WO1997037967A1 - Calcilytic compounds - Google Patents
Calcilytic compounds Download PDFInfo
- Publication number
- WO1997037967A1 WO1997037967A1 PCT/US1997/005558 US9705558W WO9737967A1 WO 1997037967 A1 WO1997037967 A1 WO 1997037967A1 US 9705558 W US9705558 W US 9705558W WO 9737967 A1 WO9737967 A1 WO 9737967A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- phenyl
- compound
- alk
- unsubstituted
- Prior art date
Links
- WWUMMJHVAOHEOA-UHFFFAOYSA-N CC(C)(Cc(cc1)ccc1OC)NC/C(/O)=[O](\C)/CC1(C2)C(C3)CC2CC3C1 Chemical compound CC(C)(Cc(cc1)ccc1OC)NC/C(/O)=[O](\C)/CC1(C2)C(C3)CC2CC3C1 WWUMMJHVAOHEOA-UHFFFAOYSA-N 0.000 description 1
- XSQNZUXNBTZEKA-UHFFFAOYSA-N CC(C)(Cc(cc1)ccc1OC)NCC(COc1cccc(Cl)c1)O Chemical compound CC(C)(Cc(cc1)ccc1OC)NCC(COc1cccc(Cl)c1)O XSQNZUXNBTZEKA-UHFFFAOYSA-N 0.000 description 1
- KCCUCFUQSYGPBH-GOSISDBHSA-N CC(C)(Cc(cc1)ccc1OC)NC[C@H](COc(cccc1)c1C#N)O Chemical compound CC(C)(Cc(cc1)ccc1OC)NC[C@H](COc(cccc1)c1C#N)O KCCUCFUQSYGPBH-GOSISDBHSA-N 0.000 description 1
- BUBALKWCMYBRJI-UHFFFAOYSA-N CC(C)CCC(CC1)=CC=C1OC Chemical compound CC(C)CCC(CC1)=CC=C1OC BUBALKWCMYBRJI-UHFFFAOYSA-N 0.000 description 1
- 0 CCCCC(CC)COC*CNC(C)(C)Cc(cc1)ccc1OC Chemical compound CCCCC(CC)COC*CNC(C)(C)Cc(cc1)ccc1OC 0.000 description 1
- OMPKFMJLRFWAOG-ROPPNANJSA-N CCCCC(CC)COC[C@@H](CNC(C)(C)Cc(cc1)ccc1OC)O Chemical compound CCCCC(CC)COC[C@@H](CNC(C)(C)Cc(cc1)ccc1OC)O OMPKFMJLRFWAOG-ROPPNANJSA-N 0.000 description 1
- JZEHWMUIAKALDN-UHFFFAOYSA-N NCC(COc1ccccc1)O Chemical compound NCC(COc1ccccc1)O JZEHWMUIAKALDN-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/22—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound oxygen atoms
- C07C311/29—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound oxygen atoms having the sulfur atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
- A61K31/137—Arylalkylamines, e.g. amphetamine, epinephrine, salbutamol, ephedrine or methadone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/18—Sulfonamides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/275—Nitriles; Isonitriles
- A61K31/277—Nitriles; Isonitriles having a ring, e.g. verapamil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/18—Drugs for disorders of the endocrine system of the parathyroid hormones
- A61P5/22—Drugs for disorders of the endocrine system of the parathyroid hormones for decreasing, blocking or antagonising the activity of calcitonin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/02—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C217/04—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated
- C07C217/28—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having one amino group and at least two singly-bound oxygen atoms, with at least one being part of an etherified hydroxy group, bound to the carbon skeleton, e.g. ethers of polyhydroxy amines
- C07C217/30—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having one amino group and at least two singly-bound oxygen atoms, with at least one being part of an etherified hydroxy group, bound to the carbon skeleton, e.g. ethers of polyhydroxy amines having the oxygen atom of at least one of the etherified hydroxy groups further bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/54—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C217/56—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by singly-bound oxygen atoms
- C07C217/60—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by singly-bound oxygen atoms linked by carbon chains having two carbon atoms between the amino groups and the six-membered aromatic ring or the condensed ring system containing that ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/54—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C217/56—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by singly-bound oxygen atoms
- C07C217/62—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by singly-bound oxygen atoms linked by carbon chains having at least three carbon atoms between the amino groups and the six-membered aromatic ring or the condensed ring system containing that ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/45—Carboxylic acid nitriles having cyano groups bound to carbon atoms of rings other than six-membered aromatic rings
- C07C255/46—Carboxylic acid nitriles having cyano groups bound to carbon atoms of rings other than six-membered aromatic rings to carbon atoms of non-condensed rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/49—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C255/54—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton containing cyano groups and etherified hydroxy groups bound to the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/23—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C323/24—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/25—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/63—One oxygen atom
- C07D213/65—One oxygen atom attached in position 3 or 5
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/44—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D317/46—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems condensed with one six-membered ring
- C07D317/48—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring
- C07D317/50—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to atoms of the carbocyclic ring
- C07D317/58—Radicals substituted by nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/50—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D333/52—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes
- C07D333/62—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the hetero ring
- C07D333/68—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
- C07D333/70—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2603/00—Systems containing at least three condensed rings
- C07C2603/56—Ring systems containing bridged rings
- C07C2603/58—Ring systems containing bridged rings containing three rings
- C07C2603/70—Ring systems containing bridged rings containing three rings containing only six-membered rings
- C07C2603/74—Adamantanes
Definitions
- the present invention relates to compounds able to inhibit calcium receptor activity and the use of such compounds.
- the compounds described herein are administered to patients to achieve a therapeutic effect.
- Extracellular Ca 2 * is under tight homeostatic control and regulates various processes such as blood clotting, nerve and muscle excitability, and proper bone formation.
- Calcium receptor proteins enable certain specialized cells to respond to changes in extracellular Ca 2 * concentration. For example, extracellular Ca 2 * inhibits the secretion of parathyroid hormone (PTH) from parathyroid cells, inhibits bone resorption by osteoclasts, and stimulates secretion of calcitonin from C-cells.
- PTH parathyroid hormone
- PTH is the principal endocrine factor regulating Ca 2 * homeostasis in the blood and extracellular fluids. PTH, by acting on bone and kidney cells, increases the level of Ca 2 * in the blood. This increase in extracellular Ca 2 * then acts as a negative feedback signal, depressing PTH secretion. The reciprocal relationship between extracellular Ca 2 * and PTH secretion forms an important mechanism maintaining bodily Ca 2 * homeostasis.
- Extracellular Ca 2 * acts directly on parathyroid cells to regulate PTH secretion.
- the existence of a parathyroid cell surface protein which detects changes in extracellular Ca 2 * has been confirmed. (Brown et al . , Nature 366: 574 , 1993.)
- this protein the calcium receptor, acts as a receptor for extracellular Ca 2 *, detects changes in the ion concentration of extracellular Ca 2 *, and initiates a functional cellular response, PTH secretion.
- Extracellular Ca 2 * can exert effects on different cell functions, reviewed in Nemeth et al . , Cell Calcium 11 : 319 , 1990.
- the role of extracellular Ca 2 * in parafollicular (C- cells) and parathyroid cells is discussed in Nemeth, Cell Calcium 11:323, 1990. These cells were shown to express similar calcium receptors.
- C- cells parafollicular cells
- C- cells parafollicular cells
- parathyroid cells were shown to express similar calcium receptors.
- These cells were shown to express similar calcium receptors.
- the role of extracellular Ca 2 * on bone osteoclasts is discussed by Zaidi, Bioscience Reports 10:493, 1990.
- Applicant is also the first to describe methods by which molecules active at these Ca 2 * receptors can be identified and used as lead molecules in the discovery, development, design, modification and/or construction of useful calcimimetics or calcilytics which are active at Ca 2 * receptors.
- Such calcimimetics or calcilytics are useful in the treatment of various disease states characterized by abnormal levels of one or more components, e.g.. polypeptides such as hormones, enzymes or growth factors, the expresssion and/or secretion of which is regulated or affected by activity at one or more Ca 2 * receptors.
- Calcilytic compounds refer to compounds able to inhibit calcium receptor activity.
- the ability of a compound to "inhibit calcium receptor activity” means that the compound causes a decrease in one or more calcium receptor activities evoked by extracellular Ca 2 *.
- calcilytic compounds to inhibit calcium receptor activity and/or achieve a beneficial effect in a patient are described below. Also described below are techniques which can be used to obtain additional calcilytic compounds.
- R 2 where Rj is selected from the group consisting of: aryl, longer-length alk, and cycloalk;
- R 3 and R 4 is each independently lower alk or together cyclopropyl
- Yj is either covalent bond, alkylene, or alkenylene
- Y 2 is alkylene
- Y 3 is alkylene
- Z is selected from the group consisting of: covalent bond, 0, S, NH, N-lower alk, alkylene, alkenylene, and alkynylene, provided that if Z is either 0, S, NH, or N-lower alk, then Y x is not a covalent bond, further provided that Y x and Z may together be a covalent bond; and pharmaceutically acceptable salts and complexes thereof.
- Section II, infra also provides definitions for other chemical groups described in the present application.
- Preferred calcilytic compounds have an IC 50 ⁇ . 50 ⁇ M, more preferably an IC 50 ⁇ 10 ⁇ M, and even more preferably an IC 50 ⁇ 1 ⁇ M, as measured using the "Calcium Receptor Inhibitor Assay" described in Example 1, infra.
- a first aspect of the present invention features a method of treating a patient by administering to the patient a therapeutically effective amount of a Structure I ⁇ , ⁇ -disubstituted arylalkylamine derivative. Treatment can be carried out, for example, to retard the disease in a patient having a disease or to prophylactically retard or prevent the onset of a disease.
- a therapeutically effective amount is the amount of compound which achieves a therapeutic effect by retarding a disease in a patient having a disease or prophylactically retarding or preventing the onset of a disease.
- it is an amount which relieves to some extent one or more symptoms of a disease or disorder in a patient; returns to normal either partially or completely one or more physiological or biochemical parameters associated with or causative of the disease or disorder; and/or reduces the likelihood of the onset of the disease of disorder.
- a "patient” refers to a mammal in which compounds characterized by their ability to inhibit calcium receptor activity, in vivo or in vitro, will have a beneficial effect.
- the patient is a human being.
- Patients benefiting from the administration of a therapeutic amount of a calcilytic compound can be identified using standard techniques known to those in the medical profession.
- Diseases or disorders which can be treated by inhibiting one or more calcium receptor activities include one or more of the following types: (1) those characterized by an abnormal bone and mineral homeostasis; (2) those characterized by an abnormal amount of an extracellular or intracellular messenger whose production can be affected by one or more calcium receptor activities; (3) those characterized by an abnormal effect (e. g.
- an intracellular or extracellular messenger which can itself be ameliorated by one or more calcium receptor activities; and (4) other diseases or disorders where inhibition of one or more calcium receptor activities exerts a beneficial effect, for example, in diseases or disorders where the production of an intracellular or extracellular messenger stimulated by receptor activity compensates for an abnormal amount of a different messenger.
- extracellular messengers whose secretion and/or effect can be affected by inhibiting calcium receptor activity are believed to include inorganic ions, hormones, neurotransmitters, growth factors, and chemokines .
- intracellular messengers include cAMP, cGMP, IP 3 , calcium, magnesium, and diacylglycerol.
- a patient is a human having a disease or disorder characterized by one or more of the following: (1) an abnormal bone or mineral homeostasis; (2) an abnormal amount of an extracellular or intracellular messenger which is ameliorated by a compound able to effect one or more calcium receptor activities; and (3) an abnormal effect of an intracellular or extracellular messenger which is ameliorated by a compound able to effect one or more calcium receptor activities.
- the disease or disorder is characterized by an abnormal bone and mineral homeostasis, more preferably calcium homeostasis.
- Abnormal calcium homeostasis is characterized by one or more of the following activities: (1) an abnormal increase or decrease in serum calcium; (2) an abnormal increase or decrease in urinary excretion of calcium; (3) an abnormal increase or decrease in bone calcium levels, for example, as assessed by bone mineral density measurements; (4) an abnormal absorption of dietary calcium; (5) an abnormal increase or decrease in the production and/or release of messengers which affect serum calcium levels such as PTH and calcitonin; and (6) an abnormal change in the response elicited by messengers which affect serum calcium levels.
- the abnormal increase or decrease in these different aspects of calcium homeostasis is relative to that occurring in the general population and is generally associated with a disease or disorder.
- the calcilytic compounds are used to treat diseases or disorders selected from the group consisting of: hypoparathyroidism, osteosarcoma, periodontal disease, fracture healing, osteoarthritis, rheumatoid arthritis, Paget's disease, humoral hypercalcemia malignancy, and osteoporosis.
- Another aspect of the present invention describes a method of treating a patient comprising the step of administering to the patient an amount of a calcilytic compound sufficient to increase serum PTH level.
- the method is carried out by administering an amount of the compound effective to cause an increase in duration and/or quantity of serum PTH level sufficient to have a therapeutic effect.
- Increasing serum PTH may be used to achieve a therapeutic effect by retarding a disease in a patient having the disease or prophylactically retarding or preventing the onset of a disease.
- Prophylactic treatment can be performed, for example, on a person with an abnormally low serum PTH; or on a person without a low serum PTH, but were increasing PTH has a beneficial effect.
- An abnormally low serum PTH is a serum PTH level lower than that occurring in the general population, and is preferably an amount associated with a disease or the onset of a disease.
- Increasing serum PTH levels can be used to treat different types of diseases including bone and mineral related diseases.
- the compound is administered to a patient to cause an increase in serum PTH having a duration up to one hour, about one to about twenty-four hours, about one to about twelve hours, about one to about six hours, about one to about five hours, about one to about four hours, about two to about five hours, about two to about four hours, or about three to about six hours.
- the compound is administered to a patient to cause an increase in serum PTH up to 0.5 fold, 0.5 to 5 fold, 5 fold to 10 ten fold, and at least 10 fold, greater than peak serum PTH in the patient.
- the peak serum level is measured with respect to the patient not undergoing treatment.
- Another aspect of the present invention features Structure I calcilytic compounds.
- compositions comprising a pharmaceutically acceptable carrier and a calcilytic compound described herein.
- the pharmaceutical composition contains the calcilytic compound in a form suitable for administration into a mammal, preferably, a human being.
- the pharmaceutical composition contains an amount of a calcilytic compound in a proper pharmaceutical dosage form sufficient to exert a therapeutic effect on a human being.
- multiple doses of pharmaceutical compositions may be used to treat a patient. Considerations and factors concerning dosage forms suitable for administration are known in the art and include potential toxic effects, solubility, route of administration, and maintaining activity.
- pharmaceutical compositions injected into the bloodstream should be soluble.
- Another aspect of the present invention features a method of screening for Structure I ⁇ , -disubstituted arylalkylamine derivatives able to inhibit calcium receptor activity.
- the method involves the steps of contacting a cell having a calcium receptor with a Structure I ⁇ , ⁇ -di- substituted arylalkylamine derivative and measuring the ability of the compound to inhibit calcium receptor activity.
- the screening method can be carried out in vivo or in vitro and is particularly useful to identify those Structure I ⁇ , -disubstituted arylalkylamine derivatives most able to act as calcilytic compounds.
- In vivo assays include measuring a physiological parameter related to calcium receptor activity, such as serum hormone levels or serum calcium ion concentration.
- In vitro assays include measuring the ability of the calcilytic compound to affect intracellular calcium concentration, or cellular hormone secretion.
- hormones levels which can be affected by calcilytic compounds include PTH and calcitonin.
- the calcilytic compounds described herein can be used as part of in vivo or in vi tro methods. Preferably, the compounds are used in vivo to achieve a beneficial effect in a patient.
- Examples of in vitro uses, and other in vivo uses include use in a method to identify other calcilytic compounds and use as a tool to investigate calcium receptor activity or the physiological effects of inhibiting calcium receptor activity in different organisms.
- the present application demonstrates the ability of calcilytic compounds to exert a physiologically relevant effect on a cell by illustrating the ability of such compounds to increase PTH secretion and also identifies a target site for calcilytic compounds.
- the present application is believed to be the first to demonstrate that calcilytic compounds can increase PTH secretion.
- Calcium receptors are present on different cell types and can regulate different responses in different cell types. While the calcilytic compounds described herein are believed to act at a calcium receptor through a calcium receptor- activity modulating site, unless otherwise explicitly stated in the claims that a compound exerts an effect by acting at a calcium receptor through such a site, there is no intention to limit the claimed methods or compound to requiring inhibition of calcium receptor activity or any particular mode of action. Rather, the present application demonstrates that compounds able to inhibit calcium receptor activity, whose calcilytic activity can be measured in vivo or in vitro, exert significant physiological effects. For example, the present application demonstrates the ability of different calcilytic compounds to prevent Ca 2 * inhibition of PTH and, thereby, result in an increase in PTH release.
- Compounds binding at the calcium receptor-activity modulating site can be identified using a labeled compound binding to the site in a competition-binding assay format.
- Preferred calcilytic compounds described herein are Structure I ⁇ , ⁇ -disubstituted arylalkylamine derivatives able to inhibit calcium receptor activity.
- Other aspects of the present invention include assays which can be used to identify those Structure I ⁇ , ⁇ -disubstituted arylalkylamine derivatives expected to be effective in inhibiting calcium receptor activity, and/or exerting a therapeutic effect in a patient; preferred groups of Structure I a, ⁇ -disubstituted arylalkylamine derivatives; and the use of the compounds described herein to treat different diseases or disorders.
- Calcium receptors respond to changes in extracellular calcium levels. The exact changes resulting from calcium receptor activity depend on the particular receptor and the cell containing the receptor. For example, the in vi tro effect of calcium on the calcium receptor in a parathyroid cell includes the following:
- An increase in internal calcium [Ca 2 *] ⁇ The increase is due to the influx of external calcium and/or to the mobilization of internal calcium. Characteristics of the increase in internal calcium include the following:
- the transient increase is abolished by pretreatment for 10 minutes with 10 mM sodium fluoride;
- the transient increase is diminished by pretreatment with an activator of protein kinase C (PKC) , such as phorbol myristate acetate (PMA), mezerein or (-)- indolactam V.
- PKC protein kinase C
- PMA phorbol myristate acetate
- mezerein mezerein
- - indolactam V The overall effect of the protein kinase C activator is to shift the concentration-response curve of calcium to the right without affecting the maximal response;
- Pretreatment with pertussis toxin does not affect this increase; 3.
- the inhibition of dopamine- and isoproterenol- stimulated cyclic AMP formation This effect is blocked by pretreatment with pertussis toxin (100 ng/ml for > 4 hours) ; and 4.
- the inhibition of PTH secretion Pretreatment with pertussis toxin (100 ng/ml for > 4 hours) does not affect the inhibition of PTH secretion.
- Calcilytic activity of a compound can be determined using techniques such as those described in the examples below and those described in publications such as Nemeth et al . , PCT/US92/07175, International Publication Number WO 93/04373, Nemeth et al . , PCT/US93/01642, International Publication Number WO 94/18959, and Nemeth et al . , PCT/US94/12117, International Publication Number WO 95/11211 (each of which are hereby incorporated by reference herein) .
- Calcilytic activity varies depending upon the cell type in which the activity is measured. For example, calcilytic compounds possess one or more, and preferably all, of the following characteristics when tested on parathyroid cells in vitro:
- the compound blocks, either partially or completely, the ability of increased concentrations of extracellular Ca 2 * to:
- the compound blocks increases in Cl " current in Xenopus oocytes injected with poly(A) '-mRNA from bovine or human parathyroid cells elicited by extracellular Ca 2 *, but not in Xenopus oocytes injected with water;
- the compound blocks a response in Xenopus oocytes, injected with cloned nucleic acid expressing the calcium receptor, elicited by extracellular Ca 2 * or a calcimimetic compound (i.e., a compound able to mimic the effect of extracellular Ca 2 *, including compounds potentiating the effect of extracellular Ca 2 *) .
- a calcimimetic compound i.e., a compound able to mimic the effect of extracellular Ca 2 *, including compounds potentiating the effect of extracellular Ca 2 *
- Calcium receptors are present in different cells. The pharmacological effects of the following cells, in response to extracellular Ca 2 *, is consistent with the presence of a calcium receptor: parathyroid cell, bone osteoclast, juxtaglomerular kidney cell, proximal tubule kidney cell, distal tubule kidney cell, central nervous system cell, peripheral nervous system cell, cell of the thick ascending limb of Henle's loop and/or collecting duct, keratinocyte in the epidermis, parafollicular cell in the thyroid (C-cell) , intestinal cell, trophoblast in the placenta, platelet, vascular smooth muscle cell, cardiac atrial cell, gastrin- secreting cell, glucagon-secreting cell, kidney mesangial cell, mammary cell, endocrine and exocrine cells in the pancreas, fat/adipose cell, immune cell, GI tract cell, skin cell, -adrenal cell, pituitary cell, hypothalamic cell and cell of
- a calcium receptor on the following cells have been confirmed using physical data, such as hybridization with nucleic acid encoding a calcium receptor: parathyroid cell, central nervous system cell, peripheral nervous system cell, cell of the thick ascending limb of Henle's loop and/or collecting duct in the kidney, parafollicular cell in the thyroid (C-cell), intestinal cell, GI tract cell, pituitary cell, hypothalamic cell, cell of the subfornical organ, and endocrine and exocrine cells in the pancreas.
- Rj is selected from the group consisting of: aryl, longer-length alk, and cycloalk.
- Rj is either optionally substituted phenyl, optionally substituted pyridyl, optionally substituted benzothiopyranyl, optionally substituted carbazole, optionally substituted naphthyl, optionally substituted tetrahydronaphthyl, optionally substituted longer-length alkyl, optionally substituted longer-length alkenyl or optionally substituted cycloalk.
- R x is either an optionally substituted phenyl; an optionally substituted naphthyl; an optionally substituted pyridyl; an optionally substituted benzothiopyranyl; an optionally substituted carbazole; unsubstituted longer-length alkyl; unsubstituted longer- length alkenyl; or monosubstituted longer-length alkyl or alkenyl, where the monosubstituent is either an optionally substituted phenyl or an optionally substituted cycloalkyl provided that the optionally substituted phenyl or optionally substituted cycloalkyl can have one to four substituents each independently selected from the group consisting of: alkoxy, lower-haloalkyl, S-unsubstituted alkyl, lower-haloalkoxy, unsubstituted alkyl, unsubstituted alkenyl, halogen, SH, CN, N0 2 , NH 2 and OH;
- R 2 is OH or alkoxy, even more preferably, R 2 is OH or methoxy;
- R 3 and R restroom is each independently lower alk or together cyclopropyl.
- R 3 and R 4 are each independently a lower alkyl, more preferably, R 3 and R 4 are each independently methyl or ethyl;
- R 5 is aryl.
- R 5 is either optionally substituted naphthyl or optionally substituted phenyl. More preferably, R 5 is substituted phenyl having a substituent in the meta or para position and optionally containing additional substituents;
- R 6 is either hydrogen or lower alkyl, more preferably R 6 is hydrogen.
- Y 1 is either covalent bond, alkylene, or alkenylene.
- Y x is either covalent bond or lower alkylene. More preferably, Y is methylene;
- Y 2 is alkylene.
- Y 2 is lower alkylene. More preferably, Y 2 is methylene;
- Y 3 is alkylene.
- Y 3 is lower alkylene. More preferably, Y 3 is methylene;
- Z is selected from the group consisting of: covalent bond, 0, S, NH, N-lower alk, alkylene, alkenylene, and alkynylene, provided that if Z is either 0, S, NH, or N-lower alk, then Y ⁇ is not a covalent bond, further provided that Y x and Z may together be a covalent bond.
- Z is selected from the group consisting of: covalent bond, O, S, NH, N-lower alk, and alkylene. More preferably, Z is either O, S, lower alkylene, even more preferably, Z is O; and pharmaceutically acceptable salts and complexes thereof.
- Alk refers to either alkyl, alkenyl, or alkynyl.
- Lower alk refers to either lower alkyl, lower alkenyl, or lower alkynyl, preferably, lower alkyl.
- Alkenyl refers to an optionally substituted hydrocarbon group containing at least one carbon-carbon double bond between the carbon atoms and containing 2-15 carbon atoms joined together.
- the alkenyl hydrocarbon group may be straight-chain or contain one or more branches. Branched- and straight-chain alkenyl preferably have 2 to 7 carbons, each of which may be optionally substituted.
- the alkenyl is a lower alkenyl, which is an unsubstituted branched- or straight-chain alkenyl having 2 to 4 carbons.
- Alkyl refers to an optionally substituted hydrocarbon group joined by single carbon-carbon bonds and having 1-15 carbon atoms joined together.
- the alkyl hydrocarbon group may be straight-chain or contain one or more branches. Branched- and straight-chain alkyl preferably have 1 to 7 carbons, each of which may be optionally substituted.
- Alkyl substituents are each independently selected from the substituents described above for alkenyl. Preferably, no more than three substituents are present. More preferably, the alkyl is a lower alkyl, which is an unsubstituted branched- or straight-chain alkyl 1 to 4 carbons in length.
- Alkynyl refers to an optionally substituted hydrocarbon group containing at least one carbon-carbon triple bond between the carbon atoms and containing 2-15 carbon atoms joined together.
- the alkynyl hydrocarbon group may be straight-chain or contain one or more branches. Branched- and straight-chain alkynyl preferably have 2 to 7 carbons, each of which may be optionally substituted.
- Alkynyl substituents are each independently selected from the substituents described above for alkenyl. Preferably, no more than three substituents are present. More preferably, the alkynyl is a lower alkynyl, which is an unsubstituted branched- or straight-chain alkynyl having 2 to 4 carbons.
- Alkenylene refers to an optionally substituted hydrocarbon chain containing at least one carbon-carbon double bond between the carbon atoms. The alkenylene chain has 2 to 6 carbons and is attached at two locations to other functional groups or structural moieties.
- the alkenylene substituents are each independently selected from the substituents described above for alkenyl. Preferably, no 18 more than three substituents are present.
- the alkenylene is a "lower alkenylene,” which is an unsubstituted branched- or straight-chain alkenylene having 2 to 3 carbons.
- Alkoxy refers to oxygen joined to an unsubstituted alkyl 1 to 12 carbon atoms in length, preferably 1 to 2 carbons in length. More preferably, the alkoxy is methoxy.
- Alkylene refers to an optionally substituted hydrocarbon chain containing only carbon-carbon single bonds between the carbon atoms.
- the alkylene chain has 1 to 6 carbons and is attached at two locations to other functional groups or structural moieties.
- the alkylene substituents are each independently selected from the substituents described above for alkenyl. Preferably, no more than three substituents are present.- More preferably, the alkylene is a "lower alkylene,” which is an unsubstituted branched- or straight-chain alkylene having 1 to 3 carbons.
- Alkynylene refers to an optionally substituted hydrocarbon chain containing at least one carbon-carbon triple bond between the carbon atoms.
- the alkynylene chain has 2 to 6 carbons and is attached at two locations to other functional groups or structural moieties.
- the alkynylene substituents are each independently selected from the substituents described above for alkenyl. More preferably, the alkynylene is a "lower alkynylene,” which is an unsubstituted branched- or straight-chain alkynylene having 2 to 3 carbons.
- Aryl refers to an optionally substituted aromatic group with at least one ring having a conjugated pi- electron system, containing up to two conjugated or fused ring systems.
- Aryl includes carbocyclic aryl, heterocyclic aryl and biaryl groups, all of which may be optionally substituted.
- the aryl is either optionally substituted phenyl, optionally substituted pyridyl, optionally substituted benzothiopyranyl, optionally substituted carbazole, optionally substituted naphthyl, optionally substituted tetrahydronaphthyl.
- the aryl has no more than five independently selected substituents.
- R is an aryl
- the aryl is either optionally substituted phenyl, optionally substituted pyridyl, optionally substituted benzothiopyranyl, optionally substituted carbazole, optionally substituted naphthyl, or optionally substituted tetrahydronaphthyl.
- R aryl substituents are each independently selected from the group consisting of: alkoxy, methylene dioxy, N(CH 3 ) 2/ C(0)OCH 3 , phenyl, lower-haloalkyl,
- each R L aryl substituent is independently selected from the group consisting of: unsubstituted C j. -C 7 alkyl, Ci-C 7 alkoxy, lower haloalkoxy, CF 3 , F, Cl, Br, CN, and N0 2 .
- R is either 2-CN- phenyl, 2,3-dichloro phenyl, 2-nitro-phenyl, or 2-cyano-3- chloro-phenyl.
- R 5 right hand aryl substituents are each independently selected from the substituents described above for alkenyl .
- the R 5 aryl substituents are each independently selected from the group consisting of: methoxy, lower alkyl, lower haloalkoxy, CFH 2 , CHF 2 , CF 3 , OCH 2 CF 3 , F, Cl, Br, I, OH, SH, CN, N0 2 , NH 2 , methylene dioxy, NH-lower alkyl, Ndower alkyl) 2 , C(O) lower alkyl, S-lower alkyl, S(0) lower alkyl, S(0) 2 lower alkyl, 0C(0) lower alkyl, SC(O) lower alkyl, 0C(S) lower alkyl, NHC(O) lower alkyl, Ndower alkyl) C(O) lower alkyl, NHC(S) lower alkyl, Ndower alkyl) C(S) lower alkyl, NHS(0)lower alkyl, Ndower alkyl) S (O) lower alkyl,
- R 5 aryl substituents are each independently selected from the group consisting of: methylene dioxy, methoxy, lower-haloalkyl, S-lower alkyl, lower-haloalkoxy, lower alkyl, halogen, SH, CN, OH, Cl, F, and Br. Preferred halogens are Cl, F, and Br.
- Carbocyclic aryl refers to an aromatic ring or ring system having all carbon atoms. The carbon atoms are optionally substituted.
- Cycloalk refers to an optionally substituted cyclic alkyl or an optionally substituted non-aromatic cyclic alkenyl and includes monocyclic and multiple ring structures such as bicyclic and tricyclic.
- the cycloalk has 3 to 15 carbon atoms, preferably, 5 to 12 carbon atoms.
- Optional substituents for the cycloalk are each independently selected from the group described above for alkenyl. Preferably, no more than three substituents are present. More preferably, the cycloalk is unsubstituted, even more preferably it is an unsubstituted cyclic alkyl.
- Preferred cycloalkyl groups include cyclohexyl and adamantyl.
- Haloalk refers to substituted alkyl or substituted alkenyl, having no more than 4 carbons, where the substituents are halogens and at least one halogen is present.
- the haloalk is an alkyl 1 to 3 carbons in length and the halogens are each independently either Cl or F, more preferably the alkyl has 2 carbons, more preferably the haloalkyl is a lower haloalkyl which has 1 carbon.
- Heterocyclic aryl refers to an aryl having 1 to 3 heteroatoms as ring atoms in the aromatic ring and the remainder of the ring atoms are carbon atoms. Suitable heteroatoms include oxygen, sulfur, and nitrogen. Examples of heterocyclic aryl include indolyl, pyridyl, quinolinyl, and isoquinolinyl.
- Longer-length alk refers to either longer-length alkyl, longer-length alkenyl, or longer-length alkynyl; preferably, longer-length alkyl or longer-length alkenyl. More preferably a longer-length alk is 4 to 20 carbon atoms.
- Longer-length alkenyl refers to an optionally substituted hydrocarbon group containing at least one carbon-carbon double bond between the carbon atoms, and which contains 2-20 carbon atoms joined together.
- the longer-length alkenyl is 4 to 20 carbon atoms.
- the longer- length alkenyl hydrocarbon group may be straight-chain or contain one or more branches.
- Longer-length alkenyl substituents are each independently selected from the alkenyl substituent list described above.
- the longer- length alkenyl is either unsubstituted or has one cycloalk or phenyl substituent. More preferably, the cycloalk substituent, if present, is unsubstituted, and more preferably the cycloalk substituent, if present, is either cyclohexyl or adamantyl.
- “Longer-length alkyl” refers to an optionally substituted hydrocarbon group joined by single carbon-carbon bonds and which contains 1-20 carbon atoms joined together. Preferably, the longer-length alkyl is 4 to 20 carbon atoms.
- the longer-length alkyl hydrocarbon group may be straight-chain or contain one or more branches.
- Longer- length alkyl substituents are each independently selected from the alkenyl substituent list described above.
- the longer-length alkyl is either unsubstituted or has one cycloalk or phenyl substituent. More preferably, the cycloalk substituent, if present, is unsubstituted, and more preferably the cycloalk substituent, if present, is either cyclohexyl or adamantyl.
- Longer-length alkynyl refers to an optionally substituted hydrocarbon group containing at least one carbon-carbon triple bond between the carbon atoms, and which contains 2-20 carbon atoms joined together.
- the longer-length alkynyl is 4 to 20 carbon atoms.
- the longer- length alkynyl hydrocarbon group may be straight-chain or contain one or more branches.
- Longer-length alkynyl substituents are each independently selected from the alkenyl substituent list described above.
- the longer- length alkynyl is either unsubstituted or has one cycloalk or phenyl substituent substituent.
- the cycloalk substituent if present, is unsubstituted, and more preferably the cycloalk substituent, if present, is either cyclohexyl or adamantyl.
- “Haloalkoxy” refers to oxygen joined to a "haloalk.”
- the haloalkoxy is a "lower-haloalkoxy,” which is an oxygen joined to a lower-haloalkyl.
- More preferred calcilytic compounds are Structure I derivatives where R 3 , R 2 , R 3 , R 4 , R 6 , Z, Y x and Y 2 are as described above for Structure I ⁇ , ⁇ -disubstituted arylalkylamine derivatives, including preferred groups (see, Section II, supra) ; and
- R 5 is either phenyl substituted with one to four independently selected substituents or an optionally substituted naphthyl having up to four independently selected substituents.
- R 5 substituents are provided in Section II, supra . , including preferred embodiments. More preferably R 5 is either a substituted phenyl comprising a substituent in a meta or para position, more preferably, the substituent present in a meta or para position is either methyl, ethyl, isopropyl, methoxy, Cl, F, Br, or lower haloalkoxy.
- Examples of compounds having an IC 50 ⁇ 50 ⁇ include compounds 1, 9, 17, 25, 29, 42, 56, 79, 90, 101 and 164; examples of preferred compounds having an IC 50 ⁇ 10 ⁇ M include compounds 2, 3, 7, 8, 26, 27, 32, 33, 35, 37, 39, 41, 45, 48, 49, 59, 61, 66, 68, 71, 75, 93, 98, 103, 104, 110, 111, 114, 123, 124, 125, 128, 132, 144, 147, 152, 155, 158, 161, 162, 169 and 170; and examples of more preferred compounds having an IC 50 ⁇ than 1 ⁇ M include compounds 5, 6, 19, 20, 21, 28, 38, 40, 43, 44, 46, 47, 50, 51, 63, 64, 65, 67, 69, 72, 74, 96, 105, 106, 109, 112, 113, 115, 116
- Structure II compounds have the following structure:
- R 1( R 2 , R 3 , and R 4 are as described above for Structure I ⁇ , ⁇ -disubstituted arylalkylamine derivatives, including preferred groups (see, Section II, supra) ; and R 5 is either an optionally substituted naphthyl having one to four substituents independently selected from the group consisting of methyl, ethyl, isopropyl, methoxy, Cl, F, Br, or lower haloalkoxy, preferably the naphthyl is unsubstituted; or a substituted phenyl having one to four substituent with at least one substituent in a meta or para position selected from the group consisting of: lower alkyl, methoxy, Cl, F, Br, and lower haloalkoxy, more preferably a methoxy is present in the para or meta position; even more preferably, the remaining R 5 substituents are independently selected from the group consisting of: methoxy, lower-haloalkyl, S-lower al
- R x is not 6-CN-2-pyridyl; and further provided that if R 5 is 3,4 dimethoxy-phenyl, then R ⁇ is not CH 3 (CH 2 ) 5 0-phenyl; 2-cyciopentyl-phenyl; 2-Cl-phenyl; 2-CN-phenyl; 2- (3-furanyl)phenyl; or 4- (1, 2, -benzisothiazol) ; preferably, R 5 is not 3,4 dimethoxy phenyl; further provided that if R 5 is 4-methoxy-phenyl, then R is not 2-cyciopentyl-phenyl; 2-CH 3 -phenyl; 2-benzyl-phenyl; 3- CH 3 , 4-CH 3 S0 2 -phenyl; or 4- (1,2, -benzisothiazol) ; further provided that if R 5 is 4-C1-phenyl, then R x is not 2-CH 3 -phenyl , 5-iso-propyl
- R 2 , R 3 , and R 4 are as described above for Structure I ⁇ , ⁇ -disubstituted arylalkylamine derivatives, including preferred groups ( see, Section II, supra) , -
- R 5 is either an optionally substituted naphthyl having one to four substituents independently selected from the group consisting of methyl, ethyl, isopropyl, methoxy, Cl, F, Br, and lower haloalkoxy, preferably the naphthyl is unsubstituted; or a substituted phenyl having one to four substituent with at least one substituent in a meta or para position selected from the group consisting of: methyl, ethyl, isopropyl, methoxy, Cl, F, Br, and lower haloalkoxy, more preferably a methoxy is present in the para or meta position; even more preferably, the remaining R 5 substituents are independently selected from the group consisting of: methoxy, lower-haloalkyl, S-lower alkyl, lower-haloalkoxy, lower alkyl, halogen, SH, CN, OH, Cl, F, and Br; and
- R x is either 2-CN-phenyl, 2,3-dichloro phenyl, 2-nitro- phenyl, 2-cyano-3-chloro-phenyl, an optionally substituted pyridyl, an optionally substituted benzothiopyranyl, or an optionally substituted carbazole, where the optionally present substituents for the pyridyl, benzothiopyranyl, and carbazole as described in Section II supra, for aryl R x substituents, including preferred substituents, and are even more preferably independently selected from the group consisting of: methoxy, lower-haloalkyl, S-lower alkyl, lower-haloalkoxy, lower alkyl, halogen, SH, CN, OH, Cl, F, and Br.
- calcilytic compounds described herein can be formulated as a pharmaceutical composition to facilitate the administration of the compound to a patient.
- Preferred formulations contain a pharmaceutically acceptable carrier and a calcilytic compound as described in Section II, supra . , including the different embodiments.
- Suitable carriers include calcium carbonate, calcium phosphate, lactose, glucose, sucrose, gelatin, vegetable oils, polyethylene glycols and physiologically compatible solvents.
- Compounds inhibiting calcium receptor activity can be used to confer beneficial effects to patients suffering from a variety of diseases or disorders.
- Diseases or disorders which can be treated using a calcilytic compound are known in the art and can be identified using the present application as a guide.
- diseases or disorders can be identified based on the functional responses of cells regulated by calcium receptor activity.
- Diseases and disorders which can be treated using the calcilytic compounds described herein include those due to different cellular defects related to calcium receptor activity in different cells, such as a defective calcium receptor or an abnormal number of calcium receptors, a defective intracellular protein acted on by a calcium receptor, or a defective protein or an abnormal number of proteins acting on a calcium receptor.
- Functional responses of cells regulated by the calcium receptor are known in the art, including PTH secretion by parathyroid cells, calcitonin secretion by C-cells, bone resorption by osteoclasts, and Ca 2 * secretion by kidney cells. Such functional responses are associated with different diseases or disorders.
- isolated osteoclasts respond to increases in the concentration of extracellular Ca 2 * with corresponding increases in [Ca 2 *]i arising partly from the mobilization of intracellular Ca 2 *.
- Increases in [Ca 2 *] x in osteoclasts are associated with the inhibition of bone resorption.
- Renin secretion from juxtaglomerular cells in the kidney is depressed by increased concentrations of extracellular Ca 2 *.
- Extracellular Ca 2 * causes the mobilization of intracellular Ca 2 * in these cells.
- Other kidney cells respond to extracellular Ca 2 * as follows: elevated Ca 2 * inhibits formation of 1,25(OH) 2 -vitamin D by proximal tubule cells, stimulates production of calcium-binding protein in distal tubule cells, and inhibits tubular reabsorption of Ca 2 * and Mg 2 * in the thick ascending limb of Henle's loop (MTAL) , and reduces vasopressin action in the cortical collecting duct.
- MTAL Henle's loop
- Other examples of functional responses affected by extracellular Ca 2 * include promoting differentiation of intestinal goblet cells, mammary cells, and skin cells; inhibiting atrial natriuretic peptide secretion from cardiac atria; reducing cAMP accumulation in platelets; altering gastrin and glucagon secretion; acting on perivascular nerves to modify cell secretion of vasoactive factors; and affecting cells of the central nervous and peripheral nervous systems.
- Diseases and disorders which might be treated or prevented, based upon the affected cells include bone and mineral-related diseases or disorders; hypoparathyroidism; those of the central nervous system such as seizures, stroke, head trauma, spinal cord injury, hypoxia-induced nerve cell damage, such as occurs in cardiac arrest or neonatal distress, epilepsy, neurodegenerative diseases such as Alzheimer's disease, Huntington's disease and Parkinson's disease, dementia, muscle tension, depression, anxiety, panic disorder, obsessive-compulsive disorder, post-traumatic stress disorder, schizophrenia, neuroleptic malignant syndrome, and Tourette's syndrome; diseases involving excess water reabsorption by the kidney, such as syndrome of inappropriate ADH secretion (SIADH) , cirrhosis, congestive heart failure, and nephrosis; hypertension; preventing and/or decreasing renal toxicity from cationic antibiotics (e.
- SIADH syndrome of inappropriate ADH secretion
- GI ulcer diseases g. , aminoglycoside antibiotics
- gut motility disorders such as diarrhea and spastic colon,- GI ulcer diseases
- GI diseases with excessive calcium absorption such as sarcoidosis
- autoimmune diseases and organ transplant rejection squamous cell carcinoma
- pancreatitis g. , aminoglycoside antibiotics
- calcilytic compounds of the present invention will typically be used to treat human patients, they may also be used to treat similar or identical diseases or disorders in other warm-blooded animal species, such as other primates, farm animals such as swine, cattle, and poultry; and sports animals and pets such as horses, dogs and cats.
- calcilytic compounds are used in the treatment of bone and mineral-related diseases or disorders.
- Bone and mineral-related diseases or disorders comprise a diverse class of disorders affecting nearly every major organ system in the body. Examples of bone and mineral-related diseases or disorders include osteosarcoma, periodontal disease, fracture healing, osteoarthritis, rheumatoid arthritis, Paget's disease, humoral hypercalcemia malignancy, and osteoporosis.
- calcilytic compounds are used to treat osteoporosis, a disease characterized by reduced bone density and an increased susceptibility to fractures. Osteoporosis is associated with aging, especially in women.
- PTH can have a catabolic or an anabolic effect on bone. Whether PTH causes a catabolic effect or an anabolic effect seems to depend on how plasma levels of PTH are altered.
- plasma levels of PTH are chronically elevated, as in hyperparathyroid states, there is a net loss of bone.
- intermittent increases in plasma PTH levels, as achieved by administration of exogenous hormone result in new bone formation.
- Anabolic action of PTH on bone is described, for example, by Dempster et al . , Endocrin . Rev. 14:690-709, 1993.
- calcilytic compounds stimulate secretion of PTH.
- Such calcilytic compounds can be used to increase bone formation in a patient, for example, by intermittent dosing, thus achieving intermittent increases in the circulating levels of PTH.
- calcilytic compounds described by the present invention can be formulated for a variety of modes of administration, including systemic and topical or localized administration. Techniques and formulations generally may be found in Remington's Pharmaceutical Sciences. 18 th ed. , Mack Publishing Co., Easton, PA, 1990 (hereby incorporated by reference herein) . Suitable dosage forms, in part, depend upon the use or the route of entry, for example, oral, transdermal, trans ⁇ mucosal, or by injection (parenteral). Such dosage forms should allow the compound to reach a target cell whether the target cell is present in a multicellular host or in culture. For example, pharmacological compounds or compositions injected into the blood stream should be soluble. Other factors are known in the art, and include considerations such as toxicity and dosage forms which retard the compound or composition from exerting its effect.
- Compounds can also be formulated as pharmaceutically acceptable salts and complexes thereof.
- Pharmaceutically acceptable salts are non-toxic salts in the amounts and concentrations at which they are administered. The preparation of such salts can facilitate the pharmacological use by altering the physical characteristics of the compound without preventing it from exerting its physiological effect. Useful alterations in physical properties include lowering the melting point to facilitate transmucosal administration and increasing the solubility to facilitate administering higher concentrations of the drug.
- the pharmaceutically acceptable salt of the different compounds may be present as a complex.
- complexes include an 8-chlorotheophylline complex (analogous to, e.g., dimenhydrinate:diphenhydramine 8-chlorotheophylline (1:1) complex; Dramamine) and various cyclodextrin inclusion complexes.
- Pharmaceutically acceptable salts include acid addition salts such as those containing sulfate, hydrochloride, fumarate, maleate, phosphate, sulfamate, acetate, citrate, lactate, tartrate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate, cyclohexylsulfamate and quinate.
- Pharmaceutically acceptable salts can be obtained from acids such as hydrochloric acid, maleic acid, sulfuric acid, phosphoric acid, sulfamic acid, acetic acid, citric acid, lactic acid, tartaric acid, malonic acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, cyclohexylsulfamic acid, fumaric acid, and quinic acid.
- acids such as hydrochloric acid, maleic acid, sulfuric acid, phosphoric acid, sulfamic acid, acetic acid, citric acid, lactic acid, tartaric acid, malonic acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, cyclohexylsulfamic acid, fumaric acid, and quinic acid.
- Pharmaceutically acceptable salts also include basic addition salts such as those containing benzathine, chloroprocaine, choline, diethanolamine, ethylenediamine, meglumine, procaine, aluminum, calcium, lithium, magnesium, potassium, sodium, ammonium, alkylamine, and zinc, when acidic functional groups, such as carboxylic acid or phenol are present.
- basic addition salts such as those containing benzathine, chloroprocaine, choline, diethanolamine, ethylenediamine, meglumine, procaine, aluminum, calcium, lithium, magnesium, potassium, sodium, ammonium, alkylamine, and zinc.
- acidic functional groups such as carboxylic acid or phenol are present.
- Such salts can be prepared using the appropriate corresponding bases.
- salts can be prepared by standard techniques. For example, the free-base form of a compound is dissolved in a suitable solvent, such as an aqueous or aqueous-alcohol in solution containing the appropriate acid and then isolated by evaporating the solution. In another example, a salt is prepared by reacting the free base and acid in an organic solvent. (See, e . g. , PCT/US92/03736, hereby incorporated by reference herein.) Carriers or excipients can also be used to facilitate administration of the compound.
- a suitable solvent such as an aqueous or aqueous-alcohol in solution containing the appropriate acid and then isolated by evaporating the solution.
- a salt is prepared by reacting the free base and acid in an organic solvent.
- Carriers or excipients can also be used to facilitate administration of the compound.
- Examples of carriers include calcium carbonate, calcium phosphate, various sugars such as lactose, glucose, or sucrose, or types of starch, cellulose derivatives, gelatin, vegetable oils, polyethylene glycols and physiologically compatible solvents.
- physiologically compatible solvents include sterile solutions of water for injection (WFI) , saline solution and dextrose.
- WFI water for injection
- the calcilytic compounds can be administered by different routes including intravenous, intraperitoneal, subcutaneous, intramuscular, oral, topical (transdermal), or transmucosal administration. For systemic administration, oral administration is preferred.
- the compounds can be formulated into conventional oral dosage forms such as capsules, tablets, and liquid preparations such as syrups, elixirs, and concentrated drops.
- injection parenteral administration
- the compounds of the invention are formulated in liquid solutions, preferably, in physiologically compatible buffers or solutions, such as saline solution, Hank's solution, or Ringer's solution.
- the compounds may be formulated in solid form and redissolved or suspended immediately prior to use. Lyophilized forms can also be produced.
- Systemic administration can also be by transmucosal or transdermal means.
- penetrants appropriate to the barrier to be permeated are used in the formulation.
- penetrants are generally known in the art, and include, for example, for transmucosal administration, bile salts and fusidic acid derivatives.
- detergents may be used to facilitate permeation.
- Transmucosal administration for example, may be through nasal sprays, rectal suppositories, or vaginal suppositories.
- the compounds of the invention can be formulated into ointments, salves, gels, or creams, as is generally known in the art.
- the amounts of various calcilytic compounds to be administered can be determined by standard procedures taking into account factors such as the compound IC 50 , EC 50 , the biological half-life of the compound, the age, size and weight of the patient, and the disease or disorder associated with the patient. The importance of these and other factors to be considered are known to those of ordinary skill in the art. Generally, it is an amount between about 0.1 and 50 mg/kg, preferably 0.01 and 20 mg/kg of the animal to be treated.
- EXAMPLE 1 Calcium Receptor Inhibitor Assay This example illustrates the use of the Calcium Receptor Inhibitor Assay. Calcilytic activity was measured by determining the IC 50 of the test compound for blocking increases of intracellular Ca 2 * elicited by extracellular Ca 2 * in HEK 293 4.0-7 cells stably expressing the human calcium receptor. HEK 293 4.0-7 cells were constructed as described by Rogers et al . , J. Bone Miner. Res . 10 Suppl. 1:S483, 1995 (hereby incorporated by reference herein) . Intracellular Ca 2 * increases were elicited by increasing extracellular Ca 2 * from 1 to 1.75 mM. Intracellular Ca 2 * was measured using fluo-3, a fluorescent calcium indicator.
- SPF-PCB contains 20 mM Na-Hepes, pH 7.4, 126 mM NaCl, 5 mM KCI, and 1 mM MgCl 2 .
- SPF-PCB was made up and stored at 4 °C. On the day of use, SPF-PCB was supplemented with 1 mg/mL of D-glucose and 1 mM CaCl 2 and then split into two fractions. To one fraction, bovine serum albumin (BSA; fraction V, ICN) was added at 5 mg/mL (SPF-PCB+) . This buffer was used for washing, loading and maintaining the cells. The BSA-free fraction was used for diluting the cells in the cuvette for measurements of fluorescence. 4. The pellet was resuspended in 10 mL of SPF-PCB+ containing 2.2 ⁇ M fluo-3 (Molecular Probes) and incubated at room temperature for 35 minutes.
- BSA bovine serum albumin
- test compound or vehicle as a control
- Cal ⁇ cilytic compounds were detected by their ability to block, in a concentration-dependent manner, increases in the concentration of intracellular Ca 2 * elicited by extracellular Ca 2 *.
- those compounds having lower IC 50 values in the Calcium Receptor Inhibitor Assay are more preferred compounds.
- Compounds having an IC 50 greater than 50 ⁇ M were considered to be inactive.
- Preferred compounds are those having an IC S0 10-50 ⁇ M, more preferred compounds have an IC 50 1-10 ⁇ M, and most preferred compounds have an IC 50 less than 1 ⁇ M.
- Examples of compounds having an IC 50 greater than 50 ⁇ M include compounds 22, 24, 34, 36, 52, 53, 54, 55, 58, 60, 62, 70, 84, 92, 99, and 102.
- ⁇ , ⁇ -disubstituted arylalkylamine derivatives include compounds which have both calcilytic activity and ⁇ -adrenergic receptor activity. If desired, ⁇ - adrenergic activity can be reduced using appropriate functional groups and structural modifications.
- Modifications which can be carried out to reduce ⁇ -adrenergic receptor activity include using alternative R 2 groups and using absolute stereochemistry opposite to that which occurs in active ⁇ -adrenergic receptor antagonists, which provides compounds corresponding to the R enantiomer when R 2 is OH.
- ⁇ - adrenergic receptor activity and binding to the ⁇ -adrenergic receptor can be measured using standard techniques. For example, see Riva et al . , Mol . Pharmacol . 35:201-210, 1989.
- the calcilytic compounds have an IC 50 .> 1.0 nM, at the ⁇ - adrenergic receptor as measured using the " ⁇ -Adrenergic Receptor Binding Assay" described below.
- using the ⁇ -Adrenergic Receptor Assay calcilytic compounds have an IC 50 >. 1.0 ⁇ M, and IC 50 >. 10.0 ⁇ M.
- ⁇ -Adrenergic Receptor Binding Assay is carried out as follows: Incubations are performed in polypropylene reaction tubes in a 37 °C water bath. To each tube 50 ⁇ L of test sample is added, followed by 300 ⁇ L of assay buffer (50 mM Tris-HCI, pH 7.5), and 50 ⁇ L of 20 nM [ 3 H] - dihydroalprenolol. The binding reaction is initiated by the addition of 100 ⁇ L of 3.75 mg/mL well-washed rat cortex membranes in assay buffer, and allowed to incubate at 37 °C for 30 minutes. Non-specific binding is determined in the presence of 10 ⁇ M alprenolol.
- the final concentration of reactants is : 2 nM [ 3 H] -dihydroalprenolol , and 75 mg/mL rat cortex membrane in a reaction volume of 0.5 mL.
- the binding reaction is terminated by rapid filtration with ice-cold assay buffer onto GF/C filters (Brandel, Gaithersburg, MD) which have been soaked for 15 minutes in assay buffer.
- the reaction is first diluted with 3 mL of cold assay buffer (4 °C) , then aspirated onto the filter followed by 3 x 3 mL washes.
- PTH secretion was determined using dissociated bovine parathyroid cells as described below for Compounds 32, 33, and 38.
- Compounds 32, 33, and 38 all stimulated PTH secretion with an EC 50 of less than 10 ⁇ M.
- PCB Parathyroid cell buffer
- BSA fraction V bovine serum albumin
- ICN Biomedicals, Inc. Costa Mesa, CA; catalog #81-003
- D-glucose reagent grade
- Percoll purification buffer was prepared immediately before use by mixing 8 mL of Percoll (Pharmacia LKB, Alameda, CA; catalog #17-0891-01) and 7 mL of a twice- concentrated PCB solution without phosphate and containing 2 mM CaCl 2 .
- Parathyroid glands were obtained from calves within minutes of slaughter at an abattoir and shipped via overnight express on ice in PCB containing 1.25 mM CaCl 2 . The glands were trimmed and minced in ice-cold PCB containing 1.25 mM CaCl 2 , 1 mg/mL of D-glucose, and 2% BSA. Dissociated cells were obtained by collagenase digestion by vigorously shaking the minced tissue at 37 °C in PCB containing 0.5 to 1.0% Collagenase P (Boehringer Mannheim, Indianapolis, IN; catalog #1249 002), 2 to 5 units of deoxyribonuclease (Sigma, St.
- Cells were pooled at 1-hour intervals by filtering the cell suspension through a 250- ⁇ m Nitex screen into 15-mL polystyrene centrifuge tubes and spinning at 100 x g for 5 minutes at 22 °C.
- the pooled cell pellet was resuspended in Percoll purification buffer and purified by centrifugation at 14,500 x g for 20 minutes at 4 °C.
- Dissociated parathyroid cells equilibrated within a buoyant density of 1.048-1.062 above a dense band of red blood cells and below a diffuse band that contains adipocytes, strands of collagen, and damaged cells.
- the dissociated parathyroid cells were removed with a sterile pipette and washed 3 to 4 times under sterile conditions in a 1:1 mixture of Ham's F-12 and Dulbecco's modified Eagle's medium (F-12/DMEM, Sigma, St. Louis, MO; catalog #D 8900) supplemented with 0.5% BSA, 100 U/mL of penicillin, 100 ⁇ g/mL of streptomycin (Gibco BRL, Grand Island, NY; catalog #15140-031) , and 20 ⁇ g/mL of gentamicin (Sigma, St. Louis, MO; catalog #G 1397) .
- the cells were finally resuspended in F-12/DMEM supplemented with lower antibiotic concentrations (10 U/mL of penicillin, 10 ⁇ g/mL of streptomycin, and 4 ⁇ g/mL of gentamicin) .
- This latter medium lacks serum and contained ITS* (insulin, transferrin, selenous acid, BSA, and linoleic acid; Collaborative Biomedical Products, Bedford, MA; catalog #40352) .
- PTH Parathyroid Hormone
- Sulfate, phosphate-free parathyroid cell buffer contains (mM) : NaCl, 126; KCI, 5; MgCl 2 , 1; Na-Hepes, pH 7.45, and variable amounts of CaCl 2 as specified (reagent grade) .
- SPF-PCB was typically supplemented with bovine serum albumin (BSA fraction V; ICN Biomedicals, Inc., Costa Mesa, CA; catalog #81-003) and 1 mg/mL of D-glucose (reagent grade) as indicated.
- BSA fraction V bovine serum albumin
- Incubations were performed in triplicate in 12 x 75 mm polypropylene or polystyrene tubes to which were added 2.5 ⁇ L of test compound.
- the tubes were kept on ice until the drug additions were completed, then were transferred to a water bath at 37 °C, and the reactions were initiated by the addition of 0.2 mL of a suspension of dissociated cells at a density of 1 to 2 million cells/mL in SPF-PCB containing 0.5 mM Ca 2 *, 1 mg/mL of D-glucose, and 0.1% BSA.
- Incubation was for 30 minutes and the reaction was terminated by placing the tubes on ice. Cells were pelleted by gentle centrifugation (500 x g for 10 minutes at 4 °C) and 0.1 mL of supernatant was removed and stored at -20 °C.
- Amino-terminal bovine PTH was measured by radioimmunoassay (RIA) using goat anti-hPTH antiserum H 2 , HPLC-purified 125 I-hPTH (1-34) and bovine PTH (1-84) standards.
- Serial dilutions of bPTH standards (1,000 pg/25 ⁇ L to 3.8 pg/25 ⁇ L) were done in 50 mM Tris, pH 7.4, containing 0.5 mM Na azide and 2% bovine serum albumin (diluent) .
- Standards and samples were incubated for 2-3 days at 4 °C in the presence of antiserum after which 1,500-2,000 cpm label/tube was added.
- dextran-coated charcoal was added to separate bound vs . free label.
- the contents of each tube were mixed and the charcoal was pelleted by centrifugation.
- the supematants were decanted into 12 x 75 mm polystyrene tubes and counted in a Packard Cobra gamma counter.
- the calcilytic compounds described by the present invention can be prepared using standard techniques. For example, an overall strategy for preparing preferred compounds described herein can be carried out as described in this section. The examples which follow illustrate the synthesis of specific compounds. Using the protocols described herein as a model, one of ordinary skill in the art can readily produce other Structure I compounds. All reagents and solvents were obtained from commercial vendors. Starting materials ( e . g. , amines and epoxides) were synthesized using standard techniques and procedures.
- GC/EI- MS Gas Chromatographic/Electron-Impact Mass Spectrometric analyses were performed on HP-5890 Series II gas chromato- graphs equipped with HP-Ultra-2 or HP-5MS columns (30 mm x 0.25 mm ID) and HP-5971 or HP-5972 Mass Selective Spectrometric Detectors (MSD's) were used.
- MPLC Medium- Pressure Liquid Chromatography separations were carried out on silica gel (400 mesh) using an FMI pump, ISCO UV-6 detector (254 nm) and FOXY 200 fraction collector.
- Chiral HPLC separations were carried out using a Beckman System Gold HPLC and UV detector (254 nm) on Diacel ® ChiralCel OD columns (Chiral Technologies, Inc., Exton, PA 19341).
- NMR Nuclear Magnetic Resonance
- Proton and carbon spectra were taken at 300 MHz and 75 MHz, respectively.
- NMR resonances are reported in ppm relative to tetramethylsilane (TMS) with the following descriptors for the observed multiplicities: s (singlet), d (doublet), t (triplet) , q (quartet) , dd (doublet of doublets) and m (multiplet) .
- J ⁇ coupling constants are reported in Hz. Elemental analyses were performed and FT-IR data were acquired by Oneida Research Services, Inc., Whitesboro, NY 13492.
- the product is purified by one of three general methods: (1) conversion to the hydrochloride salt, followed by Reversed-Phase High-Performance Liquid Chromatography (RP- HPLC, 0.1% HCl/acetonitrile) ; (2) conversion to the hydrochloride salt, followed by recrystallization from water- methanol or acetonitrile; and (3) purification by normal- phase chromatography (column chromatography or preparative, thin-layer chromatography (TLC)) .
- Hydrochloride salts were also prepared by treatment of the corresponding free base in diethyl ether with IM HCl (in diethyl ether) .
- the reaction was stirred for 48 hours at room temperature and treated with 10% HCl (200 mL) .
- the reaction was concentrated to 300 mL and diluted to 600 mL with water.
- the resulting solution was extracted with diethyl ether (2 x 300 mL) and the combined ether extracts were washed with 10% HCl (2 x 200 mL) .
- the ether extract was then extracted with IN NaOH (3 x 200 mL) .
- the combined IN NaOH washes were made acidic (pH 1) by the addition of concentrated HCl, and the resulting solution was extracted with diethyl ether (3 x 300 mL) .
- Triethylamine (16.8 g, 166 mmol) and 2, 2-dimethyl-3- (4- methoxyphenyl)propionic acid (32.6 g, 157 mmol) were dissolved in 30 mL of water and enough acetone to maintain solubility at 0 °C.
- a solution of ethyl chloroformate (20.1 g, 185 mmol) in acetone (100 mL) was then added dropwise.
- An aqueous solution (95 mL) of sodium azide (12.9 g, 198 mmol) was then added dropwise and the resulting reaction mixture stirred 45 minutes at room temperature.
- the intermediate acyl azide was then extracted into toluene (200 mL) .
- the organic extract was washed with water, dried over anhydrous magnesium sulfate, and heated at 100 °C until the evolution of nitrogen ceased (-45 min) .
- the toluene was removed under vacuum and replaced with benzyl alcohol .
- the solution was then heated at 100 °C for 16 hours.
- the excess benzyl alcohol was removed under vacuum.
- the resulting benzyl carbamate was dissolved in absolute ethanol (200 mL) and reduced in the presence of palladium hydroxide (2 g) under 90 p.s.i. hydrogen for 4 hours at room temperature.
- the reaction was filtered and concentrated to a yellow oil.
- hydrochloride salt of each enantiomer was prepared by treatment of the free amine in diethyl ether with excess IM HCl (diethyl ether) . Evaporation of the solvent yielded the hydrochloride product as a solid.
- the crude mixture (1.47 g, 6 mmol) was dissolved in 6 mL of acetonitrile, to which was added 50% KF-Celite (0.7 g, 12 mmol) and 1, l-dimethyl-2- (4-methoxyphenyl) - ethylamine (0.54 g, 3 mmole) .
- the mixture was refluxed under nitrogen for 48 hours, and then cooled and filtered. The filtrate was evaporated to dryness, and the residue was taken up in water and ether. The ether layer was dried over sodium sulfate and concentrated to dryness.
- the reaction was diluted with water, and the product extracted into diethyl ether.
- the ether layer was washed with saturated NaCl and dried over sodium sulfate.
- the ether solution was treated with Amberlyst 15 ion-exchange resin to absorb the 2, 4, 6-triphenylpyridine. The resin was filtered and the filtrate evaporated to yield 1.3 g of pure 1- (4- methylphenyl) -2-methyl-2-nitropropane.
- the 1- (4-methylphenyl) -2-methyl-2-nitropropane (1.3 g) was hydrogenated for 5 hours at 65 p.s.i. hydrogen in ethanol (30 mL) using 1.4 g of Raney nickel as catalyst. Removal of the catalyst by filtration and evaporation of the solvent yielded 1.15 g of 1, l-dimethyl-2- (4-me hoxyphenyl) ethylamine as a clear oil.
- the aqueous layer was then made acidic with HCl, and the product extracted into ether.
- the ether layer was dried over anhydrous sodium sulfate, filtered, and concentrated.
- the resulting solid was crystallized from water/methanol to yield 1.11 g of 3-chloro-2-cyanophenol .
- Nickel chloride monohydrate (0.107 g, 0.404 mmol) was dissolved in 5 mL of methanol, followed by the addition of sodium borohydride (0.05 g, 1.2 mmol) .
- sodium borohydride 0.05 g, 1.2 mmol
- 1-ethyl-l-methyl-2- ( -methoxyphenyl)nitroethane (0.18 g, 0.807 mmol) in 3 mL of methanol was added, and stirred for 5 minutes.
- Sodium borohydride (0.11 g, 2.83 mmol) was then added in portions over 5 minutes.
- the reaction was then stirred overnight under a hydrogen balloon.
- the reaction mixture was filtered, and the methanol was removed on a rotary evaporator.
- the residue was taken up in ether and dilute sodium hydroxide.
- the ether layer was separated, washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated.
- Safrole oxide was prepared by stirring a solution of safrole (1.48 mL, 10 mmol) and m-chloroperoxybenzoic acid (2.70 g, 11 mmol) in methylene dichloride (25 L) overnight. The reaction was quenched by pouring into water (50 mL) . The aqueous was extracted with ether (3 x 25 mL) . The organic layers were combined and washed with 10% aqueous sodium sulfate (2 x 25 mL) , saturated aqueous sodium bicarbonate (3 x 25 mL) , and brine (25 mL) . The organic phase was dried over magnesium sulfate and the solvents were removed in vacuo. The resulting yellow oil was used without further purification.
- the crude orange oil was purified (MPLC, silica gel, 1% MeOH/CHCl 3 ) and dissolved in methylene dichloride (5 mL) .
- the hydrochloride salt was prepared by adding an excess of 1 M HCl/ether.
- the solvents were removed in vacuo to yield 139 mg of thick oil: GC/EI-MS, /z (rel. int.) 342 (M + , .l) , 237 (15) , 236 (100) , 163 (5), 136 (6), 135 (61), 121 (23) , 78 (7), 77 (13), 70 (15) , 58 (7) .
- phenyl glycidyl ether 150 mg, 1.1 mmol
- 1, l-dimethyl-2- (3-fluoro- 4-methoxyphenyl) ethylamine 197 mg, 1 mmol
- the title compound crystallized on standing to yield 169 mg of small crystals: GC/EI-MS, m/z (rel. int.) 332 (M* - 15, .1) , 209 (16) , 208 (100), 139 (13) , 133 (5), 114 (7), 107 (6), 77 (11), 71 (12) , 70 (20) , 58 (9) .
- Potassium carbonate 13 mmol was added to a solution of 1, 5-dihydroxy napthalene (6.2 mmol) in acetone in a sealed vacuum tube. The tube was heated to 70 °C for 30 minutes. lodomethane (9.4 mmol) was added and the tube heated to 70 °C overnight .
- the reaction mixture was partitioned between ether and 10% aqueous HCl.
- the ether layer was separated and extracted with 0.5 M KOH.
- the water layer was separated and acidified with 10% aqueous HCl and extracted into ether.
- the ether layer was separated and dried over magnesium sulfate and evaporated to a solid.
- the solid was purified using reverse phase HPLC using a acetonitrile/0.1% HCl gradient yielding 179 mg 1-hydroxy-5-methoxy napthalene.
- Sodium hydride (60% suspension in mineral oil, 1 mmol) was added to a solution of 1-hydroxy-5-methoxy napthalene (1 mmol) and stirred 10 minutes.
- EXAMPLE 86 Additional compounds Additional compounds were synthesized using techniques along lines as those described above. Examples of such compounds include the following:
- Compound 22 N- (3-phenylpropyl) -1, l-dimethyl-2 (4- methoxyphenyl)ethylamine.
- Compound 24 iV- (4-phenylbutyl) -1, l-dimethyl-2- (4- methoxyphenyl)ethylamine.
- Compound 54 iV-2-hydroxy-3- (4- tert-butylphenoxy)propyl] - 1, 1-dimethyl-2-phenylethylamine.
- Compound 55 N- [2-hydroxy-3- (1-naphthoxy)propyl] -1, 1- dimethyl-2-phenylethylamine.
- Compound 102 N- (2-hydroxy-3-phenoxypropyl) -1, 1- dimethyl-2- (4-ethoxyphenyl)ethylamine.
- Compound 132 N- [2-hydroxy-3- (3, 4-dimethoxy- phenoxy)propyl] -1, l-dimethyl-2- (4-methoxyphenyl) ethylamine Hydrochloride; GC/EI-MS, m/z (rel. int.) 390 (M+,.0) , 269 (17), 268 (100), 163 (6), 153 (5), 121 (21) , 114 (17) , 77 (5) , 71 (19) , 70 (17) , 58 (7) .
- Compound 152 N- [2-hydroxy-3- (4-trifluoro- methoxyphenoxy)propyl] -1, l-dimethyl-2- (4-methoxy- phenyl) ethylamine Hydrochloride; GC/EI-MS, m/z (rel. int.) 398 (M-15,2) , 293(15), 292 (100) , 121 (20) , 77 (5) .
- Compound 153 N- [2-hydroxy-3- (2 -iso-propyl- phenoxy)propyl] -1, l-dimethyl-2- (4-methoxyphenyl) ethylamine Hydrochloride; GC/EI-MS, m/z (rel.
- Compound 160 N- [2-hydroxy-3- (3-cyanophenoxy)propyl] - 1, l-dimethyl-2- (4-methoxyphenyl) ethylamine Hydrochloride; GC/EI-MS, m/z (rel. int.) 339 (M-15, .l) , 234 (15) , 233 (100) , 121 (21) , 102 (7) , 90 (6) .
- Compound 161 N- [2-hydroxy-3- (4-cyanophenoxy)propyl] - 1, l-dimethyl-2- (4-methoxyphenyl)ethylamine Hydrochloride; GC/EI-MS, m/z (rel.
Abstract
Description












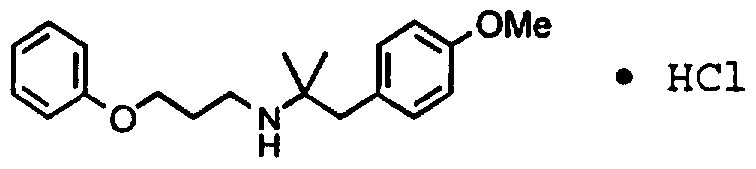
























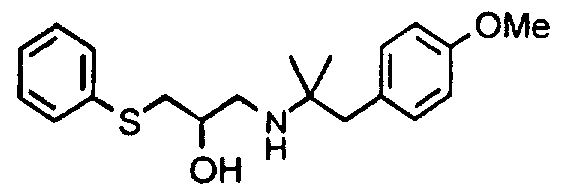


















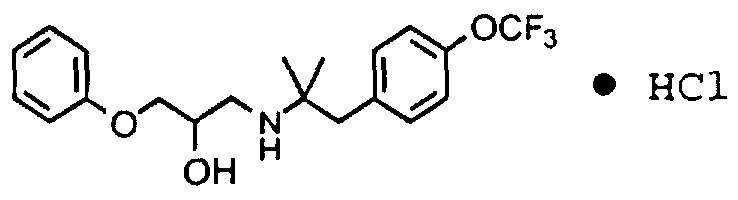



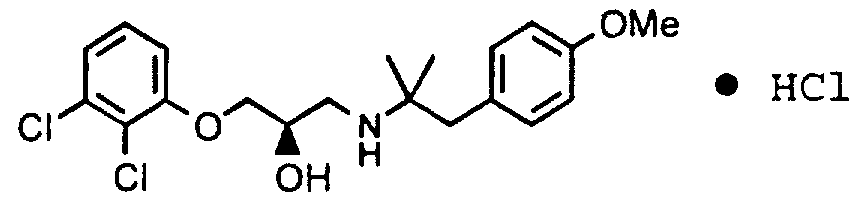

















Claims





Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| NZ332068A NZ332068A (en) | 1996-04-09 | 1997-04-04 | Alpha, alpha-disubstituted arylalkylamine derivatives as calcilytic compounds |
| EP97917848A EP0901459B1 (en) | 1996-04-09 | 1997-04-04 | Calcilytic compounds |
| PL329314A PL191027B1 (en) | 1996-04-09 | 1997-04-04 | α,α-disubstituted arylalkylamine derivative, pharmaceutical composition and use of α,α-disubstituted arylalkylamine |
| EA199800913A EA002984B1 (en) | 1996-04-09 | 1997-04-04 | Calcilytic compounds, pharmaceutical compositions and method of screening for calcilytic compounds |
| CA002251331A CA2251331C (en) | 1996-04-09 | 1997-04-04 | Calcilytic compounds |
| JP09536327A JP2001501584A (en) | 1996-04-09 | 1997-04-04 | Calcilytic compound |
| AT97917848T ATE298739T1 (en) | 1996-04-09 | 1997-04-04 | CALCYLITIC COMPOUNDS |
| BR9708632-0A BR9708632A (en) | 1996-04-09 | 1997-04-04 | Calcilic compounds |
| AU26070/97A AU726659B2 (en) | 1996-04-09 | 1997-04-04 | Calcilytic compounds |
| IL12645897A IL126458A (en) | 1996-04-09 | 1997-04-04 | Calcium receptor inhibiting calcilytic compounds |
| DE69733649T DE69733649T2 (en) | 1996-04-09 | 1997-04-04 | CALCYLITIC COMPOUNDS |
| HK99103964A HK1018899A1 (en) | 1996-04-09 | 1999-09-11 | Calcilytic compounds |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US62960896A | 1996-04-09 | 1996-04-09 | |
| US08/629,608 | 1996-04-09 | ||
| US3226396P | 1996-12-03 | 1996-12-03 | |
| US60/032,263 | 1996-12-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1997037967A1 true WO1997037967A1 (en) | 1997-10-16 |
Family
ID=26708193
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1997/005558 WO1997037967A1 (en) | 1996-04-09 | 1997-04-04 | Calcilytic compounds |
Country Status (19)
| Country | Link |
|---|---|
| US (2) | US6022894A (en) |
| EP (1) | EP0901459B1 (en) |
| JP (1) | JP2001501584A (en) |
| CN (1) | CN1254465C (en) |
| AR (1) | AR008586A1 (en) |
| AT (1) | ATE298739T1 (en) |
| AU (1) | AU726659B2 (en) |
| BR (1) | BR9708632A (en) |
| CA (1) | CA2251331C (en) |
| CO (1) | CO4820431A1 (en) |
| DE (1) | DE69733649T2 (en) |
| EA (1) | EA002984B1 (en) |
| HK (1) | HK1018899A1 (en) |
| IL (1) | IL126458A (en) |
| NZ (1) | NZ332068A (en) |
| PL (1) | PL191027B1 (en) |
| SA (1) | SA97180135B1 (en) |
| SG (1) | SG99290A1 (en) |
| WO (1) | WO1997037967A1 (en) |
Cited By (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999038500A2 (en) * | 1998-01-30 | 1999-08-05 | The Brigham And Women's Hospital, Inc. | The human calcium-sensing receptor in the detection and treatment of cancer |
| EP0973730A1 (en) * | 1997-04-08 | 2000-01-26 | Smithkline Beecham Corporation | Calcilytic compounds |
| EP1070048A1 (en) * | 1998-04-08 | 2001-01-24 | Smithkline Beecham Corporation | Calcilytic compounds |
| WO2001053254A1 (en) * | 2000-01-24 | 2001-07-26 | Smithkline Beecham Corporation | Calcilytic compounds |
| EP1148876A1 (en) * | 1999-02-02 | 2001-10-31 | SmithKline Beecham Corporation | Calcilytic compounds |
| EP1308436A1 (en) * | 2000-08-11 | 2003-05-07 | Japan Tobacco Inc. | Calcium receptor antagonists |
| EP1368318A2 (en) * | 2000-07-21 | 2003-12-10 | SmithKline Beecham Corporation | Calcilytic compounds |
| EP1383511A2 (en) * | 2000-10-25 | 2004-01-28 | Smithkline Beecham Corporation | Calcilytic compounds |
| EP1420008A1 (en) * | 2002-11-15 | 2004-05-19 | L'oreal | Composition, in particular cosmetic, comprising a secondary or tertiary amine |
| US6908935B2 (en) | 2002-05-23 | 2005-06-21 | Amgen Inc. | Calcium receptor modulating agents |
| WO2005115975A1 (en) | 2004-05-28 | 2005-12-08 | Tanabe Seiyaku Co., Ltd. | Arylalkylamines and process for production thereof |
| US7105537B2 (en) | 2003-01-28 | 2006-09-12 | Bristol-Myers Squibb Company | 2-substituted cyclic amines as calcium sensing receptor modulators |
| US7109238B2 (en) | 2000-01-24 | 2006-09-19 | Smith Kline Beecham Corporation | Calcilytic compounds |
| EP1713767A1 (en) * | 2004-02-06 | 2006-10-25 | Smithkline Beecham Corporation | Calcilytic compounds |
| US7157498B2 (en) | 2000-08-08 | 2007-01-02 | Centre National De La Recherche Scientifique (Cnrs) | Diamines having a CaSR modulating activity |
| US7176322B2 (en) | 2002-05-23 | 2007-02-13 | Amgen Inc. | Calcium receptor modulating agents |
| US7205322B2 (en) | 2003-02-12 | 2007-04-17 | Bristol-Myers Squibb Company | Thiazolidine compounds as calcium sensing receptor modulators |
| WO2007060026A1 (en) * | 2005-11-25 | 2007-05-31 | Galapagos Sas | Urea derivatives useful as calcium receptor modulators |
| US7262280B1 (en) | 1998-04-03 | 2007-08-28 | Nps Pharmaceuticals, Inc. | G-protein fusion receptors and constructs encoding same |
| US7265145B2 (en) | 2003-05-28 | 2007-09-04 | Bristol-Myers Squibb Company | Substituted piperidines and pyrrolidines as calcium sensing receptor modulators and method |
| US7285572B2 (en) | 2003-05-28 | 2007-10-23 | Japan Tobacco Inc. | CaSR antagonist |
| US7304174B2 (en) | 2003-04-23 | 2007-12-04 | Japan Tobacco Inc. | CaSR antagonist |
| US7338772B2 (en) | 2002-04-12 | 2008-03-04 | Schering Corporation | G-protein coupled receptor ligands and methods |
| US7393837B2 (en) | 1998-09-28 | 2008-07-01 | Mcgill University | Inhibition of PEX in the treatment of metabolic bone diseases |
| US7514473B2 (en) | 2002-11-26 | 2009-04-07 | Smithkline Beecham, Corp. | Calcilytic compounds |
| US7582658B2 (en) | 2003-12-25 | 2009-09-01 | Asahi Kasei Pharma Corporation | Bicyclic compound |
| WO2009148052A1 (en) | 2008-06-05 | 2009-12-10 | 旭化成ファーマ株式会社 | Sulfonamide compound and application thereof |
| EP2196461A1 (en) | 2008-12-15 | 2010-06-16 | Bayer CropScience AG | 4-Amino-1,2,3-benzoxathiazine-derivatives as pesticides |
| WO2010103429A1 (en) | 2009-03-10 | 2010-09-16 | Pfizer Inc. | 1,1-(Dimethyl-Ethylamino)-2-Hydroxy-Propoxy]-Ethyl}-3-Methyl-Biphenyl-4- Carboxylic Acid Derivatives As Calcium Receptor Antagonists |
| US8247412B2 (en) | 2005-04-29 | 2012-08-21 | Galapagos Sasu | Urea derivatives methods for their manufacture and uses thereof |
| US8486381B2 (en) | 2005-09-02 | 2013-07-16 | Amgen Inc. | Methods of modulating intestinal fluid balance |
| US9861606B2 (en) | 2012-09-28 | 2018-01-09 | King's College London | Therapeutic for treating inflammatory lung disorders |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE4426245A1 (en) * | 1994-07-23 | 1996-02-22 | Gruenenthal Gmbh | 1-phenyl-3-dimethylamino-propane compounds with pharmacological activity |
| US7202261B2 (en) * | 1996-12-03 | 2007-04-10 | Nps Pharmaceuticals, Inc. | Calcilytic compounds |
| CO5180628A1 (en) * | 1999-07-31 | 2002-07-30 | Smithkline Beecham Corp | CALCIOLITIC COMPOUNDS |
| US7648965B2 (en) * | 2004-05-14 | 2010-01-19 | Unigene Laboratories Inc. | Method for fostering bone formation and preservation |
| US7531518B2 (en) | 2004-05-14 | 2009-05-12 | Unigene Laboratories Inc. | Method for fostering bone formation and preservation |
| US20080069814A1 (en) * | 2005-01-05 | 2008-03-20 | Novacea, Inc. | Prevention of Thrombotic Disorders with Active Vitamin D Compounds or Mimics Thereof |
| MX2007013029A (en) * | 2005-04-22 | 2008-01-11 | Novacea Inc | Treatment, prevention and amelioration of pulmonary disorders associated with chemotherapy or radiotherapy with active vitamin d compounds or mimics thereof. |
| US20060293667A1 (en) * | 2005-05-19 | 2006-12-28 | Agnes Vignery | Bone implant device and methods of using same |
| WO2009055631A1 (en) * | 2007-10-25 | 2009-04-30 | Smithkline Beecham Corporation | Calcilytic compounds |
| CN103601645B (en) * | 2013-11-07 | 2016-05-25 | 上海适济生物科技有限公司 | The preparation method of 1-(phenethyl amino) propane-2-alcohol compound or its salt |
| WO2019153216A1 (en) * | 2018-02-09 | 2019-08-15 | 北京梅尔森医药技术开发有限公司 | Substituted arylamino alcohol compound, preparation method therefor, and application thereof |
| EP4069669A1 (en) * | 2019-12-05 | 2022-10-12 | Syngenta Crop Protection AG | One step process for the preparation of phenyl ethyl amine derivatives |
| CN116199605A (en) * | 2023-02-27 | 2023-06-02 | 上海睿腾医药科技有限公司 | Synthesis method of 5-chloro-2-hydroxy-dimethylbenzenesulfonamide |
Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2502993A1 (en) * | 1974-02-14 | 1975-09-04 | Haessle Ab | AMINES, THE METHOD OF MANUFACTURING THEIR PRODUCTS AND THE MEDICINAL PRODUCTS CONTAINING THEY |
| EP0002792A1 (en) * | 1978-01-03 | 1979-07-11 | Sandoz Ag | 3-Aminopropoxyaryl derivatives, their preparation and pharmaceutical compositions containing them |
| ES480066A1 (en) * | 1979-04-28 | 1980-04-01 | Especialid Latin Medicam Unive | Procedure for obtaining 1- (4-chloro-alpha, alpha-dimethyl-benzene-etanalline) -2-oxy-3- (aricoxi) propanes. (Machine-translation by Google Translate, not legally binding) |
| EP0009075A1 (en) * | 1978-06-27 | 1980-04-02 | Merck & Co. Inc. | Pyridyloxy-propanol amines and esters thereof, process for preparing the same and pharmaceutical compositions containing them |
| DE2943406A1 (en) * | 1979-10-26 | 1981-05-07 | Basf Ag, 6700 Ludwigshafen | AMINO DERIVATIVES OF 2-METHYL-5- (2-HYDROXYSTYRYL) -1,3,4-THIADIAZOL |
| DE2944222A1 (en) * | 1979-11-02 | 1981-05-14 | Basf Ag, 6700 Ludwigshafen | Amino derivs. of 1,2-benzisothiazole - beta:sympatholytics suitable for the treatment of coronary heart disease, hypertonia and cardiac arrhythmia |
| CH636856A5 (en) * | 1978-07-17 | 1983-06-30 | Sandoz Ag | 4-(3-Aminopropoxy)indole derivatives, their preparation and medicines containing them |
| DE3301198A1 (en) * | 1983-01-15 | 1984-07-19 | Hoechst Ag, 6230 Frankfurt | N-Arylalkylamine-3-propoxypyridine derivatives, process for their preparation, pharmaceutical preparations containing them and their use |
| EP0188361A1 (en) * | 1985-01-15 | 1986-07-23 | Glaxo Group Limited | Amine derivatives |
| DE4040186A1 (en) * | 1989-12-20 | 1991-06-27 | Hoechst Ag | Use of 3-amino-propan-1,2-di:ol derivs. - as racemate, enantiomeric mixt. or pure enantiomer in treatment of diabetes mellitus |
| WO1993004373A1 (en) * | 1991-08-23 | 1993-03-04 | Nps Pharmaceuticals, Inc. | Calcium receptor active molecules |
| WO1994018959A1 (en) * | 1993-02-23 | 1994-09-01 | Brigham And Women's Hospital, Inc. | Calcium receptor-active molecules |
Family Cites Families (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE298506C (en) * | ||||
| GB1129072A (en) * | 1966-02-01 | 1968-10-02 | Ici Ltd | Benzofuran and indole derivatives |
| GB1058822A (en) * | 1963-07-30 | 1967-02-15 | Ici Ltd | 3-amino-2-hydroxypropoxy heterocyclic derivatives |
| GB1199037A (en) * | 1967-09-27 | 1970-07-15 | Ici Ltd | Alkanolamine Derivatives |
| ES410417A1 (en) * | 1973-01-08 | 1976-01-01 | Andreu Sa Dr | Procedure for the obtaining of aminoalcohols derived from p-acetamidophenol. (Machine-translation by Google Translate, not legally binding) |
| DE2530613A1 (en) * | 1974-11-01 | 1976-05-06 | Haessle Ab | AMINE DERIVATIVES, PROCESS FOR THEIR MANUFACTURING AND MEDICINAL PRODUCTS CONTAINING THEM |
| US4067904A (en) * | 1975-12-19 | 1978-01-10 | Mead Johnson & Company | Alkylsulfonylphenoxypropanolamine derivatives |
| JPS5936617B2 (en) * | 1976-03-05 | 1984-09-05 | 大塚製薬株式会社 | carbostyril derivatives |
| CH619453A5 (en) * | 1976-03-17 | 1980-09-30 | Otsuka Pharma Co Ltd | |
| US4235919A (en) * | 1977-07-21 | 1980-11-25 | Sandoz Ltd. | 1-(Indol-4-yloxy)-3-(2-substituted amino)-2-propanols and pharmaceutical use thereof |
| DE2830211A1 (en) * | 1977-07-21 | 1979-02-01 | Sandoz Ag | SUBSTITUTED INDOL DERIVATIVES, THEIR PRODUCTION AND USE |
| DE2914166A1 (en) * | 1979-04-07 | 1980-10-23 | Beiersdorf Ag | ARYL-SUBSTITUTED FURANE AND METHOD FOR THEIR PRODUCTION |
| JPS568319A (en) * | 1980-03-26 | 1981-01-28 | Otsuka Pharmaceut Co Ltd | Antihistamine |
| US4332787A (en) * | 1980-06-23 | 1982-06-01 | The Massachusetts General Hospital | Assay for beta-adrenergic antagonists and antibody therefor |
| JPS5780321A (en) * | 1980-11-05 | 1982-05-19 | Otsuka Pharmaceut Co Ltd | Cardiotonic agent |
| US4376125A (en) * | 1980-11-05 | 1983-03-08 | University Of Virginia Alumni Patents Foundation | Aminobenzlpropranolol and pharmaceutical preparation thereof |
| EP0095454A3 (en) * | 1982-05-13 | 1985-04-03 | Gerot-Pharmazeutika Gesellschaft m.b.H. | Nuclens-substituted pyrogallol derivatives |
| US4517188A (en) * | 1983-05-09 | 1985-05-14 | Mead Johnson & Company | 1-Pyrimidinyloxy-3-hetaryl-alkylamino-2-propanols |
| GB2192394A (en) * | 1986-07-11 | 1988-01-13 | Glaxo Group Ltd | Amine derivatives |
| US5053514A (en) * | 1988-08-10 | 1991-10-01 | Otsuka Pharmaceutical Company, Limited | Cardiotonics |
| DE4017019A1 (en) * | 1990-05-26 | 1991-11-28 | Hoechst Ag | USE OF SUBSTITUTED SS-HYDROXYETHYLAMINES AS POTENTIAL INGREDIENTS OF THE EXOENZYME OF MUSHROOMS |
| CA2102780C (en) * | 1991-05-10 | 2007-01-09 | Alfred P. Spada | Bis mono-and bicyclic aryl and heteroaryl compounds which inhibit egf and/or pdgf receptor tyrosine kinase |
| CA2173747C (en) * | 1991-08-23 | 2006-05-23 | Edward F. Nemeth | Calcium receptor-active arylalkyl amines |
| US5763569A (en) * | 1991-08-23 | 1998-06-09 | The Brigham And Women's Hospital, Inc | Calcium receptor-active molecules |
| GB9312893D0 (en) * | 1993-06-22 | 1993-08-04 | Boots Co Plc | Therapeutic agents |
| ZA967892B (en) * | 1995-09-21 | 1998-03-18 | Lilly Co Eli | Selective β3 adrenergic agonists. |
-
1997
- 1997-04-04 CN CNB971953686A patent/CN1254465C/en not_active Expired - Lifetime
- 1997-04-04 WO PCT/US1997/005558 patent/WO1997037967A1/en active IP Right Grant
- 1997-04-04 NZ NZ332068A patent/NZ332068A/en not_active IP Right Cessation
- 1997-04-04 DE DE69733649T patent/DE69733649T2/en not_active Expired - Lifetime
- 1997-04-04 EP EP97917848A patent/EP0901459B1/en not_active Expired - Lifetime
- 1997-04-04 AU AU26070/97A patent/AU726659B2/en not_active Expired
- 1997-04-04 US US08/832,984 patent/US6022894A/en not_active Expired - Fee Related
- 1997-04-04 EA EA199800913A patent/EA002984B1/en not_active IP Right Cessation
- 1997-04-04 AT AT97917848T patent/ATE298739T1/en not_active IP Right Cessation
- 1997-04-04 PL PL329314A patent/PL191027B1/en unknown
- 1997-04-04 BR BR9708632-0A patent/BR9708632A/en not_active Application Discontinuation
- 1997-04-04 JP JP09536327A patent/JP2001501584A/en not_active Ceased
- 1997-04-04 IL IL12645897A patent/IL126458A/en not_active IP Right Cessation
- 1997-04-04 CA CA002251331A patent/CA2251331C/en not_active Expired - Lifetime
- 1997-04-04 SG SG9905132A patent/SG99290A1/en unknown
- 1997-04-08 CO CO97017849A patent/CO4820431A1/en unknown
- 1997-04-09 AR ARP970101400A patent/AR008586A1/en active IP Right Grant
- 1997-06-16 SA SA97180135A patent/SA97180135B1/en unknown
-
1998
- 1998-08-11 US US09/132,179 patent/US6521667B1/en not_active Expired - Fee Related
-
1999
- 1999-09-11 HK HK99103964A patent/HK1018899A1/en not_active IP Right Cessation
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2502993A1 (en) * | 1974-02-14 | 1975-09-04 | Haessle Ab | AMINES, THE METHOD OF MANUFACTURING THEIR PRODUCTS AND THE MEDICINAL PRODUCTS CONTAINING THEY |
| EP0002792A1 (en) * | 1978-01-03 | 1979-07-11 | Sandoz Ag | 3-Aminopropoxyaryl derivatives, their preparation and pharmaceutical compositions containing them |
| EP0009075A1 (en) * | 1978-06-27 | 1980-04-02 | Merck & Co. Inc. | Pyridyloxy-propanol amines and esters thereof, process for preparing the same and pharmaceutical compositions containing them |
| CH636856A5 (en) * | 1978-07-17 | 1983-06-30 | Sandoz Ag | 4-(3-Aminopropoxy)indole derivatives, their preparation and medicines containing them |
| ES480066A1 (en) * | 1979-04-28 | 1980-04-01 | Especialid Latin Medicam Unive | Procedure for obtaining 1- (4-chloro-alpha, alpha-dimethyl-benzene-etanalline) -2-oxy-3- (aricoxi) propanes. (Machine-translation by Google Translate, not legally binding) |
| DE2943406A1 (en) * | 1979-10-26 | 1981-05-07 | Basf Ag, 6700 Ludwigshafen | AMINO DERIVATIVES OF 2-METHYL-5- (2-HYDROXYSTYRYL) -1,3,4-THIADIAZOL |
| DE2944222A1 (en) * | 1979-11-02 | 1981-05-14 | Basf Ag, 6700 Ludwigshafen | Amino derivs. of 1,2-benzisothiazole - beta:sympatholytics suitable for the treatment of coronary heart disease, hypertonia and cardiac arrhythmia |
| DE3301198A1 (en) * | 1983-01-15 | 1984-07-19 | Hoechst Ag, 6230 Frankfurt | N-Arylalkylamine-3-propoxypyridine derivatives, process for their preparation, pharmaceutical preparations containing them and their use |
| EP0188361A1 (en) * | 1985-01-15 | 1986-07-23 | Glaxo Group Limited | Amine derivatives |
| DE4040186A1 (en) * | 1989-12-20 | 1991-06-27 | Hoechst Ag | Use of 3-amino-propan-1,2-di:ol derivs. - as racemate, enantiomeric mixt. or pure enantiomer in treatment of diabetes mellitus |
| WO1993004373A1 (en) * | 1991-08-23 | 1993-03-04 | Nps Pharmaceuticals, Inc. | Calcium receptor active molecules |
| WO1994018959A1 (en) * | 1993-02-23 | 1994-09-01 | Brigham And Women's Hospital, Inc. | Calcium receptor-active molecules |
Cited By (65)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0973730A1 (en) * | 1997-04-08 | 2000-01-26 | Smithkline Beecham Corporation | Calcilytic compounds |
| EP0973730A4 (en) * | 1997-04-08 | 2000-08-23 | Smithkline Beecham Corp | Calcilytic compounds |
| US6294531B1 (en) | 1997-04-08 | 2001-09-25 | Smithkline Beecham Corporation | Calcilytic compounds |
| WO1999038500A3 (en) * | 1998-01-30 | 2000-04-13 | Brigham & Womens Hospital | The human calcium-sensing receptor in the detection and treatment of cancer |
| WO1999038500A2 (en) * | 1998-01-30 | 1999-08-05 | The Brigham And Women's Hospital, Inc. | The human calcium-sensing receptor in the detection and treatment of cancer |
| US6296833B1 (en) | 1998-01-30 | 2001-10-02 | The Brigham And Women's Hospital, Inc. | Human calcium-sensing receptor in the detection and treatment of cancer |
| US7262280B1 (en) | 1998-04-03 | 2007-08-28 | Nps Pharmaceuticals, Inc. | G-protein fusion receptors and constructs encoding same |
| EP1070048A1 (en) * | 1998-04-08 | 2001-01-24 | Smithkline Beecham Corporation | Calcilytic compounds |
| EP1070048A4 (en) * | 1998-04-08 | 2001-11-07 | Smithkline Beecham Corp | Calcilytic compounds |
| US6395919B1 (en) | 1998-04-08 | 2002-05-28 | Smithkline Beecham Corporation | Calcilytic compounds |
| US7393837B2 (en) | 1998-09-28 | 2008-07-01 | Mcgill University | Inhibition of PEX in the treatment of metabolic bone diseases |
| EP1148876A1 (en) * | 1999-02-02 | 2001-10-31 | SmithKline Beecham Corporation | Calcilytic compounds |
| EP1148876A4 (en) * | 1999-02-02 | 2003-07-23 | Smithkline Beecham Corp | Calcilytic compounds |
| AU764746B2 (en) * | 2000-01-24 | 2003-08-28 | Nps Pharmaceuticals, Inc. | Calcilytic compounds |
| US7109238B2 (en) | 2000-01-24 | 2006-09-19 | Smith Kline Beecham Corporation | Calcilytic compounds |
| EP2141146A1 (en) * | 2000-01-24 | 2010-01-06 | SmithKline Beecham Corporation | Calcilytic compounds |
| WO2001053254A1 (en) * | 2000-01-24 | 2001-07-26 | Smithkline Beecham Corporation | Calcilytic compounds |
| EP1368318A2 (en) * | 2000-07-21 | 2003-12-10 | SmithKline Beecham Corporation | Calcilytic compounds |
| EP1368318A4 (en) * | 2000-07-21 | 2006-08-23 | Smithkline Beecham Corp | Calcilytic compounds |
| US7157498B2 (en) | 2000-08-08 | 2007-01-02 | Centre National De La Recherche Scientifique (Cnrs) | Diamines having a CaSR modulating activity |
| EP1308436A4 (en) * | 2000-08-11 | 2005-10-05 | Japan Tobacco Inc | Calcium receptor antagonists |
| US6916956B2 (en) | 2000-08-11 | 2005-07-12 | Japan Tobacco, Inc. | Calcium receptor antagonist |
| US7211685B2 (en) | 2000-08-11 | 2007-05-01 | Japan Tobacco Inc. | Calcium receptor antagonists |
| EP1308436A1 (en) * | 2000-08-11 | 2003-05-07 | Japan Tobacco Inc. | Calcium receptor antagonists |
| EP1383511A4 (en) * | 2000-10-25 | 2005-05-18 | Smithkline Beecham Corp | Calcilytic compounds |
| EP1383511A2 (en) * | 2000-10-25 | 2004-01-28 | Smithkline Beecham Corporation | Calcilytic compounds |
| US7807390B2 (en) | 2002-04-12 | 2010-10-05 | Schering Corporation | G-protein coupled receptor ligands and methods |
| US7338772B2 (en) | 2002-04-12 | 2008-03-04 | Schering Corporation | G-protein coupled receptor ligands and methods |
| US6908935B2 (en) | 2002-05-23 | 2005-06-21 | Amgen Inc. | Calcium receptor modulating agents |
| US7176322B2 (en) | 2002-05-23 | 2007-02-13 | Amgen Inc. | Calcium receptor modulating agents |
| US7196102B2 (en) | 2002-05-23 | 2007-03-27 | Amgen Inc. | Calcium receptor modulating agents |
| EP1420008A1 (en) * | 2002-11-15 | 2004-05-19 | L'oreal | Composition, in particular cosmetic, comprising a secondary or tertiary amine |
| FR2847250A1 (en) * | 2002-11-15 | 2004-05-21 | Oreal | COMPOSITION, IN PARTICULAR COSMETIC, COMPRISING SECONDARY OR TERTIARY AMINE |
| US7829594B2 (en) | 2002-11-26 | 2010-11-09 | GlaxoSmithKline, LLC | Calcilytic compounds |
| US8399517B2 (en) | 2002-11-26 | 2013-03-19 | GlaxoSmithKline, LLC | Calcilytic compounds |
| US9227914B2 (en) | 2002-11-26 | 2016-01-05 | GlaxoSmithKline, LLC | Calcilytic compounds |
| US8586631B2 (en) | 2002-11-26 | 2013-11-19 | GlaxoSmithKline, LLC | Calcilytic compounds |
| US8980950B2 (en) | 2002-11-26 | 2015-03-17 | GlaxoSmithKline, LLC | Calcilytic compounds |
| US7514473B2 (en) | 2002-11-26 | 2009-04-07 | Smithkline Beecham, Corp. | Calcilytic compounds |
| US7105537B2 (en) | 2003-01-28 | 2006-09-12 | Bristol-Myers Squibb Company | 2-substituted cyclic amines as calcium sensing receptor modulators |
| US7205322B2 (en) | 2003-02-12 | 2007-04-17 | Bristol-Myers Squibb Company | Thiazolidine compounds as calcium sensing receptor modulators |
| EP2308828A2 (en) | 2003-04-23 | 2011-04-13 | Japan Tobacco Inc. | CaSR antagonist |
| US7304174B2 (en) | 2003-04-23 | 2007-12-04 | Japan Tobacco Inc. | CaSR antagonist |
| EP2189439A2 (en) | 2003-04-23 | 2010-05-26 | Japan Tobacco, Inc. | CaSR antagonist |
| US7285572B2 (en) | 2003-05-28 | 2007-10-23 | Japan Tobacco Inc. | CaSR antagonist |
| US7265145B2 (en) | 2003-05-28 | 2007-09-04 | Bristol-Myers Squibb Company | Substituted piperidines and pyrrolidines as calcium sensing receptor modulators and method |
| US7582658B2 (en) | 2003-12-25 | 2009-09-01 | Asahi Kasei Pharma Corporation | Bicyclic compound |
| EP1713767A1 (en) * | 2004-02-06 | 2006-10-25 | Smithkline Beecham Corporation | Calcilytic compounds |
| EP1713767A4 (en) * | 2004-02-06 | 2008-01-16 | Smithkline Beecham Corp | Calcilytic compounds |
| US8492111B2 (en) | 2004-05-28 | 2013-07-23 | Mitsubishi Tanabe Pharma Corporation | Arylalkylamine compound and process for preparing the same |
| WO2005115975A1 (en) | 2004-05-28 | 2005-12-08 | Tanabe Seiyaku Co., Ltd. | Arylalkylamines and process for production thereof |
| US8778628B2 (en) | 2004-05-28 | 2014-07-15 | Mitsubishi Tanabe Pharma Corporation | Arylalkylamine compound and process for preparing the same |
| US8703721B2 (en) | 2004-05-28 | 2014-04-22 | Mitsubishi Tanabe Pharma Corporation | Arylalkylamine compound and process for preparing the same |
| US8362274B2 (en) | 2004-05-28 | 2013-01-29 | Mitsubishi Tanabe Pharma Corporation | Arylalkylamine compound and process for preparing the same |
| US8759387B2 (en) | 2004-05-28 | 2014-06-24 | Mitsubishi Tanabe Pharma Corporation | Arylalkylamine compound and process for preparing the same |
| US8247412B2 (en) | 2005-04-29 | 2012-08-21 | Galapagos Sasu | Urea derivatives methods for their manufacture and uses thereof |
| US8486381B2 (en) | 2005-09-02 | 2013-07-16 | Amgen Inc. | Methods of modulating intestinal fluid balance |
| US7875609B2 (en) | 2005-11-25 | 2011-01-25 | Galapagos Sasu | Urea derivatives, processes for their preparation, their use as medicaments, and pharmaceutical compositions containing them |
| WO2007060026A1 (en) * | 2005-11-25 | 2007-05-31 | Galapagos Sas | Urea derivatives useful as calcium receptor modulators |
| WO2009148052A1 (en) | 2008-06-05 | 2009-12-10 | 旭化成ファーマ株式会社 | Sulfonamide compound and application thereof |
| US8039514B2 (en) | 2008-06-05 | 2011-10-18 | Asahi Kasei Pharma Corporation | Sulfonamide compounds and use thereof |
| US8173641B2 (en) | 2008-12-15 | 2012-05-08 | Bayer Cropscience Ag | 4-amino-1,2,3-benzoxathiazine-derivatives as pesticides |
| EP2196461A1 (en) | 2008-12-15 | 2010-06-16 | Bayer CropScience AG | 4-Amino-1,2,3-benzoxathiazine-derivatives as pesticides |
| WO2010103429A1 (en) | 2009-03-10 | 2010-09-16 | Pfizer Inc. | 1,1-(Dimethyl-Ethylamino)-2-Hydroxy-Propoxy]-Ethyl}-3-Methyl-Biphenyl-4- Carboxylic Acid Derivatives As Calcium Receptor Antagonists |
| US9861606B2 (en) | 2012-09-28 | 2018-01-09 | King's College London | Therapeutic for treating inflammatory lung disorders |
Also Published As
| Publication number | Publication date |
|---|---|
| NZ332068A (en) | 2001-03-30 |
| IL126458A0 (en) | 1999-08-17 |
| HK1018899A1 (en) | 2000-01-07 |
| EP0901459B1 (en) | 2005-06-29 |
| IL126458A (en) | 2004-06-20 |
| CA2251331C (en) | 2009-06-09 |
| AR008586A1 (en) | 2000-02-09 |
| AU726659B2 (en) | 2000-11-16 |
| CN1221401A (en) | 1999-06-30 |
| US6521667B1 (en) | 2003-02-18 |
| PL191027B1 (en) | 2006-03-31 |
| EP0901459A1 (en) | 1999-03-17 |
| SG99290A1 (en) | 2003-10-27 |
| EA002984B1 (en) | 2002-12-26 |
| US6022894A (en) | 2000-02-08 |
| EA199800913A1 (en) | 1999-06-24 |
| CA2251331A1 (en) | 1997-10-16 |
| DE69733649D1 (en) | 2005-08-04 |
| BR9708632A (en) | 2000-01-18 |
| ATE298739T1 (en) | 2005-07-15 |
| CO4820431A1 (en) | 1999-07-28 |
| JP2001501584A (en) | 2001-02-06 |
| PL329314A1 (en) | 1999-03-15 |
| AU2607097A (en) | 1997-10-29 |
| DE69733649T2 (en) | 2006-05-18 |
| SA97180135B1 (en) | 2006-06-12 |
| CN1254465C (en) | 2006-05-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6521667B1 (en) | Calcilytic compounds | |
| JP4431609B2 (en) | Inorganic ion active compound | |
| US6432656B1 (en) | Calcilytic compounds | |
| US6001884A (en) | Calcium receptor-active molecules | |
| EP0787122B9 (en) | Calcium receptor-active compounds | |
| US6335338B1 (en) | Calcilytic compounds | |
| US7202261B2 (en) | Calcilytic compounds | |
| US6818660B2 (en) | Calcilytic compounds | |
| US20070249702A1 (en) | Calcilytic compounds | |
| US6291459B1 (en) | Calcilytic compounds | |
| WO1998044925A1 (en) | Calcilytic compounds | |
| EP1258471B1 (en) | Inorganic ion receptor-active compounds | |
| EP1770083B1 (en) | Inorganic ion receptor-active compounds | |
| AU2004202208B2 (en) | Inorganic Ion Receptor-active Compounds | |
| CA2253407C (en) | Inorganic ion receptor-active compounds | |
| WO2006042007A2 (en) | Chromenone compounds as calcilytics | |
| AU2007221967A1 (en) | Inorganic ion receptor-active compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 97195368.6 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH HU IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK TJ TM TR TT UA UG UZ VN YU AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH KE LS MW SD SZ UG AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 332068 Country of ref document: NZ |
|
| ENP | Entry into the national phase |
Ref document number: 2251331 Country of ref document: CA Ref document number: 2251331 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/1998/008327 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 1997 536327 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1997917848 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 199800913 Country of ref document: EA |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1997917848 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1997917848 Country of ref document: EP |