WO1994019321A1 - Inhibitors of hiv reverse transcriptase - Google Patents
Inhibitors of hiv reverse transcriptase Download PDFInfo
- Publication number
- WO1994019321A1 WO1994019321A1 PCT/US1994/001694 US9401694W WO9419321A1 WO 1994019321 A1 WO1994019321 A1 WO 1994019321A1 US 9401694 W US9401694 W US 9401694W WO 9419321 A1 WO9419321 A1 WO 9419321A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- carboxamide
- chloro
- alkyl
- substituted
- unsubstituted
- Prior art date
Links
- 0 N*1CCOCC1 Chemical compound N*1CCOCC1 0.000 description 4
- DZQSKXVGGYBKGO-UHFFFAOYSA-N C(c([nH]c1c2cccc1)c2Sc1ccccc1)=C/c1nc2ccccc2[o]1 Chemical compound C(c([nH]c1c2cccc1)c2Sc1ccccc1)=C/c1nc2ccccc2[o]1 DZQSKXVGGYBKGO-UHFFFAOYSA-N 0.000 description 1
- DQTXYMZMNGNTKN-UHFFFAOYSA-N CN(C(c([nH]c1ccccc11)c1Sc1ccccc1)=O)OC Chemical compound CN(C(c([nH]c1ccccc11)c1Sc1ccccc1)=O)OC DQTXYMZMNGNTKN-UHFFFAOYSA-N 0.000 description 1
- URJBJLXZJZPBPZ-LLVKDONJSA-N C[C@H](CNC(c([nH]c(cc1)c2cc1Cl)c2Sc1ccccc1)=O)O Chemical compound C[C@H](CNC(c([nH]c(cc1)c2cc1Cl)c2Sc1ccccc1)=O)O URJBJLXZJZPBPZ-LLVKDONJSA-N 0.000 description 1
- AYQRKTDZGBJKIT-OAHLLOKOSA-N C[C@H]1Nc(ccc(Cl)c2)c2C(Sc2ccccc2)=C1C(N(C)Cc1ccc[o]1)=O Chemical compound C[C@H]1Nc(ccc(Cl)c2)c2C(Sc2ccccc2)=C1C(N(C)Cc1ccc[o]1)=O AYQRKTDZGBJKIT-OAHLLOKOSA-N 0.000 description 1
- KFQBSPAPUUVFBQ-UHFFFAOYSA-N NC(c([nH]c(cc1)c2cc1Cl)c2S(c1ccccc1)=O)=O Chemical compound NC(c([nH]c(cc1)c2cc1Cl)c2S(c1ccccc1)=O)=O KFQBSPAPUUVFBQ-UHFFFAOYSA-N 0.000 description 1
- SEAXPFZOFZLHLQ-UHFFFAOYSA-N NC(c([nH]c(cc1)c2cc1Cl)c2Sc1ccccc1)=O Chemical compound NC(c([nH]c(cc1)c2cc1Cl)c2Sc1ccccc1)=O SEAXPFZOFZLHLQ-UHFFFAOYSA-N 0.000 description 1
- QUTSOBJECDVGKU-UHFFFAOYSA-N O=C(c([nH]c(cc1)c2cc1Cl)c2Sc1ccccc1)NCc1ccncc1 Chemical compound O=C(c([nH]c(cc1)c2cc1Cl)c2Sc1ccccc1)NCc1ccncc1 QUTSOBJECDVGKU-UHFFFAOYSA-N 0.000 description 1
- DIJUJUYSLCHBQA-UHFFFAOYSA-N O=Cc([nH]c1ccccc11)c1Sc1ccccc1 Chemical compound O=Cc([nH]c1ccccc11)c1Sc1ccccc1 DIJUJUYSLCHBQA-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/12—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/30—Indoles; Hydrogenated indoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/30—Indoles; Hydrogenated indoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to carbon atoms of the hetero ring
- C07D209/42—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention is concerned with compounds which inhibit the reverse transcriptase encoded by human immunodeficiency virus ( HIV) or pharmaceutically acceptable salts thereof and are of value in the prevention of infection by HIV, the treatment of infection by HIV and the treatment of the resulting acquired immune deficiency syndrome (AIDS). It also relates to pharmaceutical compositions containing the compounds and to a method of use of the present compounds and other agents for the treatment of AIDS and viral infection by HIV.
- HIV human immunodeficiency virus
- AIDS acquired immune deficiency syndrome
- a retroviras designated human immunodeficiency virus is the etiological agent of the complex disease that includes progressive destruction of the immune system (acquired immune deficiency syndrome; AIDS) and degeneration of the central and peripheral nervous system. This vims was previously known as LAV, HTLV- III, or ARV.
- retrovirus replication is reverse transcription of the RNA genome by a virally encoded reverse transcriptase to generate DNA copies of HIV sequences, a required step in viral replication. It is known that some compounds are reverse transcriptase inhibitors and are effective agents in the treatment of AIDS and similar diseases, e.g., azidothymidine or AZT.
- Nucleotide sequencing of HIV shows the presence of a pd. gene in one open reading frame [Ratner, L. et al., Nature, 313,
- Amino acid sequence homology provides evidence that the pol sequence encodes reverse transcriptase, an endonuclease and an HIV protease [Toh, H. et al., EMBO J. 4, 1267 (1985); Power, M.D. et al., Science, 231, 1567 (1986); Pearl, L.H. et al., Nature 329, 351 (1987)].
- the compounds of this invention are inhibitors of HIV reverse transcriptase. Furthermore, the compounds of the present invention do not require bioactivation to be effective.
- X is -H, -Cl, -F, -Br, -NO 2 , -CN, -OR 2 , -NR 2 R 2 , -NHSO 2 - C 1 -3 alkyl, or -NHCO-C 1 -3 alkyl;
- Y is -S(O) n - or -O-, wherein n is zero, 1 or 2;
- heterocycle unsubstituted or substituted with one or more of: a) -C 1 -5 alkyl, unsubstituted or substit uted with one or more of:
- W is O, S, -N-CN, or -N-OR 2 ,
- aryl unsubstituted or substituted with one or more of: a) -C 1 -5 alkyl, unsubstituted or substituted with one or more of: i) -OH or
- heterocycle unsubstituted or substituted with one or more of: i) -C 1 -5 alkyl,
- aryl unsubstituted or substituted with one or more of: i) -C 1 -5 alkyl,
- R 2 is hydrogen or C 1 -3 alkyl
- Z is not -CH 2 -SO-Ph, -COH, or a pharmaceutically acceptable salt or ester thereof.
- X is -H, -Cl or -F
- Y is -S(O) n -;
- R is -Ph, -tolyl, 3-Cl-phenyl, 2-pyridyl or 2-thiazolyl;
- R 6 is -H; and Z is
- X is -H or -Cl
- Y is -S(O) n -;
- R is -Ph, -tolyl, 3-Cl-phenyl or 2-thiazolyl
- R 6 is -H
- R 2 is -H and W is -O, -S or -NCN, or
- a sub-class of compounds within this class is further limited to compounds wherein
- X is -Cl
- Y is -S(O) n -;
- n 1 or 2;
- R is -Ph, 3-Cl-phenyl or 2-thiazolyl
- R 6 is -H
- R 2 is -H, and W is -O, -S or -NCN; and R 3 is
- a second embodiment of this invention encompasses compounds of formula A wherein X is selected from the group consisting of:
- R 3 is selected from the group consisting of:
- a fourth embodiment of this invention encompasses compounds of formula A wherein Y is -S(O) n - and R is -NR 2 R 3 .
- the compounds of the present invention may have asymmetric centers and occur as racemates, racemic mixtures, individual diastereomers, or enantiomers, with all isomeric forms being included in the present invention.
- variable e.g., aryl, heterocycle, R 1 , R 2 , R 3 , etc.
- its definition on each occurrence is independent of its definition at every other occurrence.
- combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- alkyl is intended to include both branched- and straight-chain saturated aliphatic
- alkoxy represents an alkyl group of indicated number of carbon atoms attached through an oxygen bridge.
- Halogen or “halo” as used herein, means fluoro, chloro, bromo and iodo.
- aryl is intended to mean any stable monocyclic, bicyclic or tricyclic carbon ring of up to
- heterocycle or heterocyclic represents a stable 5- to 7-membered monocyclic or stable 8- to 11-membered bicyclic heterocyclic ring which is either saturated or unsaturated, and which consists of carbon atoms and from one to three heteroatoms selected from the group consisting of N, O and S, and wherein the nitrogen and sulfur heteroatoms may optionally be oxidized, and the nitrogen heteroatom may optionally be quaternized, and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- the heterocyclic ring may be attached at any heteroatom or carbon atom which results in the creation of a stable structure.
- heterocyclic elements include piperidinyl, piperazinyl, 2-oxopiperazinyl, 2-oxopiperidinyl, 2- oxopyrrolidinyl, 2-oxoazepinyl, azepinyl, pyrrolyl, 4-piperidonyl, pyrrolidinyl, pyrazolyl, pyrazolidinyl, imidazolyl, imidazolinyl, imidazolidinyl, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, oxazolyl, oxazolidinyl, isoxazolyl, isoxazolidinyl, morpholinyl, thiazolyl, thiazolidinyl, isothiazolyl, quinuclidinyl, isothiazolid
- the pharmaceutically-acceptable salts of the novel compounds of this invention that are capable of salt formation include the conventional non-toxic salts or the quaternary ammonium salts of these compounds, which are formed, e.g., from inorganic or organic acids or bases.
- acid addition salts include acetate, adipate, alginate, aspartate, benzoate, benzenesulfonate, bissulfate, butyrate, citrate, camphorate, camphorsulfonate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, fumarate, glucoheptanoate, glycerophosphate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, lactate, maleate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, oxalate, pamoate, pectinate, persulfate, 3-phenylpropionate, picrate, pivalate, propionate, succinate, tartrate, thiocyanate, to
- the basic nitrogen-containing groups may be quatemized with such agents as lower alkyl halides, such as methyl, ethyl, propyl, and butyl chloride, bromides and iodides; dialkyl sulfates like dimethyl, diethyl, dibutyl; and diamyl sulfates, long chain halides such as decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides, aralkyl halides like benzyl and phenethyl bromides and others.
- Esters are also encompassed by the present invention, and include those which would readily occur to the skilled artisan, for example, C 1-4 alkyl esters.
- Schemes I-VIII for preparing the novel compounds of this invention are presented below.
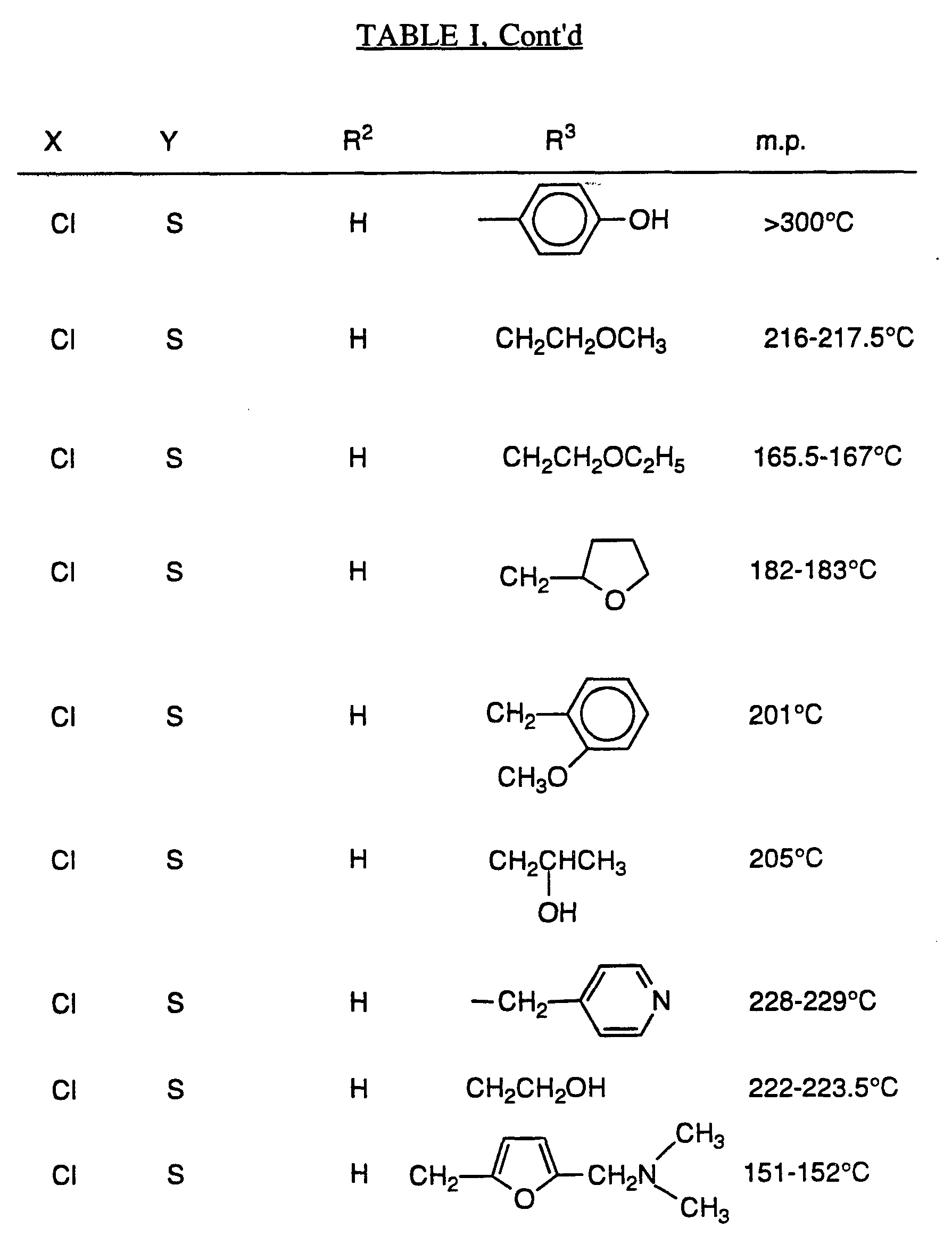
- Tables I - VII which follow the schemes illustrate the compounds that can be synthesized by Schemes I-VIII, but Schemes I - VIII are not limited by the compounds in the tables nor by any particular substituents employed in the schemes for illustrative purposes.
- the examples specifically illustrate the application of the following schemes to specific compounds.
- Scheme I, below, is a general route for synthesizing, e.g., the
- amide in can be produced directly from II by treatment with BOP reagent (benzotriazol-1- yloxytris-(dimethylamino)-phosphonium hexafluorophosphate) in the presence of the desired primary or secondary amine and triethylamine, in a solvent such as dimethyl-formamide.
- BOP reagent benzotriazol-1- yloxytris-(dimethylamino)-phosphonium hexafluorophosphate
- carboxyl group activating reagents such as 1,1'-carbonyl-diimidazole can also be used for this step.
- Saponification of ethyl 5-chloro-3-benzylindole-2- carboxylate (prepared as described below) by methods familiar to those skilled in the art, yields 5-chloro-3-benzylindole-2-carboxylic acid, which can be converted to the desired amides in the manner described for the synthesis of amides III.
- 3-phenylthioindole-2-carboxylic acid II can be converted to the corresponding acid chloride with oxalyl chloride in refluxing chloroform, and reacted with an alcohol to give the ester IV.
- la may be converted to IV by reaction with N-phenylmio-succinimide in chloroform at room
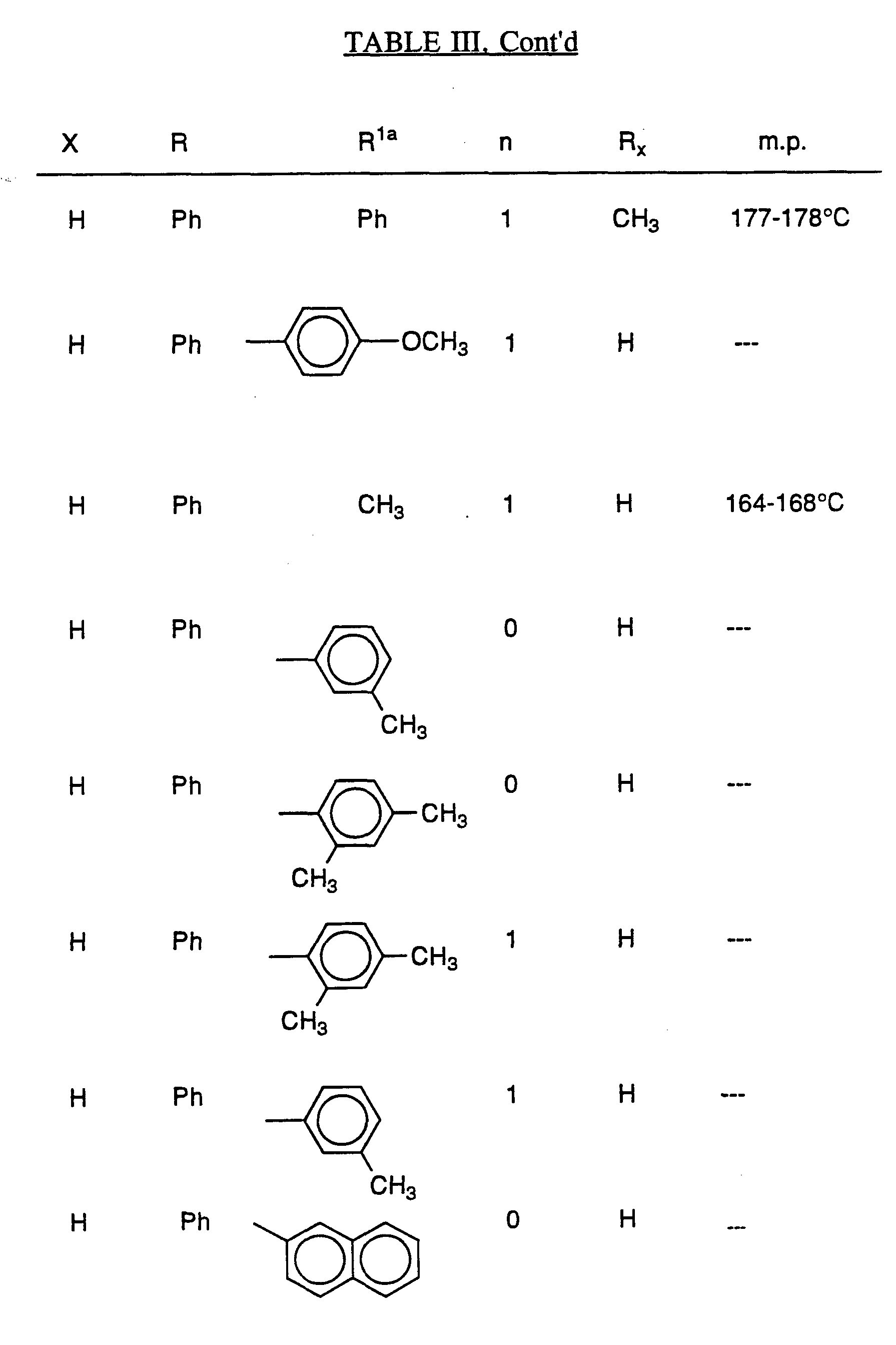
- Scheme III is a general route for synthesizing, e.g., the compounds shown in Table III, infra.
- the substituent groups employed in Scheme III correspond to the substituent groups as defined in Table III, but Scheme m is not limited by the defined substituents or compounds of Table III .
- Compound VIII can be N- alkylated, if desired, by methods familiar to those trained in the art, e.g., by treatment with sodium hydride in dimethylformamide at 0°C in the presence of an alkylating agent such as iodomethane, to give compound IX. Thereafter, compound VIII (or IX) is treated with one equivalent of peracid such as monoperoxyphthahc acid, magnesium salt (MMPP), or meta-chloroperoxy-benzoic acid in methanol or
- Schemes IV-A and IV-B show a general route for synthesizing, e.g., the compounds shown in Tables IV-A and IV-B, infra.
- the substituent groups employed in Schemes IV-A and IV-B, respectively, correspond to the substituent groups as defined in Tables IV-A and IV-B, respectively, but Schemes IV-A and IV-B are not limited by the defined substituents or compounds of Tables IV-A and
- 3-phenylthioindole- 2-carboxamides XI can be reduced to primary or secondary amines XII by reaction with an excess of borane-dimethylsulfide complex in refluxing tetrahydrofuran for 6-24 hours.
- the primary amine XII-A can be acylated with an acid chloride, such as benzoyl chloride, in chloroform in the presence of pyridine, to give the amide XIII.
- Scheme V is a general route for synthesizing, e.g., the compounds shown in Table V-A and Table V-B, infra.
- the substituent groups employed in Scheme V correspond to the substituent groups as defined in Tables V-A and V-B, but Scheme V is not limited by the defined substituents or compounds of Tables V-A and V-B.
- commercially available 2-methyl- indole XTV can be treated with sodium hydride in dimethylformamide in the presence of an aryldisulfide such as phenyldisulfide to give compound XV.
- Compound XV can be converted to the monoanion with n-butyl-lithium in tetrahydrofuran at -78°C, and then reacted with carbon dioxide to give carboxylate XVI.
- the dianion formed by the reaction of XVI with t-butyllithium could be reacted with an isocyanate, such as phenyliso-cyanate, to give a mixture of monoacylated product and diacylated product XVII (see Table V-B).
- the dianion formed by the reaction of XVI with t-butyllithium could be reacted with an N-methoxy-N-methyl amide such as N-methoxy-N- methyl-furan-2-carboxamide (prepared in a manner familiar to those skilled in the art, e.g., by the methods described in Scheme 1) to produce ketones XVIII.
- N-methoxy-N-methyl amide such as N-methoxy-N- methyl-furan-2-carboxamide (prepared in a manner familiar to those skilled in the art, e.g., by the methods described in Scheme 1) to produce ketones XVIII.
- the methodology described above is essentially that used by A. J. Katritsky and K. Akutagawa to prepare 2-indoleacetic acids, and is published in J. Am. Chem. Soc., 108, 6808 (1986).
- Scheme VI is a general route for synthesizing, e.g., the compounds shown in Table VI, infra.
- the substituent groups employed in Scheme VI correspond to the substituent groups as defined in Table VI, but Scheme VI is not limited by the defined substituents or compounds of Table VI.
- N-methoxy-N-methyl-3-phenyl- thioindole-2-carboxamide XIX (or N-methoxy-N-methyl-5-chloro-3- phenylthioindole-2-carboxamide) (prepared as in Scheme 1) can be reacted with Grignard reagents (wherein R 1 is not hydrogen) such as phenylmagnesium chloride, in tetrahyrodrofuran at -78°C to 20°C for 18-48 hours, or XIX can be reacted with other organometallic reagents well known in the art to one of ordinary skill, to produce ketones XX.
- Grignard reagents such as phenylmagnesium chloride, in tetrahyrodrofuran at -78°C to 20°C for 18-48 hours
- XIX can be reacted with other organometallic reagents well known in the art to one of ordinary skill, to produce
- N-methoxy-N-methyl-3- phenylthioindole-2-carboxamide XXI can be reduced to aldehyde XXII with lithium aluminum hydride in tetrahydrofuran at 0°C to 20°C for 2- 4 hours.
- Aldehyde XXII could be reacted with the hthium salt of [(benzoxal-2-yl)methyl]diethyl-phosphonate to produce olefin XXEI, which is then hydrogenated in the presence of 10% palladium on charcoal in methanol under one atmosphere of hydrogen to give compound XXIV.
- the compound 5-chloro-2-cyano-3-phenylthioindole in Table VII can be prepared by dehydro-sulfurization of 5-chloro-3- phenyl-thioindole-2-thiocarboxamide with, e.g., Hg(OAc) 2 .
- compounds of formula A where Y is -SO- or -SO 2 - can be synthesized by treatment of compounds where Y is -S- with a suitable oxidizing agent such as, for example, meta-chloroperoxybenzoic acid (MCPBA), sodium periodate or hydrogen peroxide in an appropriate solvent such as MeOH, CHCl 3 or acetic acid, or potassium persulfate in a solvent such as MeOH/H 2 O.
- MCPBA meta-chloroperoxybenzoic acid
- MCPBA meta-chloroperoxybenzoic acid
- sodium periodate or hydrogen peroxide in an appropriate solvent such as MeOH, CHCl 3 or acetic acid
- potassium persulfate in a solvent such as MeOH/H 2 O.
- the compounds of the present invention are useful in the inhibition of HIV reverse transcriptase, the prevention or treatment of infection by the human immunodeficiency virus (HIV) and the treatment of consequent pathological conditions such as ADDS.
- Treating AIDS or preventing or treating infection by HIV is defined as including, but not limited to, treating a wide range of states of HIV infection: AIDS, ARC (AIDS related complex), both symptomatic and asymptomatic, and actual or potential exposure to HIV.
- the compounds of this invention are useful in treating infection by HIV after suspected past exposure to HIV by, e.g., blood transfusion, organ transplant, exchange of body fluids, bites, accidental needle stick, or exposure to patient blood during surgery.
- the compounds of this invention are also useful in the preparation and execution of screening for antiviral compounds.
- the compounds of this invention are useful for isolating enzyme mutants, which are excellent screening tools for more powerful antiviral compounds.
- the compounds of this invention are useful in establishing or determining the binding site of other antivirals to HIV reverse transcriptase e.g., by competitive inhibition.
- the compounds of this invention are commercial products to be sold for these purposes.
- the compounds of the present invention may be administered orally, parenterally (including subcutaneous injections, intravenous,
- inhalation spray or rectally, in dosage unit formulations containing conventional non-toxic pharmaceutically-acceptable carriers, adjuvants and vehicles.
- a pharmaceutical composition comprising a pharmaceutical carrier and a therapeutically- effective amount of a compound of the present invention.
- compositions may be in the form of orally-administrable suspensions or tablets; nasal sprays; sterile injectable preparations, for example, as sterile injectable aqueous or oleagenous suspensions or suppositories.
- compositions When administered orally as a suspension, these compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may contain microcrystalline cellulose for imparting bulk, alginic acid or sodium alginate as a suspending agent, methylcellulose as a viscosity enhancer, and
- these compositions may contain microcrystalline cellulose, dicalcium phosphate, starch, magnesium stearate and lactose and/or other excipients, binders, extenders, disintegrants, diluents and lubricants known in the art.
- compositions When administered by nasal aerosol or inhalation, these compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other solubilizing or dispersing agents known in the art.
- the injectable solutions or suspensions may be formulated according to known art, using suitable non-toxic, parenterally- acceptable diluents or solvents, such as mannitol, 1,3-butanediol, water, Ringer's solution or isotonic sodium chloride solution, or suitable dispersing or wetting and suspending agents, such as sterile, bland, fixed oils, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- suitable non-toxic, parenterally- acceptable diluents or solvents such as mannitol, 1,3-butanediol, water, Ringer's solution or isotonic sodium chloride solution, or suitable dispersing or wetting and suspending agents, such as sterile, bland, fixed oils, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- these compositions When rectally administered in the form of suppositories, these compositions may be prepared by mixing the drug with a suitable non-irritating excipient, such as cocoa butter, synthetic glyceride, esters, or polyethylene glycols, which are solid at ordinary temperatures, but liquidify and/or dissolve in the rectal cavity to release the drug.
- a suitable non-irritating excipient such as cocoa butter, synthetic glyceride, esters, or polyethylene glycols, which are solid at ordinary temperatures, but liquidify and/or dissolve in the rectal cavity to release the drug.
- the compounds of this invention can be administered orally to humans in a dosage range of 1 to 100 mg/kg body weight in divided doses.
- One preferred dosage range is 1 to 10 mg/kg body weight orally in divided doses.
- Another preferred dosage range is 1 to 20 mg/kg body weight orally in divided doses.
- the present invention is also directed to combinations of the HIV reverse transcriptase inhibitor compounds with one or more agents useful in the treatment of AIDS.
- the compounds of this invention can be administered in combination with other compounds that are HIV reverse transcriptase inhibitors, and/or with compounds that are HIV protease inhibitors.
- the compounds of this invention may be effectively administered, whether at periods of preexposure and/or post-exposure, in combination with effective amounts of the AIDS antivirals, such as those in the following Table VIII.
- dosage levels of HIV protease inhibitors of the order of 0.02 to 5.0 or 10.0 grams-per-day are useful in the treatment or prevention of the above-indicated conditions, with oral doses two-to- five time higher.
- infection by HIV is effectively treated by the administration of from 10 to 50 milligrams of the HIV protease inhibitor per kilogram of body weight from one to three times per day. It will be understood, however, that the specific dose level and
- frequency of dosage for any particular patient may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the host undergoing therapy.
- Dosages of HIV reverse transcriptase inhibitors when used in a combination treatment with compounds of the instant invention, are comparable to those dosages specified above for the instant compounds.
- L-697,661 is 3-([(4,7-dichloro-1,3-benzoxazol-2-yl)-methyl]-amino)-5-ethyl-6- methyl-pyridin-2(1H)-one
- L-696,229 is 3-[2-(1,3-benzoxazol-2- yl)ethyl]-5-ethyl-6-methyl-pyridin-2(1H)-one
- L-735,524 is an HIV protease inhibitor with the chemical name N-(2(R)-hydroxy-1(S)- indanyl)-2(R)-phenylmethyl-4-(S)-hydroxy-5-(1-(4-(3-pyridyl-methyl)- 2(S)-N'-(t-butylcarboxamido)piperazinyl))pentaneamide
- L-738,372 is 6-chloro-4(S)-cyclopropyl-3,4-d
- the assay measures the incorporation of tritiated deoxy- guanosine monophosphate by recombinant HIV reverse transcriptase ( HIV RTR) (or other RT) into acid-precipitable cDNA at the Km values of dGTP and poly r(C)•oligo d(G)12-18.
- HIV RTR HIV reverse transcriptase
- the inhibitors of the present invention inhibit this incorporation.
- the tubes were cooled in ice for 5 minutes. Ice-cold 13% TCA containing 10 mM NaPPi (200 ⁇ l) are added and the mixture incubated on ice for 30 minutes. The precipitated cDNA is removed by filtration using presoaked glass filters [TCA, NaPP i ]. The precipitate is then washed with IN HCI, 10 mM NaPP i .
- the filter discs are then counted in a scintillation counter.
- Calculated IC 50 values for the tested compounds of this invention vary from about 3 nM to more than 300 ⁇ M.
- the IC 50 values of the most preferred compounds range from about 3 nM to about 35 nM.
- MT cells were infected at Day O at a concentration of
- the mixture was incubated overnight at 37 °C in 5% CO 2 atmosphere.
- the settled cells were resuspended and a 125 ⁇ l harvested into a separate microtiter plate. After the settling of the cells, the plates were frozen for subsequent assay of the supernatant for HIV p24 antigen.
- the concentration of HIV p24 antigen was measured by an enzyme immunoassay, described as follows. Aliquots of p24 antigen to be measured were added to microwells coated with a monoclonal antibody specific for HIV core antigen. The microwells were washed at this point, and at other appropriate steps that follow. Biotinylated HIV- specific antibody was then added, followed by conjugated streptavidin- horseradish peroxidase. A color reaction occurs from the added hydrogen peroxide and tetramethylbenzidine substrate. Color intensity is proportional to the concentration of HIV p24 antigen.
- the cell culture inhibitory concentration (CIC 95 ) for each compound is defined as that concentration which inhibited by greater than 95% the spread of infection, as assessed by a greater than 95% reduction in p24 antigen production relative to untreated controls.
- the tested compounds of the present invention were found to have CIC 95 values ranging from about 3 nM to about 400 nM for preferred species, and up to about 40 ⁇ M for others.
- the aqueous layer was separated and the pH adjusted to pH1 with 10% aqueous hydrochloric acid.
- the aqueous phase was extracted with ethyl acetate, and the ethyl acetate extract was washed with water and saturated brine, and dried over magnesium sulfate.
- the crude product was recrystallized from ethyl acetate in hexane to afford the title compound as an off-white solid.
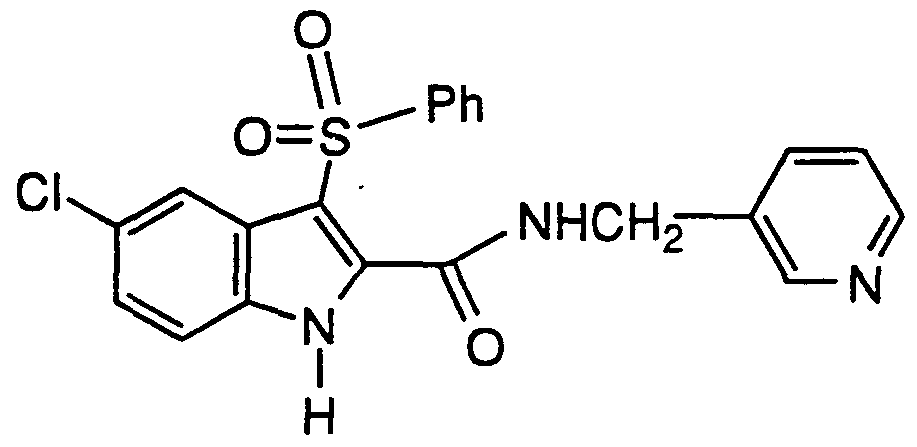
- Step B N-(3-pyridylmethyl)-5-chloro-3-phenylthioindole-2- carboxamide
- Benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorphosphate (0.73 g, 1.6 mmol) was added to a solution of 5- chloro-3-phenylthio-indole-2-carboxylic acid (0.50 g, 1.6 mmol), 3- aminomethylpyridine (0.35 g, 3.2 mmol) and triethylamine (0.50 mL, 3.2 mmol) in degassed dimethylformamide (25 mL).
- the reaction was stirred at room temperature overnight.
- the precipitated product was filtered and the filter cake washed well with water.
- the solid was triturated with 30% ethyl acetate in hexane, filtered and dried at 60°C in vacuo for 72 h.
- the title compound was obtained as an off-white solid, mp 240-241°C.
- Oxalyl chloride (0.70 mL, 9.6 mmol) was added to a solution of 5-chloro-3-phenylthioindole-2-carboxylic acid (0.97 g, 3.2 mmol) in chloroform (50 mL) under nitrogen. The reaction was refluxed for 3 h, cooled and reduced to dryness in vacuo. The resulting solid was dissolved in chloroform and added to methanol at 0°C. The methanol was removed in vacuo and the crude product chromato- graphed on silica gel with 20% ethyl acetate in hexane. The title compound was obtained as a solid, mp 193-196°C.
- Step D 2-Phenylsulfinylmethyl-3-phenylthioindole
- the title compound was prepared from 3-phenylthioindole- 2-carboxylic acid (prepared according to the procedure described by Atkinson, J.G. et al., Synthesis, p. 480-481 (1988), (4.01 g, 0.015 mol), ammonia (large excess), and benzotriazol-1-yloxytris(dimethyl- amino)phosphonium hexafluorphosphate (7.2 g, 0.016 mol) in
- the title compound was prepared from furan-3-carboxylic acid (3.4 g, 0.030 mol), N,0-dimethylhydroxylamine hydrochloride hydrochloride (2.9 g, 0.030 mol) triethylamine (8.3 mL, 0.060 mol) and benzotriazol-1-yloxytris(dimethylamino)phosphonium
- Step B 2-(2-Oxo-2-furan-3-yl)ethyl-3-phenylthioindole
- the title compound was prepared from N-methoxy-N- methylfuran-3-carboxamide (0.32 g, 2.1 mmol), and 2-methyl-3- phenylthioindole (0.50 g, 2.1 mmol) according to the general procedure described in Example 6 for the preparation of 2-(N-phenylacetamido)- 3-phenyl-thioindole.
- the crude product was chromatographed on silica gel with chloroform. The title compound was obtained as a pale yellow solid, mp 127-129°C.
- Step A N-Methoxy-N-methyl-5-chloro-3-phenylthioindole-2- carboxamide
- the title compound was prepared from 5-chloro-3-phenyl- thioindole-2-carboxylic acid (1.0 g, 3.30 mmol)) N,0-dimethyl- hydroxylamine hydrochloride (0.64 g, 6.6 mmol), triethylamine (1.0 mL, 7 mmol) and benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorphosphate (1.64 g, 3.6 mmol) in dimethylformamide according to the general procedure described in Example 1 for the preparation of N-(3-pyridylmethyl)-5-chloro-3-phenylthio-2- carboxamide.
- Step B 2-Benzoyl-5-chloro-3-phenylthioindole
- N-Methoxy-N-methyl-5-chloro-3-phenylthioindole-2- carboxamide (0.24 g, 0.69 mmol) was dissolved in dry tetrahydrofuran (5 mL) and cooled to -78°C under nitrogen.
- phenylmagnesium chloride in tetrahydrofuran (0.81 mL, 2M) was added via syringe and the reaction warmed to 20°C overnight. Water and ethyl acetate were added to the reaction and then separated. The organic phase was washed with water, 5% aqueous hydrochloric acid, saturated sodium bicarbonate, saturated brine, and dried over
- the title compound was prepared from 3-phenylthioindole- 2-carboxylic acid (1.0 g, 3.7 mmol), N,O-dmiethylhydroxylamine hydrochloride (0.54 g, 5.5 mmol), triethylamine (1.5 mL, 11 mmol) and benzotriazol-1-yloxytris(dimethylamino)phosphonium
- Step B 3-Phenylthioindole-2-carboxaldehyde
- N-Methoxy-N-methyl-3-phenylthioindole-2-carboxamide (1.57 g, 5.26 mmol) was dissolved in tetrahydrofuran (150 mL) and cooled to 0°C under nitrogen.
- a solution of lithium aluminum hydride in tetrahydrofuran (5.76 mL, IM) was added slowly via syringe and the reaction stirred a total of 1.5 h.
- Ethyl acetate (30 mL) was added, followed by saturated sodium potassium tartrate solution. The layers were separated and the organic phase washed with saturated brine and dried over magnesium sulfate. Filtration and evaporation of solvent gave the title compound as a yellow solid.
- Step C trans-2-(2-Benzoxazol-2-ylethenyl)-3-phenylthioindole
- the title compound was prepared according to the procedure described in Example 1, Step B, except substituting 3- aminopyridine for 3-aminomethyl-pyridine.
- the dimethylformamide was removed in vacuo, and the residue triturated with 1:1 ethyl acetate- hexane and filtered. Chromatography on silica gel with 40% ethyl acetate in hexane gave the title compound, mp 255-256°C.
- the title compound was prepared according to the procedure described in ⁇ xample 1, Step B; except substituting an excess of ammonia gas for 3-aminomethylpyridine and triethylamine.
- the dimethylformamide and excess ammonia were removed in vacuo and the residue partitioned between ethyl acetate and 10% hydrochloric acid.
- the organic phase was washed with water, 5% sodium hydroxide and saturated brine, and then dried over magnesium sulfate. Filtration and evaporation gave a crude product which was chromatographed on silica gel with 30% ethyl acetate in hexane.
- the title compound was obtained as a white solid mp 213-215°C.
- the title compound was prepared according to the procedure described for 5-chloro-3-phenyl-thioindole-2- thiocarboxamide except substituting N-2-furanylmethyl-5-chloro-3- phenylthioindole-2-carboxamide for 5-chloro-3-phenylthioindole-2- carboxamide.
- the crude product was chromatographed on silica gel with 3% ethyl acetate in hexane. The title compound was obtained as a bright yellow solid, mp 143-144°C.
- the title compound was prepared according to the procedure described in Example 1, Step B, except substituting 2- pyridylmethylamine for 3-aminomethylpyridine.
- the dimethylformamide was removed in vacuo and the residue triturated first with 30% ethyl acetate in hexane, then with acetonitrile.
- the title compound was obtained as a white solid, mp 209-210°C.
- Step 3 Preparation of N-(3-methoxy-4-pyridylmethyl)-5-chloro-3- phenylthioindole-2-carboxamide
- Step A 5-chloro-3-phenylsulfonylindole-2-carboxylic acid
- Step B Product of reaction of 5-chloro-3-phenyl-sulfonylindole-
- Step C N-(2,6-difluorobenzyl)-5-chloro-3-phenyl-sulfonylindole-
- 2,6-Difluorobenzylamine (0.430 g, 3.0 mmol) was added dropwise to the solution of the "acid chloride equivalent" from Step B (0.354 g, 1.0 mmol) in tetrahydrofuran solution (10 mL) cooled in an ice-acetone bath. The reaction mixture was allowed to warm to room temperature and left overnight. For work-up, ethyl acetate and water were added. The ethyl acetate phase was washed well with dilute hydrochloride acid, saturated aqueous sodium bicarbonate, and brine. After drying over magnesium sulfate and evaporation of the solvent, the residue was slurried with ethyl acetate and filtered to give the title compound, mp 274-280°C.
- Step A Preparation of 5-chloro-3-phenylsulfinyl-indole-2- carboxylic acid
- Step B N-(4-pyridylmethyl)-5-chloro-3-phenylsulfinylindole-2- carboxamide
- Step C Product of Reaction of 5-Chloro-3-(2-thiazolyl)sulfonyl- indole-2-carboxylic Acid with Oxalyl Chloride
- Step D N-(3-methoxybenzyl)-5-chloro-3-(2-thiazolyl-sulfonyl)- indole-2-carboxamide
- Step C (0.50 g, 1.4 mmol) in tetrahydrofuran (25 mL) was added slowly to a solution of tetrahydrofuran saturated with ammonia at
- N-chlorosuccinimide (3.34 g, 25 mmol) in dry methylene chloride (30 mL), cooled in an ice bath and under an inert atmosphere, was added thiophenol (2.05 mL, 20 mmol) via syringe. After stirring for 1 hour, additional N-chlorosuccinimide (0.40 g, 3 mmol) was added. After 2.5 hours total, triethylamine (3.9 mL, 28 mmol) was added drop wise. Within 15 minutes the reaction mixture was diluted with methylene chloride, the solvent washed with dilute aq. HCI and the solvent then dried (Na2S ⁇ 4), filtered through a pad of charcoal and evaporated.
- Step B Ethyl 3-phenylthio-5-chloroindole-2-carboxylate
- Step C Ethyl 3-phenylsulfonyl-5-chloroindole-2-carboxylate
- Ethyl 3-phenylthio-5-chloroindole-2-carboxylate (642 mg, 1.94 mmol) was dissolved in chloroform (35 mL) and a dried (Na2S04) solution of m-chloroperoxybenzoic acid (55% pure, 1.30 g, 4.1 mmol) in chloroform (20 mL) was added dropwise. The progress of the oxidation was monitored by tic until complete. After 5 hours, the reaction was diluted with chloroform and some methanol and the solution washed with aq. NaHC03 and aq. Na2C ⁇ 3. The dried
- Step B N-[(1-methylimidazol-2-yl)methyl]-3-phenyl-sulfonyl-5- chloroindole-2-carboxamide
- the hydrochloride salt was obtained by addition of one equivalent of ethanolic HCI to the free base, mp 284-285°C with decomposition.
- iodomethane (.05 mL, 0.8 mmol). The reaction was stirred for three days at room temperature. The solvents were removed under reduced pressure, aq. NaHCO 3 added and the product extracted into ethyl acetate/methanol. The organic layer was dried (Na 2 SO 4 ), filtered, and the solvents evaporated. The residue was purified by chromatography and the product eluted with 3% methanol/chloroform. Appropriate fractions were combined, the solvents evaporated, and the residue triturated with methylene chloride to give pure product.
Abstract
Description




































































Claims


Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BR9405737A BR9405737A (en) | 1993-02-24 | 1994-02-15 | Compound process of inhibiting HIV transcriptase, process for preventing HIV infection or treating HIV infection, pharmaceutical composition useful for inhibiting HIV transcriptase, and useful for preventing or treating HIV infection or for the treatment of AIDS or ARC |
| AU62542/94A AU6254294A (en) | 1993-02-24 | 1994-02-15 | Inhibitors of hiv reverse transcriptase |
| EP94909663A EP0686148A4 (en) | 1993-02-24 | 1994-02-15 | Inhibitors of hiv reverse transcriptase |
| PL94310410A PL175788B1 (en) | 1993-02-24 | 1994-02-15 | Inhibitors of reverse hiv transcriptase |
| JP6519119A JPH08507067A (en) | 1993-02-24 | 1994-02-15 | HIV reverse transcriptase inhibitor |
| KR1019950703549A KR960701010A (en) | 1993-02-24 | 1995-08-23 | Inhibitors of HIV reverse transcriptase |
| NO953308A NO953308L (en) | 1993-02-24 | 1995-08-23 | Inhibitors of HIV reverse transcriptase |
| BG99879A BG62089B1 (en) | 1993-02-24 | 1995-08-23 | Inhibitors of hiv reversive transcriptase |
| FI953954A FI953954A (en) | 1993-02-24 | 1995-08-23 | Inhibitors of HIV reverse transcriptase |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US2192593A | 1993-02-24 | 1993-02-24 | |
| US021,925 | 1993-02-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994019321A1 true WO1994019321A1 (en) | 1994-09-01 |
Family
ID=21806886
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1994/001694 WO1994019321A1 (en) | 1993-02-24 | 1994-02-15 | Inhibitors of hiv reverse transcriptase |
Country Status (13)
| Country | Link |
|---|---|
| EP (1) | EP0686148A4 (en) |
| JP (1) | JPH08507067A (en) |
| KR (1) | KR960701010A (en) |
| CN (1) | CN1119856A (en) |
| AU (1) | AU6254294A (en) |
| BG (1) | BG62089B1 (en) |
| BR (1) | BR9405737A (en) |
| CA (1) | CA2156420A1 (en) |
| FI (1) | FI953954A (en) |
| HU (1) | HUT74614A (en) |
| NO (1) | NO953308L (en) |
| PL (1) | PL175788B1 (en) |
| WO (1) | WO1994019321A1 (en) |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5811425A (en) * | 1997-03-04 | 1998-09-22 | Abbott Laboratories | Heterocyclic compounds as COX-2 inhibitors |
| WO2001032621A1 (en) * | 1999-10-29 | 2001-05-10 | Wakunaga Pharmaceutical Co., Ltd. | Novel indole derivatives and drugs containing the same as the active ingredient |
| WO2003101961A1 (en) * | 2002-05-30 | 2003-12-11 | Astrazeneca Ab | Novel substituted indoles |
| EP1390029A1 (en) * | 2001-04-11 | 2004-02-25 | Idenix (Cayman) Limited | Phenylindoles for the treatment of hiv |
| EP1545510A1 (en) * | 2002-08-07 | 2005-06-29 | Idenix (Cayman) Limited | Substituted phenylindoles for the treatment of hiv |
| US6989263B1 (en) * | 1994-09-23 | 2006-01-24 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Method for identifying and using compounds that inactivate HIV-1 and other retroviruses by attacking highly conserved zinc fingers in the viral nucleocapsid protein |
| WO2006023590A1 (en) * | 2004-08-19 | 2006-03-02 | Aventis Pharmaceuticals Inc. | 3-ARYLTHIOINDOLE-2-CARBOXAMIDE DERIVATIVES AND ANALOGS THEREOF AS INHIBITORS OF CASEIN KINASE Iϵ |
| US7402672B2 (en) | 2003-12-11 | 2008-07-22 | Aventis Pharmaceuticals Inc. | Substituted 1H-pyrrolo[3,2-b, 3,2-c, and 2,3-c]pyridine-2-carboxamides and related analogs as inhibitors of casein kinase lepsilon |
| US7534809B2 (en) | 2004-09-17 | 2009-05-19 | Idenix Pharmaceuticals, Inc. | Phospho-indoles as HIV inhibitors |
| US7687535B2 (en) | 2003-05-27 | 2010-03-30 | Astrazeneca Ab | Substituted 3-sulfur indoles |
| US7709521B2 (en) | 2003-08-18 | 2010-05-04 | Astrazeneca Ab | Substituted indole derivatives for pharmaceutical compositions for treating respiratory diseases |
| US7723373B2 (en) | 2002-07-17 | 2010-05-25 | Astrazeneca Ab | Indole-3-sulphur derivatives |
| US7741360B2 (en) | 2006-05-26 | 2010-06-22 | Astrazeneca Ab | Bi-aryl or aryl-heteroaryl substituted indoles |
| US7754735B2 (en) | 2002-05-30 | 2010-07-13 | Astrazeneca Ab | Substituted indoles |
| US7781598B2 (en) | 2005-01-13 | 2010-08-24 | Astrazeneca Ab | Process for the preparation of substituted indoles |
| US7960428B2 (en) | 2006-09-29 | 2011-06-14 | Idenix Pharmaceuticals, Inc. | Enantiomerically pure phosphoindoles as HIV inhibitors |
| CN112375027A (en) * | 2020-12-07 | 2021-02-19 | 中国药科大学 | Indolesulfonamide derivative and medical application thereof |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9902455D0 (en) * | 1999-02-05 | 1999-03-24 | Zeneca Ltd | Chemical compounds |
| AU2003261415C1 (en) * | 2002-08-09 | 2010-01-14 | Merck Sharp & Dohme Corp. | Tyrosine kinase inhibitors |
| WO2004014300A2 (en) * | 2002-08-09 | 2004-02-19 | Merck & Co., Inc. | Tyrosine kinase inhibitors |
| US7642277B2 (en) | 2002-12-04 | 2010-01-05 | Boehringer Ingelheim International Gmbh | Non-nucleoside reverse transcriptase inhibitors |
| CA2716332A1 (en) * | 2008-02-22 | 2009-08-27 | Irm Llc | Compounds and compositions as modulators of gpr119 activity |
| US20130059850A1 (en) * | 2010-05-06 | 2013-03-07 | Merck Sharp & Dohme Corp. | Aza-indole derivatives useful as modulators of faah |
| CN106892854B (en) * | 2015-12-21 | 2019-04-30 | 中国海洋大学 | A kind of indole alkaloids compound and its preparation method and application |
| CN107151223B (en) * | 2016-03-10 | 2020-03-31 | 中国医学科学院药物研究所 | Application of N-alkyl indole compounds in preparation of anti-influenza virus drugs |
| CN106995400B (en) * | 2017-04-10 | 2019-08-06 | 湘潭大学 | A kind of compound and its salt and its synthetic method |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1935671A1 (en) * | 1968-04-26 | 1970-03-05 | Sumitomo Chemical Co | 2-aminomethylindoles and their salts |
| EP0000972A2 (en) * | 1977-08-29 | 1979-03-07 | Technobiotic Ltd. | 2-Substituted-3-phenylindoles, their preparation, and pharmaceutical compositions containing them |
| US4255442A (en) * | 1977-08-29 | 1981-03-10 | Schering Corporation | 2-[(Methylsulfinyl)]-3-phenylindoles |
| EP0275667A1 (en) * | 1986-12-17 | 1988-07-27 | Merck Frosst Canada Inc. | 3-hetero-substituted-N-benzyl-indoles |
| WO1988008424A1 (en) * | 1987-04-27 | 1988-11-03 | The Upjohn Company | Pharmaceutically active amines |
| WO1991005761A1 (en) * | 1989-10-16 | 1991-05-02 | Uniroyal Chemical Ltd./Uniroyal Chemical Ltee | Treatment of hiv infections and compounds useful therein |
| WO1991009849A1 (en) * | 1989-12-28 | 1991-07-11 | The Upjohn Company | Diaromatic substituted anti-aids compounds |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993005020A1 (en) * | 1991-09-06 | 1993-03-18 | Merck & Co., Inc. | Indoles as inhibitors of hiv reverse transcriptase |
-
1994
- 1994-02-15 PL PL94310410A patent/PL175788B1/en unknown
- 1994-02-15 CA CA002156420A patent/CA2156420A1/en not_active Abandoned
- 1994-02-15 WO PCT/US1994/001694 patent/WO1994019321A1/en not_active Application Discontinuation
- 1994-02-15 BR BR9405737A patent/BR9405737A/en not_active Application Discontinuation
- 1994-02-15 JP JP6519119A patent/JPH08507067A/en active Pending
- 1994-02-15 HU HU9502468A patent/HUT74614A/en unknown
- 1994-02-15 AU AU62542/94A patent/AU6254294A/en not_active Abandoned
- 1994-02-15 EP EP94909663A patent/EP0686148A4/en not_active Withdrawn
- 1994-02-15 CN CN94191586A patent/CN1119856A/en active Pending
-
1995
- 1995-08-23 NO NO953308A patent/NO953308L/en unknown
- 1995-08-23 BG BG99879A patent/BG62089B1/en unknown
- 1995-08-23 FI FI953954A patent/FI953954A/en not_active Application Discontinuation
- 1995-08-23 KR KR1019950703549A patent/KR960701010A/en not_active Application Discontinuation
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1935671A1 (en) * | 1968-04-26 | 1970-03-05 | Sumitomo Chemical Co | 2-aminomethylindoles and their salts |
| EP0000972A2 (en) * | 1977-08-29 | 1979-03-07 | Technobiotic Ltd. | 2-Substituted-3-phenylindoles, their preparation, and pharmaceutical compositions containing them |
| US4255442A (en) * | 1977-08-29 | 1981-03-10 | Schering Corporation | 2-[(Methylsulfinyl)]-3-phenylindoles |
| EP0275667A1 (en) * | 1986-12-17 | 1988-07-27 | Merck Frosst Canada Inc. | 3-hetero-substituted-N-benzyl-indoles |
| WO1988008424A1 (en) * | 1987-04-27 | 1988-11-03 | The Upjohn Company | Pharmaceutically active amines |
| WO1991005761A1 (en) * | 1989-10-16 | 1991-05-02 | Uniroyal Chemical Ltd./Uniroyal Chemical Ltee | Treatment of hiv infections and compounds useful therein |
| WO1991009849A1 (en) * | 1989-12-28 | 1991-07-11 | The Upjohn Company | Diaromatic substituted anti-aids compounds |
Non-Patent Citations (9)
| Title |
|---|
| CHEMICAL ABSTRACTS, Vol. 110, No. 25, issued 19 June 1989, UHLENDORF et al., "Preparation of N-Substituted Indole Carboxamides and Indolemethyl Amines as Nervous Systems Agents", see Formula 1, Abstract No. 231432y. * |
| Chemical Pharmaceutical Bulletin, Vol. 24, issued 1976, INABA et al., "Benzodiazepine. XIII. Synthesis of 1, 4-Benzodiazepine Derivatives", pages 1076-1082. * |
| J. Medicinal Chemistry, Vol. 12, issued 12 April 1969, SAVENA et al., "Synthesis of 3-Hydroxy and 3-Methoxy-Indole-2-Carboxamides and Esters", pp. 1120-1122, see Table 1, compound 6. * |
| See also references of EP0686148A4 * |
| Synthesis, issued February 1983, NAGARATHINAM et al., "1-Benzenesulfonyl-2-Bromoethyl-3-Phenylthi oindole as a Synthon for 2-Substituted Indoles: A New Synthesis of 2-Aminomethy Indoles", pp. 156-157. * |
| Synthesis, issued February 1985, MOHAN et al., "Synthesis of 2-(2-Arylethyl)-indoles", pp. 188-190. * |
| Synthesis, issued June 1988, ATKINSON et al., "A New Synthesis of 3-Arylthiandoles", pp. 480-481. * |
| Synthesis, issued November 1982, NAGARATHRAM et al., "A Convienent Synthesis of 2-(2-Arylethyl)-3-ethoxycarbony)-5-methoxyi ndoles via the Wittig-Horner Reaction", pp. 926-927. * |
| Tetrahedron Letters, Vol. 24, No. 33, issued 1983, VEDACHALAM et al., "A Facile Preparation of 2-Alkyl-indoles - Potential Intermediates for Alkoids", pp. 3531-3532. * |
Cited By (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6989263B1 (en) * | 1994-09-23 | 2006-01-24 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Method for identifying and using compounds that inactivate HIV-1 and other retroviruses by attacking highly conserved zinc fingers in the viral nucleocapsid protein |
| US5811425A (en) * | 1997-03-04 | 1998-09-22 | Abbott Laboratories | Heterocyclic compounds as COX-2 inhibitors |
| WO2001032621A1 (en) * | 1999-10-29 | 2001-05-10 | Wakunaga Pharmaceutical Co., Ltd. | Novel indole derivatives and drugs containing the same as the active ingredient |
| EP1390029A1 (en) * | 2001-04-11 | 2004-02-25 | Idenix (Cayman) Limited | Phenylindoles for the treatment of hiv |
| US6710068B2 (en) | 2001-04-11 | 2004-03-23 | Idenix Pharmaceuticals, Inc. | Phenylindoles for the treatment of HIV |
| EP1390029A4 (en) * | 2001-04-11 | 2005-11-30 | Idenix Cayman Ltd | Phenylindoles for the treatment of hiv |
| CN100404505C (en) * | 2002-05-30 | 2008-07-23 | 阿斯利康(瑞典)有限公司 | Novel substituted indoles |
| WO2003101961A1 (en) * | 2002-05-30 | 2003-12-11 | Astrazeneca Ab | Novel substituted indoles |
| US7754735B2 (en) | 2002-05-30 | 2010-07-13 | Astrazeneca Ab | Substituted indoles |
| US8093278B2 (en) | 2002-05-30 | 2012-01-10 | Astrazeneca Ab | Substituted indoles |
| US7166607B2 (en) | 2002-05-30 | 2007-01-23 | Astrazeneca Ab | Substituted indoles |
| US7723373B2 (en) | 2002-07-17 | 2010-05-25 | Astrazeneca Ab | Indole-3-sulphur derivatives |
| EP1545510A1 (en) * | 2002-08-07 | 2005-06-29 | Idenix (Cayman) Limited | Substituted phenylindoles for the treatment of hiv |
| US7365090B2 (en) | 2002-08-07 | 2008-04-29 | Idenix Pharmaceuticals, Inc. | Substituted phenylindoles for the treatment of HIV |
| EP1545510A4 (en) * | 2002-08-07 | 2006-11-15 | Idenix Cayman Ltd | Substituted phenylindoles for the treatment of hiv |
| US7687535B2 (en) | 2003-05-27 | 2010-03-30 | Astrazeneca Ab | Substituted 3-sulfur indoles |
| US7709521B2 (en) | 2003-08-18 | 2010-05-04 | Astrazeneca Ab | Substituted indole derivatives for pharmaceutical compositions for treating respiratory diseases |
| US7402672B2 (en) | 2003-12-11 | 2008-07-22 | Aventis Pharmaceuticals Inc. | Substituted 1H-pyrrolo[3,2-b, 3,2-c, and 2,3-c]pyridine-2-carboxamides and related analogs as inhibitors of casein kinase lepsilon |
| US7626027B2 (en) | 2003-12-11 | 2009-12-01 | Aventis Pharmaceuticals Inc | Substituted 1H-pyrrolo[2,3-c]pyridine-2-carboxamides and related analogs as inhibitors of casein kinase Iε |
| WO2006023590A1 (en) * | 2004-08-19 | 2006-03-02 | Aventis Pharmaceuticals Inc. | 3-ARYLTHIOINDOLE-2-CARBOXAMIDE DERIVATIVES AND ANALOGS THEREOF AS INHIBITORS OF CASEIN KINASE Iϵ |
| US8008340B2 (en) | 2004-08-19 | 2011-08-30 | Aventis Pharmaceuticals Inc. | 3-arylthioindole-2-carboxamide derivatives and analogs thereof as inhibitors of casein kinase I |
| US7534809B2 (en) | 2004-09-17 | 2009-05-19 | Idenix Pharmaceuticals, Inc. | Phospho-indoles as HIV inhibitors |
| EP2256124A1 (en) | 2004-09-17 | 2010-12-01 | IDENIX Pharmaceuticals, Inc. | Phospho-indoles as HIV inhibitors |
| US8044091B2 (en) | 2004-09-17 | 2011-10-25 | Idenix Pharmaceuticals, Inc. | Phospho-indoles as HIV inhibitors |
| US7781598B2 (en) | 2005-01-13 | 2010-08-24 | Astrazeneca Ab | Process for the preparation of substituted indoles |
| US7741360B2 (en) | 2006-05-26 | 2010-06-22 | Astrazeneca Ab | Bi-aryl or aryl-heteroaryl substituted indoles |
| US7960428B2 (en) | 2006-09-29 | 2011-06-14 | Idenix Pharmaceuticals, Inc. | Enantiomerically pure phosphoindoles as HIV inhibitors |
| US8486991B2 (en) | 2006-09-29 | 2013-07-16 | Idenix Pharmaceuticals, Inc. | Enantiomerically pure phosphoindoles as HIV inhibitors |
| CN112375027A (en) * | 2020-12-07 | 2021-02-19 | 中国药科大学 | Indolesulfonamide derivative and medical application thereof |
| CN112375027B (en) * | 2020-12-07 | 2023-03-31 | 中国药科大学 | Indolesulfonamide derivative and medical application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| PL310410A1 (en) | 1995-12-11 |
| BG62089B1 (en) | 1999-02-26 |
| EP0686148A1 (en) | 1995-12-13 |
| BR9405737A (en) | 1995-12-05 |
| EP0686148A4 (en) | 1996-02-07 |
| KR960701010A (en) | 1996-02-24 |
| CA2156420A1 (en) | 1994-09-01 |
| FI953954A0 (en) | 1995-08-23 |
| FI953954A (en) | 1995-08-23 |
| JPH08507067A (en) | 1996-07-30 |
| BG99879A (en) | 1996-02-28 |
| PL175788B1 (en) | 1999-02-26 |
| HU9502468D0 (en) | 1995-10-30 |
| CN1119856A (en) | 1996-04-03 |
| NO953308L (en) | 1995-10-24 |
| HUT74614A (en) | 1997-01-28 |
| AU6254294A (en) | 1994-09-14 |
| NO953308D0 (en) | 1995-08-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5527819A (en) | Inhibitors of HIV reverse transcriptase | |
| WO1994019321A1 (en) | Inhibitors of hiv reverse transcriptase | |
| AU656615B2 (en) | Inhibitors of HIV reverse transcriptase | |
| EP0934307B1 (en) | Substituted azabicylic compounds and their use as inhibitors of the production of tnf and cyclic amp phosphodiesterase | |
| EP1725544B1 (en) | 3-[4-heterocyclyl-1,2,3-triazol-1-yl]-n-aryl-benzamides as inhibitors of the cytokines production for the treatment of chronic inflammatory diseases | |
| JP4500161B2 (en) | Phthalazinone derivatives | |
| JP4986853B2 (en) | Benzimidazole compounds that inhibit prostaglandin D synthase | |
| KR19990022574A (en) | Imidazole compound | |
| IE904146A1 (en) | Indole Derivatives | |
| JPH10512264A (en) | New compound | |
| WO1998006715A1 (en) | Novel piperazine containing compounds | |
| BG63769B1 (en) | 1,4,53-trisubstituted imidazoles, useful as cytokines | |
| JPH02300182A (en) | Benzopyran derivative and production thereof | |
| WO2003048152A2 (en) | Inflammation modulators | |
| JP2002534385A (en) | New compound | |
| SK9162001A3 (en) | Substituted (aminoiminomethyl or aminomethyl)benzoheteroaryl compounds as factor xa inhibitors | |
| RU2135484C1 (en) | Quinoxaline derivatives, method of their synthesis and pharmaceutical composition | |
| EP3154941A1 (en) | Small molecule lfa-1 inhibitors | |
| HU193918B (en) | Process for preparing dihydropyridine derivatives | |
| US4965266A (en) | Heteroarylcarboxamide derivatives, process for preparing the same and pharmaceutical composition containing the same | |
| JPH02311479A (en) | Quinoline derivative | |
| BG63362B1 (en) | New substituted imidazoles |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 94191586.7 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU BB BG BR BY CA CN CZ FI HU JP KR KZ LK LV MG MN MW NO NZ PL RO RU SD SK UA UZ |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 262641 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1994909663 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2156420 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV1995-2137 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 103795 Country of ref document: SK Ref document number: 953954 Country of ref document: FI Ref document number: 95-01503 Country of ref document: RO |
|
| WWP | Wipo information: published in national office |
Ref document number: 1994909663 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: PV1995-2137 Country of ref document: CZ |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1994909663 Country of ref document: EP |