C1.NCC1=CC=CC(I)=C1 Chemical compound CCCC(CC)C1=CC=CC=C1.CCCC[Sn](CCC)(CCCC)C1=CC=CC(C[NH3+])=C1.NCC1=CC=CC(I)=C1 OSZHESZVXRFOFL-UHFFFAOYSA-O 0.000 description 1
- FMYAKXODOXKLEV-UHFFFAOYSA-N CCCC(CC)C1=CC=CC=C1.[Cl-].[H]N(CC1=CC([Sn](CCC)(CCCC)CCCC)=CC=C1)C(=N)N.[H]N(CC1=CC=CC(I)=C1)C(=N)N Chemical compound CCCC(CC)C1=CC=CC=C1.[Cl-].[H]N(CC1=CC([Sn](CCC)(CCCC)CCCC)=CC=C1)C(=N)N.[H]N(CC1=CC=CC(I)=C1)C(=N)N FMYAKXODOXKLEV-UHFFFAOYSA-N 0.000 description 1
- FMYAKXODOXKLEV-MNRDFGGWSA-N CCCC(CC)C1=CC=CC=C1.[H]N(CC1=CC([Sn](CCC)(CCCC)CCCC)=CC=C1)C(=N)N.[H]N(CC1=CC=CC([131I])=C1)C(=N)N Chemical compound CCCC(CC)C1=CC=CC=C1.[H]N(CC1=CC([Sn](CCC)(CCCC)CCCC)=CC=C1)C(=N)N.[H]N(CC1=CC=CC([131I])=C1)C(=N)N FMYAKXODOXKLEV-MNRDFGGWSA-N 0.000 description 1
- NFBMOFGOQHVYKF-UHFFFAOYSA-N [H]N(CC1=CC=CC([InH2])=C1)C(=N)N Chemical compound [H]N(CC1=CC=CC([InH2])=C1)C(=N)N NFBMOFGOQHVYKF-UHFFFAOYSA-N 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C277/00—Preparation of guanidine or its derivatives, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups
- C07C277/08—Preparation of guanidine or its derivatives, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups of substituted guanidines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/0404—Lipids, e.g. triglycerides; Polycationic carriers
- A61K51/0406—Amines, polyamines, e.g. spermine, spermidine, amino acids, (bis)guanidines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/05—Isotopically modified compounds, e.g. labelled
Definitions
- This invention relates to the preparation of a radiopharmaceutical.
- this invention relates to a process for the preparation a radiolabelled haloaromatic, as well as intermediate precursor compounds used in the process.
- MIBG meta-iodobenzylguanidine
- iodine atom when radiolabelled with the iodine atom, is used in nuclear medicine as either an imaging agent for diagnosis, or as a therapeutic agent for neural crest tumors such as neuroblastoma.
- [ 123 I]MIBG provides diagnostic cardiac images as well as images of tumors.
- the longer-lived [ 131 I]MIBG is used at much higher radiation and chemical doses for the treatment of tumors.
- no-carrier-added [ 131 I]MIBG routes to no-carrier-added [ 131 I]MIBG have been developed which could reduce the chemical dose of MIBG by about a factor of 100.
- Precursors to no-carrier-added [ 131 I]MIBG such as 3-tributylstannylbenzylamine 2 , 3-trimethylsilylbenzylguanidine 2 , and 3-trimethylstannylguanidinium 3 , have not found widespread application. These compounds have a short shelf life, and must be stored in a freezer shielded from light.
- U.S. Pat. No. 5,565,185 discloses a no-carrier-added process of radiolabelling MIBG by halodestannylation.
- the process is disadvantageous in that a number of impurities remain in solution with the radiolabelled MIBG.
- toxic tin by-products remain in solution and must be separated before the radiolabelled MIBG is ready for use.
- R 2 is selected from an alkyl group, an aryl group, a hydrogen atom, a halogen atom, a substituted oxygen atom, a substituted nitrogen atom, a substituted sulfur atom, a carbonyl group, a cyano group, an amino group, and a guanidino group.
- *X is selected from any suitable radiohalide, and is preferably selected from 123 I, 125 I, and 131 I.
- the process comprises reacting an insoluble polymer compound (II) with a radiohalogen ion in the presence of an oxidant and a preferably organic solvent, where compound (II) has the formula:
- R 1 is an alkyl group, and is preferably a butyl group.
- R 2 is as described above.
- an intermediate insoluble polymer compound comprises the repeating unit formula:
- R 1 is selected from an alkyl group
- R 2 is selected from an alkyl group, an aryl group, a hydrogen atom, a halogen atom, a substituted oxygen atom, a substituted nitrogen atom, a substituted sulfur atom, a carbonyl group, a cyano group, an amino group, and a guanidino group.
- the process comprises reacting the compound of formula:
- n is selected from 123, 125, and 131; the process comprising contacting a compound having the formula:
- FIG. 1A shows the 119 Sn MAS NMR spectrum for Polymer 1
- FIG. 1B shows the 119 Sn MAS NMR spectrum for Polymer 1 after hydrolysis
- FIG. 1C shows the 119 Sn MAS NMR spectrum for Polymer 2
- FIG. 1D shows the 119 Sn MAS NMR spectra for Polymer 3 recovered after iodination
- FIG. 2 shows the HPLC analysis of the iodination products of reaction of Polymer 2 with cyanamide at selected reaction times.
- the process provides for the radiohalogenation of a haloaromatic compound using a polymer-supported pharmaceutical, as follows:
- R 1 is an alkyl group, and is preferably a butyl group
- R 2 is selected from an alkyl group, an aryl group, a hydrogen atom, a halogen atom, a substituted oxygen atom, a substituted nitrogen atom, a substituted sulfur atom, a carbonyl group, a cyano group, an amino group, a guanidino group
- Nu is a nucleophile provided by the solvent or oxidant
- *X is selected from any suitable radiohalide, such as, for example, 131 I or 123 I.
- a process is provided to synthesize no-carrier-added [ 131 I]MIBG or [ 123 I]MIBG using polymer-supported radiopharmaceutical precursors 4 .
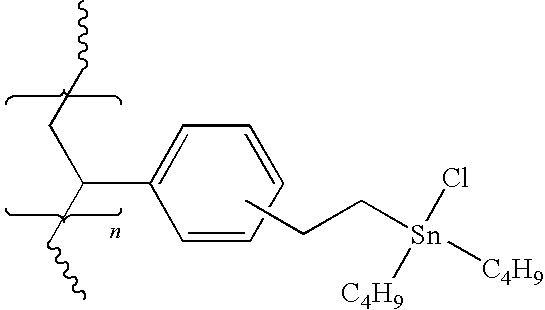
- An insoluble polymer is prepared in which the pharmaceutical to be radiohalogenated is bound to the polymer through a tin-aryl bond as illustrated above. Treatment with the radiohalide and an appropriate oxidant results in the release of the radiohalogenated pharmaceutical while unreacted polymer and polymer side-products remain insoluble and easily removed by filtration.
- the polymer above is derived by reaction of the appropriate organolithium with a polymeric chlorostannane.
- Polymer 1 described in detail below, is used as an intermediate compound to derive the polymers to be used in the radiohalogenation of a haloaromatic compound.
- Polymer 2 is one such reagent derived from Polymer 1
- Polymer 3 is the preferred polymer-supported 3-benzylguanidinium reagent for the preparation of no-carrier-added [ 131 I]MIBG or [ 123 I]MIBG. Polymers 2 and 3 are also described in detail below.
- Polymer 1 has the Following Structural Formula
- Polymer 1 The characteristics of Polymer 1 are preferably as follows:
- Polymer 1 Samples of Polymer 1 were prepared as indicated above. Polymer 1 was characterized both chemically and spectroscopically. The availability of Sn—Cl bonds was assessed by treatment of Polymer 1 with KOH in ethanol/THF at room temperature. Polymer 1 (201 mg) was soaked in 7 mL of absolute ethanol, 3 mL (3 mmol) of 1 M potassium hydroxide, and 1 mL of tetrahydrofuran for 24 h at room temperature. The polymer was filtered and washed with 70% ethanol (5 ⁇ 5 mL). The ethanol and THF in the filtrate were removed on rotary evaporator and the residual liquid was transferred into a 50 mL volumetric flask and topped up to the mark with water. Then 20 mL of this solution was diluted to 50 mL Erlenmeyer by addition of 1 M nitric acid. This solution was then titrated for Cl ⁇ following the Mohr procedure. 6
- the amount of chloride ion released was found to be 1.7 ⁇ 0.1 mmol/g of polymer. If the polymer was composed exclusively of the units indicated in the structure shown for Polymer 1, the maximum amount of chloride released would be 2.5 mmol/g of polymer. However, significant amounts of divinylbenzene had been used when forming the polymer and the yield of material was 32%.
- Polymer 1 was also investigated by MAS NMR spectroscopy on samples that had been preswollen with chloroform which had the effect of sharpening the signals.
- the 13 C NMR spectrum was consistent with the anticipated structure showing signals for all of the carbons.
- the signals for the backbone CH and CH 2 and phenyl carbons were quite broad and indistinct.
- the CH 2 —CH 2 carbons and the carbons on the butyl chains were readily assigned by analogy with non-polymeric species.
- the 119 Sn NMR spectrum ( FIG. 1A ) showed but one peak at 148 ppm consistent with species of the general structure R 3 Sn—Cl.
- Polymer 1 was preferably converted in two steps first into Polymer 2 which bears the 3-benzylammonium moiety and then into Polymer 3 functionalised with a 3-benzylguanidinium species.
- Compound 4 preferably had the following characteristics:
- Polymer 2 The characteristics of Polymer 2 were preferably as follows:
- the synthesis of Polymer 3 involved treatment of Polymer 2 with a large excess of cyanamide to convert the ammonium group into a guanidinium group, as illustrated below.
- Polymer 3 The characteristics of Polymer 3 are preferably as follows:
- 3-iodobenzylamine hydrochloride (2.0 g, 7.4 mmol) was combined with 0.62 g (7.4 mmol) of dicyandiamide and an argon atmosphere was introduced. The mixture was melted at 154° C. for 30 min and water (20 mL) was added to dissolve the brownish solid. HPLC analysis showed 8 mol % of 3-iodobenzylamine hydrochloride, 17 mol % of MIBG and 75 mol % of 3-iodobenzylbiguanidinium nitrate (excluding dicyandiamide at 9.6 min) with a retention times of 6.1 min, 11.2 min and 12.7 min respectively.
- the characteristics of the 3-iodobenzylbiguanidinium nitrate were preferably as follows:
- the filtrate was analyzed by HPLC.
- the UV detector trace showed a large peak at the solvent front and several smaller peaks well before MIBG peak.
- the corresponding radioactivity detector trace showed two peaks at 1.9 and 15.6 min which were confirmed by coinjection to be 131 I-(2.4%) and [ 131 I]MIBG (97.6%) respectively.
- Table 1 shows the effect on the radiochemical yield of [ 131 I]MIBG of increasing the concentration of H 2 O 2 and acetic acid at various reaction times. As the first column indicates, the exclusion of acetic acid from the reaction mixture led to low yields and apparently slow reactions. The third column indicates a general increase in yield with reaction time. At two hour reaction times, increasing concentrations of H 2 O 2 and acetic acid led to increased yields which approached near quantitative conversion. It was these conditions that were chosen for preparation of [ 131 I]MIBG, as illustrated.
- Purification of the product from the radiolabelling reaction involved two steps. The first was simple filtration through a syringe tip filter. An HPLC analysis of this product using a UV detector showed a large peak at the solvent front and several minor peaks. The radioactivity detector showed two peaks corresponding to 131 I— at about 2% and the other to [ 131 I]MIBG at 98%.
- the second step in the purification involved selective absorption and desorption of the [ 131 I]MIBG onto a C18 Sep-PakTM cartridge. When the primarily aqueous solution from the filtration step was passed through the C18 Sep-PakTM cartridge, essentially all of the radioactivity was absorbed onto the cartridge with the small amount of iodide passing through. When the cartridge was washed with ethanol/water, 82% of the radioactivity was released. A further 10% was released when methanol was used as eluant.
- [ 123 I]MIBG was produced using IodobeadsTM as the oxidant, which resulted in a radiochemical yield of about 55%. Since later experiments with Na 131 I showed that about 45% of the radioactivity remained absorbed to the insoluble polymeric materials, this experiment demonstrates that Polymer 3 is an effective precursor to [ 123 I]MIBG. Similar results can also be expected with [ 125 I]MIBG.
- the process produces no-carrier-added [ 131 I]MIBG in ⁇ 90% radiochemical yield and high chemical purity. Isolation and purification are simple, involving just filtration and absorption and desorption onto a C18 Sep-PakTMcartridge. No-carrier-added material should avoid the potential pharmacological side effects accompanying the current method of production.
- the process and precursor compounds, according to the present invention is useful in the field of nuclear medicine for the production of radiopharmaceutical compounds.
Abstract
According to a first aspect of the invention, a process is disclosed for the preparation of radiolabelled haloaromatic compounds. According to a second aspect of the invention, intermediate precursor insoluble polymer compounds used in the preparation of the radiolabelled haloaromatics are disclosed, as well as processes for the preparation of the intermediate precursor insoluble polymer compounds.
Description
- This invention relates to the preparation of a radiopharmaceutical. In particular, this invention relates to a process for the preparation a radiolabelled haloaromatic, as well as intermediate precursor compounds used in the process.
- A number of radiolabelled haloaromatic compounds have found application in nuclear medicine. For example, meta-iodobenzylguanidine (“MIBG”), when radiolabelled with the iodine atom, is used in nuclear medicine as either an imaging agent for diagnosis, or as a therapeutic agent for neural crest tumors such as neuroblastoma. When labelled with the shorter-lived iodine-123, [123I]MIBG provides diagnostic cardiac images as well as images of tumors. The longer-lived [131I]MIBG is used at much higher radiation and chemical doses for the treatment of tumors.
- By far the most common method of producing either [123I]MIBG or [131I]MIBG is by a Cu+ catalyzed isotopic exchange process which commences with 1-2 mg of MIBG and the desired amount of radioiodide. Because isotopic exchange is an equilibrium process, the product obtained by this process necessarily contains a significant amount of carrier MIBG. Considerable effort1 has been placed towards developing a convenient procedure that proceeds in near quantitative radiochemical yields. However, this method has the drawback of producing [131I]MIBG of low specific activity resulting in chemical doses of 1-5 mg when therapeutic samples are prepared. Doses of this magnitude carry potential hypertensive side effects.
- Accordingly, routes to no-carrier-added [131I]MIBG have been developed which could reduce the chemical dose of MIBG by about a factor of 100. Precursors to no-carrier-added [131I]MIBG, such as 3-tributylstannylbenzylamine2, 3-trimethylsilylbenzylguanidine2, and 3-trimethylstannylguanidinium3, have not found widespread application. These compounds have a short shelf life, and must be stored in a freezer shielded from light.
- U.S. Pat. No. 5,565,185 discloses a no-carrier-added process of radiolabelling MIBG by halodestannylation. However, the process is disadvantageous in that a number of impurities remain in solution with the radiolabelled MIBG. In particular, toxic tin by-products remain in solution and must be separated before the radiolabelled MIBG is ready for use.
- Accordingly, there is a need for a process for no-carrier-added synthesis of radiolabelled haloaromatic compounds, which can be easily and practically separated from possibly toxic impurities.
- It is an object of the invention to provide a process for no-carrier-added synthesis of radiolabelled haloaromatic compounds, where the impurities can be removed by simple filtration, thereby making the process suitable for “kit” formulation.
- It is also an object of the invention to provide intermediate insoluble polymer precursors which have a long shelf life and can be stored at room temperature without special conditions, and to which the unlabelled compounds and side products are bound, thereby facilitating removal of these undesirable impurities by filtration.
- According to a first aspect of the present invention, a process of preparing a radiolabelled haloaromatic compound (I) of the formula:
-

- is provided, wherein,
R2 is selected from an alkyl group, an aryl group, a hydrogen atom, a halogen atom, a substituted oxygen atom, a substituted nitrogen atom, a substituted sulfur atom, a carbonyl group, a cyano group, an amino group, and a guanidino group. *X is selected from any suitable radiohalide, and is preferably selected from 123I, 125I, and 131I. The process comprises reacting an insoluble polymer compound (II) with a radiohalogen ion in the presence of an oxidant and a preferably organic solvent, where compound (II) has the formula: -

- wherein,
R1 is an alkyl group, and is preferably a butyl group. R2 is as described above. - According to a second aspect of the invention, an intermediate insoluble polymer compound is provided. The compound comprises the repeating unit formula:
-

- wherein,
R1 is selected from an alkyl group; and
R2 is selected from an alkyl group, an aryl group, a hydrogen atom, a halogen atom, a substituted oxygen atom, a substituted nitrogen atom, a substituted sulfur atom, a carbonyl group, a cyano group, an amino group, and a guanidino group. - According to a third aspect of the invention, a process of preparing an intermediate insoluble polymer compound of formula:
-

- is provided. The process comprises reacting the compound of formula:
-

- with a 1-(3-bomobenzyl)-2,2,5,5-tetramethyl-1,2,5-azadisilolidine compound of the structural formula:
-

- According to a fourth aspect of the invention, a process is provided for preparing an intermediate insoluble polymer compound of formula:
-

- comprising the steps of:
- (a) reacting a first insoluble polymer compound of formula:
-
- with a 1-(3-bomobenzyl)-2,2,5,5-tetramethyl-1,2,5-azadisilolidine compound of the structural formula:
-

- to produce the compound having the formula:
-

- (b) reacting a compound having the formula:
-

- with toluene and NCNH2 to convert the ammonium group to a guanidinium group.
- According to a fifth aspect of the invention, a process is provided for preparing a compound of the formula:
-

- comprising contacting a compound of the formula:
-

- with iodine in an organic solvent.
- According to a sixth aspect of the invention, a process is provided for preparing a compound of the formula:
-

- wherein n is selected from 123, 125, and 131;
the process comprising contacting a compound having the formula: -

- with a solution of NanI and an oxidizing agent in the presence of a buffering agent; wherein n is as described above.
- Further features of the invention will be described or will become apparent in the course of the following detailed description.
- In order that the invention may be more clearly understood, the preferred embodiment thereof will now be described in detail by way of example, with reference to the accompanying drawings, in which:
-
FIG. 1A shows the 119Sn MAS NMR spectrum forPolymer 1; -
FIG. 1B shows the 119Sn MAS NMR spectrum forPolymer 1 after hydrolysis; -
FIG. 1C shows the 119Sn MAS NMR spectrum forPolymer 2; -
FIG. 1D shows the 119Sn MAS NMR spectra forPolymer 3 recovered after iodination; and -
FIG. 2 shows the HPLC analysis of the iodination products of reaction ofPolymer 2 with cyanamide at selected reaction times. - The process, according to the present invention, provides for the radiohalogenation of a haloaromatic compound using a polymer-supported pharmaceutical, as follows:
-

- wherein:
R1 is an alkyl group, and is preferably a butyl group;
R2 is selected from an alkyl group, an aryl group, a hydrogen atom, a halogen atom, a substituted oxygen atom, a substituted nitrogen atom, a substituted sulfur atom, a carbonyl group, a cyano group, an amino group, a guanidino group;
Nu is a nucleophile provided by the solvent or oxidant; and
*X is selected from any suitable radiohalide, such as, for example, 131I or 123I. - Although the preferred embodiment of the present invention refers to isotopes of Iodine only, such references are intended by way of example, and are not intended to be limiting. The present invention may be applied to any suitable radiohalide.
- According to a preferred embodiment of the present invention, a process is provided to synthesize no-carrier-added [131I]MIBG or [123I]MIBG using polymer-supported radiopharmaceutical precursors4. An insoluble polymer is prepared in which the pharmaceutical to be radiohalogenated is bound to the polymer through a tin-aryl bond as illustrated above. Treatment with the radiohalide and an appropriate oxidant results in the release of the radiohalogenated pharmaceutical while unreacted polymer and polymer side-products remain insoluble and easily removed by filtration.
- The polymer above is derived by reaction of the appropriate organolithium with a polymeric chlorostannane.
Polymer 1, described in detail below, is used as an intermediate compound to derive the polymers to be used in the radiohalogenation of a haloaromatic compound.Polymer 2 is one such reagent derived fromPolymer 1, andPolymer 3 is the preferred polymer-supported 3-benzylguanidinium reagent for the preparation of no-carrier-added [131I]MIBG or [123I]MIBG.Polymers -

- and was prepared by a known procedure5, as shown schematically below.
-

- After being run through a short silica column to remove antioxidant, commercial divinylbenzene (36 g, 225 mmol) and di-n-butyldichlorostannane (35 g, 115 mmol) were reacted overnight under nitrogen with dibutylstannane (27.5 g, 115 mmol) in the presence of AIBN (0.75 g, 4.6 mmol) near or below 30° C. Then divinylbenzene (6.5 g, 40.6 mmol), AIBN (1.25 g, 7.6 mmol), 1-octanol (85.5 g) and methyl-cellulose (0.63 g) dissolved in water (250 mL) were added to the previously prepared monomer. This mixture was refluxed for 8 h in a resin kettle under nitrogen with rapid stirring (1000 rpm).
- After cooling, water was added to the resin kettle and the solution was decanted from the granular polymer particles. Washing with water was continued until the supernatant ran clear. After a similar treatment with acetone (5×200 mL), the polymer was filtered with a coarse sintered-glass funnel and washed with methanol (2×200 mL), toluene (3×200 mL) and THF (3×200 mL). After drying overnight under vacuum at room temperature, 66.5 g (73% yield by weight) of white grainy solid was obtained.
- The characteristics of
Polymer 1 are preferably as follows: -
- Solid-state MAS 13C NMR (swollen with CHCl3), δ ppm: 14.6 (CH3), 18.6 (Sn—CH2—), 27.7 (—CH2CH2CH3), 28.6 (—CH2CH3), 42 (broad, Ar—CH—, Ar—CH2— and backbone —CH2—), 128 (broad, aryl CH), 144 (broad, Aryl C).
- Solid-state MAS 119Sn NMR (swollen with CHCl3), δ ppm.: 148.
- DRIFT spectrum, cm−1: 3036, 3060 (aromatic C—H); 2968, 2938, 2879, 2860 (aliphatic C—H); 1604, 1510 (aromatic C═C vibrations), 1486, 1450.2968, 2938, 2879, 2860 (aliphatic C—H); 1604, 1510 (aromatic C═C vibrations), 1486, 1450.
- Samples of
Polymer 1 were prepared as indicated above.Polymer 1 was characterized both chemically and spectroscopically. The availability of Sn—Cl bonds was assessed by treatment ofPolymer 1 with KOH in ethanol/THF at room temperature. Polymer 1 (201 mg) was soaked in 7 mL of absolute ethanol, 3 mL (3 mmol) of 1 M potassium hydroxide, and 1 mL of tetrahydrofuran for 24 h at room temperature. The polymer was filtered and washed with 70% ethanol (5×5 mL). The ethanol and THF in the filtrate were removed on rotary evaporator and the residual liquid was transferred into a 50 mL volumetric flask and topped up to the mark with water. Then 20 mL of this solution was diluted to 50 mL Erlenmeyer by addition of 1 M nitric acid. This solution was then titrated for Cl− following the Mohr procedure.6 - The amount of chloride ion released was found to be 1.7±0.1 mmol/g of polymer. If the polymer was composed exclusively of the units indicated in the structure shown for
Polymer 1, the maximum amount of chloride released would be 2.5 mmol/g of polymer. However, significant amounts of divinylbenzene had been used when forming the polymer and the yield of material was 32%. -
Polymer 1 was also investigated by MAS NMR spectroscopy on samples that had been preswollen with chloroform which had the effect of sharpening the signals. The 13C NMR spectrum was consistent with the anticipated structure showing signals for all of the carbons. The signals for the backbone CH and CH2 and phenyl carbons were quite broad and indistinct. However, the CH2—CH2 carbons and the carbons on the butyl chains were readily assigned by analogy with non-polymeric species. The 119Sn NMR spectrum (FIG. 1A ) showed but one peak at 148 ppm consistent with species of the general structure R3Sn—Cl. This confirmed that all of the tin atoms were bonded to chlorine in spite of the apparent discrepancy in the hydrolysis results mentioned above. This was particularly reassuring since IR spectra invariably gave a broad absorption near 3500 cm−1 which was consistent with hydrolysis of the Sn—Cl bond to Sn—OH. The 119Sn NMR spectrum of hydrolysed material (FIG. 1B ) was obtained and showed the complete absence of the peak at 148 ppm with a group of peaks appearing at about 90 ppm which confirmed thatPolymer 1 has tin bonded exclusively to chlorine and that all of these bonds are available for hydrolysis. - As illustrated in detail below,
Polymer 1 was preferably converted in two steps first intoPolymer 2 which bears the 3-benzylammonium moiety and then intoPolymer 3 functionalised with a 3-benzylguanidinium species. - The conversion of
Polymer 1 intoPolymer 2 was preceded by the preparation of 1-(3-bomobenzyl)-2,2,5,5-tetramethyl-1,2,5-azadisilolidine. 3-Bromobenzylamine hydrochloride (25.0 g, 112.3 mmol) was converted to the free base by neutralization with sodium hydroxide and extraction into methylene chloride. Drying and solvent removal resulted in 20.3 g of a brown liquid which was used as such. To the above 3-bromobenzylamine (20.3 g, 109 mmol), in a round bottom flask equipped with a condenser and under argon atmosphere, was added 150 mL of methylene chloride and 30.3 mL (217 mmol) of triethylamine. To this stirred mixture cooled with an ice-bath was added slowly 109 mL (109 mmol) of 1M 1,2-bis(chlorodimethylsilyl)ethane dissolved in methylene chloride. During the addition, considerable precipitate formed. The ice-bath was removed and the mixture was stirred for another 6.5 h at room temperature. The white solid was removed by filtration and washed with methylene chloride (2×30 mL). The filtrate was evaporated using a rotary evaporator resulting in further precipitate formation. Hexanes (100 mL) were added to this mixture. The solid was removed by filtration and washed with hexanes (2×20 mL). After hexane removal from the filtrate, a yellowish liquid was obtained which was distilled to yield 29.0 g (82% yield) of light yellowish liquid (bp. 93-5oC/0.05 mm Hg). - Compound 4 preferably had the following characteristics:
-
- 1H NMR spectrumδ (acetone-d6): δ 0 (s, 12H, CH3); 0.79 (s, 4H, Si—CH2—CH2—Si); 4.05 (s, 2H, Ar—CH2—N); 7.25 (t, 1H, 5-H), 7.30 (d, 1H, 6-H), 7.38 (d, 1H, 4-H), 7.47 (s, 1H, 2-H).
- 13C NMR spectrum (chloroform-d): δ 0 (CH3); 8.2 (Si—CH2—CH2—Si); 45.8 (Ar—CH2—N); 122.4 (C-3), 126.3 (C-6), 129.5 (C-5), 129.7 (C-2), 130.9 (C-4), 146.2 (C-1). IR (neat): cm−1 3068 (aromatic C—H stretch); 2949, 2903, 2857 (aliphatic C—H stretch); 1604, 1484 (aromatic C═C vibrations); 849 (vs asymmetric (Si—N—Si stretching), 784 (pCH3), 678 (νasSi—C). (aromatic C—H bending). MS: M/
Z
- Following the preparation of 1-(3-bomobenzyl)-2,2,5,5-tetramethyl-1,2,5-azadisilolidine, the preparation of
Polymer 2 proceeded as follows: -

- To 25.0 g (76.1 mmol) of Compound 4 in a three-neck flask equipped with an argon inlet, a serum cap and a powder addition sidearm containing Polymer 1 (28.6 g) was added 250 mL of freshly distilled dried tetrahydrofuran. To this flask cooled to −78° C. was added slowly 30.5 mL (76.1 mmol) of 2.5 M n-butyllithium. The initially colourless solution turned purple and then brownish at 25 min.
Polymer 1 was then tipped into the reaction solution and the mixture was stirred gently in the dry-ice-acetone bath for 7 h. After the temperature was allowed to rise to room temperature over 2 h and then held at room temperature for another 1 h, methanol (10 mL) was added followed by sufficient 1M hydrogen chloride to give a pH of 4-5. The mixture was stirred overnight and allowed to stand unstirred for 15 min. - After the upper cloudy solution was decanted, 200 mL of methanol was added. Again the cloudy upper layer was decanted and this process was repeated 4 times. The polymer was filtered through a coarse sintered glass funnel and washed with 50% methanol/water solution (100 mL), methanol (3×100 mL), and 95% ethanol (50 mL). After vacuum drying at room temperature, 30.1 g (84 wt % yield) of a white grainy material was obtained.
- The characteristics of
Polymer 2 were preferably as follows: -
- Solid-state MAS 13C NMR spectrum: δ 14.6 (CH3), 18.6 (Sn—CH2-), 27.7 (—CH2CH2CH3), 28.6 (—CH2CH3), 42 (broad, Ar—CH—, Ar—CH2— and backbone —CH2-), 128 (broad, aryl CH), 144 (broad, Aryl C)
- Solid-state MAS 119Sn NMR spectrum: δ −43.6 ppm
- IR spectrum (KBr): cm−1 3435 (broad, —NH3 + stretches); 3027 (aromatic C—H stretch); 2927, 2857 (aliphatic C—H stretches); 1611, 1498 (aromatic C═C vibrations).
- Because 3-bromobenzyl amine has reactive NH bonds, the amino group was first protected as an azadisilolidine7 and then reacted with one equivalent of n-butyllithium.
Polymer 1 was added to the presumed monolithium species so prepared.Polymer 2 was characterized by 13C and 119Sn MAS NMR spectroscopy. - The 13C NMR spectrum was consistent with the proposed structure. Again, the signals attributed to the backbone CH and CH2 carbons and the phenyl carbons appeared as broad peaks. The peaks due to the Sn—CH2—CH2 carbon was identifiable as well as the carbons of the n-butyl group. The benzylic carbons were not readily identified. Perhaps most characteristic was the shift in the Sn—CH2—C3H7 carbon signals from 18.6 ppm for
Polymer 1 to 10.5 ppm forPolymer 2. Again the IR spectrum gave a broad absorption near 3500 cm−1 indicating that considerable hydrolysis accompanied this reaction. However as indicated inFIG. 1C , the 119Sn spectrum showed primarily one peak at −43.6 ppm with a small peak at 90 ppm consistent with SnOH and representing about 10% of the tin signals. Thus it would seem that all of the Sn—Cl bonds had reacted with the organolithium reagent with a small amount of concurrent hydrolysis. - The iodination of
Polymer 2 proceeded in accordance with the following reaction: -

- To the previously prepared Polymer 2 (100 mg) was added 10 mL of methanol and 1 mL of 0.2 M iodine in acetonitrile solution. After stirring gently at room temperature for 16.5 h, 1 mL of 1.0 M sodium metabisulphite solution was added. The mixture was transferred into a 100 mL volumetric flask and topped up to 100 mL with 0.01 M KH2PO4. An aliquot was filtered through a syringe filter, and the filtrate was analyzed by HPLC. The retention time of 3-iodobenzylammonium peak was 8.8 min and the amount was determined by comparison to a standard 3-iodobenzylammonium hydrochloride solution (0.4 mM).
- The extent of reaction in forming
Polymer 2 and the availability of the 3-benzyl ammonium species inPolymer 2 was probed by reaction with iodine as illustrated above. This was anticipated to yield the 3-iodobenzyl ammonium ion (“MIBA”) which was identified and quantified by HPLC. It was found that about 0.95±0.05 mmol/g of 3-iodobenzyl ammonium ion was released per gram ofPolymer 2. Again this was somewhat less that the maximum calculated amount ifPolymer 2 had the structure indicated above. However, 119Sn NMR spectroscopy proved to be of considerable value as shown inFIG. 1D . A comparison ofFIG. 1C forPolymer 2 andFIG. 10 forpolymer 2 recovered after iodination shows a complete loss of the signal at −43.6 ppm consistent with complete release of the 3-benzyl ammonium group. - Preferably, the synthesis of
Polymer 3 involved treatment ofPolymer 2 with a large excess of cyanamide to convert the ammonium group into a guanidinium group, as illustrated below. -

- Under an argon atmosphere, 20.0 g of
Polymer 2, 15.1 g (360 mmol) of cyanamide, 100 μL (0.72 mmol) of triethylamine and 250 mL of toluene were added to a flask equipped with a reflux condenser. The mixture was heated for 25 h at 54° C. and the hot reaction mixture was filtered through a coarse sintered glass funnel. The polymer was washed with acetonitrile (4×100 mL), methanol (4×100 mL) and acetonitrile (2×100 mL). After vacuum drying at room temperature overnight, 20.7 g of white grainy material was obtained. - The characteristics of
Polymer 3 are preferably as follows: -
- IR spectrum (KBr): cm−1 3340, 3271, 3174 (N—H stretch); 3066, 3027 (aromatic C—H stretch); 2978, 2929, 2880, 2860 (aliphatic C—H stretch); 1677, 1658 (C═N stretch); 1521, 1496 (aromatic C═C vibrations); 719 (aromatic C—H bend).
- The iodination of
Polymer 3 proceeded in accordance with the following reaction: -

- To 26.4 mg of
Polymer 3 suspended in 4 mL of methanol was added 300 μL of 0.2 M iodine in acetonitrile solution. After stirring for 13.5 h at room temperature, 0.1 mL to of 1 M sodium metabisulphite solution was added. The mixture was transferred into a 100 mL volumetric flask and topped up to 100 mL with 0.01 M KH2PO4 buffer. After filtering through a syringe filter, the filtrate was analysed by HPLC by comparison to standard 3-iodobenzylammonium chloride solutions (0.4 mM) and 3-iodobenzylguanidinium chlorideref solutions (0.2 mM) which had a retention times of 8.7 min and 15.6 min respectively. - Unreacted ammonium groups resulted in the release of the 3-iodobenzyl ammonium ion while ammonium groups that had reacted with cyanamide would result in the release of the 3-iodobenzyl guanidinium ion, as illustrated above. Thus, at selected time intervals, polymeric material was isolated from the reaction mixture and treated with an excess of iodine. After reduction of the iodine, aliquots were analyzed by HPLC and the results are presented in
FIG. 3 . At 75° C. after 1 h, 80% of the ammonium groups had been converted to guanidinium groups and by 2 h conversion had progressed to 96.2%. By 5 h, there was less than 0.3% of unreacted ammonium groups. - However, a new peak with a retention time slightly longer than the 3-iodobenzyl guanidinium was observed. This material was identified to be the 3-iodobenzylbiguanidinium ion in two ways. First, an authentic sample of 3-iodobenzylbiguanidinium nitrate was produced8, as described in detail below. The sample was found to have an identical retention time with the new peak. Secondly, a small sample of the new material was isolated from a reaction that was allowed to run considerably longer and at a higher temperature than those shown in
FIG. 2 . Although not enough material could be isolated in a sufficiently pure form to allow a mixed mp., the 13C NMR spectrum was identical with the authentic sample of 3-iodobenzylbiguanidinium nitrate that had been produced. The guanidinium group reacted further with cyanamide, but at a rate considerably slower than the ammonium group so that the biguanidinium species does not start to appear until very late in the conversion process. - With this time course in mind, a reasonably large batch of
polymer 2 was reacted for 25 h. at 54° C. After the usual washing,Polymer 3, so produced, was analyzed by iodination to yield 3-iodobenzyl guanidinium at a level of 1.1±0.05 mmol/g ofPolymer 3. - In a 25 mL flask, 3-iodobenzylamine hydrochloride (2.0 g, 7.4 mmol) was combined with 0.62 g (7.4 mmol) of dicyandiamide and an argon atmosphere was introduced. The mixture was melted at 154° C. for 30 min and water (20 mL) was added to dissolve the brownish solid. HPLC analysis showed 8 mol % of 3-iodobenzylamine hydrochloride, 17 mol % of MIBG and 75 mol % of 3-iodobenzylbiguanidinium nitrate (excluding dicyandiamide at 9.6 min) with a retention times of 6.1 min, 11.2 min and 12.7 min respectively. After decolorizing with charcoal, the solution was adjusted to pH˜9 with saturated sodium carbonate. The upper aqueous solution was decanted from the oil which was separated. Water (5 mL) was added to the oil followed by 1 M nitric acid to a pH˜5. A lot of bubbles formed during the addition of nitric acid and the mixture turned into a clear solution at pH˜5. White crystals formed about 1 minute later.
- After 20 min. at room temperature, the crystals were collected by filtration and washed with water (3×3 mL). After drying, 1.22 g of white crystals were obtained. Concentration of the filtrates to about 10 mL yielded 0.23 g more of white crystals. The two batches of crystals were combined and recrystallized using acetonitrile (25 mL) to provide 0.25 g of white mushroom shape crystals. HPLC analysis showed 99 mol % of 3-iodobenzylbiguanidinium nitrate, 1 mol % of MIBG and dicyandiamide. The 0.25 g of crystals were recrystallized again from 6 mL of acetonitrile to yield 99.3 mg (4% yield) of white mushroom shape crystals of mp 133.5-134.5° C.
- The characteristics of the 3-iodobenzylbiguanidinium nitrate were preferably as follows:
-
- 1H NMR spectrum (methanol-d4): δ 4.34 (d, 2H, CH2), 7.05 (t, 1H, 5-H), 7.28 (d, 1H, 6-H), 7.56 (d, 1H, 4-H), 7.63 (s, 1H, 2-H).
- 13C NMR spectrum (methanol-d4): δ 45.5 (CH2), 95.3 (C-3), 127.9 (C-6), 131.5 (C-5), 137.4 (C-2), 137.5 (C-4), 142.4 (C-2), 160.3 (middle C═N), 162.1 (terminal C═N).
- FT-IR spectrum (KBr): cm−1 3506, 3467, 3428, 3330, 3301, 3213 (N—H stretch); 2968.1, 2938.9 (aliphatic C—H stretch); 2440 (—NH2+ stretch); 1658, 1638 (C═N stretch); 1555 (asymmetric NO3− stretch); 1428.0, 1394 (symmetric NO3− stretches); 782 (aromatic C—H bend).
- UV spectrum: δmax=232 nm, εmax=2.49×104. Anal. calc'd for C9H13IN6O3: C, 28.44; H, 3.45; N, 22.11; I, 33.38. Found: C, 28.61; H, 3.44; N, 22.07; I, 33.19.
- Following the successful completion of the iodination of
Polymer 3, the radioiodination ofPolymer 3 using sodium [131I]iodide, proceeded in accordance with the following reaction: -

- Into a 2 mL vial was placed 0.5 mg of
Polymer 3, 300 μL of methanol, 100 μl of 0.1 M potassium dihydrogen orthophosphate, 45.5 MBq of a Na131I solution and a 100 μL aliquot of a solution prepared from 2 mL of acetic acid and 2 mL of 50% hydrogen peroxide solution diluted to 50 mL with water. The mixture was occasionally shaken for 2 h at room temperature and then 200 μL of 0.1 M sodium metabisulphite was added to the reaction mixture. - After filtration through a syringe filter, the filtrate was analyzed by HPLC. The UV detector trace showed a large peak at the solvent front and several smaller peaks well before MIBG peak. The corresponding radioactivity detector trace showed two peaks at 1.9 and 15.6 min which were confirmed by coinjection to be 131I-(2.4%) and [131I]MIBG (97.6%) respectively.
- An aliquot of the reaction mixture, 25.4 MBq, was passed through a Sep-Pak™ cartridge and washed with 5 mL of water. Radioactivity was found in the washes, 1.0 MBq, and on the cartridge, 24.4 MBq. Washing of the cartridge with 1.3 mL of 42% ethanol, released 20.0 MBq of radioactivity into the washes. A wash with 1 mL of methanol released a further 3.1 MBq of [131I]MIBG for a total of 23.1 MBq (95%). The washes were analyzed by HPLC. The trace from the UV detector showed two small peaks at the solvent front and a peak at 15.6 min which, by coinjection, was confirmed to be MIBG. The corresponding radioactivity trace showed a single peak at 15.6 min.
- The success of the radioiodination was assessed by HPLC using both an UV detector and a γ-ray detector. The oxidant, H2O2/HOAc, or its by-products are readily separable from any [131I]MIBG produced, and results of this oxidizing system are presented in Table 1. Labelling reactions were run at room temperature on about one or two grains of
Polymer 3 in a mixture of methanol and 0.1 M KH2PO4 using Na131I. Under these conditions after filtration, essentially all of the radioactivity was found in solution either as 131I− or as [131I]MIBG as evidenced by their HPLC retention times using a radioactivity detector. Table 1 shows the effect on the radiochemical yield of [131I]MIBG of increasing the concentration of H2O2 and acetic acid at various reaction times. As the first column indicates, the exclusion of acetic acid from the reaction mixture led to low yields and apparently slow reactions. The third column indicates a general increase in yield with reaction time. At two hour reaction times, increasing concentrations of H2O2 and acetic acid led to increased yields which approached near quantitative conversion. It was these conditions that were chosen for preparation of [131I]MIBG, as illustrated. -
TABLE 1 Radiochemical yield of [131I]MIBG from treatment of Polymer 3 with Na 131I and H2O2/HOAc as oxidant asa function of reaction time and oxidant concentration Reactiona time [H2O2], mM/[HOAc], mM (min) 50/0 20/24 37/42 50/57 60/68 30 0 30 60 30 70 63 120 45 80 90 98 150 56 180 85 aReactions were run at room temperature in a mixture of methanol and 0.1M KH2PO4. - Purification of the product from the radiolabelling reaction involved two steps. The first was simple filtration through a syringe tip filter. An HPLC analysis of this product using a UV detector showed a large peak at the solvent front and several minor peaks. The radioactivity detector showed two peaks corresponding to 131I— at about 2% and the other to [131I]MIBG at 98%. The second step in the purification involved selective absorption and desorption of the [131I]MIBG onto a C18 Sep-Pak™ cartridge. When the primarily aqueous solution from the filtration step was passed through the C18 Sep-Pak™ cartridge, essentially all of the radioactivity was absorbed onto the cartridge with the small amount of iodide passing through. When the cartridge was washed with ethanol/water, 82% of the radioactivity was released. A further 10% was released when methanol was used as eluant.
- An HPLC analysis of these solutions, using an UV detector, showed two small peaks near the solvent front and a small peak at the retention time of MIBG. The area of this peak was rather too small to allow a reliable calculation of the specific activity of the [131I]MIBG. The corresponding radioactivity trace showed but one peak at the retention time of MIBG. Thus following this procedure, the no-carrier-added [131I]MIBG was produced in about 92% radiochemical yield with a specific activity of ≧500 Ci/mmol.
- [123I]MIBG was produced using Iodobeads™ as the oxidant, which resulted in a radiochemical yield of about 55%. Since later experiments with Na131I showed that about 45% of the radioactivity remained absorbed to the insoluble polymeric materials, this experiment demonstrates that
Polymer 3 is an effective precursor to [123I]MIBG. Similar results can also be expected with [125I]MIBG. - The process, according to the present invention, produces no-carrier-added [131I]MIBG in ≧90% radiochemical yield and high chemical purity. Isolation and purification are simple, involving just filtration and absorption and desorption onto a C18 Sep-Pak™cartridge. No-carrier-added material should avoid the potential pharmacological side effects accompanying the current method of production.
- Although the above description refers to radioiodination of MIBG, such references are not intended to be limiting. The process and intermediate compound according to the present invention can be used for synthesis of any number radiolabelled haloaromatic compounds.
- It will be appreciated that the above description relates to the preferred embodiment by way of example only. Many variations on the invention will be obvious to those knowledgeable in the field, and such obvious variations are within the scope of the invention as described and claimed, whether or not expressly described.
- The process and precursor compounds, according to the present invention, is useful in the field of nuclear medicine for the production of radiopharmaceutical compounds.
-
- 1) Wafelman, A. R., Konings, M. C. P., Hoefnagel, C. A., Maes, R. A. A. and Beijnen, J. H., Appl Radiat. Isot., 997 (1994) and references therein.
- 2) Vaidyanathan, G. and Zalutsky, M. R., Appl Radiat. Isot., 621 (1993).
- 3) Flanagan, R. J., Goel, A., Charleson, F. P. and Hunter, D. H., J. Label. Comp. Radiopharm., 636 (1995).
- Hunter, D. H., Goel, A. and Flanagan, R. J., “Process for the Preparation of Radiolabelled meta-Halobenzylguanidine”, U.S. Pat. No. 5,585,185.
- 4) Culbert, P. A., and Hunter, D. H., Polymer Supported Radiopharmaceuticals: 123 I and 131I-labelled N-Isopropyl-4-Iodoamphetamine, Reactive Polymers, 19, 247-253 (1993).
- 5) Gerigk, U., Gerlach, M., Neumann, W. P., Veiler, R. and Weintritt, V., Synthesis, 448 (1990).
- 6) R. A. Day, Jr.; A. L. Underwood, Quantitative Analysis Laboratory Manual (Prentice-Hall, Inc.), 2nd ed., p. 115-116, Englewood Cliffs, N.J., 1967.
- 7) Djuric, S., Venit, J., and Magnus, P., Tetrahedron Lett., 22, 1787-1790 (1981).
- 8) Shapiro, S. L., Parino, V. A., Rogow, E., Freedman, L., J. Am. Chem. Soc., 81, 3728 (1959).
Claims (2)
1. A process of preparing a radiolabelled haloaromatic compound (I) of the formula:

wherein,
R2 is selected from an alkyl group, an aryl group, a hydrogen atom, a halogen atom, a substituted oxygen atom, a substituted nitrogen atom, a substituted sulfur atom, a carbonyl group, a cyano group, an amino group, and a guanidino group;
*X is selected from any suitable radiohalide;
said process comprising reacting a radiohalogen ion with a insoluble polymer compound (II) in the presence of a solvent and an oxidant, said polymer compound (II) having the formula:

wherein,
R1 is an alkyl group; and
R2 is as described above.
2-23. (canceled)
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/702,009 US20100274052A1 (en) | 1997-10-02 | 2010-02-08 | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US6088697P | 1997-10-02 | 1997-10-02 | |
| PCT/CA1998/000933 WO1999018053A1 (en) | 1997-10-02 | 1998-10-02 | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
| US09/529,017 US6461585B1 (en) | 1998-10-02 | 1998-10-02 | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
| US10/224,261 US7273601B2 (en) | 2000-07-18 | 2002-08-20 | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
| US11/850,306 US7658910B2 (en) | 1997-10-02 | 2007-09-05 | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
| US12/702,009 US20100274052A1 (en) | 1997-10-02 | 2010-02-08 | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/850,306 Continuation US7658910B2 (en) | 1997-10-02 | 2007-09-05 | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20100274052A1 true US20100274052A1 (en) | 2010-10-28 |
Family
ID=24108161
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/224,261 Expired - Fee Related US7273601B2 (en) | 1997-10-02 | 2002-08-20 | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
| US11/850,306 Expired - Fee Related US7658910B2 (en) | 1997-10-02 | 2007-09-05 | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
| US12/702,009 Abandoned US20100274052A1 (en) | 1997-10-02 | 2010-02-08 | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/224,261 Expired - Fee Related US7273601B2 (en) | 1997-10-02 | 2002-08-20 | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
| US11/850,306 Expired - Fee Related US7658910B2 (en) | 1997-10-02 | 2007-09-05 | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
Country Status (1)
| Country | Link |
|---|---|
| US (3) | US7273601B2 (en) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7273601B2 (en) | 2000-07-18 | 2007-09-25 | The University Of Western Ontario | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
| US20030192585A1 (en) * | 2002-01-25 | 2003-10-16 | Konarka Technologies, Inc. | Photovoltaic cells incorporating rigid substrates |
| JP2012532833A (en) | 2009-07-11 | 2012-12-20 | バイエル ファーマ アクチエンゲゼルシャフト | Nonpolar and polar leaving groups |
| EP3888694A1 (en) | 2014-10-24 | 2021-10-06 | Molecular Insight Pharmaceuticals, Inc. | Preparations of meta-iodobenzylguanidine and precursors thereof |
Citations (61)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5565185A (en) * | 1994-07-20 | 1996-10-15 | Merck Frosst Canada, Inc. | Process for the preparation of radiolabeled meta-halobenzylguanidine |
| US5818842A (en) * | 1994-01-21 | 1998-10-06 | Newbridge Networks Corporation | Transparent interconnector of LANs by an ATM network |
| US5938732A (en) * | 1996-12-09 | 1999-08-17 | Sun Microsystems, Inc. | Load balancing and failover of network services |
| US5986662A (en) * | 1996-10-16 | 1999-11-16 | Vital Images, Inc. | Advanced diagnostic viewer employing automated protocol selection for volume-rendered imaging |
| US6052733A (en) * | 1997-05-13 | 2000-04-18 | 3Com Corporation | Method of detecting errors in a network |
| US6061348A (en) * | 1995-12-22 | 2000-05-09 | Cisco Technology, Inc. | Method and apparatus for dynamically allocating bandwidth for a time division multiplexed data bus |
| US6111880A (en) * | 1997-12-05 | 2000-08-29 | Whittaker Corporation | Hybrid packet/cell switching, linking, and control system and methodology for sharing a common internal cell format |
| US6229538B1 (en) * | 1998-09-11 | 2001-05-08 | Compaq Computer Corporation | Port-centric graphic representations of network controllers |
| US6272113B1 (en) * | 1998-09-11 | 2001-08-07 | Compaq Computer Corporation | Network controller system that uses multicast heartbeat packets |
| US6308282B1 (en) * | 1998-11-10 | 2001-10-23 | Honeywell International Inc. | Apparatus and methods for providing fault tolerance of networks and network interface cards |
| US20020012352A1 (en) * | 1998-12-18 | 2002-01-31 | Goran Hansson | Internet protocol handler for telecommunications platform with processor cluster |
| US20020016926A1 (en) * | 2000-04-27 | 2002-02-07 | Nguyen Thomas T. | Method and apparatus for integrating tunneling protocols with standard routing protocols |
| US6363497B1 (en) * | 1997-05-13 | 2002-03-26 | Micron Technology, Inc. | System for clustering software applications |
| US6381218B1 (en) * | 1998-09-11 | 2002-04-30 | Compaq Computer Corporation | Network controller system that uses directed heartbeat packets |
| US20020051464A1 (en) * | 2000-09-13 | 2002-05-02 | Sin Tam Wee | Quality of transmission across packet-based networks |
| US6461585B1 (en) * | 1998-10-02 | 2002-10-08 | The University Of Western Ontario | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
| US20020191612A1 (en) * | 2001-01-26 | 2002-12-19 | Placeware, Inc. | Method and apparatus for automatically determining an appropriate transmission method in a network |
| US20030012730A1 (en) * | 2000-07-18 | 2003-01-16 | Hunter Duncan H. | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
| US6512774B1 (en) * | 1999-03-18 | 2003-01-28 | 3Com Corporation | Fail over with multiple network interface cards |
| US20030118039A1 (en) * | 2001-12-17 | 2003-06-26 | Hidetaka Nishi | Gateway |
| US20030142795A1 (en) * | 2002-01-31 | 2003-07-31 | Gavette Sherman L. | Home network telephone answering system and method for same |
| US20030174729A1 (en) * | 2002-03-01 | 2003-09-18 | Matthias Heink | ATM-Port with integrated ethernet switch interface |
| US6633563B1 (en) * | 1999-03-02 | 2003-10-14 | Nortel Networks Limited | Assigning cell data to one of several processors provided in a data switch |
| US20040030757A1 (en) * | 2002-06-11 | 2004-02-12 | Pandya Ashish A. | High performance IP processor |
| US6714535B1 (en) * | 1999-11-08 | 2004-03-30 | Broadmedia, Inc. | Method and system for unlimited use of telephony services over a data network without incurring long distance calling tolls |
| US20040066782A1 (en) * | 2002-09-23 | 2004-04-08 | Nassar Ayman Esam | System, method and apparatus for sharing and optimizing packet services nodes |
| US20040071142A1 (en) * | 2002-10-11 | 2004-04-15 | Hitachi, Ltd. | Packet communication device |
| US6728780B1 (en) * | 2000-06-02 | 2004-04-27 | Sun Microsystems, Inc. | High availability networking with warm standby interface failover |
| US6738826B1 (en) * | 2000-02-24 | 2004-05-18 | Cisco Technology, Inc. | Router software upgrade employing redundant processors |
| US6741585B1 (en) * | 2000-05-05 | 2004-05-25 | Lucent Technologies Inc. | Interworking of addressing in an internetwork |
| US6754745B1 (en) * | 1999-08-06 | 2004-06-22 | Accelerated Networks | Method and apparatus for distributing a clock in a network |
| US20040131064A1 (en) * | 1994-01-21 | 2004-07-08 | Alcatel Canada Inc. | Digital communications system |
| US6763479B1 (en) * | 2000-06-02 | 2004-07-13 | Sun Microsystems, Inc. | High availability networking with alternate pathing failover |
| US6766482B1 (en) * | 2001-10-31 | 2004-07-20 | Extreme Networks | Ethernet automatic protection switching |
| US6771673B1 (en) * | 2000-08-31 | 2004-08-03 | Verizon Communications Inc. | Methods and apparatus and data structures for providing access to an edge router of a network |
| US6778491B1 (en) * | 2000-03-31 | 2004-08-17 | Alcatel | Method and system for providing redundancy for signaling link modules in a telecommunication system |
| US6850531B1 (en) * | 1999-02-23 | 2005-02-01 | Alcatel | Multi-service network switch |
| US6856591B1 (en) * | 2000-12-15 | 2005-02-15 | Cisco Technology, Inc. | Method and system for high reliability cluster management |
| US6862564B1 (en) * | 2000-10-26 | 2005-03-01 | Sycamore Networks, Inc. | Network emulator |
| US20050053073A1 (en) * | 2003-09-03 | 2005-03-10 | Andiamo Systems, Inc. A Delaware Corporation | Switch port analyzers |
| US6879667B1 (en) * | 2001-05-07 | 2005-04-12 | General Bandwidth Inc. | System and method for interfacing telephony voice signals with a broadband access network |
| US6891836B1 (en) * | 1999-06-03 | 2005-05-10 | Fujitsu Network Communications, Inc. | Switching complex architecture and operation |
| US6895528B2 (en) * | 2000-08-07 | 2005-05-17 | Computer Network Technology Corporation | Method and apparatus for imparting fault tolerance in a switch or the like |
| US6910148B1 (en) * | 2000-12-07 | 2005-06-21 | Nokia, Inc. | Router and routing protocol redundancy |
| US6928482B1 (en) * | 2000-06-29 | 2005-08-09 | Cisco Technology, Inc. | Method and apparatus for scalable process flow load balancing of a multiplicity of parallel packet processors in a digital communication network |
| US20050185577A1 (en) * | 1998-09-11 | 2005-08-25 | Kenichi Sakamoto | IP packet communication apparatus |
| US6938092B2 (en) * | 2001-03-07 | 2005-08-30 | Alacritech, Inc. | TCP offload device that load balances and fails-over between aggregated ports having different MAC addresses |
| US20050243716A1 (en) * | 2004-05-03 | 2005-11-03 | Bitar Nabil N | Systems and methods implementing 1‘and N:1 line card redundancy |
| US6975587B1 (en) * | 2000-08-25 | 2005-12-13 | Nortel Networks Limited | Mechanism for automatic protection switching in a router |
| US20050281190A1 (en) * | 2004-06-17 | 2005-12-22 | Mcgee Michael S | Automated recovery from a split segment condition in a layer2 network for teamed network resources of a computer systerm |
| US20060143309A1 (en) * | 2004-12-29 | 2006-06-29 | Mcgee Michael S | Verifying network connectivity |
| US7177943B1 (en) * | 2001-12-27 | 2007-02-13 | Cisco Technology, Inc. | System and method for processing packets in a multi-processor environment |
| US7185094B2 (en) * | 2001-03-30 | 2007-02-27 | Sandcherry, Inc. | Media session framework using a control module to direct and manage application and service servers |
| US20070083528A1 (en) * | 2000-09-13 | 2007-04-12 | Fortinet, Inc. | Switch management system and method |
| US7212519B2 (en) * | 1999-07-14 | 2007-05-01 | Ericsson Inc. | Combining narrowband applications with broadband transport |
| US7233567B1 (en) * | 2000-09-22 | 2007-06-19 | Nortel Networks Limited | Apparatus and method for supporting multiple traffic redundancy mechanisms |
| US7239605B2 (en) * | 2002-09-23 | 2007-07-03 | Sun Microsystems, Inc. | Item and method for performing a cluster topology self-healing process in a distributed data system cluster |
| US7263060B1 (en) * | 2001-06-28 | 2007-08-28 | Network Appliance, Inc. | Multiple switch protected architecture |
| US7424025B2 (en) * | 2003-10-01 | 2008-09-09 | Santera Systems, Inc. | Methods and systems for per-session dynamic management of media gateway resources |
| US20080317055A1 (en) * | 2003-02-03 | 2008-12-25 | Jerker Mattias Zetterlund | Shared risk group handling within a media gateway |
| US20090092044A1 (en) * | 2001-10-10 | 2009-04-09 | Juniper Networks, Inc. | Selector in switching matrix, line redundant method, and line redundant system |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999018053A1 (en) | 1997-10-02 | 1999-04-15 | The University Of Western Ontario | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
-
2002
- 2002-08-20 US US10/224,261 patent/US7273601B2/en not_active Expired - Fee Related
-
2007
- 2007-09-05 US US11/850,306 patent/US7658910B2/en not_active Expired - Fee Related
-
2010
- 2010-02-08 US US12/702,009 patent/US20100274052A1/en not_active Abandoned
Patent Citations (63)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5818842A (en) * | 1994-01-21 | 1998-10-06 | Newbridge Networks Corporation | Transparent interconnector of LANs by an ATM network |
| US20040131064A1 (en) * | 1994-01-21 | 2004-07-08 | Alcatel Canada Inc. | Digital communications system |
| US5565185A (en) * | 1994-07-20 | 1996-10-15 | Merck Frosst Canada, Inc. | Process for the preparation of radiolabeled meta-halobenzylguanidine |
| US6061348A (en) * | 1995-12-22 | 2000-05-09 | Cisco Technology, Inc. | Method and apparatus for dynamically allocating bandwidth for a time division multiplexed data bus |
| US5986662A (en) * | 1996-10-16 | 1999-11-16 | Vital Images, Inc. | Advanced diagnostic viewer employing automated protocol selection for volume-rendered imaging |
| US5938732A (en) * | 1996-12-09 | 1999-08-17 | Sun Microsystems, Inc. | Load balancing and failover of network services |
| US6363497B1 (en) * | 1997-05-13 | 2002-03-26 | Micron Technology, Inc. | System for clustering software applications |
| US6052733A (en) * | 1997-05-13 | 2000-04-18 | 3Com Corporation | Method of detecting errors in a network |
| US7658910B2 (en) * | 1997-10-02 | 2010-02-09 | The University Of Western Ontario | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
| US6111880A (en) * | 1997-12-05 | 2000-08-29 | Whittaker Corporation | Hybrid packet/cell switching, linking, and control system and methodology for sharing a common internal cell format |
| US6229538B1 (en) * | 1998-09-11 | 2001-05-08 | Compaq Computer Corporation | Port-centric graphic representations of network controllers |
| US20050185577A1 (en) * | 1998-09-11 | 2005-08-25 | Kenichi Sakamoto | IP packet communication apparatus |
| US6272113B1 (en) * | 1998-09-11 | 2001-08-07 | Compaq Computer Corporation | Network controller system that uses multicast heartbeat packets |
| US6381218B1 (en) * | 1998-09-11 | 2002-04-30 | Compaq Computer Corporation | Network controller system that uses directed heartbeat packets |
| US6461585B1 (en) * | 1998-10-02 | 2002-10-08 | The University Of Western Ontario | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
| US6308282B1 (en) * | 1998-11-10 | 2001-10-23 | Honeywell International Inc. | Apparatus and methods for providing fault tolerance of networks and network interface cards |
| US20020012352A1 (en) * | 1998-12-18 | 2002-01-31 | Goran Hansson | Internet protocol handler for telecommunications platform with processor cluster |
| US6850531B1 (en) * | 1999-02-23 | 2005-02-01 | Alcatel | Multi-service network switch |
| US6633563B1 (en) * | 1999-03-02 | 2003-10-14 | Nortel Networks Limited | Assigning cell data to one of several processors provided in a data switch |
| US6512774B1 (en) * | 1999-03-18 | 2003-01-28 | 3Com Corporation | Fail over with multiple network interface cards |
| US6891836B1 (en) * | 1999-06-03 | 2005-05-10 | Fujitsu Network Communications, Inc. | Switching complex architecture and operation |
| US7212519B2 (en) * | 1999-07-14 | 2007-05-01 | Ericsson Inc. | Combining narrowband applications with broadband transport |
| US6754745B1 (en) * | 1999-08-06 | 2004-06-22 | Accelerated Networks | Method and apparatus for distributing a clock in a network |
| US6714535B1 (en) * | 1999-11-08 | 2004-03-30 | Broadmedia, Inc. | Method and system for unlimited use of telephony services over a data network without incurring long distance calling tolls |
| US6738826B1 (en) * | 2000-02-24 | 2004-05-18 | Cisco Technology, Inc. | Router software upgrade employing redundant processors |
| US6778491B1 (en) * | 2000-03-31 | 2004-08-17 | Alcatel | Method and system for providing redundancy for signaling link modules in a telecommunication system |
| US20020016926A1 (en) * | 2000-04-27 | 2002-02-07 | Nguyen Thomas T. | Method and apparatus for integrating tunneling protocols with standard routing protocols |
| US6741585B1 (en) * | 2000-05-05 | 2004-05-25 | Lucent Technologies Inc. | Interworking of addressing in an internetwork |
| US6763479B1 (en) * | 2000-06-02 | 2004-07-13 | Sun Microsystems, Inc. | High availability networking with alternate pathing failover |
| US6728780B1 (en) * | 2000-06-02 | 2004-04-27 | Sun Microsystems, Inc. | High availability networking with warm standby interface failover |
| US6928482B1 (en) * | 2000-06-29 | 2005-08-09 | Cisco Technology, Inc. | Method and apparatus for scalable process flow load balancing of a multiplicity of parallel packet processors in a digital communication network |
| US7273601B2 (en) * | 2000-07-18 | 2007-09-25 | The University Of Western Ontario | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
| US20030012730A1 (en) * | 2000-07-18 | 2003-01-16 | Hunter Duncan H. | Preparation of radiolabelled haloaromatics via polymer-bound intermediates |
| US6895528B2 (en) * | 2000-08-07 | 2005-05-17 | Computer Network Technology Corporation | Method and apparatus for imparting fault tolerance in a switch or the like |
| US6975587B1 (en) * | 2000-08-25 | 2005-12-13 | Nortel Networks Limited | Mechanism for automatic protection switching in a router |
| US6771673B1 (en) * | 2000-08-31 | 2004-08-03 | Verizon Communications Inc. | Methods and apparatus and data structures for providing access to an edge router of a network |
| US20070083528A1 (en) * | 2000-09-13 | 2007-04-12 | Fortinet, Inc. | Switch management system and method |
| US20020051464A1 (en) * | 2000-09-13 | 2002-05-02 | Sin Tam Wee | Quality of transmission across packet-based networks |
| US7233567B1 (en) * | 2000-09-22 | 2007-06-19 | Nortel Networks Limited | Apparatus and method for supporting multiple traffic redundancy mechanisms |
| US6862564B1 (en) * | 2000-10-26 | 2005-03-01 | Sycamore Networks, Inc. | Network emulator |
| US6910148B1 (en) * | 2000-12-07 | 2005-06-21 | Nokia, Inc. | Router and routing protocol redundancy |
| US6856591B1 (en) * | 2000-12-15 | 2005-02-15 | Cisco Technology, Inc. | Method and system for high reliability cluster management |
| US20020191612A1 (en) * | 2001-01-26 | 2002-12-19 | Placeware, Inc. | Method and apparatus for automatically determining an appropriate transmission method in a network |
| US6938092B2 (en) * | 2001-03-07 | 2005-08-30 | Alacritech, Inc. | TCP offload device that load balances and fails-over between aggregated ports having different MAC addresses |
| US7185094B2 (en) * | 2001-03-30 | 2007-02-27 | Sandcherry, Inc. | Media session framework using a control module to direct and manage application and service servers |
| US6879667B1 (en) * | 2001-05-07 | 2005-04-12 | General Bandwidth Inc. | System and method for interfacing telephony voice signals with a broadband access network |
| US7263060B1 (en) * | 2001-06-28 | 2007-08-28 | Network Appliance, Inc. | Multiple switch protected architecture |
| US20090092044A1 (en) * | 2001-10-10 | 2009-04-09 | Juniper Networks, Inc. | Selector in switching matrix, line redundant method, and line redundant system |
| US6766482B1 (en) * | 2001-10-31 | 2004-07-20 | Extreme Networks | Ethernet automatic protection switching |
| US20030118039A1 (en) * | 2001-12-17 | 2003-06-26 | Hidetaka Nishi | Gateway |
| US7177943B1 (en) * | 2001-12-27 | 2007-02-13 | Cisco Technology, Inc. | System and method for processing packets in a multi-processor environment |
| US20030142795A1 (en) * | 2002-01-31 | 2003-07-31 | Gavette Sherman L. | Home network telephone answering system and method for same |
| US20030174729A1 (en) * | 2002-03-01 | 2003-09-18 | Matthias Heink | ATM-Port with integrated ethernet switch interface |
| US20040030757A1 (en) * | 2002-06-11 | 2004-02-12 | Pandya Ashish A. | High performance IP processor |
| US20040066782A1 (en) * | 2002-09-23 | 2004-04-08 | Nassar Ayman Esam | System, method and apparatus for sharing and optimizing packet services nodes |
| US7239605B2 (en) * | 2002-09-23 | 2007-07-03 | Sun Microsystems, Inc. | Item and method for performing a cluster topology self-healing process in a distributed data system cluster |
| US20040071142A1 (en) * | 2002-10-11 | 2004-04-15 | Hitachi, Ltd. | Packet communication device |
| US20080317055A1 (en) * | 2003-02-03 | 2008-12-25 | Jerker Mattias Zetterlund | Shared risk group handling within a media gateway |
| US20050053073A1 (en) * | 2003-09-03 | 2005-03-10 | Andiamo Systems, Inc. A Delaware Corporation | Switch port analyzers |
| US7424025B2 (en) * | 2003-10-01 | 2008-09-09 | Santera Systems, Inc. | Methods and systems for per-session dynamic management of media gateway resources |
| US20050243716A1 (en) * | 2004-05-03 | 2005-11-03 | Bitar Nabil N | Systems and methods implementing 1‘and N:1 line card redundancy |
| US20050281190A1 (en) * | 2004-06-17 | 2005-12-22 | Mcgee Michael S | Automated recovery from a split segment condition in a layer2 network for teamed network resources of a computer systerm |
| US20060143309A1 (en) * | 2004-12-29 | 2006-06-29 | Mcgee Michael S | Verifying network connectivity |
Also Published As
| Publication number | Publication date |
|---|---|
| US7273601B2 (en) | 2007-09-25 |
| US7658910B2 (en) | 2010-02-09 |
| US20030012730A1 (en) | 2003-01-16 |
| US20080085984A1 (en) | 2008-04-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Hunter et al. | Polymer‐supported radiopharmaceuticals:[131I] MIBG and [123I] MIBG | |
| Pirkle et al. | Useful and easily prepared chiral stationary phases for the direct chromatographic separation of the enantiomers of a variety of derivatized amines, amino acids, alcohols, and related compounds | |
| Gahan et al. | Encapsulated metal ions: synthesis, structure, and spectra of nitrogen-sulfur ligand atom cages containing cobalt (III) and cobalt (II) | |
| TWI640496B (en) | Method for producing 2-fluoro-4-borono-l-phenylalanine, and a derivative of 2-fluoro-4-borono-l-phenylalanine | |
| Shiue et al. | No-carrier-added fluorine-18-labeled N-methylspiroperidol: synthesis and biodistribution in mice | |
| US9550704B2 (en) | Method for synthesizing radiopharmaceuticals using a cartridge | |
| CA2345710C (en) | Preparation of radiolabelled haloaromatics via polymer-bound intermediates | |
| US20100274052A1 (en) | Preparation of radiolabelled haloaromatics via polymer-bound intermediates | |
| AU640350B2 (en) | Novel tc-99m complexes | |
| WO2006127024A1 (en) | Reagents and methods for labeling terminal olefins | |
| US6461585B1 (en) | Preparation of radiolabelled haloaromatics via polymer-bound intermediates | |
| EP2192928A2 (en) | Perfluoro-aryliodonium salts in nucleophilic aromatic 18f-fluorination | |
| CN106536490A (en) | Radioiodinated compounds | |
| Culbert et al. | Polymer-supported radiopharmaceuticals: 123I-and 131I-labelled N-isopropyl-4-iodoamphetamine | |
| US20110020206A1 (en) | Adsorbent, preparation method thereof and sr-90/y-90 generator using the same | |
| CN105330664B (en) | A kind of synthetic method of Xi Gelieting impurity | |
| Jeong et al. | Synthesis of no‐carrier‐added [18F] fluoroacetate | |
| CN110698515B (en) | Marked fatty acid derivative containing isocyano group and application thereof | |
| CN109320500B (en) | A kind of18F-labeled benzimidazole compound and preparation method and application thereof | |
| Koren et al. | Synthesis and initial in vitro characterization of 6‐[18F] fluoro‐3‐(2 (S)‐azetidinylmethoxy) pyridine, a high‐affinity radioligand for central nicotinic acetylcholine receptors | |
| EP0546233B1 (en) | Method for synthesis and 99mTc labelling of 2-alkoxyisobutylisonitrile | |
| Hörnfeldt et al. | Synthesis of 11 C-Labelled Haloalkanonitriles and Examples of their Use in Some Alkylation Reactions | |
| Kowai et al. | Synthesis of polymer-bound 6-mercuric carboxylate DOPA precursors and solid phase labelling method of 6-radioiodinated L-DOPA | |
| WO2006121035A1 (en) | Process for producing organic compound labeled with radioactive halogen | |
| Stephan et al. | Binding and extraction of pertechnetate and perrhenate by azacages |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| STCB | Information on status: application discontinuation |
Free format text: ABANDONED -- FAILURE TO RESPOND TO AN OFFICE ACTION |