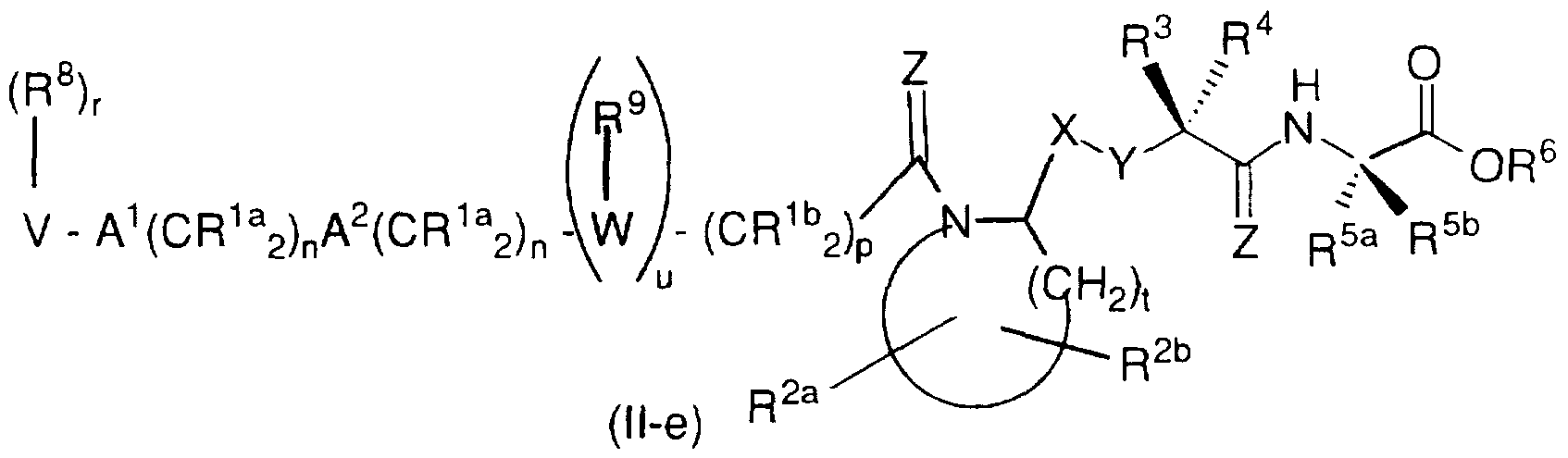TTTLE OF THE INVENTION
A METHOD OF TREATING CANCER
BACKGROUND OF THE INVENTION
The present invention relates to a method of treating cancer using a combination of a compound which is a inhibitor of geranylgeranyl-protein transferase-type I and a compound which is a inhibitor of farnesyl-protein transferase. The invention further relates to a method of treating cancer using a combination of a compound which is a selective inhibitor of geranylgeranyl-protein transferase-type I and a compound which is a selective inhibitor of farnesyl-protein transferase.
Prenylation of proteins by intermediates of the isoprenoid biosynthetic pathway represents a new class of post-translational modification (Glomset, J. A., Gelb, M. H., and Farnsworth, C. C.
(1990). Trends Biochem. Sci. 15, 139-142; Maltese, W. A. ( 1990). FASEB J. 4, 3319-3328). This modification typically is required for the membrane localization and function of these proteins. Prenylated proteins share characteristic C-terminal sequences including CaaX (C, Cys; a, usually aliphatic amino acid; X, another amino acid), XXCC, or XCXC. Three post-translational processing steps have been described for proteins having a C-terminal CaaX sequence: addition of either a 15 carbon (farnesyl) or 20 carbon (geranylgeranyl) isoprenoid to the Cys residue, proteolytic cleavage of the last 3 amino acids, and methylation of the new C-terminal carboxylate (Cox, A. D. and Der, C. J. (1992a). Critical Rev. Oncogcnesis 3:365-400; Newman, C. M. H. and Magee, A. I. (1993). Biochim. Biυphys. Ada 1 155:79-96). Some proteins may also have a fourth modification: palmitoylation of one or two Cys residues N-terminal to the farnesylated Cys. Proteins terminating with a XXCC or XCXC motif are modified by geranylgeranylation on the Cys residues and do not require an endoproteolytic processing step. While some mammalian cell proteins terminating in XCXC are carboxymethylated, it is not clear whether carboxymethylation follows prenylation of proteins terminating with a XXCC motif (Clarke, S.
(1992). Annu. Rev. Biochem. 61 , 355-386). For all of the prenylated proteins, addition of the isoprenoid is the first step and is required for the subsequent steps (Cox, A. D. and Der, C. J. (1992a). Critical Rev. Oncogenesis 3:365-400; Cox, A. D. and Der, C. J. (1992b) Current Opinion Cell Biol. 4:1008-1016).
Three enzymes have been described that catalyze protein prenylation: farnesyl-protein transferase (FPTase), geranylgeranyl- protein transferase type I (GGPTase-I), and geranylgeranyl-protein transferase type-II (GGPTase-II, also called Rab GGPTase). These enzymes are found in both yeast and mammalian cells (Clarke, 1992; Schafer, W. R. and Rine, J. ( 1992) Annu. Rev. Genet. 30:209-237). FPTase and GGPTase-I are α/β heterodimeric enzymes that share a common α subunit; the β subunits are distinct but share approximately 30% amino acid similarity (Brown, M. S. and Goldstein, J. L. (1993). Nature 366, 14-15; Zhang, F. L., Diehl, R. E., Kohl, N. E., Gibbs, J. B., Giros, B., Casey, P. J., and Omer, C. A. (1994). J. Biol. Chem. 269, 3175-3180). GGPTase-II has different α and β subunits and complexes with a third component (REP, Rab Escort Protein) that presents the protein substrate to the α/β catalytic subunits. Each of these enzymes selectively uses farnesyl diphosphate or geranylgeranyl diphosphate as the isoprenoid donor and selectively recognizes the protein substrate. FPTase farnesylates CaaX-containing proteins that end with Ser, Met, Cys, Gin or Ala. GGPTase-I geranylgeranylates CaaX-containing proteins that end with Leu or Phe. For FPTase and GGPTase-I, CaaX tetrapeptides comprise the minimum region required for interaction of the protein substrate with the enzyme. GGPTase-II modifies XXCC and XCXC proteins; the interaction between GGPTase-II and its protein substrates is more complex, requiring protein sequences in addition to the C-terminal amino acids for recognition. The enzymological characterization of these three enzymes has demonstrated that it is possible to selectively inhibit one with little inhibitory effect on the others (Moores, S. L., Schaber, M. D., Mosser, S. D., Rands, E.,
O'H ara, M. B., Garsky, V. M., Marshall, M. S., Pompliano, D. L.,
and Gibbs, J. B., J. Biol. Chem., 266: 17438 (1991 ), U.S. Pat. No.
5,470,832).
The characterization of protein prenylation biology and enzymology has opened new areas for the development of inhibitors which can modify physiological processes. The prenylation reactions have been shown genetically to be essential for the function of a variety of proteins (Clarke, 1992; Cox and Der, 1992a; Gibbs, J. B. (1991 ). Cell 65: 1-4; Newman and Magee, 1993; Schafer and Rine, 1992). This requirement often is demonstrated by mutating the CaaX Cys acceptors so that the proteins can no longer be prenylated. The resulting proteins are devoid of their central biological activity. These studies provide a genetic "proof of principle" indicating that inhibitors of prenylation can alter the physiological responses regulated by prenylated proteins.
The Ras protein is part of a signalling pathway that links cell surface growth factor receptors to nuclear signals initiating cellular proliferation. Biological and biochemical studies of Ras action indicate that Ras functions like a G-regulatory protein. In the inactive state, Ras is bound to GDP. Upon growth factor receptor activation, Ras is induced to exchange GDP for GTP and undergoes a conformational change. The GTP-bound form of Ras propagates the growth stimulatory signal until the signal is terminated by the intrinsic GTPase activity of Ras, which returns the protein to its inactive GDP bound form (D.R. Lowy and D.M. Willumsen, Ann. Rev. Biochem. 62:851 -891 (1993)). Activation of Ras leads to activation of multiple intracellular signal transduction pathways, including the MAP Kinase pathway and the Rho/Rac pathway (Joneson et al., Science 277 :810-812).
Mutated ras genes are found in many human cancers, including colorectal carcinoma, exocrine pancreatic carcinoma, and myeloid leukemias. The protein products of these genes are defective in their GTPase activity and constitutively transmit a growth stimulatory signal.
The Ras protein is one of several proteins that are known to undergo post-translational modification. Farnesyl-protein transferase utilizes farnesyl pyrophosphate to covalently modify the Cys thiol group
of the Ras CAAX box with a farnesyl group (Reiss et al., Cell, 62:81 -88 (1990); Schaber et al., J. Biol. Chem., 265: 14701 -14704 (1990); Schafer et al., Science, 249:1 133-1 139 (1990); Marine et al., Proc. Natl. Acad. Sci USA, 87:7541 -7545 (1990)).
Ras must be localized to the plasma membrane for both normal and oncogenic functions. At least 3 post-translational modifications are involved with Ras membrane localization, and all 3 modifications occur at the C-terminus of Ras. The Ras C-terminus contains a sequence motif termed a "CAAX" or "Cys-Aaa1-Aaa2-Xaa" box (Cys is cysteine, Aaa is an aliphatic amino acid, the Xaa is any amino acid) (Willumsen et al., Nature 310 :583-586 (1984)). Depending on the specific sequence, this motif serves as a signal sequence for the enzymes farnesyl-protein transferase or geranylgeranyl-protein transferase, which catalyze the alkylation of the cysteine residue of the CAAX motif with a C 15 or C20 isoprenoid, respectively. (S. Clarke., Ann. Rev. Biochem. 61 :355-386 (1992); W.R. Schafer and J. Rine, Ann. Rev. Genetics 30:209-237 (1992)). It has been shown that the N- Ras and K-Ras proteins' sequences do not exhibit the absolute substrate specificity for farnesyl-protein transferase that is found for the H-Ras C-terminus sequence, which allows N-Ras and K-Ras proteins to be processed bygeranylgeranyl-protein transferase as well (Moores, S. L. et al., J. Biol. Chem., 266:17438 ( 1991 ) and James, G. et al., J. Biol. Chem., 270:6221 -6226 (1995)). However, direct inhibition of farnesyl- protein transferase would be more specific and attended by fewer side effects than would occur with the required dose of a general inhibitor of isoprene biosynthesis.
Other farnesylated proteins include the Ras-related
GTP-binding proteins such as RhoB, fungal mating factors, the nuclear lamins, and the gamma subunit of transducin. James, et al., J. Biol.
Chem. 269, 14182 ( 1994) have identified a peroxisome associated protein Pxf which is also farnesylated. James, et al., have also suggested that there are farnesylated proteins of unknown structure and function in addition to those listed above.
Protein geranylgeranyltransferase type-I (GGTase-I) transfers a geranylgeranyl group from the prenyl donor geranylgeranyl diphosphate to the cysteine residue of substrate proteins containing a C-terminal CAAX -motif in which the "X" residue is leucine or phenyl- alanine (Clark, 1992; Newman and Magee, 1993). Known targets of GGTase-I include the gamma subunits of brain heterotrimeric
G proteins and Ras-related small GTP-binding proteins such as RhoA, RhoB, RhoC, CDC42Hs, Rac1 , Rac2, R-Ras, TC21 , Rap1 A and RaplB (Newman and Magee, 1993; Cox and Der, 1992a). The proteins RhoA, RhoB, RhoC, Rac1 , Rac2 and CDC42Hs have roles in the regulation of cell shape (Ridley, A. J. and Hall, A. (1992). Cell 70:389-399; Ridley, A. J., Paterson, H. F., Johnston, C. L., Keikmann, D., and Hall, A.
(1992). Cell 70:401 -410; Bokoch, G. M. and Der, C. J. (1993). FASEB J. 7:750-759). Rac and Rap proteins have roles in neutrophil activation (Bokoch and Der, 1993).
Activation of growth factor function and Ras function can cause tumor formation. Recently, it was demonstrated that the Rho and Rac proteins transmit intracellular signals initiated by growth factors and by Ras protein (Prendergast, G. C. and Gibbs, J. B. (1993). Adv. Cancer Res. 62, 19-64; Ridley and Hall, 1992; Ridley et al., 1992).
Specifically, experiments demonstrated that the function of Rho and Rac proteins was required by Ras and growth factors to change cell shape, a biological parameter indicative of cellular transformation and cancer. Activated forms of Rho and Rac proteins have also been shown to cause cellular transformation in cell culture (Symons, M., Current Opinion Biotechnology, 6:668-674 (1995)). Since Rho and Rac proteins require geranylgeranylation for function, an inhibitor of GGPTase-I would block the functions of these proteins and be useful as an anticancer agent.
Inhibitors of farnesyl-protein transferase (FPTase) have been described in two general classes. The first class includes analogs of farnesyl diphosphate (FPP), while the second is related to protein substrates (e.g., Ras) for the enzyme. The peptide derived inhibitors that have been described are generally cysteine containing molecules that
are related to the CAAX motif that is the signal for protein prenylation. (Schaber et al., ibid; Reiss et. al, ibid', Reiss et al., PNAS, 88:732-736 (1991 )). Such inhibitors may inhibit protein prenylation while serving as alternate substrates for the farnesyl-protein transferase enzyme, or may be purely competitive inhibitors (U.S. Patent 5,141 ,851 , University of Texas; N.E. Kohl et al. , Science, 260 : 1934-1937 (1993); Graham, et al., J. Med. Chem., 37, 725 (1994)).
Mammalian cells express four types of Ras proteins (H-, N, K4A-, and K4B-Ras) among which K-Ras4B is the most frequently mutated form of Ras in human cancers. Inhibition of farnesyl-protein transferase has been shown to block the growth of H-ras -transformed cells in soft agar and to modify other aspects of their transformed phenotype. It has also been demonstrated that certain inhibitors of farnesyl-protein transferase selectively block the processing of the H-Ras oncoprotein intracellularly (N.E. Kohl et al., Science, 260: 1934- 1937 (1993) and G.L. James et al., Science, 260: 1937-1942 ( 1993).
Recently, it has been shown that an inhibitor of farnesyl-protein transferase blocks the growth of H-ras-dependent tumors in nude mice (N.E. Kohl et al., Proc. Natl. Acad. Sci U.S.A., 91 :9141 -9145 ( 1994) and induces regression of mammary and salivary carcinomas in H-ras transgenic mice (N.E. Kohl et al, Nature Medicine, 1 :792-797 ( 1995). In vivo inhibition of K-Ras4B processing using a non-selective inhibitor of FPTase and GGTase has recently been demonstrated (E.C. Lerner et al., J. Biol. Chemistry 270:26770-26773 (1995)).
Indirect inhibition of farnesyl-protein transferase in vivo has been demonstrated with lovastatin (Merck & Co., Rahway, NJ) and compactin (Hancock et al., ibid; Casey et al., ibid; Schafer et al., Science 245:379 (1989)). These drugs inhibit HMG-CoA reductase, the rate limiting enzyme for the production of polyisoprenoids includ- ing farnesyl pyrophosphate. Inhibition of farnesyl pyrophosphate biosynthesis by inhibiting HMG-CoA reductase blocks Ras membrane localization in cultured cells.
A pharmaceutically effective combination of
geranylgeranyl-protein transferase-type I inhibitor and a farnesyl-
protein transferase inhibitor are used in the present invention to treat cancer, such as in tumor cells that are less susceptable to treatment by one of the selective inhibitors whin administered alone. SUMMARY OF THE INVENTION
A method of treating cancer is disclosed which is
comprised of administering to a mammalian patient in need of such treatment an effective amount of a combination of a geranylgeranyl- protien transferase-type I inhibitor and a farnesyl protein transferase inhibitor. Preferably a selective geranylgeranyl-protein transferase- type I inhibitor and a selective farnesyl protein transferase inhibitor are used in such a combination.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1. Autoradiograph of the 13% acrylamide gel chromatography of the immunoprecipitates from Assay Set No. 1.
Assays that are illustrated include in vivo prenylation inhibition by a selective FPTase inhibitor (Compound 5), a selective geranylgeranyl- protein transferase-type I inhibitor (Compound 1 ) and a non-selective inhibitor (Compound 7). The intensities of the bands corresponding to prenylated and nonprenylated Ras proteins are compared to determine the percent inhibition of prenyl transfer to protein. Figure 2. Autoradiograph of the 13% acrylamide gel chromatography of the immunoprecipitates from Assay Set No. 2, which includes in vivo prenylation inhibition by a combination of a selective FPTase inhibitor (Compound 5) and a selective
geranylgeranyl-protein transferase-type I inhibitor (Compound 1 ). The intensities of the bands corresponding to prenylated and nonprenylated Ras proteins are compared to determine the percent inhibition of prenyl transfer to protein.
Figure 3. Autoradiograph of the 13% acrylamide gel chromatography of the immunoprecipitates from Assay Set No. 3, which includes in vivo prenylation inhibition by a consecutive combination of a selective FPTase inhibitor (Compound 3) and a selective geranylgeranyl-protein transferase-type I inhibitor
(Compound 1). The intensities of the bands corresponding to
prenylated and nonprenylated Ras proteins are compared to
determine the percent inhibition of prenyl transfer to protein. DETAILED DESCRIPTION OF THE INVENTION
The present invention relates to a method of treating cancer which is comprised of admininstering to a mammalian patient in need of such treatment an effective amount of a combination of a geranylgeranyl-protein transferase-type I inhibitor and a farnesyl- protein transferase inhibitor. The present method of treating cancer by simultaneously inhibiting farnesyl-protein transferase and
geranylgeranyl-protein transferase-type I offers advantages over previously disclosed methods utilizing prenyl-protein transferase inhibitors, in that the inhibitory activity of the instant combination of inhibitors against FPTase and/or GGTase can be varied by formulation depending on the nature of the cancer cells to be treated. In particular, such variable inhibitory potency combinations are useful in treatment of human cancers associated with the K-Ras4B and N-Ras mutated forms of Ras whose processing are not blocked by a potent selective FPTase inhibitor alone. Any compound which inhibits geranylgeranyl-protein transferase-type I and any compound which inhibits farnesyl protein transferase can be used in the instant method. Preferably the
compounds utilized in the instant combination are a selective
geranylgeranyl-protein transferase-type I inhibitor and a selective farnesyl-protein transferase inhibitor.
As used herein the term geranylgeranyl-protein transferase- type I inhibiting compound refers to compounds which antagonize, inhibit or counteract the activity of the gene coding geranylgeranyl- protein transferase-type I or the protein produced in response thereto.
The term farnesyl protein transferase inhibiting compound likewise refers to compounds which antagonize, inhibit or counteract the activity of the gene coding farnesyl-protein transferase or the protein produced in response thereto.
In general, a geranylgeranyl-protein transferase-type I inhibitor may be distinquished from a farnesyl-protein transferase inhibitor by having greater inhibitory activity against geranylgeranyl- protein transferase-type I than against farnesyl-protein transferase. In general, a farnesyl-protein transferase inhibitor may be distinquished from a geranylgeranyl-protein transferase-type I inhibitor by having greater inhibitory activity against farnesyl-protein transferase than against geranylgeranyl-protein transferase-type I.
The term selective as used herein refers to the inhibitory activity of the particular compound against geranylgeranyl-protein transferase-type I activity when compared to the inhibitory activity of the compound against farnesyl-protein transferase activity. A compound is considered a selective inhibitor of geranylgeranyl-protein transferase-type I, for example, when its in vitro activity, as assessed by the assay described in Example 16, is at least 10 times greater that the in vitro acitivity of the same compound against farnesyl-protein transferase in that assay. A compound is considered a selective inhibitor of farnesyl-protein transferase, for example, when its in vitro farnesyl- protein transferase inhibitory activity, as assessed by the assay described in Example 16, is at least 10 times greater that the in vitro acitivity of the same compound against geranylgeranyl-protein transferase-type I in that assay. Preferably, a selective compound exhibits at least 20 times greater activity against one of the enzymatic activities when comparing geranylgeranyl-protein transferase-type I inhibition and farnesyl-protein transferase inhibition. More preferably the selectivity is at least 100 times or more. It is understood that the greater the selectivity of a geranylgeranyl-protein transferase-type I inhibitor or farnesyl-protein transferase inhibitor, the more preferred such a compound is in the instant combination.
The extent of selectivity of the two inhibitors that comprise the method of the instant invention effects the advantages that the method of treatment claimed herein offers over previously disclosed non-selective inhibitors of prenyl -transferase enzymes. In particular, use of two independent inhibitor components that have complementary, essentially non-overlapping inhibitory activities allows the person utilizing the instant method of treatment to independently and accurately vary the inhibitory activity of the combination without having to synthesize a single drug having a particular GGTase-type I/FPTase inhibitory profile.
Cancers which are treatable in accordance with the invention described herein include cancers of the brain, breast, colon, genitourinary tract, lymphatic system,pancreas, rectum, stomach, larynx, liver and lung, and chronic myelogenous leukemia. More particularly, such cancers include histiocytic lymphoma, lung
adenocarcinoma, pancreatic carcinoma, colo-rectal carcinoma
and small cell lung cancers.
The pharmaceutical composition of this invention may be administered to mammals, preferably humans, either alone or, preferably, in combination with pharmaceutically acceptable carriers or diluents, optionally with known adjuvants, such as alum, in a pharmaceutical composition, according to standard pharmaceutical practice. The compounds can be administered orally or parenterally, including the intravenous, intramuscular, intraperitoneal, subcutaneous, rectal and topical routes of administration.
For oral use of a chemotherapeutic combination according to this invention, the selected combination or compounds may be administered, for example, in the form of tablets or capsules, or as an aqueous solution or suspension. In the case of tablets for oral use, carriers which are commonly used include lactose and corn starch, and lubricating agents, such as magnesium stearate, are commonly added. For oral administration in capsule form, useful diluents include lactose and dried com starch. When aqueous suspensions are required for oral use, the active ingredients are combined with emulsifying and
suspending agents. If desired, certain sweetening and/or flavoring agents may be added. For intramuscular, intraperitoneal, subcutaneous and intravenous use, sterile solutions of the active ingredient are usually prepared, and the pH of the solutions should be suitably adjusted and buffered. For intravenous use, the total concentration of solutes should be controlled in order to render the preparation isotonic.
The combinations of the instant invention may also be co- administered with other well known therapeutic agents that are selected for their particular usefulness against the condition that is being treated. For example, the instant combinations may be useful in combination with other known anti-cancer and cytotoxic agents.
If formulated as a fixed dose, such combination products employ the combinations of this invention within the dosage range described below and the other pharmaceutically active agent(s) within its approved dosage range. Combinations of the instant invention may alternatively be used sequentially with known pharmaceutically acceptable agent(s) when a multiple combination formulation is inappropriate.
The present invention also encompasses a pharmaceutical composition useful in the treatment of cancer, comprising the
administration of a therapeutical ly effective amount of the combinations of this invention, with or without pharmaceutically acceptable carriers or diluents. Suitable compositions of this invention include aqueous solutions comprising compounds of this invention and pharmacolo- gically acceptable carriers, e.g., saline, at a pH level, e.g., 7.4. The solutions may be introduced into a patient's blood-stream by local bolus injection.
When a combination according to this invention is
administered into a human subject, the daily dosage will normally be determined by the prescribing physician with the dosage generally varying according to the age, weight, and response of the individual patient, as well as the severity of the patient's symptoms.
In one exemplary application, a suitable amount of a geranylgeranyl-protein transferase-type I inhibitor and a farnesyl-
protein transferase inhibitor are administered to a mammal undergoing treatment for cancer. Administration occurs in an amount of each type of inhibitor of between about 0.1 mg/kg of body weight to about 60 mg/kg of body weight per day, preferably of between 0.5 mg/kg of body weight to about 40 mg/kg of body weight per day.
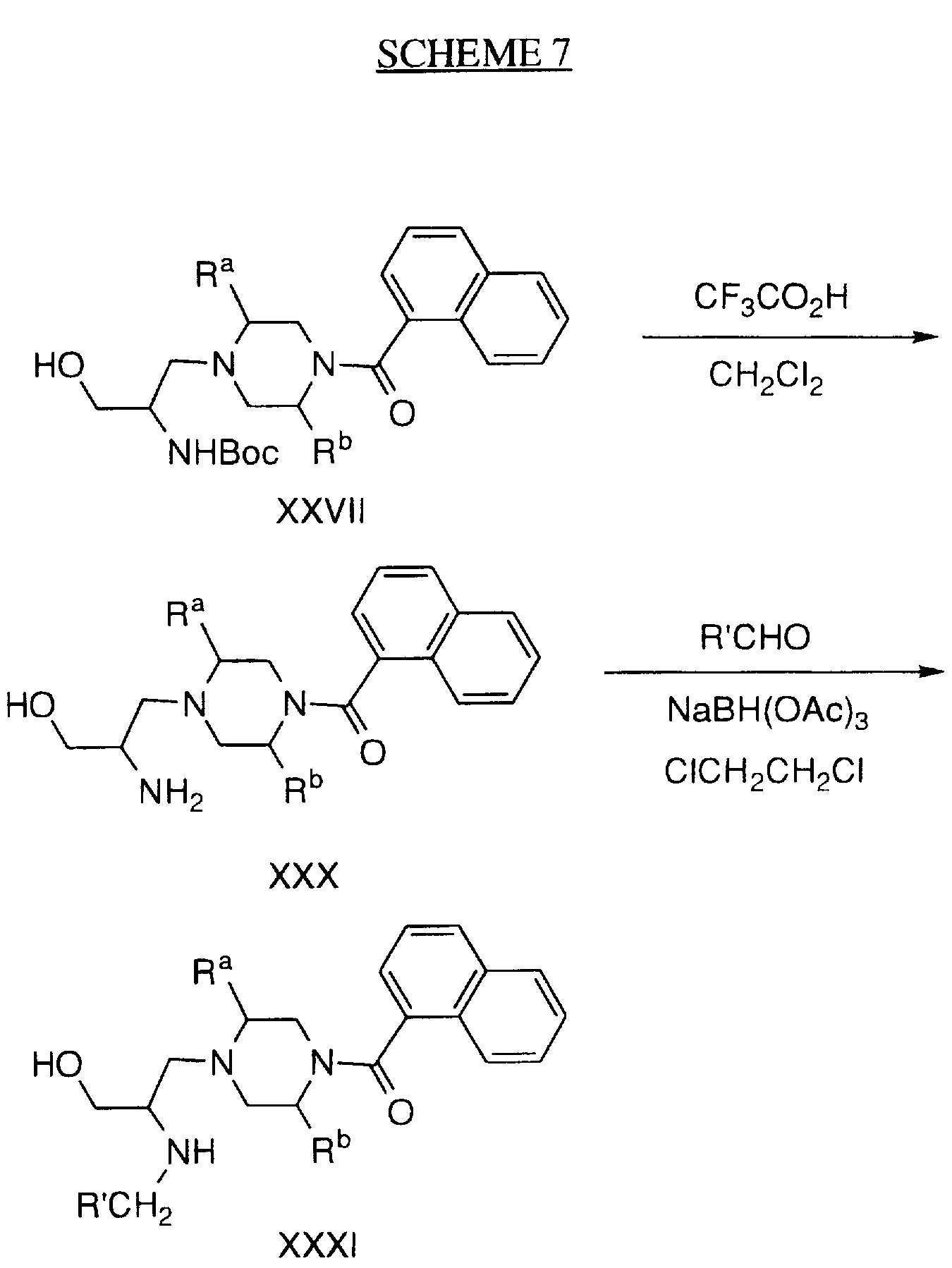
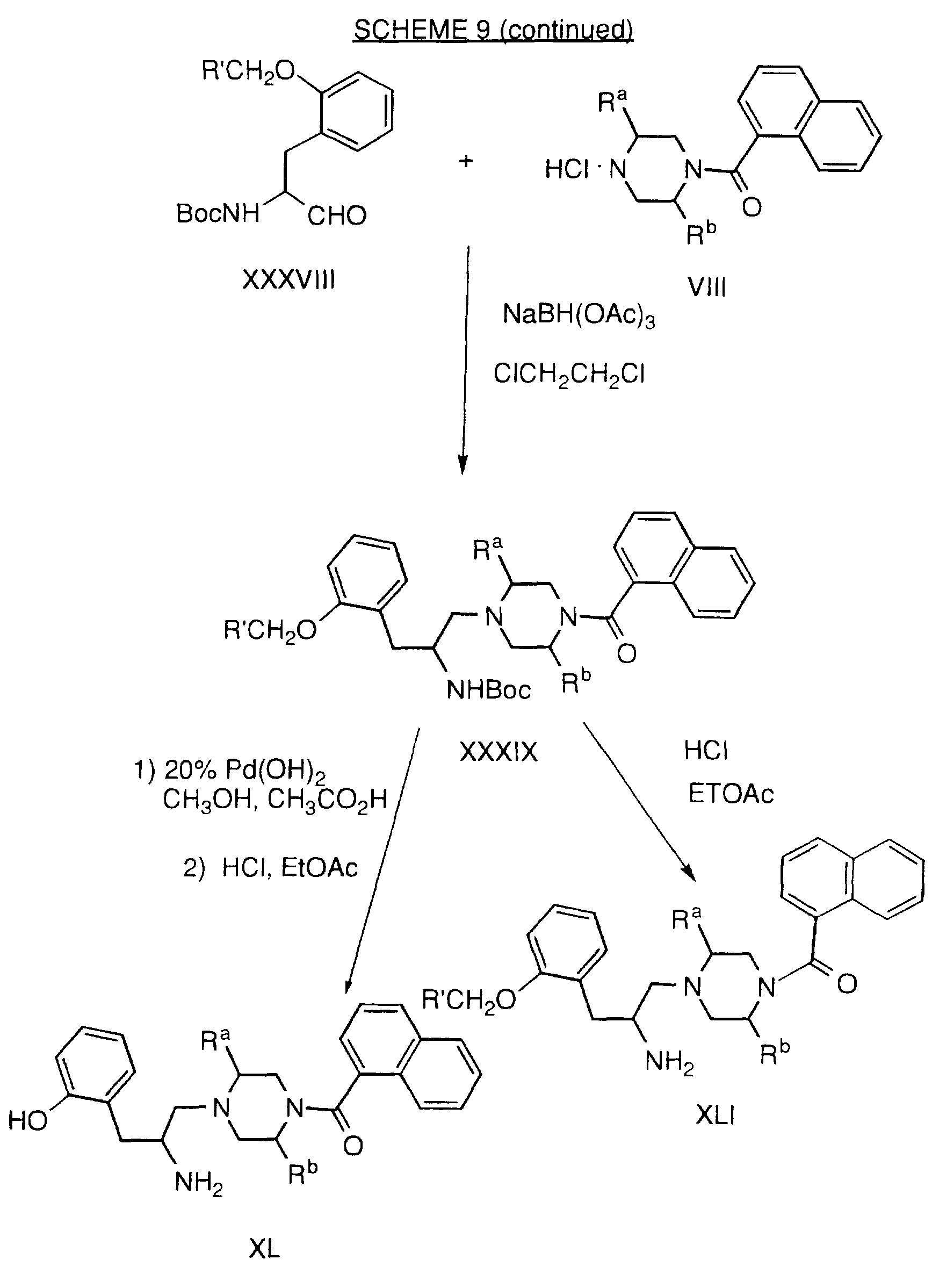
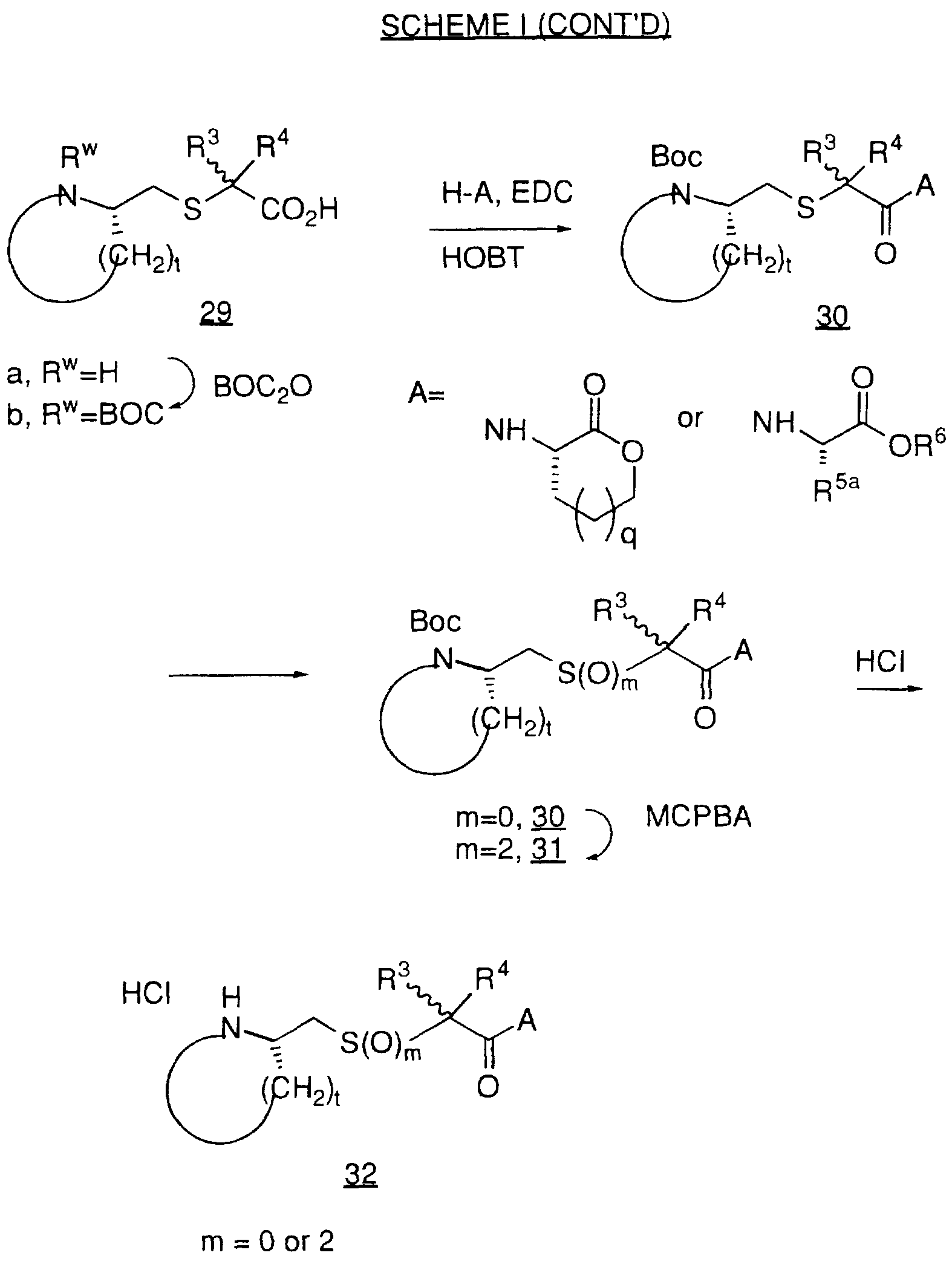
Examples of geranylgeranyl-protein transferase-type I inhibiting compounds and in particular selective geranylgeranyl-protein transferase-type I inhibiting compounds, have been described in U.S. Pat. No. 5,470,832 (Merck) and include the following:
wherein:
R1 and R2 are independently selected from:
a) C2 - C8 alkyl;
b) C2 - C8 alkenyl;
c) C2 - C8 alkynyl;
d) substituted C1 - C8 alkyl;
e) aryl;
f) substituted aryl;
g) heteroaryl;
h) substituted heteroaryl; and
i) the side chain of a naturally occurring amino acid;
R
3 is selected from alkyl, alkenyl and alkynyl of 1 to 6 carbon atoms, either branched or straight chain, which is unsubstituted or substituted with a phenyl group;
and Z is H
2 or O;
or the pharmaceutically acceptable salts or disulfides thereof.
Examples of farnesyl protein transferase inhibiting compounds and in particular selective farnesyl protein transferase inhibiting compounds include the following:
(a) a compound represented by formula (Il-a) through (II- c):
wherein with respect to formula (ll-a):
or a pharmaceutically acceptable salt thereof,
R1a and R1b are independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)N R10-, CN, NO2,(R10)2N-C(NR10)-,R10C(O)-, R10OC(O)-, N3, -N(R10)2, or R10OC(O)NR10-,
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocyclyl, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or R11OC(O)-NR10-; R2 and R3 are independently selected from: H; unsubstituted or
substituted C1-8 alkyl, unsubstituted or substituted C2-8 alkenyl,
unsubstituted or substituted C
2-8 alkynyl, unsubstituted or substituted aryl, unsubstituted or substituted heterocycle,
wherein the substituted group is substituted with one or more of:
1) aryl or heterocycle, unsubstituted or substituted with: a) C1 -4 alkyl,
b) (CH2)pOR6,
c) (CH2)PNR6R7,
d) halogen,
2) C3-6 cycloalkyl,
3) OR6,
4) SR6, S(O)R6, SO2R6,
R
2 and R
3 are attached to the same C atom and are combined to form (CH
2)
u - wherein one of the carbon atoms is optionally replaced by a moiety selected from: O, S(O)
m, -NC(O)-, and -N(COR
1 0)- ;
R4 and R5 are independently selected from H and CH3; and any two of R2, R3, R4 and R5 are optionally attached to the same carbon atom; R6, R7 and R7a are independently selected from: H; C1 -4 alkyl, C3-6 cycloalkyl, heterocycle, aryl, aroyl, heteroaroyl, arylsulfonyl, heteroarylsulfonyl, unsubstituted or substituted with:
a) C1 -4 alkoxy,
b) aryl or heterocycle,
c) halogen,
f) — SO2R1 1 , or
g) N(R 1 0)2; or
R6 and R7 may be joined in a ring;
R7 and R7a may be joined in a ring;
R8 is independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R10O-,
R11S(O)m-, R10C(O)NR10-, CN, NO2, R10 2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or
R11OC(O)NR10-, and
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R10O-, R11S(O)m-, R10C(O)NH-, CN, H2N-C(NH)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, orR10OC(O)NH-;
R9 is selected from:
a) hydrogen,
b) C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C-(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-, and
c) C1-C6 alkyl unsubstituted or substituted by perfluoroalkyl, F, Cl, Br, R10O-, R11S(O)m-, R10C(O)N R10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-; R10 is independently selected from hydrogen, C1-C6 alkyl, benzyl and aryl;
R11 is independently selected from C1-C6 alkyl and aryl;
A1 and A2 are independently selected from: a bond, -CH=CH-, -C≡ C-, -C(O)-, -C(O)NR 10-, -NR 10C(O)-, O, -N(R 10)-, -S(O)2N(R 10)-,
-N(R 10)S(O)2-, or S(O)m; V is selected from:
a) hydrogen,
b) heterocycle,
c) aryl,
d) C1 -C20 alkyl wherein from 0 to 4 carbon atoms are
replaced with a a heteroatom selected from O, S, and N, and
e) C2-C20 alkenyl,
provided that V is not hydrogen if A1 is S(O)m and V is not hydrogen if A 1 is a bond, n is 0 and A2 is S(O)m;
W is a heterocycle;
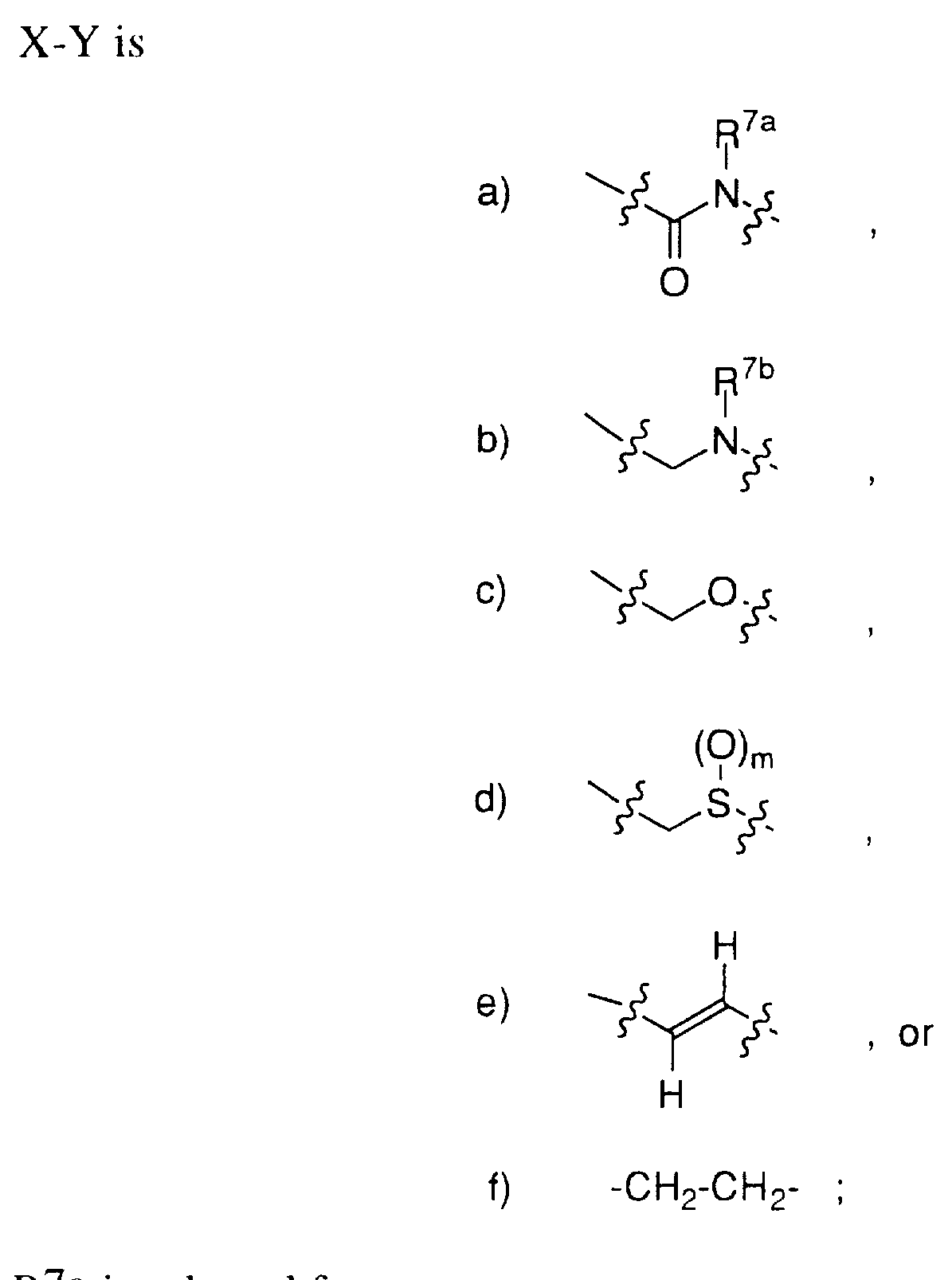
X is -CH2-, -C(=O)-, or -S(=O)m-; Y is aryl, heterocycle, unsubstituted or substituted with one or
more of:
1 ) C1 -4 alkyl, unsubstituted or substituted with:
a) C1 -4 alkoxy,
b) NR6R7,
c) C3-6 cycloalkyl,
d) aryl or heterocycle,
e) HO,
f) -S(O)mR6 or
g) -C(O)NR6R7,
2) aryl or heterocycle,
3) halogen,
4) OR6,
5) NR6R7,
6) CN,
7) NO2,
8) CF3;
9) -S(O)mR6,
10) -C(O)NR6R7, or
11) C3-C6 cycloalkyl; m is 0, 1 or 2;
n is 0, 1, 2, 3 or 4;
p is 0, 1 , 2, 3 or 4;
r is 0 to 5, provided that r is 0 when V is hydrogen;
s is 0 or 1 ;
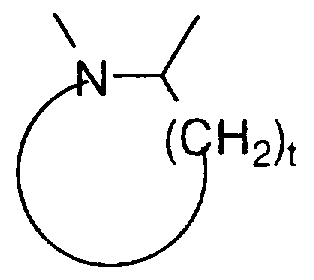
t is 0 or 1 ; and
u is 4 or 5; with respect to formula (Il-b):
or a pharmaceutically acceptable salt thereof,
R1 a, R1 b, R10, R1 1 , m, R2, R3, R6, R7, p, R7a, u, R8, A1 , K2, V, W, X, n, p, r, s, t and u are as defined above with respect to formula (Il-a);
R4 is selected from H and CH3; and any two of R2, R3 and R4 are optionally attached to the same carbon atom;
R9 is selected from:
a) hydrogen,
b) alkenyl, alkynyl, perfluoroalkyl, F, C1, Br, R10O-,
R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C-(NR10).,R10C(O)-, R10OC(O)-, N3,
-N(R10)2,or R11OC(O)NR10-, and
c) C1-C6 alkyl unsubstituted or substituted by perfluoroalkyl, F, Cl, Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-; H2 or O; aryl, heteroaryl, arylmethyl, heteroarylmethyl,
arylsulfonyl, heteroarylsulfonyl, unsubstituted or
substituted with one or more of the following:
1) C1-4 alkyl, unsubstituted or substituted with
a) C1-4 alkoxy,
b) NR6R7,
c) C3-6 cycloalkyl,
d) aryl or heterocycle,
e) HO,
f) -S(O)mR6, or
g) -C(O)NR6R7,
2) aryl or heterocycle,
3) halogen,
4) OR6,
5) NR6R7,
6) CN,
7) NO2,
8) CF3;
9) -S(O)mR6,
10) -C(O)NR6R7, or
11) C3-C6 cycloalkyl;
with respect to formula (II-c):
or a pharmaceutically acceptable salt thereof, R1 a, R 1 b, R 10, R1 1 , m, R2, R3, R6, R7, p, u, R7a, R8, A1 , A2, V, W, X, n, r and t are as defined above with respect to formula (Il-a);
R4 is selected from H and CH3; and any two of R2, R3 and R4 are optionally attached to the same carbon atom;
G is O;
Z is aryl, heteroaryl, arylmethyl, heteroarylmethyl,
arylsulfonyl, heteroarylsulfonyl, unsubstituted or
substituted with one or more of the following:
1 ) C1 -4 alkyl, unsubstituted or substituted with:
a) C1 -4 alkoxy,
b) NR6R7,
c) C3-6 cycloalkyl,
d) aryl or heterocycle,
e) HO,
f) -S(O)mR6, or
g) -C(O)NR6R7,
2) aryl or heterocycle,
3) halogen,
4) OR6,
5) NR6R7,
6) CN,
7) NO2,
8) CF3;
9) -S(O)mR6,
10) -C(O)NR6R7, or
1 1 ) C3-C6 cycloalkyl; and s is
(b) a compound represented by formula (Il-d) through (Il-g):
or a pharmaceutically acceptable salt thereof,
R11, V, W, m, n, p and r are as defined above with respect to formula (Il-a);
R1a and R1b are independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-,
C) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocyclyl, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)-NR10-;
R2a and R2b are independently selected from:
a) hydrogen,
b) C1-C6 alkyl unsubstituted or substituted by C2-C6 alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, N3, (R10)2N- C(NR10)-, R10C(O)-, R10OC(O)-, -N(R10)2, or R11OC(O)NR10-,
c) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl, R10O, R11S(O)m-, R10C(O)NR10-, CN, NO2, ( R10)2N-C(NR10), R10C(O)-, R10OC(O)-, N3, -N(R10)2, or
R11OC(O)NR10-, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and C3-C10 cycloalkyl; R3 and R4 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone, and
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocyclyl group, wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-,
N3, -N( R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl; or R3 and R4 are combined to form - (CH2)s-;
R5a and R5b are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone,
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocycle group, wherein the substituent is selected from F, Cl, Br, CF3, N(R10)2, NO2, R10O-, R11S(O)m-,
R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-. R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl,
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl; or
R
5a and R
5b are combined to form - (CH
2)
s - wherein one of the carbon atoms is optionally replaced by a moiety selected from: O, S(O)
m, -NC(O)-, and-N(COR
10)-;
R7a is selected from
a) hydrogen,
b) unsubstituted or substituted aryl,
c) unsubstituted or substituted heterocycle,
d) unsubstituted or substituted C3-C10 cycloalkyl, and e) C1 -C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl; R7b is selected from
a) hydrogen,
b) unsubstituted or substituted aryl,
c) unsubstituted or substituted heterocycle,
d) unsubstituted or substituted C3-C10 cycloalkyl,
e) C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl,
f) a carbonyl group which is bonded to an unsubstituted or substituted group selected from aryl, heterocycle, C3-C10 cycloalkyl and C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl, and
g) a sulfonyl group which is bonded to an unsubstituted or substituted group selected from aryl, heterocycle, C3-C10 cycloalkyl and C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl;
R8 is independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R10O-,
R11S(O)m-, R10C(O)NR10-, CN, NO2, R10 2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or
R11OC(O)NR10-, and
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R10O-, R11S(O)m-, R10C(O)NH-,CN, H2N-C(NH)-, R10C(O)-, R10OC(O)-, N3, -N( R10)2, or R10OC(O)NH-;
R9 is selected from:
a) hydrogen,
b) C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F, Cl,
Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C-(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-, and
c) C1-C6 alkyl unsubstituted or substituted by perfluoroalkyl, F, Cl, Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-; R10 is independently selected from H, C1-C6 alkyl, benzyl, substituted aryl and C1-C6 alkyl substituted with substituted aryl;
A1 and A2 are independently selected from: a bond, -CH=CH-, -C≡C-, -C(O)-, -C(O)NR10-, -NR10C(O)-, O, -N(R10)-, -S(O)2N(R10)-,
-N(R10)S(O)2-, or S(O)m; Z is independently H2 or O; s is 4 or 5;
t is 3, 4 or 5; and
u is 0 or 1; with respect to formula (Il-e):
or a pharmaceutically acceptable salt thereof,
R11, W, m, n, p and r are as defined above with respect to formula (Il-a);
R1a and R1b are independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-,
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocyclyl, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)-NR10-;
R2a and R2b are independently selected from:
a) hydrogen,
b) C1-C6 alkyl unsubstituted or substituted by C2-C6
alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, N3,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, -N(R10)2, or R11OC(O)NR10-,
c) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and C3-C10 cycloalkyl; R3 and R4 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone,
c) substituted or unsubstituted C1 -C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocyclyl group,
wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R1OO-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl; or R3 and R4 are combined to form - (CH2)s-;
R5a and R5b are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone,
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocycle group, wherein the substituent is selected from F, Cl, Br,
CF3, N(R10)2, NO2, R10O-, R11S(O)m-,
R10C(O)NR10-,CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl; or
R5a and R5b are combined to form - (CH2)s - wherein one of the carbon atoms is optionally replaced by a moiety selected from: O, S(O)m, -NC(O)-, and-N(COR10)-;
R6 is
a) substituted or unsubstituted C1-C8 alkyl, substituted or unsubstituted C5-C8 cycloalkyl, or substituted or unsubstituted cyclic amine, wherein the substituted alkyl, cycloalkyl or cyclic amine is substituted with 1 or 2 substituents independently selected from:
1) C1-C6 alkyl,
2) aryl,
3) heterocycle,
4) -N(R11)2,
5) -OR10, or
a) hydrogen,
b) unsubstituted or substituted aryl,
c) unsubstituted or substituted heterocycle,
d) unsubstituted or substituted C3-C 10 cycloalkyl, and e) C 1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl; R7b is selected from
a) hydrogen,
b) unsubstituted or substituted aryl,
c) unsubstituted or substituted heterocycle,
d) unsubstituted or substituted C3-C10 cycloalkyl,
e) C1 -C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl,
f) a carbonyl group which is bonded to an unsubstituted or substituted group selected from aryl, heterocycle, C3-C10 cycloalkyl and C1 -C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C1 0 cycloalkyl, and
g) a sulfonyl group which is bonded to an unsubstituted or substituted group selected from aryl, heterocycle, C3-C10
cycloalkyl and C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl;
R8 is independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R10O-,
R11 S(O)m-, R10C(O)NR10-, CN, NO2, R10 2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or
R11OC(O)NR10-, and
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R10O-, R11S(O)m-, R10C(O)NH-, CN, H2N-C(NH)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or R10OC(O)NH-;
R9 is selected from:
a) hydrogen,
b) C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F,
Cl, Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2, (R10)2N-C-(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-, and
c) C1-C6 alkyl unsubstituted or substituted by perfluoroalkyl, F, Cl, Br, R10O-, R11 S(O)m-, R10C(O)NR10-, CN,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11 OC(O)NR10-; R10 is independently selected from H, C1-C6 alkyl, benzyl, substituted aryl and C1-C6 alkyl substituted with substituted aryl;
R12 is hydrogen or C1-C6 alkyl;
R13 is C1-C6 alkyl;
A' and A 2 are independently selected from: a bond, -CH=CH-, -C≡C-, -C(O)-, -C(O)NR10-, -NR10C(O)-, O, -N(R10)-, -S(O)2N(R10)-, -N(R10)S(O)2-,orS(O)m;
Z is independently H2 or O; s is 4 or 5;
t is 3, 4 or 5; and
u is 0 or 1; with respect to formula (II -f):
or a pharmaceutically acceptable salt thereof,
R11, V, W, m, n, p and r are as defined above with respect to formula (II-a);
R1a and R1b are independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2 orR11OC(O)NR10-,
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocyclyl, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)-NR10-;
R2a and R2b are independently selected from:
a) hydrogen,
b) C1-C6 alkyl unsubstituted or substituted by C2-C6
alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, N3, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-,
-N(R10)2, or R11OC(O)NR10-,
c) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6
alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11 OC(O)NR 10-, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and C3-C10 cycloalkyl; R3 and R4 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone, and
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocyclyl group, wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10-,
CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C
3-C
10 cycloalkyl; or R
3 and R
4 are combined to form - (CH
2)
s - ;
R7a is selected from
a) hydrogen,
b) unsubstituted or substituted aryl,
c) unsubstituted or substituted heterocycle,
d) unsubstituted or substituted C3-C 10 cycloalkyl, and e) C1 -C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl; R7b is selected from
a) hydrogen,
b) unsubstituted or substituted aryl,
c) unsubstituted or substituted heterocycle.
d) unsubstituted or substituted C3-C10 cycloalkyl,
e) C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl,
f) a carbonyl group which is bonded to an unsubstituted or substituted group selected from aryl, heterocycle, C3-C10 cycloalkyl and C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl, and
g) a sulfonyl group which is bonded to an unsubstituted or substituted group selected from aryl, heterocycle, C3-C10 cycloalkyl and C1-C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl;
R8 is independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R10O-,
R11S(O)m-,R10C(O)NR10-, CN, NO2, R10 2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or
R11OC(O)NR10-, and
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R10O-, R11 S(O)m-, R10C(O)NH-, CN, H2N-C(NH)-,R10C(O)-, R10OC(O)-, N3, -N(R10)2, or R10OC(O)NH-;
R9 is selected from:
a) hydrogen,
b) C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F,
Cl, Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2, (R10)2N-C-(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11 OC(O)NR10-, and
c) C1-C6 alkyl unsubstituted or substituted by perfluoroalkyl, F, Cl, Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11 OC(O)NR10-; R10 is independently selected from H, C1-C6 alkyl, benzyl, substituted aryl and C1-C6 alkyl substituted with substituted aryl;
R12 is hydrogen or C1-C6 alkyl;
R13 is C1-C6 alkyl;
A1 and A2 are independently selected from: a bond, -CH=CH-, -C≡C-, -C(O)-, -C(O)NR10-, -NR10C(O)-, O, -N(R10)-, -S(O)2N(R10)-, -N(R10)S(O)2-, or S(O)m; Z is independently H2 or O; q is 0, 1 or 2;
s is 4 or 5;
t is 3, 4 or 5; and
u is 0 or 1; with respect to formula (Il-g):
or a pharmaceutically acceptable salt thereof,
R11, V, W, m, n, p and r are as previously defined with respect to formula (Il-a); R1a and R1b are independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or R11OC(O)NR10-,
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)-NR10-; R2a and R2b are independently selected from:
a) hydrogen,
b) C1-C6 alkyl unsubstituted or substituted by C2-C6 alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, N3, (R10)2N- C(NR10)-, R10C(O)-, R10OC(O)-, -N(R10)2, or
R10OC(O)NR10-,
c) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6
alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2 or R11OC(O)NR10-, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and C3-C10 cycloalkyl; R3 and R4 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone,
c) substituted or unsubstituted C1 -C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocycle group, wherein the substituent is selected from F, Cl, Br, NCR10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10-
CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl; or R3 and R4 are combined to form - (CH2)s - ;
7
γ
a) hydrogen,
b) unsubstituted or substituted aryl,
c) unsubstituted or substituted heterocycle,
d) unsubstituted or substituted C3-C10 cycloalkyl, and e) C1 -C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl; R7b is selected from
a) hydrogen,
b) unsubstituted or substituted aryl,
c) unsubstituted or substituted heterocycle,
d) unsubstituted or substituted C3-C10 cycloalkyl,
e) C1 -C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl,
f) a carbonyl group which is bonded to an unsubstituted or substituted group selected from aryl, heterocycle, C3-C10 cycloalkyl and C1 -C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl, and
g) a sulfonyl group which is bonded to an unsubstituted or substituted group selected from aryl, heterocycle, C3-C10 cycloalkyl and C1 -C6 alkyl substituted with hydrogen or an unsubstituted or substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl;
R8 is independently selected from:
a) hydrogen,
b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br, R 10O-,
R11 S(O)m-, R 1 0C(O)NR 1 0-, CN, NO2, R 1 0 2N-C(NR 10)-,
R10C(O)-, R10OC(O)-, N3, -N(R10)2, or
R11OC(O)NR10-, and
c) C1-C6 alkyl unsubstituted or substituted by aryl,
heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F, Cl, Br,R10O-, R11S(O)m-, R10C(O)NH-, CN, H2N-C(NH)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or R10OC(O)NH-;
R9 is selected from:
a) hydrogen,
b) C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl,
F, Cl, Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, NO2, (R10)2N-C-(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-, and
c) C1-C6 alkyl unsubstituted or substituted by perfluoroalkyl,
F, Cl, Br, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2, or R11OC(O)NR10-; R10 is independently selected from H, C1-C6 alkyl, benzyl, substituted aryl and C1-C6 alkyl substituted with substituted aryl;
R12 is hydrogen or C1-C6 alkyl; R13 is C1-C6 alkyl;
A1 and A2 are independently selected from: a bond, -CH=CH-, -C≡C-, -C(O)-, -C(O)NR10-, -NR10C(O)-, O, -N(R10)-, -S(O)2N(R10)-, -N(R10)S(O)2-, or S(O)m;
Z is independently H2 or O; q is 0, 1 or 2;
s is 4 or 5;
t is 3, 4 or 5; and
u is 0 or 1 ;
(c) a compound represented by formula (Il-h) through (Il-k):
(
wherein with respect to formula (Il-h):
or a pharmaceutically acceptable salt thereof,
R1a, R1b, R8, R9, R10, R11, A1, A2, V, W, m, n, p and r are as previously defined with respect to formula (Il-a);
R2 and R3 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone, and
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocyclyl group, wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R10O-, R11S(O)m-,R10C(O)NR10-, CN, (R10)2N-C(NR10)-,R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl; or
R2 and R3 are combined to form - (CH2)s- ; or
R
2 or R
3 are combined with R
6 to form a ring such that
R
4a, R
4b, R
7a and R
4b are independently selected from:
a) hydrogen,
b) C1-C6 alkyl unsubstituted or substituted by alkenyl, R10O- R11S(O)m-, R10C(O)NR10-, CN, N3, (R10)2N-C(NR10). R10C(O)-, R10OC(O)-, -N(R10)2, or R11OC(O)NR10-, c) aryl, heterocycle, cycloalkyl, alkenyl, R10O-,
R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, or R11OC(O)NR10-, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and C3-C10 cycloalkyl;
R5a and R5b are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone,
c) substituted or unsubstituted C1 -C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocycle group, wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R10O-, R11S(O)m-,R10C(O)NR10-
CN, (R10)2N-C(NR10)-, R10OC(O,)- R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl; or
R5a and R5b are combined to form - (CH2)s - wherein one of the carbon atoms is optionally replaced by a moiety selected from: O, S(O)m, -NC(O)-, and -N(COR10)- ;
R6 is independently selected from hydrogen or C1 -C6 alkyl;
Q is a substituted or unsubstituted nitrogen-containing C4-C9 mono or bicyclic ring system, wherein the non-nitrogen containing ring may be an aromatic ring, a C5-C7 saturated ring or a heterocycle;
X, Y and Z are independently H2 or O; s is 4 or 5;
t is 3, 4 or 5; and
u is 0 or 1 ; with respect to formula (II-i):
or a pharmaceutically acceptable salt thereof,
wherein: R1 a, R1 b, R8, R9, R 10, R1 1 , A1 , A2, V, W, m, n, p and r are as previously defined with respect to formula (Il-a);
R2 and R3 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone, and
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocyclyl group, wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl; or
R2 and R3 are combined to form - (CH2)s - ; or R2 or R3 are combined with R6 to form a ring such that
R
4a, R
4b, R
7a and R
7b are independently selected from:
a) hydrogen,
b) C1-C6C alkyl unsubstituted or substituted by alkenyl, R10O-,
R11S(O)m -, R10C(O)NR10-, CN, N3, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, -N(R10)2, or R11 OC(O)NR10-, c) aryl, heterocycle, cycloalkyl, alkenyl, R10O-,
R11(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3,
-N(R10)2 or R11 OC(O)NR10-, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and C3-C10 cycloalkyl;
R5a and R5b are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone,
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocycle group, wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R10O-, R11S(O)m-,R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl; or
R5a and R5b are combined to foπn - (CH2)s - wherein one of the carbon atoms is optionally replaced by a moiety selected from: O, S(O)m, -NC(O)-, and -N(COR10)- ;
R6 is independently selected from hydrogen or C1-C6 alkyl;
R12 is
a) substituted or unsubstituted C1 -C8 alkyl or substituted or unsubstituted C5-C8 cycloalkyl, wherein the substituent on the alkyl or cycloalkyl is selected from:
1) aryl,
2) heterocycle,
3) -N(R11)2,
R13 is independently selected from hydrogen and C1-C6 alkyl;
R14 is independently selected from C1-C6 alkyl;
Q is a substituted or unsubstituted nitrogen-containing C4-C9 mono or bicyclic ring system, wherein the non-nitrogen containing ring may be an aromatic ring, a C5-C7 saturated ring or a heterocycle;
X, Y and Z are independently H2 or O; s is 4 or 5;
t is 3, 4 or 5; and
u is 0 or 1; with respect to formula (II-j):
or a pharmaceutically acceptable salt thereof,
R1a, R1b, R8, R9, R10, R11, A1, A2, V, W, m, n, p and r are as previously defined with respect to formula (Il-a);
R2 and R3 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone, and
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocyclyl group, wherein the substituent is selected from F, Cl, Br,
N(R10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10- CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and C3-C10 cycloalkyl; or
R2 and R3 are combined to form - (CH2)s - ; or
R2 or R3 are combined with R6 to form a ring such that
R4a, R4b, R7a and R7b are independently selected from:
a) hydrogen,
b) C1-C6 alkyl unsubstituted or substituted by alkenyl,R10O-, R11S(O)m-, R10C(O)NR10-, CN, N3, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, -N(R10)2, or R11OC(O)NR10-, c) aryl, heterocycle, cycloalkyl, alkenyl, R10O-,
R11S(O)m-, R10C(O)NR10-, CN, NO2, (R10)2N-
C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N( R10)2 or
R11OC(O)NR10-, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and C3-C10 cycloalkyl;
R6 is independently selected from hydrogen or C1-C6 alkyl;
Q is a substituted or unsubstituted nitrogen-containing C4-C9 mono or bicyclic ring system, wherein the non-nitrogen containing ring may be an aromatic ring, a C5-C7 saturated ring or a heterocycle;
X, Y and Z are independently H2 or O; q is 0, 1 or 2;
s is 4 or 5;
t is 3, 4 or 5; and
u is 0 or 1; with respect to formula (Il-k):
or a pharmaceutically acceptable salt thereof, R
1a, R
1b, R
8, R
9, R
10, R
11, A
1, A
2,V, W, m, n, p, and r are as defined above with respect to formula (Il-a);
R2 and R3 are independently selected from:
a) a side chain of a naturally occurring amino acid,
b) an oxidized form of a side chain of a naturally occurring amino acid which is:
i) methionine sulfoxide, or
ii) methionine sulfone, and
c) substituted or unsubstituted C1-C20 alkyl, C2-C20 alkenyl, C3-C10 cycloalkyl, aryl or heterocyclyl group, wherein the substituent is selected from F, Cl, Br, N(R10)2, NO2, R10O-, R11S(O)m-, R10C(O)NR10-, CN, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, N3, -N(R10)2, R11OC(O)NR10- and C1-C20 alkyl, and
d) C1-C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocycle and
C3-C10 cycloalkyl; or
R2 and R3 are combined to form - (CH2)s-; or
R2 or R3 are combined with R6 to form a ring such that
R4a, R4b, R7a and R7b are independently selected from:
a) hydrogen,
b) C1-C6 alkyl unsubstituted or substituted by alkenyl, R10O-, R11S(O)m-, R10C(O)NR10-, CN, N3, (R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-, -N(R10)2, or R11OC(O)NR10-, c) aryl, heterocycle, cycloalkyl, alkenyl, R10O-,
R11S(O)m-, R10C(O)NR10-, CN, NO2,
(R10)2N-C(NR10)-, R10C(O)-, R10OC(O)-,
N3, -N(R10)2 or R11OC(O)NR10-, and
d) C1 -C6 alkyl substituted with an unsubstituted or
substituted group selected from aryl, heterocyclyl and C3-C10 cycloalkyl;
R6 is independently selected from hydrogen or C1 -C6 alkyl;
Q is a substituted or unsubstituted nitrogen-containing C4-C9 mono or bicyclic ring system, wherein the non-nitrogen containing ring may be an aromatic ring, a C5-C7 saturated ring or a heterocycle;
X, Y and Z are independently H2 or O; q is 0, 1 or 2;
s is 4 or 5;
t is 3, 4 or 5; and
u is 0 or 1 ;
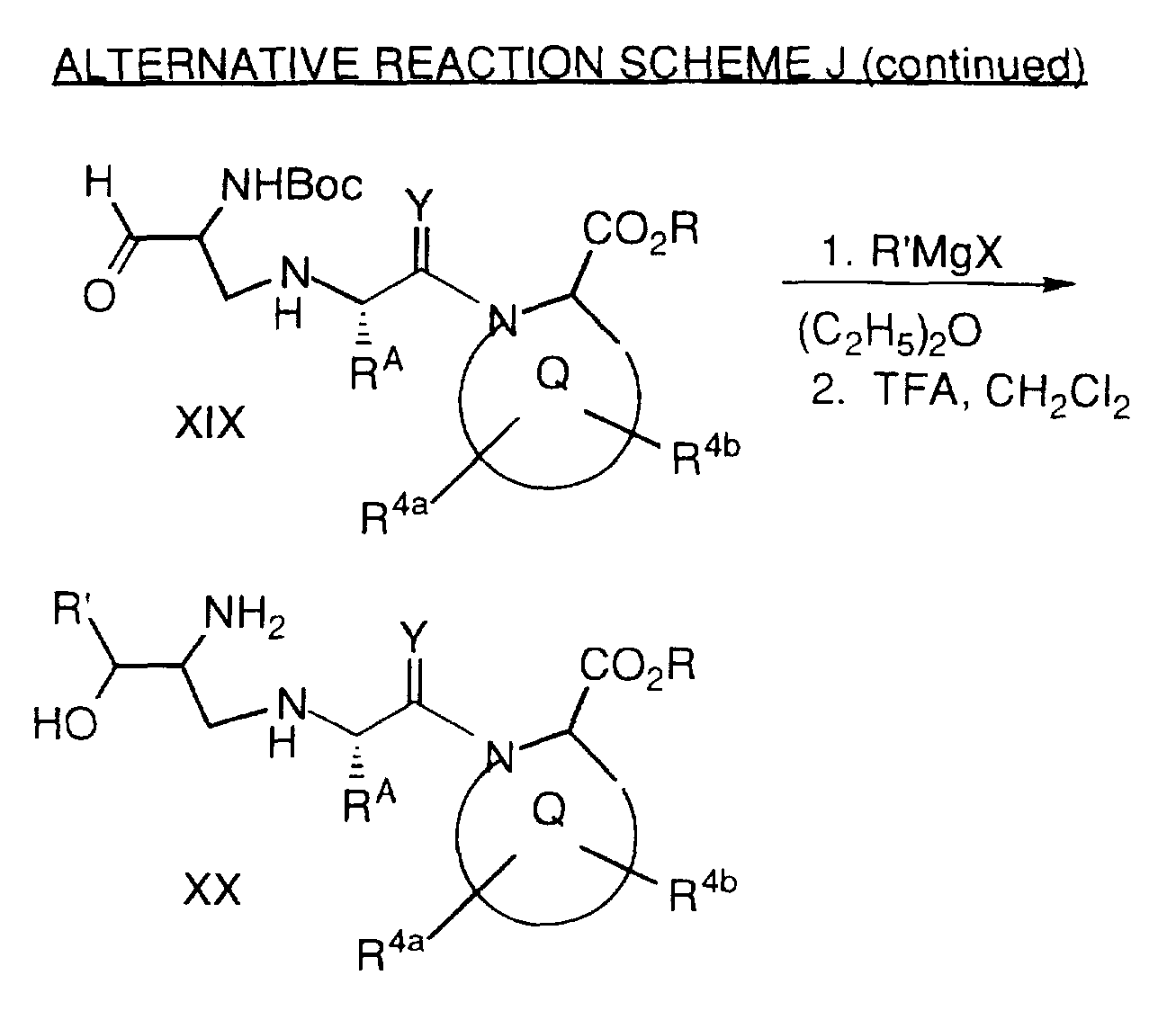
(d) a compound represented by formula (II-l) 2
wherein
Rc is selected from:
R1 is hydrogen, an alkyl group, an aralkyl group, an acyl group, an aracyl group, an aroyl group, an alkylsulfonyl group,
aralkylsulfonyl group or arylsulfonyl group, wherein alkyl and acyl groups comprise straight chain or branched chain hydrocarbons of 1 to 6 carbon atoms;
R2 and R3 are
the side chains of naturally occurring amino acids, including their oxidized forms which may be methionine sulfoxide or methionine sulfone, or in the alternative may be substituted or unsubstituted aliphatic, aromatic or heteroaromatic groups, such as allyl, cyclohexyl, phenyl, pyridyl, imidazolyl or saturated chains of 2 to 8 carbon atoms which may be branched or unbranched, wherein the aliphatic substitutents may be substituted with an aromatic or heteroaromatic ring;
R4 is hydrogen or an alkyl group, wherein the alkyl group
comprises straight chain or branched chain hydrocarbons of 1 to 6 carbon atoms; R5 is selected from:
a) a side chain of naturally occurring amino acids, b) an oxidized form of a side chain of naturally occurring amino acids selected from methionine sulfoxide and methionine sulfone,
c) substituted or unsubstituted aliphatic, aromatic or heteroaromatic groups, such as allyl, cyclohexyl, phenyl, pyridyl, imidazolyl, or saturated chains of 2 to 8 carbon atoms which may be branched or unbranched, wherein the aliphatic substituent is optionally substituted with an aromatic or heteroaromatic ring, and
d) -CH2CH2OH or -CH2CH2CH2OH;
R6 is a substituted or unsubstituted aliphatic, aromatic or
heteroaromatic group such as saturated chains of 1 to 8
carbon atoms, which may be branched or unbranched, wherein the aliphatic substituent may be substituted with an aromatic or heteroaromatic ring;
T is 0 or S(O)m;
m is 0, 1 or 2;
n is 0, 1 or 2; and the pharmaceutically acceptable salts and disulfides thereof.
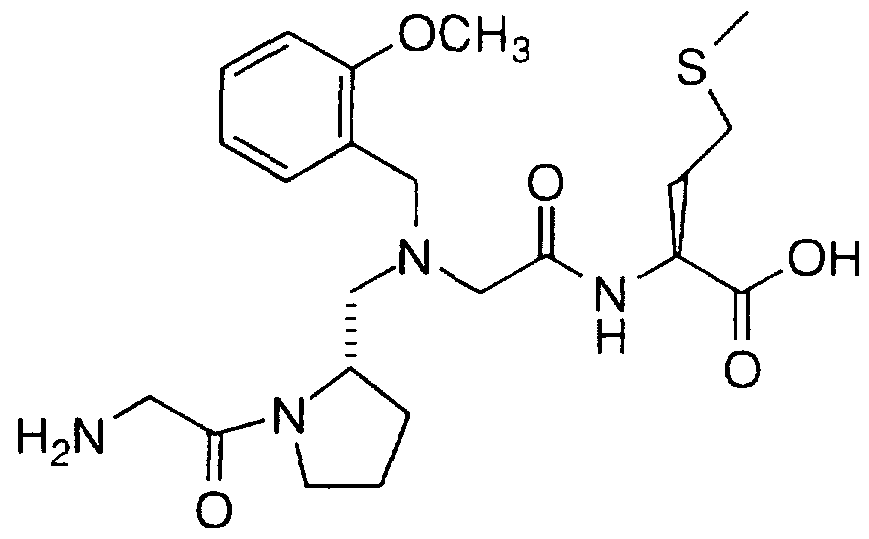
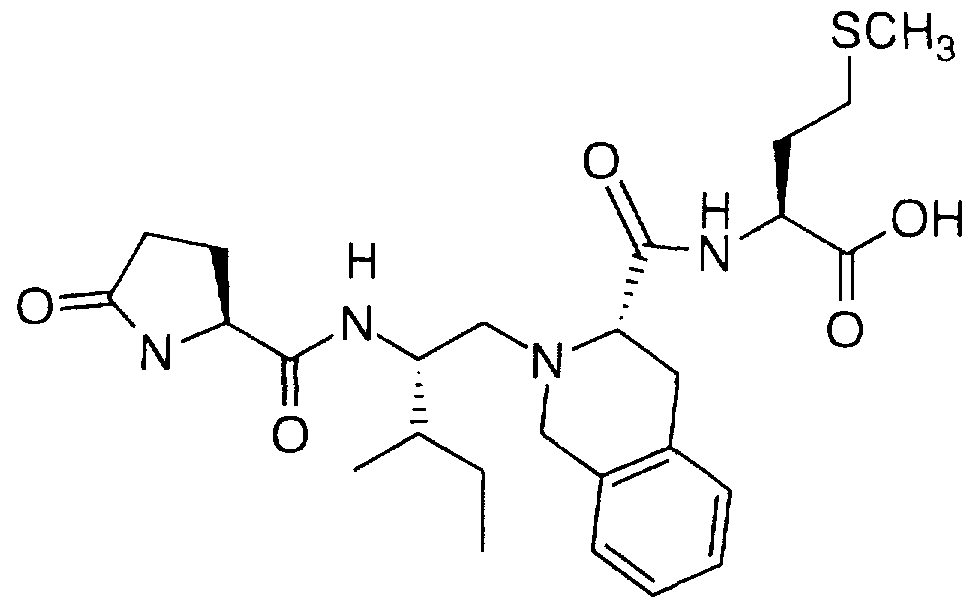
Examples of compounds that selectively inhibit geranylgeranyl-protein transferase-type I include the following: N-(2(R)-amino-3-mercaptopropyl)-valyl-isoleucyl-leucine (Compound
2);
N-(2(R)-amino-3-mercaptopropyl)-valyl-isoleucyl-leucine methyl ester (Compound 1);
N-[ 2(S)-(2(S)-(2(R)-amino-3-mercaptopropylamino)-3(S)- methylpentyloxy)-3-methylbutanoyl]-leucine; and
N-[2(S)-(2(S)-(2(R)-amino-3-mercaptopropylamino)-3(S)- methylpentyloxy)-3-methylbutanoyl]-leucine methyl ester, and the pharmaceutically acceptable salts and disulfides thereof.
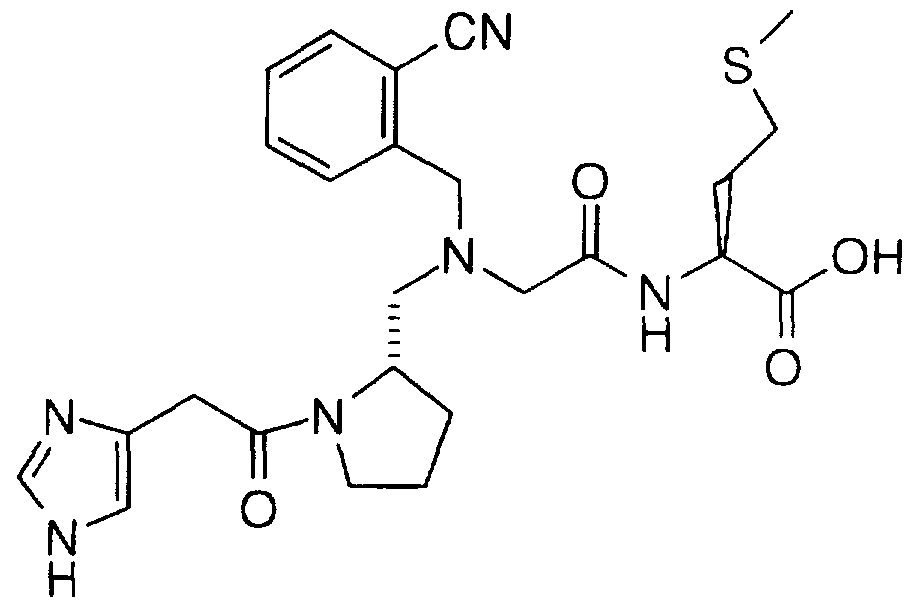
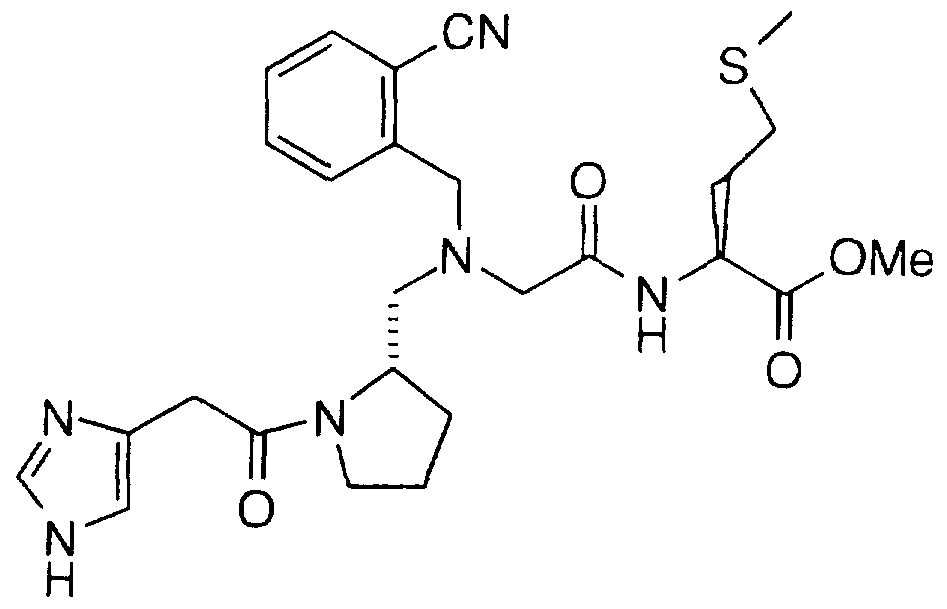
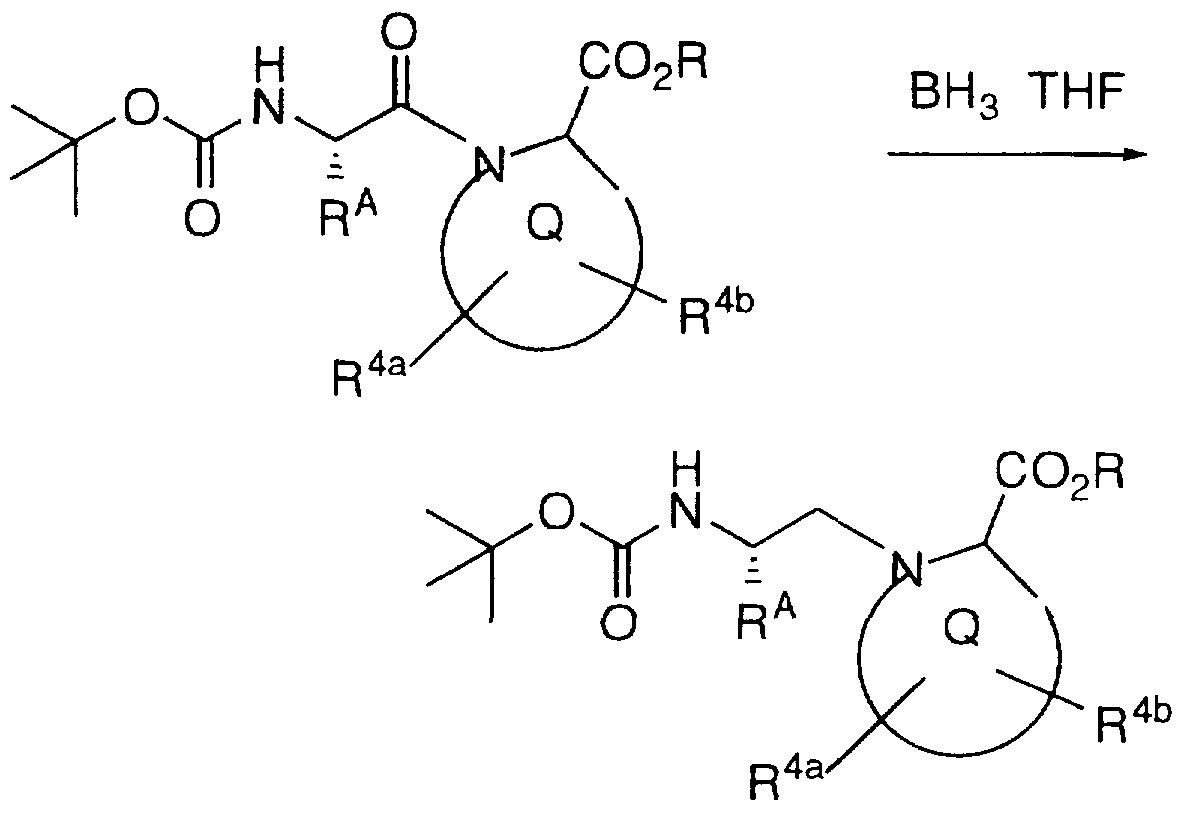
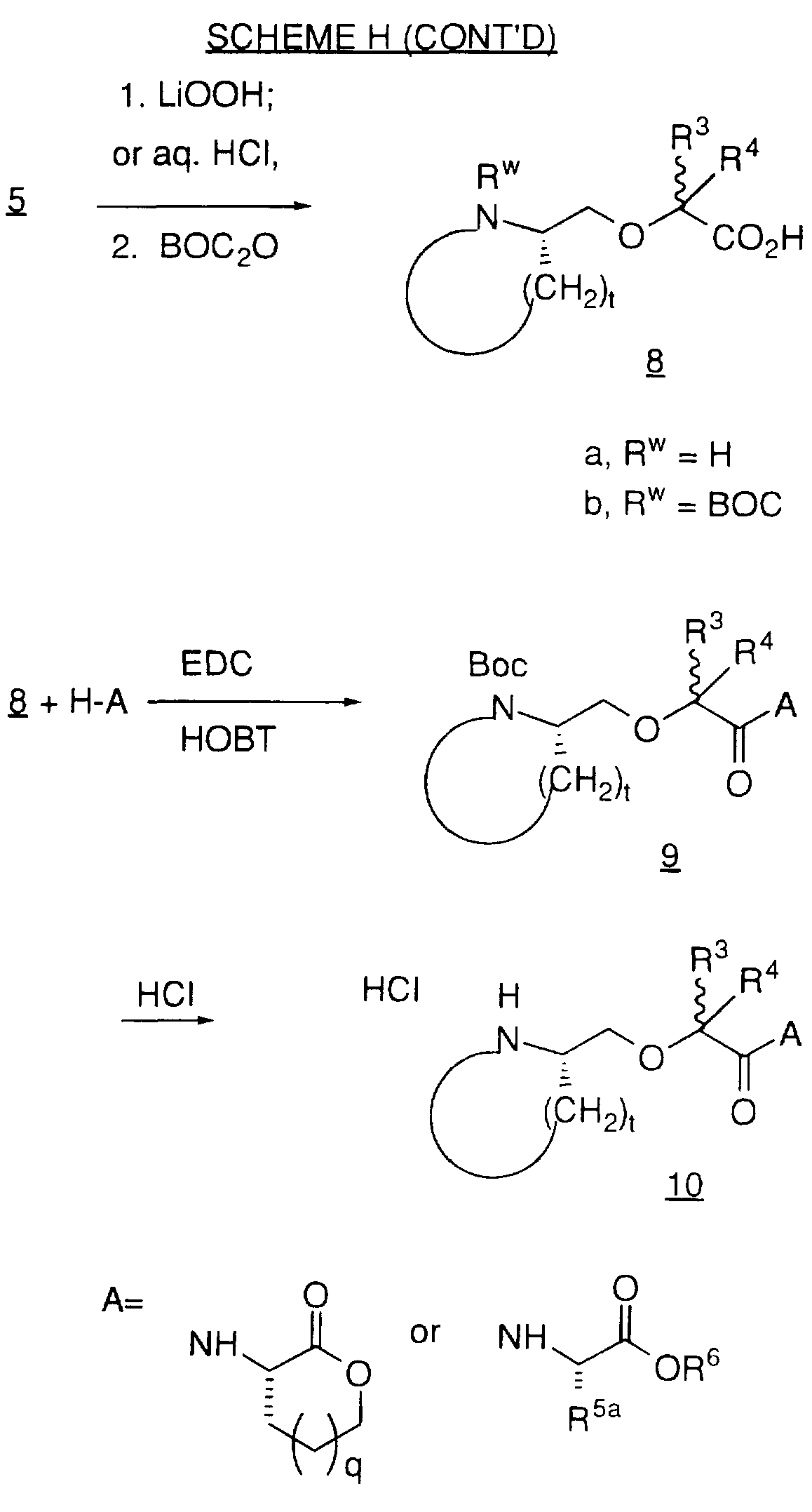
Examples of compounds which selectively inhibit farnesyl protein transferase include the following:
2(S)-Butyl-1 -(2,3-diaminoprop-1 -yl)-1 -( 1 -naphthoyl)piperazine;
1 -(3-Amino-2-(2-naphthylmethylamino)prop- 1 -yl)-2(S)-butyl-4-(1- naphthoyl)piperazine;
2(S)-Butyl-1-{5-[1-(2-naphthylmethyl)]-4,5-dihydroimidazol)methyl-4- (1-naphthoyl)piperazine; 1-[5-(1-Benzylimidazol)methyl]-2(S)-butyl-4-(1-naphthoyl)piperazine;
1-{5-[1-(4-nitrobenzyl)]imidazolylmethyl}-2(S)-butyl-4-(1- naphthoyl)piperazine; 1-(3-Acetamidomethylthio-2(R)-aminoprop-1-yl)-2(S)-butyl-4-(1- naphthoyl)piperazine;
2(S)-Butyl-1-[2-(1-imidazolyl)ethyl]sulfonyl-4-(1-naphthoyl)piperazine; 2(R)-Butyl-1-imidazolyl-4-methyl-4-(1-naphthoyl)piperazine;
2(S)-Butyl-4-(1-naphthoyl)-1-(3-pyridylmethyl)piperazine;
1-2(S)-butyl-(2(R)-(4-nitrobenzyl)amino-3-hydroxypropyl)-4-(1- naphthoyl)piperazine;
1-(2(R)-Amino-3-hydroxyheptadecyl)-2(S)-butyl-4-(1-naphthoyl)- piperazine; 2(S)-Benzyl-1-imidazolyl-4-methyl-4-(1-naphthoyl)piperazine;
1-(2(R)-Amino-3-(3-benzylthio)propyl)-2(S)-butyl-4-(1- naphthoyl)piperazine; 1-(2(R)-Amino-3-[3-(4-nitrobenzylthio)propyl])-2(S)-butyl-4-(1- naphthoyl)piperazine;
2(S)-ButyI-1-[(4-imidazolyl)ethyl]-4-(1-naphthoyl)piperazine;
2(S)-Butyl-1-[(4-imidazoIyl)methyl]-4-(1-naphthoyl)piperazine;
2(S)-Butyl-1-[(1-naphth-2-ylmethyl)-1H-imidazol-5-yl)acetyl]-4-(1- naphthoyl)ρiperazine;
2(S)-Butyl-1-[(1-naphth-2-ylmethyl)-1H-imidazol-5-yl)ethyl]-4-(1- naphthoyl)piperazine;
1-(2(R)-Amino-3-hydroypropyl)-2(S)-butyl-4-(1-naphthoyl)piperazine;
1-(2(R)-Amino-4-hydroxybutyl)-2(S)-butyl-4-(1-naphthoyl)piperazine;
1-(2-Amino-3-(2-benzyloxyphenyl)propyl)-2(S)-butyl-4-(1- naphthoyI)piperazine;
1-(2-Amino-3-(2-hydroxyphenyl)propyl)-2(S)-butyl-4-(1- naphthoyl)piperazine;
1-[3-(4-imidazolyl)propyl]-2(S)-butyl-4-(1-naphthoyl)-piperazine;
2(S)-n-Butyl-4-(2,3-dimethylphenyl)-1-(4-imidazolylmethyl)- piperazin-5-one;
2(S)-n-Butyl-1-[1-(4-cyanobenzyI)imidazol-5-ylmethyl]-4-(2,3- dimethylphenyl)piperazin-5-one;
1-[1-(4-Cyanobenzyl)imidazol-5-ylmethyl]-4-(2,3-dimethylphenyl)- 2(S)-(2-methoxyethyl)piperazin-5-one; 2(S)-n-Butyl-4-(1-naphthoyl)-1-[1-(1-naphthyImethyl)imidazol-5- ylmethyl]-piperazine;
2(S)-n-Butyl-4-(1-naphthoyl)-1-[1-(2-naphthyImethyl)imidazoI-5- ylmethyl]-piperazine;
2(S)-n-Butyl-1-[1-(4-cyanobenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)piperazine; 2(S)-n-Butyl-1-[1-(4-methoxybenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)piperazine;
2(S)-n-Butyl-1-[1-(3-methyl-2-butenyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)piperazine;
2(S)-n-Butyl-1-[1-(4-fluorobenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)piperazine;
2(S)-n-Butyl-1-[1-(4-chlorobenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)piperazine;
1-[1-(4-Bromobenzyl)imidazol-5-yImethyl)-2(S)-n-butyl-4-(1- naphthoyl)piperazine; 2(S)-n-Butyl-4-(1-naphthoyl)-1-[1-(4-trifluoromethylbenzyl)imidazol-5- ylmethyl]-piperazine;
2(S)-n-Butyl-1-[1-(4-methylbenzyl)imidazol-5-yImethyl]-4-(1
naphthoyl)-piperazine;
2(S)-n-Butyl-1-[1-(3-methylbenzyl)imidazol-5-ylmethyl]-4-(1
naphthoyl)-piperazine;
1-[1-(4-Phenylbenzyl)imidazol-5-ylmethyll-2(S)-n-butyl-4-(1- naphthoyl)-piperazine;
2(S)-n-Butyl-4-(1-naphthoyl)-1-[1-(2-phenylethyl)imidazol-5-y-methyl]- piperazine;
2(S)-n-Butyl-4-(1-naphthoyl)-1-[1-(4-trifluoromethoxy)imidazol-5- ylmethyl]piperazine;
1-{[1-(4-cyanobenzyl)-1H-imidazol-5-yl]acetyl}-2(S)-n-butyl-4-(1- naphthoyl)piperazine;
1-{5-[1-(4-nitrobenzyl)]imidazolylmethyl}-2(S)-butyl-4-(1- naphthoyl)piperazine
1-[5-(1-Benzylimidazol)methyl]-2(S)-butyl-4-(1-naphthoyl)piperazine
1-(22R)-Amino-3-(3-benzylthio)propyl)-2(S)-butyl-4-(1- naphthoyl)piperazine
1-(2(R)-Amino-3-[3-(4-nitrobenzylthio)propyl])-2(S)-butyl-4-(1- naphthoyl)piperazine
2(S)-n-Butyl-1-[1-(4-cyanobenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)piperazine
2(S)-n-Butyl-1-[1-(4-cyanobenzyl)imidazol-5-ylmethyl]-4-(2,3- dimethylphenyl)piperazin-5-onn
2(S)-n-Butyl-1-[1-(4-chlorobenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)piperazine
1-{[1-(4-cyanobenzyl)-1H-imidazoI-5-yl]acetyl}-2(S)-n-butyl-4-(1- naphthoyl)piperazine
1-[1-(4-Cyanobenzyl)imidazol-5-ylmethyl]-4-(2,3-dimethylphenyl)- 2(S)-(2-methoxyethyl)piperazin-5-one
N-[1-(4-Imidazoleacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1
naphthylmethyl)glycylmethionine
N-[1-(4-Imidazoleacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthyl- methyl)glycyl-methionine methyl ester;
N-[1-(2(S),3-Diaminopropionyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(2(S),3-Diaminopropionyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(3-Aminopropionyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine; N-[1-(3-Aminopropionyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-n -(2(S)-Amino-3-benzyloxycarbonylaminopropionyl)pyrrolidin- 2(S)-ylmethyl]-N-(1-naphthylmethyI)glycyl-methionine; N-[1-(2(S)-Amino-3-benzyloxycarbonylaminopropionyl)pyrrolidin- 2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(3-Amino-2(S)-benzyloxycarbonylaminopropionyl)pyrrolidin- 2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl-methionine;
N-[1-(3-Amino-2(S)-benzyloxycarbonylaminopropionyl)pyrrolidin- 2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(L-Glutaminyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(L-Glutaminyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(L-Histidyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine; N-[1-(L-Histidyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(D-Histidyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(D-Histidyl)pyrroIidin-2(S)-ylmethyI]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(L-Pyroglutamyl)pyrrolidin-2(S)-ylmethyl)-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(L-Pyroglutamyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
2(S)-[1-(2(S)-Pyroglutamyl)pyrrolidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine; 2(S)-[1-(2(S)-Pyroglutamyl)pyrrolidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine methyl ester;
2(S)-[1-(2(S)-Pyroglutamyl)pyrrolidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine isopropyl ester;
2(S)-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine;
2(S)-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine methyl ester;
2(S)-[1-(2(S)-Pyroglutamyl)pyrrolidin-2(S)-yImethyloxy]-3- phenylpropionyl-methionine sulfone; 2(S)-[1-(2(S)-Pyroglutamyl)pyrrolidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine sulfone methyl ester;
2(S)-[1-(Pyrid-3-ylcarboxy)pyrrolidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine;
2(S)-[1-(Pyrid-3-ylcarboxy)pyrrolidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine methyl ester;
2(R)-{2-[1-(Naphth-2-yl)-lH-imidazol-5-ylacetyl]pyrrolidin-2(S)- ylmethoxy}-3-phenylpropionyl-methionine;
2(R)-{2-[1-(Naphth-2-yl)-1H-imidazol-5-ylacetyl]pyrrolidin-2(S)- ylmethoxy}-3-phenylpropionyl-methionine methyl ester;
2(S)-[1-(Pyrid-3-ylmethyl)pyrrolidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine; 2(S)-[1-(Pyrid-3-ylmethyl)pyrroIidin-2(S)-ylmethyloxy]-3- phenylpropionyl-methionine methyl ester;
N-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine isopropyl ester;
N-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1 naphthylmethyl)glycyl-methionine sulfone isopropyl ester;
N-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine sulfone;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine methyl ester; N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine isopropyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine sulfone methyl ester;
N-[ 1 -(Glycyl) pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine sulfone;
N-[1-(Sarcosyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine methyl ester;
N-[1-(Sarcosyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine; N-[1-(N,N-Dimethylglycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(N,N-Dimethylglycyl) pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-3(S)-ethyl-2(S)-ylmethyl]-N- (1-naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-3(S)-ethyl-2(S)-ylmethyl]-N- ( 1 -naphthylmethyl)glycyl-methionine;
N-[1-(Glycyl)pyrroIidin-3(S)-ethyl-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester; N-[1-(Glycyl)pyrrolidin-3(S)-ethyl-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(4-Cyanobenzyl)-1H-imidazol-5-ylacetyl)pyrrolidin-2(S)- ylmethyl]-N-(1-naphthyImethyl)glycyl-methionine methyl ester;
N-[1-(4-Cyanobenzyl)-1H-imidazol-5-ylacetyl)pyrrolidin-2(S)- ylmethyl]-N-(1-naphthylmethyl)glycyl-methionine;
N-[1-(2-Acetylamino-3(S)- benzyloxycarbonylaminopropionyl)pyrrolidin-2(S)-yImethyl]-N-(1- naphthylmethyl)glycyl-methionine; N-[1-(2-Acetylamino-3(S)-aminopropionyl)pyrrolidin-2(S)-ylmethyl]- N-(1-naphthylmethyl)glycyl-methionine;
N-[1-(2-Amino-3(S)-acetylaminopropionyl)pyrrolidin-2(S)-ylmethyl]- N-(1-naphthylmethyl)glycyl-methionine;
2(S)-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-3(S)-ethyl-2(S)- ylmethyloxyl-3-phenylpropionyl-methionine methyl ester;
2(S)-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-3(S)-ethyl-2(S)- yImethyloxyl-3-phenylpropionyl-methionine;
2(R)-{2-[1-(4-Cyanobenzyl)-1H-imidazol-5-ylacetyl]pyrrolidin-2(S)- ylmethoxy}-3-phenyl propionylmethionine methyl ester; 2(R)-{2-[1-(4-Cyanobenzyl)-1H-imidazol-5-ylacetyllpyrrolidin-2(S)- ylmethoxy}-3-phenyl propionyl-methionine;
2(R)-{2-[1-(4-Nitrobenzyl)-1H-imidazol-5-ylacetyl]pyrrolidin-2(S)- ylmethoxy}-3-phenyl propionyl-methionine methyl ester;
2(R)-{2-[1-(4-Nitrobenzyl)-1H-imidazol-5-ylacetyI]pyrrolidin-2(S)- ylmethoxy}-3-phenyl propionyl-methionine;
2(R)-{2-[1-(4-Methoxybenzyl)-1H-imidazol-5-ylacetyl]pyrrolidin-2(S)- ylmethoxy}-3-phenyl propionyl-methionine methyl ester;
2(R)-{2-[1-(4-Methoxybenzyl)-1H-imidazol-5-ylacetyl]pyrrolidin-2(S)- ylmethoxy}-3-phenyl propionyl-methionine;
2(R)-{2-[1-(4-Cyanobenzyl)-1H-imidazol-5-ylacetyl)pyrrolidin-3(S)- ethyl-2(S)-ylmethoxy}-3-phenyl propionyl-methionine methyl ester;
2(R)-{2-[1-(4-Cyanobenzyl)-1H-imidazol-5-ylacetyIjpyrrolidin-3(S)- ethyl-2(S)-ylmethoxy}-3-phenyl propionyl-methionine;
N-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-acetylamino)alanine methyl ester; N-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-acetylamino)alanine;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyI]-N-(1-naphthylmethyl)glycyl- (β-acetylamino)alanine methyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-yImethyl]-N-(1-naphthylmethyl)glycyl- (β-acetylamino)alanine;
N-[1-(Seryl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine methyl ester;
N-[1-(D-Alanyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine methyl ester; N-[1-(1H-imidazol-4-carbonyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(Isoasparagyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(1H-Imidazol-4-propionyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(3-Pyridylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthyImethyl)glycyl-methionine methyl ester;
N-[1-(2-Pyridylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester; N-[1-(4-Pyridylglycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[ 1 -(Seryl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthyImethyl)glycyl- methionine;
N-[1-(D-Alanyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyI- methionine;
N-[1-(1H-Imidazol-4-carbonyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(Isoasparagyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine; N-[1-(1H-Imidazol-4-propionyl)pyrrolidin-2(S)-ylmethyl]-N-(1 naphthylmethyl)glycyl-methionine;
N-[1-(3-Pyridylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(2-Pyridylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(4-Pyridylglycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylmethyl)pyrrolidin-2(S)-yImethyl]-N-(1- naphthylmethyl)gIycyl-methionine;
N-[1-(2-Aminoethyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(Glycyl)pyrrolidin-2(S)-yImethyl]-N-(1-naphthyImethyl)glycyl- (2-thienyl)alanine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrroIidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(trifluoromethyl)alanine; N-[1-(1H-Imidazol-4-ylacetyl)pyrroIidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(2(S)-amino-4-acetylamino)butyric acid;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(N,N-dimethyl)glutamine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N- (benzyl)glycyl-methionine;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(benzyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylacetyl)pyriolidin-2(S)-ylmethyl]-N-(4- methoxybenzyl)glycyl-methionine;
N-[1-(Glycyl)pyrrolidin-3(S)-ethyl-2(S)-ylmethyl]-N-(benzyl)glycyl- methionine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-3(S)-ethyl-2(S)-ylmethyI]-N- (benzyl)glycyl-methionine; N-((4-Imidazolyl)methyl-(2S)-pyrrolidinylmethyl)-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- (2-thienyl)alanine methyl ester;
N-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(N,N-dimethyl)glutamine methyl ester; N-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-2(S)-yImethyl]-N-(1- naphthylmethyl)glycyl-(trifluoromethyl)alanine methyl ester;
N-[1-(1H-imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(2(S)-amino-4-acetylamino)butyric acid methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N- (benzyl)glycyl-methionine methyl ester; N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(benzyl)glycyl-methionine methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(4- methoxybenzyl)glycyl-methionine methyl ester;
N-[1-(1H -Imidazol-4-ylacetyl)pyrrolidin-3(S)-ethyl-2(S)-ylmethyl]-N- (benzyl)glycyl -methionine methyl ester;
N-[1-(Glycyl)pyrrolidin-3(S)-ethyl-2(S)-ylmethyl]-N-(benzyl)glycyl- methionine methyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyI)glycyl- methionine isopropyl ester; N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine cyclohexyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)gIycyl- methionine benzyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-yImethyl]-N-(1-naphthylmethyl)glycyl- methionine ethyl ester;
N-[1-(Sarcosyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine isopropyl ester;
N-[1-(N,N-Dimethylglycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine isopropyl ester; N-[1-(Glycyl)pyrroIidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine(2-pyridylmethyl) ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine(1-glyceryl)ester;
N-[1-L-Prolylpyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(L-Prolyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(1-Morpholinoacetyl)pyrrolidin-2(S)-yImethyl]-N-(1
naphthylmethyl)glycyl-methionine methyl ester; N-[1-(1-Morpholinoacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine;
N-[1-(4-Piperidinecarbonyl)pyrrolidin-2(S)-yImethyl]-N-(1
naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(4-Piperidinecarbonyl)pyrrolidin-2(S)-ylmethyl]-N-(1
naphthylmethyl)glycyl-methionine;
N-[1-(3-Piperidinecarbonyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
1 N-[1-(3-Piperidinecarbonyl)pyrroIidin-2(S)-ylmethyl]-N-(1 naphthylmethyl)glycyl-methionine; N-[ 1-(2-Pyridylglycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[ 1-(2-PyridylglycyI)pyrrolidin-2(S)-ylmethyl]-N-(1 - naphthylmethyl)glycyl-methionine;
N-[ 1-(4-Pyridylglycyl)pyrrolidin-2(S)-ylmethyl ]-N-(1- naphthylmethyl)glycyl-methionine methyl ester;
N-[1-(4-Pyridylglycyl)pyrrolidin-2(S)-ylmethyl]-N-( 1- naphthy Imethy l)glycyl-methionine;
N-[ 1 -(4-Pyridyl(N-methyI)glycyl)ρyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester; N-[ 1-(4-Pyridyl(N-methyl)glycyl)pyrrolidin-2(S)-ylmethyl]-N-( 1- naphthylmethyl)glycyl-methionine;
N-[ 1-(1H-Imidazol-4-ylpropionyl) pyrrolidin-2(S)-yImethyl]-N-( 1- naphthylmethyl)glycyl-(β-acetylamino)alanine;
N-[ 1-(1H-Imidazol-4-ylpropionyl) pyrrolidin-2(S)-ylmethyl]-N-(1 - naphthylmethyl)glycyl-(β-acetylamino)alanine methyl ester;
N-[1 -(4-Pyridylglycyl) pyrrolidin-2(S)-ylmethyl]-N-(1 - naphthylmethyl)glycyl-(β-acetylamino)alanine;
N-[1 -(4-Pyridylglycyl) pyrrolidin-2(S)-ylmethyl]-N-( 1 - naphthylmethyl)glycyl-(β-acetylamino)alanine methyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- (β-acetylamino)alanine cyclohexyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-yImethyl]-N-(1- naphthylmethyl)glycyl-(N-methyl)glutamine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(N-methyl)glutamine methyl ester; N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-methylcarbonylamino)alanine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-methylcarbonylamino)alanine methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-methylsulfonylamino)alanine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-methylsulfonylamino)alanine methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-propionylamino)alanine; N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-yImethyl)-N-(1- naphthylmethyl)glycyl-(β-propionylamino)alanine methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-pyrrolidinon-1-ylamino)alanine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-yImethyl]-N-(1- naphthylmethyl)glycyl-(β-pyrrolidinon-1-ylamino)alanine methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(3- methoxybenzyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(3- methoxybenzyl)glycyl-methionine methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-yImethyl]-N-(2- methoxybenzyl)glycyl-methionine; N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(2- methoxybenzyl)glycyl-methionine methyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(3-methoxybenzyl)glycyl- methionine;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(3-methoxybenzyl)glycyl- methionine methyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-yImethyl]-N-(2-methoxybenzyl)glycyl- methionine;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(2-methoxybenzyl)glycyI- methionine methyl ester; N-[1-(1H-Imidazol-4-ylpropionyl)pyrrolidin-2(S)-ylmethyl]-N-(2- methoxybenzyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylpropionyl)pyrrolidin-2(S)-ylmethyl]-N-(2- methoxybenzyl)glycyl-methionine methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrroIidin-2(S)-ylmethyI]-N-(3- cyanobenzyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(3- cyanobenzyl)glycyl-methionine methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(4- cyanobenzyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyI]-N-(2- cyanobenzyl)glycyl-methionine; N-(1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(2- cyanobenzyl)glycyl-methionine methyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(2-cyanobenzyl)glycyl- methionine;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(2-cyanobenzyl)glycyl- methionine methyl ester;
N-[1-(1H-Imidazol-4-ylpropionyl)pyrrolidin-2(S)-ylmethyl]-N-(2- cyanobenzyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylpropionyl)pyrrolidin-2(S)-ylmethyl)-N-(2- cyanobenzyl)glycyl-methionine methyl ester; N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(2- methylbenzyl)glycyl-methionine;
N-[1-(1H-lmidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(2- methylbenzyl)glycyl-methionine methyl ester;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(2- trifluoromethylbenzyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(2- trifluoromethylbenzyl)glycyl-methionine methyl ester;
N-(1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylsulfonyl)glycyl-methionine;
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylsulfonyl)glycyl-methionine methyl ester; N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine 4-N-methylpiperidinyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine tert-butyl ester;
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine 3-pentyl ester;
N-[1-(4-Pyridylglycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine isopropyl ester;
N-[1-(1H-Imidazol-4-ylpropionyl)pyrrolidin-2(S)-ylmethyl)-N-(11- naphthylmethyl)glycyl-methionine isopropyl ester; N-[1-(4-Imidazoleacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine methyl ester
N-[1-(4-Imidazoleacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine isopropyl ester
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine methyl ester
N-[1-(Glycyl)pyrrolidin-2(S)-yImethyl]-N-(1-naphthylmethyl)glycyl- methionine isopropyl ester
N-[1-(L-Pyroglutamyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine
N-[1-(L-PyrogIutamyl)pyrrolidin-2(S)-ylmethyI]-N-(1- -aphthylmethyl)glycyl-methionine methyl ester
2(S)-[1-(1H-Imidazol-4-ylacetyl)pyrroIidin-3(S)-ethyl-2(S)- ylmethyIoxy]-3-phenylpropionyl-methionine methyl ester
2(S)-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-3(S)-ethyl-2(S)- yImethyloxy]-3-phenylpropionyl-methionine
N-[1-(Sarcosyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine o
N-[1-(Sarcosyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine methyl ester
N-[1-(N,N-Dimethylglycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine
N-[1-(N,N-Dimethylglycyl)pyrrolidin-2(S)-ylmethyI]-N-(1- naphthylmethyl)glycyl-methionine methyl ester
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-yImethyl]-N-(1- naphthylmethyl)glycyl-(β-acetylamino)alanine methyl ester
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-(β-acetylamino)alanine
N-[1-(Glycyl)pyrrolidin-2(S)-yImethyl]-N-(1-naphthylmethyl)glycyl- (β-acetylamino)alanine cyclohexyl ester
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- (β-acetylamino)alanine
N-[1-(4-Pyridylglycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine isopropyl ester
N-[1-(4-Pyridylglycyl)pyrrolidin-2(S)-ylmethyl]-N-(1- naphthylmethyl)glycyl-methionine
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyI]-N-(2- methoxybenzyl)glycyl-methionine
N-[1-(1H-Imidazol-4-ylacetyl)ρyrrolidin-2(S)-ylmethyl]-N-(2- methoxybenzyl)glycyl-methionine methyl ester
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(2-methoxybenzyl)glycyl- methionine methyl ester
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(2-methoxybenzyl)glycyl- methionine
N-[1-(1H-lmidazol-4-ylpropionyl)pyrrolidin-2(S)-ylmethyl]-N-(2- methoxybenzyl)glycyl-methionine methyl ester
N-[1-(1H-Imidazol-4-ylpropionyl)pyrroIidin-2(S)-ylmethyl]-N-(2- methoxybenzyl)glycyl-methionine
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-yImethyl]-N-(2- cyanobenzyl)glycyl-methionine
N-[1-(1H-Imidazol-4-ylacetyl)pyrrolidin-2(S)-ylmethyl]-N-(2- cyanobenzyl)glycyl-methionine methyl ester
N-[1-(Glycyl)pyrrolidin-2(S)-ylmethyl]-N-(1-naphthylmethyl)glycyl- methionine 4-N-methylpiperidinyl ester
N-[1-(1H-Imidazol-4-ylpropionyl)pyrrolidin-2(S)-ylmethyl]-N-(1 naphthylmethyl)glycyl-methionine isopropyl ester
N-[(1H-imidazol-4-ylacetyl-2(S)-amino)-3(S)-methylpentyl]-1,2,3,4- tetrahydro-3(S)-isoquinolinecarbonyl-methionine methyl ester; N-1(1H-imidazol-4-ylacetyl-2(S)-amino)-3(S)-methylpentyl]-1,2,3,4- tetrahydro-3(S)-isoquinolinecarbonyl-methionine;
N-[1-(1H-imidazol-4-ylacetyl)-3(S)-ethylpyrrolidin-2(S)-ylmethyl]- prolyl-methionine methyl ester;
N-[1-(1H-imidazol-4-ylacetyI)-3(S)-ethylpyrrolidin-2(S)-ylmethyl]- prolyl-methionine;
N-[1-GlycylpyriOlidin-2(S)-ylmethyl]-3(S)-ethylprolyl-methionine methyl ester;
N-[1-Glycylpyrrolidin-2(S)-ylmethyl]-3(S)-ethylprolyl-methionine;
N-[L-Pyroglutamyl-2(S)-amino-3(S)-methylpentyl]-1,2,3,4-tetrahydro- 3(S)-isoquinolinecarbonyl-methionine
N-[L-Pyroglutamyl-2(S)-amino-3(S)-methylpentyl]-1 ,2,3,4-tetrahydro- 3(S)-isoquinolinecarbonyl-methionine methyl ester
N-[ 1 -(1H-imidazol-4-ylacetyl)-pyrrolidin-2(S)-ylmethyl]-3(S)- ethylprolyl-methionine
N-[ 1 -(1H-imidazol-4-ylacetyI)-pyrrolidin-2(S-)ylmethyl]-3(S)- ethylprolyl-methionine methyl ester
N-1 (1 H-imidazol-4-ylacetyl-2(S)-amino)-3(S)-methylpentyl )-prolyl- methionine methyl ester
N-[(1H-imidazol-4-ylacetyl-2(S)-amino)-3(S)-methylpentyl]-prolyl- methionine
N-[( 1 H-imidazol-4-ylacetyl-2(S)-amino)-3(S)-methylpentyl]-1 ,2,3,4- tetrahydro-3(S)-isoquinolinecarbonyl-methionine methyl ester
N-[( 1H-imidazol-4-ylacetyl-2(S)-amino)-3(S)-methylpentyl]-1 ,2,3,4- tetrahydro-3(S)-isoquinolinecarbonyl-methionine
N-[L-Pyroglutamyl-2(S)-amino-3(S)-methylpentyl]-1,2,3,4-tetrahydro- 3(S)-isoquinolinecarbonyl-methionine methyl ester N-[L-Pyroglutamyl-2(S)-amino-3(S)-methylpentyl]-1,2,3,4-tetrahydro- 3(S)-isoquinolinecarbonyl-methionine
N-[(1H-imidazol-4-ylacetyl-2(S)-amino)-3(S)-methylpentyl]-prolyl- methionine methyl ester
N-[(1H-imidazol-4-ylacetyl-2(S)-amino)-3(S)-methylpentyl]-prolyl- methionine
N-[1-(1H-imidazol-4-ylacetyl)-3(S)-ethylpyrrolidin-2(S)-ylmethyl]- prolyl-methionine methyl ester
N-[1-(1H-imidazol-4-ylacetyl)-3(S)-ethylpyrrolidin-2(S)-ylmethyl]- prolyl-methionine N-[1-(1H-imidazol-4-ylacetyl)-pyrrolidin-2(S)-ylmethyl]-3(S)- ethylprolyl-methionine methyl ester
N-[1-(1H-imidazol-4-ylacetyl)-pyrrolidin-2(S)-ylmethyl]-3(S)- ethylprolyl-methionine
N-[1-Glycylpyrrolidin-2(S)-ylmethyl]-3(S)-ethylprolyl-methionine methyl ester
N-[1-Glycylpyrrolidin-2(S)-ylmethyl]-3(S)-ethylprolyl-methionine 2(S)-Butyl-1-(2,3-diaminoρrop-1-yl)-1-(1-naphthoyl)piperazine
1-(3-Amino-2-(2-naphthylmethylamino)prop-1-yl)-2(S)-butyl-4-(1- naphthoyl)piperazine
2(S)-Butyl-1-{5-[1-(2-naphthyImethyl)]-4,5-dihydroimidazol}methyl-4- (1-naphthoyl)piperazine
1-[5-(1-Benzylimidazol)methyl]-2(S)-butyl-4-(1-naphthoyl)piperazine
1-{5-[1-(4-nitrobenzyl)]imidazolylmethyl}-2(S)-butyl-4-(1- naphthoyl)piperazine
1-(3-Acetamidomethylthio-2(R)-aminoprop-1-yl)-2(S)-butyl-4-(1- naphthoyl)piperazine
2(S)-Butyl-1-[2-(1-imidazolyl)ethyljsulfonyl-4-(1-naphthoyl)piperazine
2(R)-Butyl-1-imidazolyI-4-methyl-4-(1-naphthoyl)piperazine
2(S)-Butyl-4-(1-naphthoyl)-1-(3-pyridylmethyl)piperazine
1-2(S)-butyl-(2(R)-(4-nitrobenzyl)amino-3-hydroxypropyl)-4-(1- naphthoyl)piperazine
1-(2(R)-Amino-3-hydroxyheptadecyl)-2(S)-butyl-4-(1-naphthoyl)- piperazine
2(S)-Benzyl-1-imidazolyl-4-methyl-4-(1-naphthoyl)piperazine
1-(2(R)-Amino-3-(3-benzylthio)propyl)-2(S)-butyl-4-(1- naphthoyl)piperazine
1-(2(R)-Amino-3-l3-(4-nitrobenzylthio)propyl])-2(S)-butyl-4-(1- naphthoyl)piperazine
2(S)-Butyl-1-[(4-imidazolyl)ethyl]-4-(1-naphthoyl)piperazine
2(S)-Butyl-1-[(4-imidazolyl)methyl]-4-(1-naphthoyl)piperazine
2(S)-Butyl-1-[(1-naphth-2-ylmethyl)-1H-imidazol-5-yl)acetyl]-4-(1- naphthoyl)piperazine 2(S)-Butyl-1-1(1-naphth-2-ylmethyl)-1H-imidazol-5-yl)ethyl]-4-(1- naphthoyl)piperazine
1-(2(R)-Amino-3-hydroypropyl)-2(S)-butyl-4-(1-naphthoyl)piperazine 1-(2(R)-Amino-4-hydroxybutyl)-2(S)-butyl-4-(1-naphthoyl)piperazine
1-(2-Amino-3-(2-benzyloxyphenyl)propyI)-2(S)-butyl-4-(1- naphthoyl)piperazine 1-(2-Amino-3-(2-hydroxyphenyl)propyl)-2(S)-butyl-4-(1- naphthoyl)piperazine
1-[3-(4-imidazolyl)propyl]-2(S)-butyl-4-(1-naphthoyl)-piperazine 2(S)-n-Butyl-4-(2,3-dimethylphenyl)-1-(4-imidazolylmethyl)- piperazin-5-one
2(S)-n-Butyl-1-[1-(4-cyanobenzyl)imidazol-5-ylmethyl]-4-(2,3- dimethylphenyl)piperazin-5-one
1-[1-(4-Cyanobenzyl)imidazol-5-ylmethyl]-4-(2,3-dimethylphenyl)- 2(S)-(2-methoxyethyl)piperazin-5-one
2(S)-n-Butyl-4-(1-naphthoyl)-1-[1-(1-naphthyImethyl)imidazol-5- ylmethyl]-piperazine
2(S)-n-Butyl-4-(1-naphthoyl)-1-[1-(2-naphthyImethyl)imidazol-5- ylmethyl]-piperazine
2(S)-n-Butyl-1-[1-(4-cyanobenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyI)piperazine
2(S)-n-Butyl-1-[1-(4-methoxybenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)piperazine
2(S)-n-Butyl-1-[1-(3-methyl-2-butenyl)imidazol-5-ylmethyl]-4-(1 naphthoyl)piperazine 2(S)-n-Butyl-1-[1-(4-fluorobenzyl)imidazol-5-ylmethyl)-4-(1- naphthoyl)piperazine
2(S)-n-Butyl-1-[1-(4-chlorobenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)piperazine
1-[1-(4-Bromobenzyl)imidazol-5-ylmethyl]-2(S)-n-butyl-4-(1- naphthoyl)piperazine
2(S)-n-Butyl-4-(1-naphthoyl)-1-[1-(4-trifluoromethylbenzyl)imidazol-5- ylmethyl]-piperazine
2(S)-n-Butyl-1-[1-(4-methylbenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)-piperazine 2(S)-n-Butyl-1-[1-(3-methylbenzyl)imidazol-5-ylmethyl]-4-(1- naphthoyl)-piperazine
1-[1-(4-Phenylbenzyl)imidazol-5-ylmethyl]-2(S)-n-butyl-4-(1- naphthoyl)-piperazine
2(S)-n-Butyl-4-(1-naphthoyl)-1-[1-(2-phenylethyl)imidazol-5-ylmethyl] piperazine
2(S)-n -Butyl-4-(1 -naphthoyl)-1-[1 -(4-trifluoromethoxy)imidazol-5- ylmethyl]piperazine
1-{ [ 1 -(4-cyanobenzyl)-1H-imidazol-5-yl]acetyl}-2(S)-n -butyl-4-(1 - naphthoyl)piperazine
(N-[ 1 -Cyanobenzyl)-1 H-imidazol-5-yl)acetyl]pyrrolidin-2(S)-ylmethyl]- 3(S)-ethyl-proIyl methionine
(N-[ 1 -Cyanobenzyl)-1H-imidazol-5-yl)acetyl]pyrrolidin-2(S)-ylmethyl]- 3(S)-ethyl-prolyl methionine methyl ester
(N-[ 1 -Cyanobenzyl)-1H-imidazol-5-yl)acetyl]pyrrolidin-2(S)-ylmethyl]- 3(S)-ethyl-prolyl methionine isopropyl ester
N-[ 1 -(1H-Imidazol-4-propionyl) pyrrolidin-2(S)-ylmethyl]-N-(2- methoxybenzyl)glycyl-methionine isopropyl ester;
2(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)-methyl]- pentyloxy-3-phenylpropionyl-homoserine lactone (Compound 3), 2(S)-[2(S)-[2(R)-Amino-3-mercapto)propylamino-3(S)- methyl]pentyloxy-3-phenylpropionyl-homoserine (Compound 4),
2(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)- methyI]pentyloxy-2-methyl -3-phenylpropionyl-homoserine lactone,
2(S)-[2(S)-[2(R)-Amino-3-mercapto]propyIamino-3(S)- methyl ]pentyloxy-2-methyl-3-phenylpropionyl-homoserine,
2(S)-[2(S)-[2(R)-Amino-3-mercapto)propylamino-3(S)- methyl]pentyloxy-4-pentenoyl-homoserine lactone,
2(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)-methyl]- pentyloxy-4-pentenoyl-homoserine,
2(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)- methyl]pentyloxypentanoyl-homoserine lactone,
2(S)-[2(S)-[2(R)-Amino-3-mercapto]propylaniino-3(S)- methyl ]pentyloxypentanoyl-homoserine,
2(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)-methyl]5- pentyloxy-4-methylpentanoyl-homoserine lactone, 2(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)- methyl]pentyloxy-4-methylpentanoyl-homoserine,
2(S)-[2(S)-[2(R)-Amino-3-mercaptolpropylamino-3(S)- methyl]pentyloxy-3-methylbutanoyl-homoserine lactone,
2(S)-[2(S)-[2(R)-Amino-3-mercapto]ρropylamino-3(S)- methyl]pentyloxy-3-methylbutanoyl-homoserine,
2(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)- methyl]pentyloxy-3-phenylbutanoyl-homoserine lactone,
2(S)-[2(S)-[2(R)-Amino-3-mercapto ]propylamino-3(S)-methyl]- pentyloxy-3-phenylbutanoyl-homoserine, 2(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)- methyl]pentylthio-2-methyl-3-phenylpropionyl-homoserine lactone,
2(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)- methyl]pentylthio-2-methyl-3-phenylpropionyl-homoserine,
2(S)-[2(S)-[2(R)-Amino-3-mercaptolpropylamino-3(S)- methyl]pentylsulfonyl-2-methyl-3-phenylpropionyl-homoserine lactone,
2(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)-methyl]-
pentylsulfonyl-2-methyl-3-phenylpropionyl-homoserine,
2(S)-[2(S)-[2(R)-Amino-3-mercapto]propylaniino-3(S)-methyl]- pentyloxy-3-phenylpropionyl-methionine methyl ester,
2(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)- methyl]pentyloxy-3-phenylpropionyl-methionine,
2(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)- methyl]pentyloxy-3-phenylpropionyl-methionine sulfone methyl ester (Compound 5),
2(S)-[2(S)-[2(R)-Amino-3-mercapto]ρropylamino-3(S)- methyl]pentyloxy-3-phenylpropionyl-methionine sulfone (Compound 6),
2(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)-methyl]- pentyloxy-3-phenylpropionyl-methionine sulfone isopropyl ester,
2-(S)-[2(S)-[2(R)-Amino-3-mercapto]proρylamino-3(S)-methyl]- pentyloxy-3-naphth-2-yl-propionyt-methionine sulfone methyl ester,
2-(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)-methyl]- pentyloxy-3-naphth-2-yl-propionyI-methionine sulfone, 2-(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)- methyl]pentyloxy-3-naphth-1-yl-propionyl-methionine sulfone methyl ester,
2-(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)- methyl]pentyloxy-3-naphth-1 -yl-propionyl-methionine sulfone,
2-(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)- methyl]pentyloxy-3-methybutanoyI-methionine methyl ester.
2-(S)-[ 2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)- methyl]pentyloxy-3-methybutanoyl-methionine,
Disulphide of 2(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino- 3(S)methyl]pentyloxy-3-phenylpropionyl-homoserine lactone,
Disulphide of 2(S)-[ 2(S)-[2(R)-Amino-3-mercapto]propylamino-3(S)- methyl]pentyloxy-3-phenylpropionyI-homoserine, Disulphide of 2(S)-[2(S)-[2(R)-Amino-3-mercapto]propylamino- 3(S)methyl]pentyloxy-3-methylbutanoyl-methionine methyl ester and the pharmaceutically acceptable salts, disulfides or optical isomers thereof.
Compounds which are useful in the present invention, and methods of synthesis thereof, can be found in the following patents, pending applications and publications:
WO 95/32987 published on 7 December 1995.
U. S. Pat. No. 5,420,245;
European Pat. Publ. 0 618 221 ;
WO 94/26723;
WO 95/08542 ;
WO 95/1 1917;
WO 95/12612.
U. S. Pat. No. 5,238,922 granted on August 24, 1993; ;
U. S. Pat. No. 5,340,828 granted on August 23, 1994; ; U. S. Pat. No. 5,480,893 granted on January 2, 1996; ;
U. S. Pat. No. 5,352,705 granted on October 4, 1994;
U. S. Pat. No. 5,504,1 15 granted on April 2, 1996;
U. S. Pat. No. 5,326,773 granted on July 5, 1994;
U. S. Pat. No. 5,504,212 granted on April 2, 1996; ;
U. S. Pat. No. 5,439,918 granted on August 8, 1995;
USSN 08/968,025 filed on October 29, 1992 and USSN 08/143,943 filed on October 27, 1993 ;
USSN 08/080,028 filed on June 18, 1993 and USSN 08/237,586 filed on May 1 1 , 1994 ;
USSN 08/314,987 filed on September 29, 1994 USSN 08/315, 171 filed on September 29, 1994
USSN 08/315,046 filed on September 29, 1994 ;
USSN 08/315,161 filed on September 29, 1994; USSN 08/399,282 filed on March 6, 1995; USSN 472,077 filed on June 6, 1995 and
USSN 08/527,972 filed on September 14, 1995
U. S. Pat. No. 5,491 ,164 granted on February 13, 1996; ; USSN 08/314,974 filed on September 29, 1994
USSN 08/412,621 filed on March 29, 1995 and USSN 08/448,865 filed on May 24, 1995 ;
USSN 08/413,137 filed on March 29, 1995; ;
USSN 08/412,828 filed on March 29, 1995;
USSN 08/412,829 filed on March 29, 1995; and USSN 08/470,690 filed on June 6, 1995; and USSN 08/600,728 filed on February 28, 1996;
USSN 08/412,830 filed on March 29, 1995;
USSN 08/449,038 filed on May 24, 1995; ; and
USSN 08/468,160 filed on June 6, 1995. ;
All patents, publications and pending patent applications identified are hereby incorporated by reference.
The term "alkyl" refers to a monovalent alkane
(hydrocarbon) derived radical containing from 1 to 15 carbon atoms unless otherwise defined. It may be straight, branched or cyclic. Preferred straight or branched alkyl groups include methyl, ethyl, propyl, isopropyl, butyl and t-butyl. Preferred cycloalkyl groups include cyclopentyl and cyclohexyl.
When substituted alkyl is present, this refers to a straight, branched or cyclic alkyl group as defined above, substituted with 1-3 groups as defined with respect to each variable.
Heteroalkyl refers to an alkyl group having from 2-15 carbon atoms, and intermpted by from 1 -4 heteroatoms selected from O, S and N.
The term "alkenyl" refers to a hydrocarbon radical straight, branched or cyclic containing from 2 to 15 carbon atoms and at least one carbon to carbon double bond. Preferably one carbon to carbon double bond is present, and up to four non- aromatic (non-resonating) carbon-carbon double bonds may be present. Examples of alkenyl groups include vinyl, allyl,
isopropenyl, pentenyl, hexenyl, heptenyl, cyclopropenyl,
cyclobutenyl, cyclopentenyl, cyclohexenyl, 1 -propenyl, 2-butenyl, 2-methyl-2-butenyl, isoprenyl, farnesyl, geranyl, geranylgeranyl and the like. Preferred alkenyl groups include ethenyl, propenyl, butenyl and cyclohexenyl. As described above with respect to alkyl, the straight, branched or cyclic portion of the alkenyl group may contain double bonds and may be substituted when a substituted alkenyl group is provided.
The term "alkynyl" refers to a hydrocarbon radical straight, branched or cyclic, containing from 2 to 15 carbon atoms and at least one carbon to carbon triple bond. Up to three carbon- carbon triple bonds may be present. Preferred alkynyl groups
include ethynyl, propynyl and butynyl. As described above with respect to alkyl, the straight, branched or cyclic portion of the
alkynyl group may contain triple bonds and may be substituted when a substituted alkynyl group is provided.
Aryl refers to aromatic rings e.g., phenyl, substituted phenyl and like groups as well as rings which are fused, e.g., naphthyl and the like. Aryl thus contains at least one ring having at least 6 atoms, with up to two such rings being present, containing up to 10 atoms therein, with alternating (resonating) double bonds between adjacent carbon atoms. The preferred aryl groups are phenyl and naphthyl.
Aryl groups may likewise be substituted as defined below. Preferred substituted aryls include phenyl and naphthyl substituted with one or two groups. With regard to the farnesyl transferase inhibitors, "aryl" is intended to include any stable monocyclic, bicyclic or tricyclic carbon ring(s) of up to 7 members in each ring, wherein at least one ring is aromatic. Examples of aryl groups include phenyl, naphthyl,
anthracenyl, biphenyl, tetrahydronaphthyl, indanyl, phenanthrenyl and the like.
The term "heteroaryl" refers to a monocyclic aromatic hydrocarbon group having 5 or 6 ring atoms, or a bicyclic aromatic group having 8 to 10 atoms, containing at least one heteroatom,
O, S or N, in which a carbon or nitrogen atom is the point of
attachment, and in which one additional carbon atom is optionally replaced by a heteroatom selected from O or S, and in which from 1 to 3 additional carbon atoms are optionally replaced by nitrogen heteroatoms. The heteroaryl group is optionally substituted with up to three groups.
Heteroaryl thus includes aromatic and partially aromatic groups which contain one or more heteroatoms. Examples of this type are thiophene, purine, imidazopyridine, pyridine, oxazole, thiazole,
oxazine, pyrazole, tetrazole, imidazole, pyridine, pyrimidine, pyrazine and triazine. Examples of partially aromatic groups are tetrahydro- imidazo[4,5-clpyridine, phthalidyl and saccharinyl, as defined below.
With regard to the farnesyl transferase inhibitors, the term heterocycle or heterocyclic, as used herein, represents a stable 5- to 7- membered monocyclic or stable 8- to 1 1 -membered bicyclic or stable 1 1 -15 membered tricyclic heterocycle ring which is either saturated or unsaturated, and which consists of carbon atoms and from one to four heteroatoms selected from the group consisting of N, O, and S, and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring. The heterocyclic ring may be attached at any heteroatom or carbon atom which results in the creation of a stable structure. Examples of such heterocyclic elements include, but are not limited to, azepinyl, benzimidazolyl, benzisoxazolyl, benzofurazanyl, benzopyranyl, benzothiopyranyl, benzofuryl,
benzothiazolyl, benzothienyl, benzoxazolyl, chromanyl, cinnolinyl, dihydrobenzofuryl, dihydro-benzothienyl, dihydrobenzothiopyranyl, dihydrobenzothio-pyranyl sulfone, furyl, imidazolidinyl, imidazolinyl, imidazolyl, indolinyl, indolyl, isochromanyl, isoindolinyl, isoquinolinyl, isothiazolidinyl, isothiazolyl, isothiazolidinyl, morpholinyl,
naphthyridinyl, oxadiazolyl, 2-oxoazepinyl, 2-oxopiperazinyl, 2- oxopiperidinyl, 2-oxopyrrolidinyl, piperidyl, piperazinyl, pyridyl, pyridyl N-oxide, pyridonyl, pyrazinyl, pyrazolidinyl, pyrazolyl, pyrimidinyl, pyrrolidinyl, pyrrolyl, quinazolinyl, quinolinyl, quinolinyl N-oxide, quinoxalinyl, tetrahydro furyl, tetrahydroisoquinolinyl, tetrahydro-quinolinyl, thiamorpholinyl, thiamorpholinyl sulfoxide, thiazolyl, thiazolinyl, thienofuryl, thienothienyl, and thienyl.
Preferably, heterocycle is selected from imidazolyl, 2-oxopyrrolidinyl, piperidyl, pyridyl and pyrrolidinyl.
With regard to the farnesyl transferase inhibitors, the terms
"substituted aryl", "substituted heterocycle" and "substituted cycloalkyl" are intended to include the cyclic group which is substituted with 1 or 2 substitutents selected from the group which includes but is not limited to
F, Cl, Br, CF3, NH2, N(C1 -C6 alkyl)2, NO2, CN, (C1 -C6 alkyl)O-, -OH, (C1 -C6 alkyI)S(O)m-, (C1 -C6 alkyl)C(O)NH-, H2N-C(NH)-, (C1 -C6 alkyl)C(O)-, (C1 -C6 alkyl)OC(O)-, N3,(Cl -C6 alkyl)
OC(O)NH- and C1-C20 alkyl.
In the present method, amino acids which are disclosed are identified both by conventional 3 letter and single letter abbreviations as indicated below:
The compounds used in the present method may have asymmetric centers and occur as racemates, racemic mixtures, and as individual diastereomers, with all possible isomers, including optical isomers, being included in the present invention. Unless otherwise specified, named amino acids are understood to have the natural "L" stereoconfiguration
The following structure:
represents a cyclic amine moiety having 5 or 6 members in the ring, such a cyclic amine which may be optionally fused to a phenyl or cyclohexyl ring. Examples of such a cyclic amine moiety include, but are not limited to, the following specific structures:
It is also understood that substitution on the cyclic amine moiety by R2a and R2b may be on different carbon atoms or on the same carbon atom.
When R
3 and R
4 are combined to form - (CH
2)
s -, cyclic moieties are formed. Examples of such cyclic moieties include, but are not limited to:
When R
5a and R
5b are combined to form - (CH
2)
s -, cyclic moieties as described hereinabove for R
3 and R
4 are formed.
In addition, such cyclic moieties may optionally include a heteroatom(s). Examples of such heteroatom-containing cyclic moieties include, but are not limited to:
The pharmaceutically acceptable salts of the compounds of this invention include the conventional non-toxic salts of the compounds of this invention as formed, e.g., from non-toxic inorganic or organic acids. For example, such conventional non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like: and the salts
prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenyl-acetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxy-benzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isethionic, trifluoroacetic and the like.
It is intended that the definition of any substituent or variable (e.g., R 10, Z, n, etc.) at a particular location in a molecule be independent of its definitions elsewhere in that molecule. Thus, -N(R 1 0)2 represents -NHH, -NHCH3, -NHC2H5, etc. It is understood that substituents and substitution patterns on the compounds of the instant invention can be selected by one of ordinary skill in the art to provide compounds that are chemically stable and that can be readily synthesized by techniques known in the art as well as those methods set forth below.
The pharmaceutically acceptable salts of the compounds of this invention can be synthesized from the compounds of this invention
which contain a basic moiety by conventional chemical methods.
Generally, the salts are prepared by reacting the free base with stoichiometric amounts or with an excess of the desired salt-forming inorganic or organic acid in a suitable solvent or various combinations of solvents.
The compounds of formulas (Il-a) through (Il-k) can be synthesized from their constituent amino acids by conventional peptide synthesis techniques, and the additional methods described below.
Standard methods of peptide synthesis are disclosed, for example, in the following works: Schroeder et al., "The Peptides", Vol. I,
Academic Press 1965, or Bodanszky et al., "Peptide Synthesis",
Interscience Publishers, 1966, or McOmie (ed.) "Protective Groups in Organic Chemistry" , Plenum Press, 1973, or Barany et al., "The
Peptides: Analysis, Synthesis, Biology" 2, Chapter 1 , Academic Press, 1980, or Stewart et al., "Solid Phase Peptide Synthesis", Second Edition,
Pierce Chemical Company, 1984. Also useful in exemplifying syntheses of specific unnatural amino acid residues are European Pat. Appl.
No. 0 350 163 A2 (particularly page 51-52) and J. E. Baldwin et al.
Tetrahedron, 50:5049-5066 (1994). With regards to the synthesis of instant compounds containing a (β-acetylamino)alanine residue at the C-terminus, use of the commercially available Nα-Z-L-2,3- diaminopropionic acid (Fluka) as a starting material is preferred.
Abbreviations used in the description of the chemistry and in the Examples that follow are:
Ac2O Acetic anhydride;
Boc t-Butoxycarbonyl;
DBU 1 ,8-diazabicyclo[5.4.0]undec-7-ene;
DMAP 4-Dimethylaminopyridine;
DME 1,2-Dimethoxyethane;
DMF Dimethylformamide;
EDC 1 -(3-dimethylaminopropyl)-3-ethyl-carbodiimide- hydrochloride;
HOBT 1 -Hydroxybenzotriazole hydrate;
Et3N Triethylamine;
EtOAc Ethyl acetate;
FAB Fast atom bombardment;
HOOBT 3-Hydroxy-1 ,2,2-benzotriazin-4(3 H)-one;
HPLC High-performance liquid chromatography;
MCPBA m-Chloroperoxybenzoic acid;
MsCl Methanesulfonyl chloride;
NaHMDS Sodium bis(trimethylsilyl)amide;
Py Pyridine;
TFA Trifluoroacetic acid;
THF Tetrahydrofuran.
The compounds are useful in various pharmaceutically acceptable salt forms. The term "pharmaceutically acceptable salt" refers to those salt forms which would be apparent to the pharmaceutical chemist, i.e., those which are substantially non-toxic and which provide the desired pharmacokinetic properties, palatability, absorption, distribution, metabolism or excretion. Other factors, more practical in nature, which are also important in the selection, are cost of the raw materials, ease of crystallization, yield, stability, hygroscopicity and flowability of the resulting bulk drug. Conveniently, pharmaceutical compositions may be prepared from the active ingredients in
combination with pharmaceutically acceptable carriers.
Pharmaceutically acceptable salts include conventional non-toxic salts or quartemary ammonium salts formed, e.g., from non-toxic inorganic or organic acids. Non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like; and the salts
prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic,
methanesulfonic, ethane disulfonic, oxalic, isethionic, trifluoroacetic and the like.
The pharmaceutically acceptable salts of the present invention can be synthesized by conventional chemical methods.
Generally, the salts are prepared by reacting the free base or acid with stoichiometric amounts or with an excess of the desired salt- forming inorganic or organic acid or base, in a suitable solvent or solvent combination.
The farnesyl transferase inhibitors of formula (Il-a) through (II-c) can be synthesized in accordance with Schemes 1 -16, in addition to other standard manipulations such as ester hydrolysis, cleavage of protecting groups, etc., as may be known in the literature or exemplified in the experimental procedures. Substituents Ra and Rb, as shown in the Schemes, represent the substituents R2, R3 , R4 and R5; however their point of attachment to the ring is illustrative only and is not meant to be limiting.
These reactions may be employed in a linear sequence to provide the compounds of the invention or they may be used to synthesize fragments which are subsequently joined by the alkylation reactions described in the Schemes.
Synopsis of Schemes 1 -16:
The requisite intermediates are in some cases commercially available, or can be prepared according to literature procedures, for the most part. In Scheme 1 , for example, the synthesis of 2-alkyl substituted piperazines is outlined, and is essentially that described by J. S. Kiely and S. R. Priebe in Organic Preparations and Proceedings Int., 1990, 22, 761 -768. Boc-protected amino acids I, available commercially or by procedures known to those skilled in the art, can be coupled to N-benzyl amino acid esters using a variety of dehydrating agents such as DCC (dicyclohexycarbodiimide) or EDC·HCl ( 1 -ethyl-3- (3-dimethylaminopropyl)carbodiimide hydrochloride) in a solvent such as methylene chloride , chloroform, dichloroethane, or in dimethyl- formamide. The product II is then deprotected with acid, for example hydrogen chloride in chloroform or ethyl acetate, or trifluoroacetic acid in methylene chloride, and cyclized under weakly basic conditions to give the diketopiperazine III. Reduction of III with lithium aluminum hydride in refluxing ether gives the piperazine IV, which is protected
as the Boc derivative V. The N-benzyl group can be cleaved under standard conditions of hydrogenation, e.g., 10% palladium on carbon at 60 psi hydrogen on a Parr apparatus for 24-48 h. The product VI can be treated with an acid chloride, or a carboxylic acid under standard dehydrating conditions to furnish the carboxamides VII. A final acid deprotection step gives the intermediate VIII (Scheme 2). Intermediate VIII can be reductively alkylated with a variety of aldehydes, such as IX, prepared by standard procedures, such as that described by O. P. Goel, U. Krolls, M. Stier and S. Kesten in Organic Syntheses, 1988, 67, 69-75, from the appropriate amino acid (Scheme 3). The reductive alkylation can be accomplished at pH 5-7 with a variety of reducing agents, such as sodium triacetoxyborohydride or sodium cyanoboro- hydride, in a solvent such as dichloroethane, methanol or dimethyl- formamide. The product X can be deprotected to give the final compounds XI with trifluoroacetic acid in methylene chloride. The final product XI is isolated in the salt form, for example, as a trifluoro- acetate, hydrochloride or acetate salt, among others. The product diamine XI can further be selectively protected to obtain XII, which can subsequently be reductively alkylated with a second aldehyde to obtain XIII. Removal of the protecting group, and conversion to the cyclized product such as the dihydroimidazole XV, can be accomplished by literature procedures.
Alternatively, the protected piperazine intermediate VII can be reductively alkylated with other aldehydes such as 1 -trityl-4- carboxaldehyde or 1 -trityl-4-imidazolylacetaldehyde, to give products such as XVI (Scheme 4) (Tr = trityl). The trityl protecting group can be removed from XVI to give XVII, or alternatively. XVI can first be treated with an alkyl halide then subsequently deprotected to give the alkylated imidazole XVIII. Alternatively, the intermediate VIII can be acylated or sulfonylated by standard techniques. The imidazole acetic acid XIX can be converted to the acetate XXI by standard procedures, and XXI can be first reacted with an alkyl halide, then treated with refluxing methanol to provide the regiospecifically alkylated imidazole acetic acid ester XXII. Hydrolysis and reaction with piperazine VIII in
the presence of condensing reagents such as 1 -(3-dimethylaminopropyl)- 3-ethylcarbodiimide (EDC) leads to acylated products such as XXIV.
If the piperazine VIII is reductively alkylated with an aldehyde which also has a protected hydroxyl group, such as XXV in Scheme 6, the protecting groups can be subsequently removed to unmask the hydroxyl group (Schemes 6, 7). The alcohol can be oxidized under standard conditions to e.g. an aldehyde, which can then be reacted with a variety of organometallic reagents such as Grignard reagents, to obtain secondary alcohols such as XXIX. In addition, the fully deprotected amino alcohol XXX can be reductively alkylated (under conditions described previously) with a variety of aldehydes to obtain secondary amines, such as XXXI (Scheme 7), or tertiary amines.
The protected amino alcohol XXVII can also be utilized to synthesize 2-aziridinylmethylpiperazine.s such as XXXII (Scheme 8). Treating XXVII with 1 ,1 '-sulfonyldiimidazole and sodium hydride in a solvent such as dimethylformamide leads to the formation of aziridine XXXII. The aziridine reacts in the presence of a nucleophile, such as a thiol, in the presence of base to yield the ring-opened product XXXIII.
Piperazine VIII can be reacted with an aldehyde derived from an amino acid, such as an O-alkylated tyrosine, to obtain
compounds such as XXXIX. When R' is an aryl group, XXXIX can first be hydrogenated to unmask the phenol, and the amine group deprotected with acid to produce XL. Alternatively, the amine
protecting group in XXXIX can be removed, and O-alkylated phenolic amines such as XLI produced.
Depending on the identity of the amino acid I, various side chains can be incorporated onto the piperazine. For example, when I is a protected β-benzyl ester of aspartic acid, the intermediate diketo- piperazine XLII (where n=1 and R=benzyl) is obtained, as shown in Scheme 10. Subsequent reduction reduces the ester to the alcohol XLIII, which can then be reacted with a variety of alkylating agents such as an alkyl iodide, under basic conditions, for example, sodium hydride in dimethylformamide or tetrahydrofuran. The resulting ether
XLIV can then be carried on to final products as described in Schemes
3-9.
N-Aryl piperazines can be prepared as described in Scheme
1 1. An aryl amine XLV is reacted with bis -chloroethyl amine hydro- chloride (XLVI) in refluxing n -butanol to furnish compounds XL VII.
The resulting piperazines XLVII can then be carried on to final products as described in Schemes 3-9.
Piperazin-5-ones can be prepared as shown in Scheme 12.
Reductive amination of protected amino aldehydes XLIX (prepared from I as described previously) gives rise to compound L. This is then reacted with bromoacetyl bromide under Schotten-Baumann conditions.
Ring closure is effected with a base, such as sodium hydride, in a polar aprotic solvent, such as dimethylformamide, to give LI. The carbamate protecting group is removed under acidic conditions, such as trifluoro- acetic acid in methylene chloride or hydrogen chloride gas in methanol or ethyl acetate, and the resulting piperazine can then be carried on to final products as described in Schemes 3-9.
The isomeric piperazin-3-ones can be prepared as described in Scheme 13. The imine formed from arylcarboxamides LII and 2- aminoglycinal diethyl acetal (LIll) can be reduced under a variety of conditions, including sodium triacetoxyborohydride in dichloroethane, to give the amine LIV. Amino acids I can be coupled to amines LIV under standard conditions, and the resulting amide LV when treated with aqueous acid in tetrahydrofuran can cyclize to the unsaturated LVI. Catalytic hydrogenation under standard conditions gives the requisite intermediate LVII, which is elaborated to final products as described in Schemes 3-9.
Access to alternatively substituted piperazines is described in Scheme 14. Following deprotection, e.g., with trifluoroacetic acid, the N-benzyl piperazine V can be acylated with an aryl carboxylic acid.
The resulting N-benzyl aryl carboxamide LIX can be hydrogenated in the presence of a catalyst to give the piperazine carboxamide LX which can then be carried on to final products as described in Schemes 3-9.
Reaction Scheme 15 provides an example of the synthesis
of compounds wherein the substituents R2 and R3 are combined to form - (CH2)u -. For example, 1 -aminocyclohexane-1 -carboxylic acid LXI can be converted to the spiropiperazine LXVI essentially according to the procedures outlined in Schemes 1 and 2. The piperazine inter- mediate LXIX can be deprotected as before, and carried on to final products as described in Schemes 3-9. It is understood that reagents utilized to provide the substituent Y which is 2-(naphthyl) and the imidazolylalkyl substituent may be readily replaced by other reagents well known in the art and readily available to provide other N- substituents on the piperazine.
The aldehyde XLIX from Scheme 12 can also be reductively alkylated with an aniline as shown in Scheme 16. The product LXXI can be converted to a piperazinone by acylation with chloroacetyl chloride to give LXXII, followed by base-induced cyclization to LXXIII. Deprotection, followed by reductive alkylation with a protected imidazole carboxaldehyde leads to LXXV, which can be alkylation with an arylmethylhalide to give the imidazolium salt LXXVI. Final removal of protecting groups by either solvolysis with a lower alkyl alcohol, such as methanol, or treatment with triethylsilane in methylene chloride in the presence of trifluoroacetic acid gives the final product LXXVII.
XXXIV
The geranylgeranyl-protein transferase-type I inhibitors and certain of the farnesyl transferase inhibitors can be synthesized in accordance with general Reaction Schemes A-E in addition to other standard manipulations such as ester hydrolysis, cleavage of protecting groups, etc., as may be known in the literature or exemplified in the
experimental procedures. Some key bond-forming and peptide modifying reactions are:
Reaction A Amide bond formation and protecting group cleavage using standard solution or solid phase methodologies.
Reaction B Preparation of a reduced peptide subunit by reductive
alkylation of an amine by an aldehyde using sodium cyanoborohydride or other reducing agents.
Reaction C Alkylation of a reduced peptide subunit with an alkyl or aralkyl halide or, alternatively, reductive alkylation of a reduced peptide subunit with an aldehyde using sodium cyanoborohydride or other reducing agents.
Reaction D Peptide bond formation and protecting group cleavage
using standard solution or solid phase methodologies.
Reaction E Preparation of a reduced subunit by borane reduction of the amide moiety.
Reaction Schemes A-E illustrate bond-forming and peptide modifying reactions incorporating acyclic peptide units. Such reactions are equally useful when the - NHC(RA) - moiety of the reagents and compounds illustrated is replaced with the following moiety:
which can be substituted with R
4a, R
4b, R
7a and R
7b in accordance with structures (ll-d) through (Il-k). These reactions may be employed in a linear sequence to provide the compounds of the invention or they may
be used to synthesize fragments which are subsequently joined by the alkylation reactions described in the Reaction Schemes.
REACTION SCHEME A
Reaction A. Coupling of residues to form an amide bond
ALTERNATIVE REACTION SCHEME A FOR COMPOUNDS (Il-h) THROUGH (Il-k)
Coupling of residues to form an amide bond
REACTION SCHEME B
Preparation of reduced peptide subunits by reductive alkylation
ALTERNATIVE REACTION SCHEME B FOR COMPOUNDS
(Il-h) THROUGH (Il-k)
Preparation of reduced peptide subunits by reductive alkylation
REACTION SCHEME C
Alkylation/reductive alkylation of reduced peptide subunits
where R
A and R
B are R
3 , R
4, R
5a or R
5b as previously defined; R
C is
R6 as previously defined or a carboxylic acid protecting group; XL is a
leaving group, e.g., Br-, I- or MsO-; and Ry is defined such that R7b is generated by the reductive alkylation process.
ALTERNATIVE REACTION SCHEME C for COMPOUNDS
(Il-h) THROUGH (Il-k)
Deprotection of reduced peptide subunits
REACTION SCHEME D
Coupling of residues to form an amide bond
ALTERNATIVE REACTION SCHEME D FOR COMPOUNDS (Il-h) THROUGH (Il-k)
Coupling of residues to form an amide bond
REACTION SCHEME E
Preparation of reduced dipeptides from peptides
ALTERNATIVE REACTION SCHEME E FOR COMPOUNDS
(ll-h) THROUGH (ll-o)
Preparation of reduced dipeptides from peptides
All variables are as defined above.
Certain compounds wherein X-Y is an ethenylene or ethylene unit are prepared by employing the reaction sequences shown in Reaction Schemes F and G. Scheme F outlines the preparation of the alkene isosteres utilizing standard manipulations such as Weinreb amide formation, Grignard reaction, acetylation, ozonolysis, Wittig reaction, ester hydrolysis, peptide coupling reaction, mesylation, cleavage of peptide protecting groups, reductive alkylation, etc., as may be known in the literature or exemplified in the Experimental Procedure. For simplicity, substituents R2a and R2b on the cyclic amine moiety are not shown. It is, however, understood that the reactions illustrated are also applicable to appropriately substituted cyclic amine compounds as well as for acyclic amine moieties. The key reactions are: stereoselective reduction of the Boc-aminoenone to the corresponding syn aminoalcohol (Scheme F, Step B, Part 1 ), and stereospecific boron triflouride or zinc chloride activated organo-magnesio, organo-lithio, or organo-zinc copper(l) cyanide SN 2' displacement reaction (Scheme F, Step G).
Through the use of optically pure N-Boc amino acids as starting material and these two key reactions, the stereochemistry of the final
products is well defined. In Step H of Scheme F, the amino terminus sidechain, designated Rx is incorporated using coupling reaction A and RxCOOH; the alkylation reaction C using RxCHO and a reducing agent; or alkylation reaction C using RxCH2XL. Such reactions as described in Step H are described in more detail in Reaction Schemes J-X hereinbelow.
The alkane analogs are prepared in a similar manner by including an additional catalytic hydrogenation step as outlined in Reaction Scheme G.
REACTION SCHEME F
The oxa isostere compounds of this invention are prepared according to the route outlined in Scheme H. An aminoalcohol 1 is acylated with alpha-chloroacetyl chloride in the presence of trialkyl- amines to yield amide 2. Subsequent reaction of 2 with a deprotonation reagent (e.g., sodium hydride or potassium t-butoxide) in an ethereal solvent such as THF provides morpholinone 3. Alkylation of 3 with R3 XL, where XL is a leaving group such as Br, I- or Cl- in THF/DME (1 ,2-dimethoxyethane) in the presence of a suitable base, preferably NaHMDS [sodium bis(trimethylsilyl)amide], affords 4, which is retreated with NaHMDS followed by either protonation or the addition of an alkyl halide R4χ to give 5a or 5b, respectively, as a enantiomeric mixture. Alternatively, 5a can be prepared from 3 via an aldol condensation approach. Namely, deprotonation of 3 with NaHMDS
followed by the addition of a carbonyl compound RyRzCO gives the adduct 6. Dehydration of 6 can be effected by mesylation and
subsequent elimination catalyzed by DBU (1 ,8-diazabicyclo[5.4.0]undec- 7-ene) or the direct treatment of 6 with phosphorus oxychloride in pyridine to give olefin 7. Then, catalytic hydrogenation of 7 yields 5a (wherein -CHRyRz constitutes R3). Direct hydrolysis of 5 with lithium hydrogen peroxide in aqueous THF, or aqueous HCl, produces acid 8a. Compound 8a is then derivatized with BOC-ON or BOC anhydride to give 8b. The peptide coupling of acid 8b with either an alpha- aminolactone (e.g., homoserine lactone, etc.) or the ester of an amino acid is carried out under the conditions exemplified in the previously described references to yield derivative 9. Treatment of 9 with gaseous hydrogen chloride gives 10, which undergoes further elaboration as described in Reaction Schemes J- hereinbelow.
An alternative method for the preparation of the prolyl oxa isostere (compounds 23 and 24 ) is shown in Scheme H-1.
Referring to Scheme H- 1 , the aminoalcohol 1 is protected with
trifluoroacetic anhydride and the blocked compound 15 treated with diphenyl disulfide in the presence of tributylphosphine to provide the thioether 16. Chlorination of compound 16 provides compound 17 which can be reacted with the appropriate carboxylic acid alcohol in the presence of silver perchlorate and tin (II) chloride, to afford the mixed acetal 18. Removal of the phenylmercapto moiety with Raney nickel provides compound 19. Compound 19 is doubly deprotected, then selectively BOC protected to provide the acid 20, which undergoes the steps previously described for incorporating terminal amino acid. Still another alternative method for the preparation of the prolyl oxa isostere (compounds 23 and 24 ) is described in the literature [Ruth E.
TenBrink, J. Org. Chem., 52, 418-422 (1987)].
The thia, oxothia and dioxothia isostere compounds of this invention are prepared in accordance to the route depicted in Scheme I. Aminoalcohol 1 is derivatized with BOC2O to give 25. Mesylation of 25 followed by reaction with methyl alpha-mcrcaptoacetate in the presence of cesium carbonate gives sulfide 26. Removal of the BOC group in 26 with TFA followed by neutralization with di-isopropyl- ethylamine leads to lactam 27. Sequential alkylation of 27 with the alkyl halides R3X and R4X in THF/DME using NaHDMS as the deprotonation reagent produces 28. Hydrolysis of 28 in hydro-chloride to yield 29a, which is derivatized with Boc anhydride to yield 29b. The coupling of 29b with an alpha-aminolactone (e.g., homoserine lactone, etc.) or the ester of an amino acid is carried out under conventional conditions
as exemplified in the previously described references to afford 30. Sulfide 30 is readily oxidized to sulfone 31 by the use of MCPBA (m-chloroperoxybenzoic acid). The N-BOC group of either 30 or 31 is readily removed by treatment with gaseous hydrogen chloride.
Λ
J
Reaction Schemes J - R illustrate reactions wherein the non- sulfhydryl-containing moiety at the N-terminus of the compounds of the instant invention is attached to the fully elaborated cyclic amino peptide unit, prepared as described in Reaction Schemes A-I. It is understood that the reactions illustrated may also be performed on a simple cyclic
amino acid, which may then be further elaborated utilizing reactions described in Reaction Schemes A- 1 to provide the instant compounds.
The intermediates whose synthesis are illustrated in
Reaction Schemes A-I can be reductively alkylated with a variety of aldehydes, such as V, as shown in Reaction Scheme J. The aldehydes can be prepared by standard procedures, such as that described by O. P. Goel, U. Krolls, M. Stier and S. Kesten in Organic Syntheses, 1988, 67, 69-75, from the appropriate amino acid (Reaction Scheme F). The reductive alkylation can be accomplished at pH 5-7 with a variety of reducing agents, such as sodium triacetoxyborohydride or sodium cyanoborohydride in a solvent such as dichloroefhane, methanol or dimethylformamide. The product VI can be deprotected with
trifluoroacetic acid in methylene chloride to give the final compounds
VII. The final product VII is isolated in the salt form, for example, as a trifluoroacetate, hydrochloride or acetate salt, among others. The product diamine VII can further be selectively protected to obtain
VIII, which can subsequently be reductively alkylated with a second aldehyde to obtain IX. Removal of the protecting group, and conversion to cyclized products such as the dihydroimidazole XI can be accomplished by literature procedures.
Alternatively, the protected cyclic aminopeptidyl intermediate can be reductively alkylated with other aldehydes such as 1 -trityl-4-carboxaldehyde or 1 -trityl-4-imidazolylacetaldehyde, to give products such as XII (Reaction Scheme K). The trityl protecting group can be removed from XII to give XIII, or alternatively, XII can first be treated with an alkyl halide then subsequently deprotected to give the alkylated imidazole XIV. Alternatively, the dipeptidyl analog
intermediate can be acylated or sulfonylated by standard techniques.
The imidazole acetic acid XV can be converted to the protected acetate XVII by standard procedures, and XVII can be first reacted with an alkyl halide, then treated with refluxing methanol to provide the regiospecifically alkylated imidazole acetic acid ester
XVIII. Hydrolysis and reaction with the protected dipeptidyl analog intermediate in the presence of condensing reagents such as 1 -(3-
dimethylaminopropyI)-3-ethylcarbodiimide (EDC) leads to acylated products such as XIX.
If the protected dipeptidyl analog intermediate is
reductively alkylated with an aldehyde which also has a protected hydroxyl group, such as XX in Reaction Scheme N, the protecting groups can be subsequently removed to unmask the hydroxyl group (Reaction Schemes N, P). The alcohol can be oxidized under standard conditions to e.g. an aldehyde, which can then be reacted with a variety of organometallic reagents such as Grignard reagents, to obtain secondary alcohols such as XXIV. In addition, the fully deprotected amino alcohol XXV can be reductively alkylated (under conditions described previously) with a variety of aldehydes to obtain secondary amines, such as XXVI (Reaction Scheme P), or tertiary amines.
The Boc protected amino alcohol XXII can also be utilized to synthesize 2-aziridinylmethylpiperazines such as XXVII (Reaction Scheme Q). Treating XXII with 1,1'-sulfonyldiimidazole and sodium hydride in a solvent such as dimethylformamide led to the formation of aziridine XXVII . The aziridine may be reacted in the presence of a nucleophile, such as a thiol, in the presence of base to yield the ring- opened product XXVIII .
In addition, the protected dipeptidyl analog intermediate can be reacted with aldehydes derived from amino acids such as
O-alkylated tyrosines, according to standard procedures, to obtain compounds such as XXXIV, as shown in Reaction Scheme R. When R' is an aryl group, XXXIV can first be hydrogenated to unmask the phenol, and the amine group deprotected with acid to produce XXXV. Alternatively, the amine protecting group in XXXIV can be removed, and O-alkylated phenolic amines such as XXXVI produced.
w
The intermediates whose synthesis are illustrated in Reaction Schemes A and C can be reductively alkylated with a variety
of aldehydes, such as I, as shown in Reaction Scheme F. The aldehydes can be prepared by standard procedures, such as that described by O. P. Goel, U. Krolls, M. Stier and S. Kesten in Organic Syntheses, 1988, 67, 69-75, from the appropriate amino acid (Reaction Scheme F). The reductive alkylation can be accomplished at pH 5-7 with a variety of reducing agents, such as sodium triacetoxyborohydride or sodium cyanoborohydride in a solvent such as dichloroethane, methanol or dimethylformamide. The product II can be deprotected to give the final compounds III with trifluoroacetic acid in methylene chloride. The final product III is isolated in the salt form, for example, as a trifluoroacetate, hydrochloride or acetate salt, among others. The product diamine III can further be selectively protected to obtain IV, which can subsequently be reductively alkylated with a second aldehyde to obtain V. Removal of the protecting group, and conversion to cyclized products such as the dihydroimidazole VII can be accomplished by literature procedures.
Alternatively, the protected dipeptidyl analog intermediate can be reductively alkylated with other aldehydes such as 1 -trityl-4- carboxaldehyde or 1-trityl-4-imidazolylacetaldehyde, to give products such as VIII (Alternative Reaction Scheme G). The trityl protecting group can be removed from VIII to give IX, or alternatively, VIII can first be treated with an alkyl halide then subsequently deprotected to give the alkylated imidazole X. Alternatively, the dipeptidyl analog intermediate can be acylated or sulfonylated by standard techniques.
The imidazole acetic acid XI can be converted to the acetate XIII by standard procedures, and XIII can be first reacted with an alkyl halide, then treated with refluxing methanol to provide the regiospecifically alkylated imidazole acetic acid ester XIV
(Alternative Reaction Scheme H). Hydrolysis and reaction with the protected dipeptidyl analog intermediate in the presence of condensing reagents such as 1 -(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC) leads to acylated products such as XV.
If the protected dipeptidyl analog intermediate is reductively alkylated with an aldehyde which also has a protected
hydroxyl group, such as XVI in Reaction Scheme I, the protecting groups can be subsequently removed to unmask the hydroxyl group (Reaction Schemes I, J). The alcohol can be oxidized under standard conditions to e.g. an aldehyde, which can then be reacted with a variety of organometallic reagents such as Grignard reagents, to obtain secondary alcohols such as XX. In addition, the fully deprotected amino alcohol XXI can be reductively alkylated (under conditions described previously) with a variety of aldehydes to obtain secondary amines, such as XXII (Reaction Scheme K), or tertiary amines.
The Boc protected amino alcohol XVIII can also be utilized to synthesize 2-aziridinylmethylpiperazines such as XXIII (Reaction Scheme L). Treating XVIII with 1 ,1 '-sulfonyldiimidazole and sodium hydride in a solvent such as dimethylformamide led to the formation of aziridine XXIII . The aziridine reacted in the presence of a nucleophile, such as a thiol, in the presence of base to yield the ring-opened product XXIV .
In addition, the protected dipeptidyl analog intermediate can be reacted with aldehydes derived from amino acids such as
O-alkylated tyrosines, according to standard procedures, to obtain compounds such as XXX, as shown in Reaction Scheme M. When R' is an aryl group, XXX can first be hydrogenated to unmask the phenol, and the amine group deprotected with acid to produce XXXI. Alternatively, the amine protecting group in XXX can be removed, and O-alkylated phenolic amines such as XXXII produced.
Similar procedures as are illustrated in Reaction Schemes
F-M may be employed using other peptidyl analog intermediates such as those whose synthesis is illustrated in Reaction Schemes B - E.
X
)
Certain compounds used in the invention are described below.
EXAMPLES
Examples provided are intended to assist in a further understanding of the invention. Partivular materials employed, species and conditions are intended to be further illustrative of the invention and not limitative of the reasonable scope thereof.
The standard workup referred to in the examples refers to solcent extraction and washing the organic solution with 10% citric acid, 10% sodium bicarbonate and brine as appropriate. Sllutions were dried over sodium sulfate and eveporated in vacuo on a rotary evaporator.
EXAMPLE 1
(S)-1-(3-chlorophenyI)-4-[ 1-(4-cyanobenzyl)-imidazolylmethyl]-5-[2- (methanesulfonyl)ethyl ]-2-piperazinone dihydrochloride
Step A: 1 -triphenylmethyl-4-(hydroxymethyl)-imidazole
To a solution of 4-(hydroxymethyl)imidazole hydrochloride (35.0 g, 260 mmol) in 250 mL of dry DMF at room temperature was added triethylamine (90.6 mL, 650 mmol). A white solid precipitated from the solution. Chlorotriphenylmethane (76.1 g, 273 mmol) in 500 mL of DMF was added dropwise. The reaction mixture was stirred for 20 hours, poured over ice, filtered, and washed with ice water. The resulting product was slurried with cold dioxane, filtered, and dried in vacuo to provide the titled product as a white solid which was sufficiently pure for use in the next step.
Step B: 1 -triphenylmethyl-4-(acetoxymethyl)-imidazole
Alcohol from Step A (260 mmol, prepared above) was suspended in 500 mL of pyridine. Acetic anhydride (74 mL, 780 mmol) was added dropwise, and the reaction was stirred for 48 hours
during which it became homogeneous. The solution was poured into 2 L of EtOAc, washed with water (3 x 1 L), 5% aq. HCl soln. (2 x 1 L), sat. aq. NaHCO3, and brine, then dried (Na2SO4), filtered, and concentrated in vacuo to provide the crude product. The acetate was isolated as a white powder which was sufficiently pure for use in the next reaction.
Step C: 1-(4-cyanobenzyl)-5-(acetoxymethyl)-imidazole
hydrobromide
A solution of the product from Step B (85.8 g, 225 mmol) and α-bromo-p-tolunitrile (50.1 g, 232 mmol) in 500 mL of EtOAc was stirred at 60°C for 20 hours, during which a pale yellow precipitate formed. The reaction was cooled to room temperature and filtered to provide the solid imidazolium bromide salt. The filtrate was concen- trated in vacuo to a volume 200 mL, reheated at 60°C for two hours, cooled to room temperature, and filtered again. The filtrate was concentrated in vacuo to a volume 100 mL, reheated at 60°C for another two hours, cooled to room temperature, and concentrated in vacuo to provide a pale yellow solid. All of the solid material was combined, dissolved in 500 mL of methanol, and warmed to 60°C. After two hours, the solution was reconcentrated in vacuo to provide a white solid which was triturated with hexane to remove soluble materials. Removal of residual solvents in vacua provided the titled product hydrobromide as a white solid which was used in the next step without further purification.
Step D: 1 -(4-cyanobenzyl)-5-(hydroxymethyl)-imidazole
To a solution of the acetate from Step C (50.4 g, 150 mmol) in 1.5 L of 3: 1 THF/water at 0°C was added lithium hydroxide monohydrate (18.9 g, 450 mmol). After one hour, the reaction was concentrated in vacuo, diluted with EtOAc (3 L), and washed with water, sat. aq. NaHCO3 and brine. The solution was then dried
(Na2SO4), filtered, and concentrated in vacua to provide the crude
product as a pale yellow fluffy solid which was sufficiently pure for use in the next step without further purification.
Step E: 1 -(4-cyanobenzyl)-5-imidazolecarboxaldehyde
To a solution of the alcohol from Step D (21.5 g, 101 mmol) in 500 mL of DMSO at room temperature was added triethyl- amine (56 mL, 402 mmol), then SO3-pyridine complex (40.5 g, 254 mmol). After 45 minutes, the reaction was poured into 2.5 L of EtOAc, washed with water (4 x 1 L) and brine, dried (Na2SO4), filtered, and concentrated in vacuo to provide the aldehyde as a white powder which was sufficiently pure for use in the next step without further
purification.
Step F: (S)-2-(tert-butoxycarbonylamino)-N-methoxy-N-methyl-4- (methylthio)butanamide
L-N -Boc-methionine (30.0 g, 0.120 mol), N,O- dimethylhydroxylamine hydrochloride (14.1 g, 0.144 mol), EDC hydrochloride (27.7 g, 0.144 mol) and HOBT (19.5 g, 0.144 mol) were stirred in dry DMF (300 mL) at 20°C under nitrogen. More N,O- dimethylhydroxylamine hydrochloride (2.3 g, 23 mmol) was added to obtain pH 7-8. The reaction was stirred overnight, the DMF distilled to half the original volume under high vacuum, and the residue partitioned between ethyl acetate and sat. NaHCO3 soln. The organic phase was washed with saturated sodium bicarbonate, water, 10% citric acid, and brine, and dried with sodium sulfate. The solvent was removed in vacua to give the title compound.
Step G: (S)-2-(tert-butoxycarbonylamino)-4-(methylthio)butanal
A suspension of lithium aluminum hydride (5.02 g, 0.132 mol) in ether (500 mL) was stirred at room temperature for one hour. The solution was cooled to -50°C under nitrogen, and a solution of the product from Step F (39.8 g, ca. 0.120 mol) in ether (200 mL) was added over 30 min, maintaining the temperature below -40°C. When the addition was complete, the reaction was warmed to 5°C, then
recooled to -45°C. Analysis by tic revealed incomplete reaction. The solution was rewarmed to 5 °C, stirred for 30 minutes, then cooled to -50°C. A solution of potassium hydrogen sulfate (72 g, 0.529 mol) in 200 mL water was slowly added, maintaining the temperature below -20°C. The mixture was wasmed to 5 °C, filtered through Celite, and concentrated in vacuo to provide the title aldehyde.
Step H: (S)-2-(tert-butoxycarbonylamino)-N-(3-chlorophenyl)-4- (methylthio)butanamine
To a solution of 3-chloroaniline ( 10.3 mL, 97.4 mmol), the product from Step G (23.9 g, 97.4 mmol), and acetic acid (27.8 mL, 487 mmol) in dichloroethane (250 mL) under nitrogen was added sodium triacetoxyborohydride (41.3 g, 195 mmol). The reaction was stirred overnight, then quenched with saturated sodium bicarbonate solution. The solution was diluted with CHCl3, and the organic phase was washed with water, 10% citric acid and brine. The solution was dried over sodium sulfate and concentrated in vacua to provide the crude product (34.8 g) which was chromatographed on silica gel with 20% ethyl acetate in hexane to obtain the title compound .
Step I: (S)-4-(tert-butoxycarbonyl)- 1 -(3-chlorophenyl)-5-12-
(methylthio)ethyl]piperazin-2-one
A solution of the product from Step H (22.0 g, 63.8 mmol) in ethyl acetate (150 mL) was vigorously stirred at 0°C with saturated sodium bicarbonate (150 mL). Chloroacetyl chloride (5.6 mL, 70.2 mmol) was added dropwise, and the reaction stirred at 0°C for 2h. The layers were separated, and the ethyl acetate phase was washed with 10% citric acid and saturated brine, and dried over sodium sulfate. After concentration in vacua, the resulting crude product (27.6 g) was dissolved in DMF (300 mL) and cooled to 0°C under argon. Cesium carbonate (63.9 g, 196 mmol) was added, and the reaction was stirred for two days, allowing it to warm to room temperature. Another portion of cesium carbonate (10 g, 30 mmol) was added, and the reaction was stirred for 16 hours. The DMF was distilled in vacua, and
the residue partitioned between ethyl acetate and water. The organic phase was washed with saturated brine, and dried over sodium sulfate. The crude product was chromatographed on silica gel with 20-25% ethyl acetate in hexane to obtain the title compound.
Step J: (S)-4-(tert-butoxycarbonyl)-1 -(3-chlorophenyl)-5-[2-
(methanesulfonyl)ethyl]piperazin-2-one
A solution of the product from Step I (14.2 g, 37 mmol) in methanol (300 mL) was cooled to 0 °C, and a solution of magnesium monoperoxyphthalate (54.9 g, 1 1 1 mmol) in 210 mL MeOH was added over 20 minutes. The ice bath was removed, and the solution was allowed to warm to room temperature. After 45 minutes, the reaction was concentrated in vacuo to half the original volume, then quenched by the addition of 2N Na2S2O3 soln. The solution was poured into EtOAc and sat NaHCO3 solution, and the organic layer was washed with brine, dried (Na2SO4), filtered, and concentrated in vacuo to provide the crude sulfone. This material was chromatographed on silica gel with 60-100% ethyl acetate in hexane to obtain the titled compound. Step K: (S)- 1 -(3-chlorophenyl)-5-[ 2-
(methanesulfonyl)ethyl]piperazin-2-one
Through a solution of Boc-protected piperazinone from Step J (1.39 g, 3.33 mmol) in 30 mL of EtOAc at 0 °C was bubbled anhydrous HCl gas. The saturated solution was stirred for 35 minutes, then concentrated in vacuo to provide the hydrochloride salt as a white powder. This material was suspended in EtOAc and treated with dilute aqueous NaHCO3 solution. The aqueous phase was extracted with EtOAc, and the combined organic mixture was washed with brine, dried (Na2SO4), filtered, and concentrated in vacua. The resulting amine was reconcentrated from toluene to provide the titled material suitable for use in the next step.
Step L: (S)-1-(3-chlorophenyl)-4-[ 1 -(4- cyanobenzyl)imidazolylmethyl]-5-[2-(methanesulfonyl)- ethyl]-2-piperazinone dihydrochloride
To a solution of the amine from Step K (898 mg, 2.83 mmol) and imidazole carboxaldehyde from Step E (897 mg, 4.25 mmol) in 15 mL of 1 ,2-dichloroethane was added sodium triacetoxyboro- hydride (1.21 g, 5.7 mmol). The reaction was stirred for 23 hours, then quenched at 0 °C with sat. NaHCO3 solution. The solution was poured into CHCI3 , and the aqueous layer was back-extracted with CHCl3. The combined organics were washed with brine, dried
(Na2SO4), filtered, and concentrated in vacua. The resulting product was purified by silica gel chromatography (95:5:0.5-90:10:0
EtOAc: MeOH:NH4CI), and the resultant product was taken up in
EtOAc/methanol and treated with 2.1 equivalents of 1 M HCl/ether solution. After concentrated in vacua, the product dihydrochloride was isolated as a white powder.
EXAMPLE 2 1 -(3-chIorophenyl)-4-[ 1 -(4-cyanobenzyl)imidazolyl-mefhyl]-2- piperazinone dihydrochloride
Step A: N-(3-chlorophenyl)ethylenediamine hydrochloride
To a solution of 3-chloroaniline (30.0 mL, 284 mmol) in 500 mL of dichloromethane at 0 °C was added dropwise a solution of 4 N HCl in 1,4-dioxane (80 mL, 320 mmol HCl). The solution was warmed to room temperature, then concentrated to dryness in vacua to provide a white powder. A mixture of this powder with 2- oxazolidinone (24.6 g, 282 mmol) was heated under nitrogen
atmosphere at 160 °C for 10 hours, during which the solids melted, and gas evolution was observed. The reaction was allowed to cool, forming the crude diamine hydrochloride salt as a pale brown solid.
Step B: N-(tert-butoxycarbonyl)-N'-(3- chlorophenyl)ethylenediamine
The amine hydrochloride from Step A (ca. 282 mmol, crude material prepared above) was taken up in 500 mL of THF and 500 mL of sat. aq. NaHCO3 soln., cooled to 0°C, and di-tert- butylpyrocarbonate (61.6 g, 282 mmol) was added. After 30 h, the reaction was poured into EtOAc, washed with water and brine, dried (Na2SO4), filtered, and concentrated in vacua to provide the titled carbamate as a brown oil which was used in the next step without further purification.
Step C: N -[2-(tert-butoxycarbamoyl)ethyl]-N-(3-chlorophenyl)-
2-chloroacetamide
A solution of the product from Step B (77 g, ca. 282 mmol) and triethylamine (67 mL, 480 mmol) in 500 mL of CH2CI2 was cooled to 0°C. Chloroacetyl chloride (25.5 mL, 320 mmol) was added dropwise, and the reaction was maintained at 0°C with stirring. After 3 h, another portion of chloroacetyl chloride (3.0 mL) was added dropwise. After 30 min, the reaction was poured into EtOAc (2 L) and washed with water, sat. aq. NH4CI soln, sat. aq. NaHCO3 soln., and brine. The solution was dried (Na2SO4), filtered, and concentrated in vacua to provide the chloroacetamide as a brown oil which was used in the next step without further purification. Step D: 4-(tert-butoxycarbonyl)-1 -(3-chlorophenyl)-2- piperazinone
To a solution of the chloroacetamide from Step C (ca. 282 mmol) in 700 mL of dry DMF was added K2CO3 (88 g, 0.64 mol). The solution was heated in an oil bath at 70-75 °C for 20 hours, cooled to room temperature, and concentrated in vacua to remove ca. 500 mL of DMF. The remaining material was poured into 33% EtOAc/hexane, washed with water and brine, dried (Na2SO4), filtered, and concentrated in vacuo to provide the product as a brown oil. This material was purified by silica gel chromatography (25-50% EtOAc/hexane) to yield
pure product, along with a sample of product (ca. 65% pure by HPLC) containing a less polar impurity.
Step E: 1 -(3-chlorophenyl)-2-piperazinone
Through a solution of Boc-protected piperazinone from Step D (17.19 g, 55.4 mmol) in 500 mL of EtOAc at -78°C was bubbled anhydrous HCl gas. The saturated solution was warmed to 0°C, and stirred for 12 hours. Nitrogen gas was bubbled through the reaction to remove excess HCl, and the mixture was warmed to room temperature. The solution was concentrated in vacua to provide the hydrochloride as a white powder. This material was taken up in 300 mL of CH2CI2 and treated with dilute aqueous NaHCO3 solution. The aqueous phase was extracted with CH2CI2 (8 x 300 mL) until tic analysis indicated complete extraction. The combined organic mixture was dried
(Na2SO4), filtered, and concentrated in vacuo to provide the titled free amine as a pale brown oil. Step F: 1 -(3-chlorophenyl)-4-[ 1 -(4-cyanobenzyl)imidazolylmethyl]-
2-piperazinone dihydrochloride
To a solution of the amine from Step E (55.4 mmol, prepared above) in 200 mL of 1 ,2-dichloroethane at 0°C was added 4 Å powdered molecular sieves (10 g), followed by sodium triacetoxyboro- hydride (17.7 g, 83.3 mmol). The imidazole carboxaldehyde from Step E of Example 1 (1 1.9 g, 56.4 mmol) was added, and the reaction was stirred at 0°C. After 26 hours, the reaction was poured into EtOAc, washed with dilute aq. NaHCO3, and the aqueous layer was back- extracted with EtOAc. The combined organics were washed with brine, dried (Na2SO4), filtered, and concentrated in vacua. The resulting product was taken up in 500 mL of 5: 1 benzene:CH2Cl2, and propyl- amine (20 mL) was added. The mixture was stirred for 12 hours, then concentrated in vacuo to afford a pale yellow foam. This material was purified by silica gel chromatography (2-7% MeOH/CH2Cl2), and the resultant white foam was taken up in CH2CI2 and treated with 2.1 equivalents of 1 M HCl/ether solution. After concentrated in vacua, the product dihydrochloride was isolated as a white powder.
N-[ 1 -(1H-Imidazol-4-propionyl) pyrrolidin-2(S)-ylmethyl]-N-(2- methoxybenzyl)glycyl-methionine isopropyl ester
Step A: 2-Methoxybenzylglycine methyl ester
2-Methoxybenzyl alcohol (53.5 g, 0.39 mol) was dissolved in CH2CI2 (200 mL), treated with diisopropylethylamine (81 mL, 0.74 mol), cooled to 0°C. with stirring in an ice-CH3OH bath under Ar, and treated dropwise with methanesulfonyl chloride (33 mL, 0.43 mol). After 0.5 hr, the reaction mixture was left to warm to ambient temperature and stirred for 4 hr. This solution and diisopropylethylamine (202.5 mL, 1.16 mol) were added alternately portionwise with to a slurry of glycine methyl ester hydrochloride ( 146.5 g, 1.17 mol) in DMF (250 mL) with stirring at 0°C. The reaction mixture was left to stir and warm to room temperature overnight. The DMF was removed under reduced pressure, and the residue was partitioned between EtOAc (1 L) and satd NaHCO3 solution (1 L). The aqueous layer was washed with EtOAc (2 x 600 mL), the organics combined, washed with brine and dried (MgSO4). Filtration and concentration to dryness gave the title compound after chromatography (SiO2, 1 -5% CH3OH/CH2CI2).
Step B: N-[(2S)-(t-Butoxycarbonylpyrrolidinyl-methyl)-N-(2- mefhoxybenzyl)glycine methyl ester
2-Methoxybenzylglycine methyl ester (27.4 g, 0.131 mol) was dissolved in 1 ,2-dichIoroethane (500 ml), 3 Å molecular sieves (20 g) were added, and the pH of the reaction mixture adjusted to pH 5 with acetic acid (7.5 mL, 0.131 mol). N-(t-Butoxycarbonyl)-L-prolinal
(26.1 g, 0.131 mol) (J. Org. Chem. (1994) 59, [21 ], 6287-95) was added followed by sodium triacetoxyborohydride (33.2 g, 0.157 mol). The mixture was stirred at ambient temperature for 18 h, filtered through celite and concentrated. The residue was partitioned between EtOAc and sat. NaHCO3 (500 ml/100 ml). The aqueous layer was washed with EtOAc (3x100 ml). The organic layers were combined, dried with Na2SO4, filtered, and concentrated to give the title
compound. Step C: N-[(2S)-(t-Butoxycarbonylpyrrolidinyl-methyl)-N-(2- methoxybenzyl)glycine
N-[(2S)-(t-Butoxycarbonylpyrrolidinylmethyl)-N-(2- methoxybenzyl)glycine methyl ester (7.0 g, 0.018 mol) was dissolved in CH3OH (150 ml) and 1N NaOH (71 ml, 0.071 mol) was added with cooling in an ice-water bath. The mixture was stirred at ambient temperature for 2 hr, neutralized with 1N HCl (71 ml, 0.071 mol), concentrated to remove the CH3OH, and the residue extracted with EtOAc (3x200 mL). The organic layers were combined, dried with Mg2SO4, filtered, and concentrated to give the title compound as a foam.
Step D: Methionine isopropyl ester hydrochloride
N-(t-Butoxycarbonyl)methionine (25 g, 0.1 mol), isopropyl alcohol (1 1.8 mL, 0.15 mol), EDC (21.1 g, 0.1 1 mol), and 4-dimethyl- aminopyridine (DMAP) (1.22 g, 0.01 mol) were dissolved in CH2CI2 (400 mL) with stirring in an ice-water bath. After 2 h the bath was removed, and the solution was left to stir o.n. at RT. The reaction mixture was concentrated to dryness, then partitioned between EtOAc and H2O, the aqueous layer washed with EtOAc (2 x 50 mL), the organics combined, washed with NaHCO3 soln, brine, and dried
(Na2SO4). Filtration and concentration to dryness gave a yellow oil after chromatography (flash silica gel column, hexane: EtOAc, 6: 1 to 5: 1 ).
N-(t-Butoxycarbonyl)methionine isopropyl ester (20.5 g, 0.07 mol) was dissolved in EtOAc (200 mL) with stirring and cooling to -20°C in a dry ice- acetone bath. HCl gas was bubbled into the solution for 10 min, the flask was stoppered and stirred for 1 h. Tic (EtOAc: hexane, 1 :3) indicates loss of starting material. Argon was bubbled through the soln for 5 min, then it was concentrated to dryness to give the title compound as a white solid.
Step E: N-[ (2S)-(t-Butoxycarbonylpyrrolidinyl-methyI)-N-(2- methoxybenzyl)glycyl-methionine isopropyl ester
N-[(2S)-(t-Butoxycarbonylpyrrolidinylmethyl)-N-(2- methoxybenzyl)glycine (from step C) (5.98 g, 0.0158 mol), dissolved in CH2Cl2 (100 mL), was treated with HOBT (2.57 g, 0.019 mol), EDC (4.54 g, 0.024 mol), and methionine isopropyl ester hydrochloride (4.33 g, 0.019 mol). The pH was adjusted to 7.5 with Et3N (8.81 mL, 0.063 mol) and the mixture was stirred at ambient temperature for 18 h. The mixture was diluted with EtOAc (150 mL) and washed sequentially with 10% citric. acid soln, saturated NaHCO3 solution, brine, and dried (MgSO4). Filtration and concentration to dryness gave the title compound as a thick oil. This was used without further purification.
Step F: N-((2S)-Pyrrolidinylmethyl)-N-(2-methoxybenzyl)- glycyl-methionine isopropyl ester bis hydrochloride
N-[(2S)-(t-ButoxycarbonylpyrroIidinylmethyl)-N-(2- methoxybenzyl)glycyl-methionine isopropyl ester (0.85 g, 1.54 mmol) was dissolved in EtOAc (25 mL) and cooled to 0°C. HCl was bubbled through the mixture until the soln was saturated, and it was stoppered and stirred for 3 hr. Argon was bubbled through the mixture to remove excess HCl and the mixture was then concentrated to give the title compound.
Step G: N-[1 -(1 H-Imidaaol-4-propionyl) pyrrolidin-2(S)- yImethyl]-N-(2-methoxybenzyl)glycyl-methionine
isopropyl ester
N-((2S)-Pyrrolidinylmethyl)-N-(2-methoxybenzyI)glycyl methionine isopropyl ester bis hydrochloride (0.800 g, 1.53 mmol), dissolved in DMF (30 mL), was treated with 1H-imidazol-4-propionic acid (0.43 g, 3.05 mmol) (prepared by catalytic hydrogenation of urocanic acid in 20% acetic acid with Pd on carbon), and BOP reagent (1.35 g, 3.05 mmol). The pH was adjusted to 7.5 with N-methyl- morpholine (0.756 mL, 6.89 mmol), and the mixture was stirred at ambient temperature for 18 h. The mixture was concentrated to dryness, diluted with EtOAc (100 mL), washed with 5% Na2CO3 solution, brine, and dried (MgSO4). Filtration and concentration to dryness gave an oil which was purified by chromatography (silica gel, 95:5 CH2Cl2/MeOH) to give the title compound as a foam.
1 H NMR (CD3OD); δ 7.58 (d, 1H, J=1 Hz), 7.25-7.31 (m, 2H), 6.89- 7.00 (m, 2H), 6.81 (s, 1H), 5.00-5.06 (m, 1 H), 4.49-4.56 (m, 1 H), 4.23- 4.30 (m, 1H), 3.91 (d, 1 H, J=13 Hz), 3.86 (s, 3H), 3.54 (d, 1 H, J=13Hz), 3.30-3.41 (m, 2H), 3.36 (d, 1 H, J= 17 Hz), 3.15 (d, 1 H, J= 17 Hz), 2.85- 2.92 (m, 2H), 2.56-2.77 (m, 3H), 2.30-2.45 (m, 3H), 2.05-2.17 (m, 1 H), 2.04 (s, 3H), 1.69-1.86 (m, 5H), 1.24 (d, 6H, J=6 Hz).
Anal, calculated for C29H43N5O5S• 0.6 H2O:
C, 59.59; H, 7.62; N, 1 1 .98;
Found: C, 59.58; H, 7.43; N, 12.02.
EXAMPLE 4
(N-[ 1 -Cyanobenzyl)- 1H-imidazol-5-yl)acetyl]pyrrolidin-2(S)-ylmethyl]- 3(S)-ethyl-prolyl methionine isopropyl ester
Step A: Diethyl 1-Acetyl-5-hydroxy-3-ethylpyrrolidine-2,2- dicarboxylate
Sodium (4.02 g, 0.175 mol) was dissolved in a stirred solution of diethyl acetamidomalonate (235.4 g, 1 .19 mol) in abs
EtOH (1.4 L) at ambient temperature under argon. The reaction mixture was cooled to 0°C, and trans-2-pentenal (100 g, 1.08 mol) was added dropwise maintaining the reaction temperature at <5°C.
After the addition, the reaction was allowed to warm to room
temperature, stirred for 4 h, then quenched with acetic acid (28 mL). The solution was concentrated in vacua, and the residue dissolved in EtOAc (1.5 L), washed with 10% NaHCO3 solution (2 x 300 mL), brine, and dried (MgSO4). The solution was filtered and concentrated to 700 mL, then heated to reflux and treated with hexane (1 L). On cooling, the title compound precipitated and was collected, mp 106 - 109°C. 1 H NMR (CD3OD) δ 5.65 (d, 1 H, J= 5 Hz), 4.1 - 4.25 (m, 4H), 2.7-2.8 (m, 1 H), 2.21 (s, 3H), 2.10 (dd, 1H, J = 6, 13, Hz), 1.86- 1.97 (m, 2H), 1.27 (t, 3H, J= 7 Hz), 1.23 (t, 3H, J= 7 Hz), 1.1 - 1.25 (m, 1H), 0.97 (t, 3H, J= 7 Hz).
Step B: Diethyl 1 - Acetyl-3-ethylpyrrolidine-2.2-dicarboxylate
To a solution of diethyl 1 -acetyl-5-hydroxy-3-ethyl- pyrrolidine-2,2-dicarboxylate (287 g, 0.95 mol) and triethylsilane (228 mL, 1.43 mol) in CH2CI2 (3 L) under argon was added trifluoroacetic acid (735 mL, 9.53 mol) dropwise with stirring while maintaining the internal temperature at 25 °C by means of an ice bath. After stirring for 3 h at 23 °C, the solution was concentrated in vacua, , the residue diluted with CH2CI2 (1.5 L), then treated with H2O ( 1 L) and solid Na2CO3 with vigorous stirring until the solution was basic. The organic layer was separated, dried (Na2SO4), filtered, then concentrated to give the title compound as a yellow oil which was used without further purification.
Step C: 3-Ethylproline hydrochloride (Cis:Trans Mixture)
Diethyl 1-acetyl-3-ethylpyrrolidine-2,2-dicarboxylate
(373 g, 0.95 mol) was suspended in 6N HCl (2 L) and HO Ac (500 mL) and heated at reflux for 20 h. The reaction mixture was cooled, washed with EtOAc (1L), then concentrated in vacua to give an oil which crystallized upon trituration with ether to give the title compound.
1H NMR (D2O) δ 4.23 (d, 1H, J= 8 Hz), 3.84 (d, 1 H, J= 8 Hz), 3.15- 3.4 (m, 4H), 2.33- 2.44 (m, 1 H), 2.19-2.4 (m, 1H), 2.02- 2.15 (m, 2H), 1.53- 1.72 (m, 3H), 1.23- 1.43 (m, 2H), 1.0- 1.15 (m, 1H), 0.75 - 0.83 (m, 6H). Step D: N-[(tert-Butyloxy)carbonyl]-cis:trans-3-ethylproline methyl ester
3-Ethylproline hydrochloride (Cis:Trans Mixture) (20 g, 0.1 1 mol) was dissolved in CH3OH (200 mL), and the solution was saturated with HCl gas, then stirred at 23°C for 24 h. Argon was bubbled through the solution to remove excess HCl. The solution was treated with NaHCO3 (>84 g) to a pH of 8, then di-tert-butyl dicarbonate (25.1 g, 0.1 15 mol) dissolved in CH3OH (20 mL) was
added slowly. After stirring for 18 h at 23°C, the mixture was filtered, the filtrate concentrated, and the residue triturated with EtOAc, filtered again, and concentrated to give the title compound as an oil.
Step E: N-[(tert-Butyloxy)carbonyl]-trans-3-ethylproline and N- [(tert-Butyloxy)carbonyl]-cis-3-ethylproline methyl este N-[(tert-Butyloxy)carbonyl]-cis,trans-3-ethylproline methyl ester (29.1 g, 0.1 13 mol) was dissolved in CH3OH (1 14 mL) with cooling to 0°C, then treated with 1 N NaOH (1 14 mL). After stirring for 20 h at 23°C, the solution was concentrated to remove the CH3OH and then extracted with EtOAc (3 x). The organic layers were
combined, dried (MgSO4), filtered, and concentrated to give 12.8 g of N-[(tert-Butyloxy)carbonyl]-cis-3-ethylproline methyl ester as an oil. The aqueous layer was acidified with solid citric acid and extracted with EtOAc (2 x), the organic layers combined, dried (MgSO4), filtered, and concentrated to give N- [(tert-Butyloxy)carbonyl]-trans-3-ethylproline as an oil. 1H NMR (CD3OD) δ 3.86 and 3.78 (2 d, 1H, J = 6 Hz), 3.33 - 3.58 (m, 2H), 2.01 - 2.22 (m, 2H), 1.5 - 1 .74 (m, 2H), 1.33 - 1.5 (m, 1 H), 1.45 and 1.42 (2 s, 9H), 0.98 (t, 3H, J= 8 Hz).
Step F: 3(S)-Ethyl-2(S)-proline hydrochloride
N- [(tert-Butyloxy)carbonyl]-trans-3-ethylproline (15.5 g, 0.064 mol), S-α-methylbenzylamine (9.03 mL, 0.070 mol), HOBT (10.73 g, 0.70 mol), and N-methylmorpholine (8 mL, 0.076 mol) were dissolved in CH2CI2 (150 mL) with sitrring in an ice-H2O bath, treated with EDC (13.4 g, 0.070 mol) stirred at 23°C for 48 h. The reaction mixture was partitioned between EtOAc and 10% citric acid solution, the organic layer washed with satd NaHCO3 solution, brine and dried (MgSO4), filtered, and concentrated to give an oil. This oil was dissolved in a minimum amount of ether (10 mL) to crystallize the desired S,S,S diastereomer (4.2 g), mp 1 18-121 °C. A solution of this product in 8N HCl (87 mL) and glacial acetic acid (22 mL) was heated at reflux overnight. The solution was concentrated on a rotary
evaporator, and the residue taken up in H2O and extracted with ether. The aqueous layer was concentrated to dryness to give a 1 : 1 mixture of 3(S)-ethyl-2(S)-proline hydrochloride and α-methylbenzylamine.
3(S)-Ethyl-2(S)-proline containing α-methylbenzylamine (2.0 g, 0.0128 mol) was dissolved in dioxane (10 mL) and H2O (10 mL) with stirring and cooling to 0°C. N,N-diisopropylethylamine (2.2 mL, 0.0128 mol) and di-tert-butyl-dicarbonate (2.79 g, 0.0128 mol) were added and stirring was continued at 23°C for 48 h. The reaction mixture was partitioned between EtOAc (60 mL) and H2O (30 mL), the organic layer washed with 0.5N NaOH (2 x 40 mL), the aqueous layers combined and washed with EtOAc ( 30 mL) and this layer back extracted with 0.5 N NaOH (30 mL). The aqueous layers were combined and carefully acidified at 0°C with 1 N HCl to pH 3. This mixture was extracted with EtOAc (3 x 40 mL), the organics combined, dried (MgSO4), filtered and concentrated to give N-[(tert-Butyloxy) carbonyl-3(S)-ethyl-2(S)-proline as a colorless oil. N-[(tert -Butyloxy) carbonyl-3(S)-ethyl-2(S)-proline was dissolved in EtOAc (50 mL) and the solution was saturated with HCl gas with cooling in an ice-H2O bath. The solution was stoppered and stirred at 0°C. for 3 hr. Argon was bubbled through the solution to remove excess HCl, and the solution was concentrated to dryness to give 3(S)-ethyl-2(S)-proline hydrochloride.
Step G: N-(t-Butyloxycarbonyl)-pyrrolidin-2(S)-ylmethyl]-3(S)- ethyl-prollne
3(S)-Ethyl-2(S)-proline hydrochloride (2.33 g, 0.013 mol) was dissolved in CH3OH (20 mL), treated with 3A molecular sieves (2 g) and KOAc ( 1.27 g, 0.013 mol) to adjust the pH of the reaction mixture to 4.5-5, then N-[(tert-Butyloxy)carbonyl-prolinal (Pettit et al., J. Org. Chem. (1994) 59, [21 ] 6287-95) (3.36 g, 0.017 mol) was added, and the mixture was stirred for 16 hrs at room temperature. The reaction mixture was filtered, quenched with aq satd NaHCO3 (5 mL) and concentrated to dryness. The residue was extracted with CHCl3. The extract was dried (MgSO4), filtered, and concentrated to give the title compound and inorganic salts.
Step H: N-(t-Butyloxycarbonyl)-pyrrolidin-2(S)-ylmethyl]-3(S)- ethyl-prolyl methionine isopropyl ester
N-(t-Butyloxycarbonyl)-pyrrolidin-2(S)-ylmethyl]-3(S)- ethyl-proline (2.4 g, 0.008 mol), methionine isopropyl ester hydrochloride (2.21 g, 0.0097 mol), HOBT ( 1.49 g, 0.0097 mol) and EDC (1.86 g, 0.0097 mol) were dissolved in DMF ( 15 mL) at room
temperature and treated with N-methylmorpholine (3 mL, 0.024 mol). The reaction mixture was stirred overnight at room temperature, then concentrated and partitioned between EtOAc and H2O. The organic layer was washed with aq satd NaHCO3 solution, brine, and dried (MgSO4). The crude product was chromatographed on a flash silica gel column eluting with hexane: EtOAc, 7:3 to give N-(t- butyloxycarbonyl)-pyrrolidin-2(S)-ylmethyl]-3(S)-ethyl-prolyl methionine isopropyl ester.
Step I: Pyrrolidin-2(S)-ylmethyl]-3(S)-ethyl-prolyl methionine isopropyl ester hydrochloride
N-(t-butyloxycarbonyl)-pyrrolidin-2(S)-ylmethyl]-3(S)- ethyl-prolyl methionine isopropyl ester (1.38 g, 0.0028 mol) was dissolved in EtOAc (40 mL), cooled to -20°C, saturated with HCl gas, and stirred at 0°C. for 1.25 hr, and room temperature for 0.25 hr.
Concentration to dryness gave pyrrolidin-2(S)-ylmethyl]-3(S)-ethyl- prolyl methionine isopropyl ester hydrochloride.
Step J: Preparation of 1H-Imidazole-4- acetic acid methyl ester hydrochloride
A solution of 1H-imidazole-4-acetic acid hydrochloride
(4.00g, 24.6 mmol) in methanol (100 ml) was saturated with gaseous hydrogen chloride. The resulting solution was allowed to stand at room temperature (RT) for 18hr. The solvent was evaporated in vacuo to afford the title compound as a white solid.
1 H NMR(CDCl3, 400 MHz) δ 8.85(1H, s),7.45(l H, s), 3.89(2H, ss and
3.75(3H, s) ppm.
Step K: Preparation of 1 -(Triphenylmethyl)-1 H-imidazol-4- ylacetic acid methyl ester
To a solution of 1H-Imidazole-4- acetic acid methyl ester hydrochloride (24.85g, 0.141mol) in dimethyl formamide (DMF) (1 15ml) was added triethylamine (57.2 ml, 0.412mol) and triphenyl- methyl bromide(55.3g, 0.171mol) and the suspension was stirred for 24hr. After this time, the reaction mixture was diluted with ethyl acetate (EtOAc) (1 1) and water (350 ml). The organic phase was washed with sat. aq. NaHCO3 (350 ml), dried (Na2SO4) and evaporated in vacuo. The residue was purified by flash chromatography (SiO2, 0- 100% ethyl acetate in hexanes; gradient elution) to provide the title compound as a white solid.
1 H NMR (CDCl3, 400 MHz) δ 7.35( 1 H, s), 7.31 (9H, m), 7.22(6H, m), 6.76(1 H, s), 3.68(3H, s) and 3.60(2H, s) ppm.
Step L: Preparation of [ 1 -(4-Cyanobenzyl)- 1 H-imidazol-5-yl]acetic acid methyl ester
To a solution of 1 -(Triphenylmethyl)- 1H-imidazol-4- ylacetic acid methyl ester (8.00g, 20.9mmol) in acetonitrile (70 ml) was added bromo-p-toluonitrile (4.10g, 20.92 mmol) and heated at 55°C for 3 hr. After this time, the reaction was cooled to room temperature and the resulting imidazolium salt (white precipitate) was collected by filtration. The filtrate was heated at 55°C for 18hr. The reaction mixture was cooled to room temperature and evaporated in vacuo. To the residue was added EtOAc (70 ml) and the resulting white precipitate collected by filtration. The precipitated imidazolium salts were combined, suspended in methanol ( 100 ml) and heated to reflux for 30min. After this time, the solvent was removed in vacuo, the resulting residue was suspended in EtOAc (75ml) and the solid isolated by filtration and washed (EtOAc). The solid was treated with sat aq
NaHCO3 (300ml) and CH2CI2 (300ml) and stirred at room
temperature for 2 hr. The organic layer was separated, dried (MgSO4) and evaporated in vacuo to afford the title compound as a white solid :
1 HNMR(CDCl3, 400 MHz) δ 7.65(1H, d, J=8Hz), 7.53(1H, s), 7.15(1H, d, J=8Hz), 7.04(1H, s), 5.24(2H, s), 3.62(3H, s) and 3.45(2H, s) ppm.
Step M: Preparation of [1-(4-cyanobenzyl)- 1H-imidazol-5-yl ]acetic acid
A solution of [ 1 -(4-cyanobenzyl)-1H-imidazol-5-yl]acetic acid methyl ester (4.44g, 17.4mmol ) in THF (100ml) and 1 M lithium hydroxide (17.4 ml, 17.4 mmol) was stirred at RT for 18 hr. 1 M HCl ( 17.4 ml) was added and the THF was removed by evaporation in vacuo. The aqueous solution was lyophilized to afford the title
compound containing lithium chloride as a white solid.
1 H NMR(CD3OD, 400 MHz) δ 8.22( 1 H, s), 7.74(1 H, d, J=8.4Hz), 7.36(1 H, d, J=8.4Hz), 7.15(1 H, s), 5.43(2H, s) and 3.49(2H, s) ppm. Step N: Preparation of N-[( 1 -(4-Cyanobenzyl)- 1 H-imidazol-5- yl)acetyl]pyrrolidin-2(S)-ylmethyl]-3(S)-ethyl-prolyl methionine isopropyl ester
[1 -(4-Cyanobenzyl)-1H-imidazol-5-yl]acetic acid• LiCl (0.416 g, 1.47 mmol), pyrrolidin-2(S)-ylmethyl]-3(S)-ethyl-prolyl methionine isopropyl ester hydrochloride (Step I) ( 0.63 g, 1.33 mmol), HOOBT (0.239 g, 1.47 mmol), and EDC (0.281 g, 1.47 mmol) were dissolved in degassed DMF (20 mL) with stirring at room temperature, N-mefhylmorpholine (0.8 mL, 5.32 mmol) was added to achieve a pH of 7, and stirring was continued overnight. The reaction mixture was concentrated to remove most of the DMF, and the residue was
partitioned between EtOAc and aq satd NaHCO3 solution. The aq layer was washed with EtOAc, the organics combined, washed with brine and dried (MgSO4). Filtration and concentration to dryness gave the title compound after chromatography on silica gel eluting with
CH2Cl2:CH3OH, 95:5.
Anal, calcd for C33H46N6O4S• 0.7 H2O:
C, 62.38; H, 7.52; N, 13.23;
Found: C, 62.40; H, 7.17; N, 13.1 1.
FAB MS 623 (M+1 )
EXAMPLE 5
2(S)-n-Butyl-1 -[1 -(4-cyanobenzyl)imidazol-5-ylmethyl]-4-(2,3- dimethylphenyl)piperazin-5-one
1 -[ 1 -(4-Cyanobenzyl)imidazol-5-ylmethyl]-4-(2,3-dimethylphenyl)- 2(S)-(2-methoxyethyl)piperazin-5-one ditrifluoroacetic acid salt Step A: N-Methoxy-N-methyl 4-benzyloxy-2(S)-(tert- butoxycarbonylamino)butanamide
4-Benzyloxy-2(S)-(tert-butoxycarbonylamino)butanoic acid (1.00 g, 3.23 mmol) was converted to the title compound following the procedure described in Example 24, Step A, using EDC• HCl (0.680 g, 3.55 mmol). HOBT (0.436 g, 3.23 mmol) and N,O-dimethylhydroxyl- amine hydrochloride (0.473 g, 4.85 mmol) in DMF (50 mL) at pH 7. After workup, the title compound was obtained as a clear gum.
Step B: 4-(1 -Benzyloxyethyl)-2(S)-(tert-butoxycarbonylamino) butanal
The title compound was obtained by lithium aluminum hydride reduction of the product of Step A using the procedure described in Example 24, Step B.
Step C: N-(2,3-Dimethylphenyl)-4-(2-benzyloxy
butoxycarbonylamino)butanamine
The title compound was prepared from the product of Step C according to the procedure described in Example 24, Step C, using 2,3-dimethylaniline (0.505 mL, 4.14 mmol), sodium triacetoxyboro- hydride (1.20 g, 5.65 mmol) and crushed molecular sieves ( 1 g) at pH 5 in dichloroethane (20 mL). The title compound was obtained after purification on silica gel, eluting with 15% ethyl acetate in hexane. Step D: 2(S)-(2-Benzyloxyethyl)-1 -tert-butoxycarbonyl-4-(2,3- dimethylphenyl)piperazin-5-one
The title compound was prepared from the product of Step C according to the procedure described in Example 24, Step D, using chloroacetyl chloride (0.21 mL, 2.57 mmol) in 60 mL 1 :1 ethyl acetate: saturated sodium bicarbonate, followed by reaction of the crude product with sodium hydride (0.373 g, 60% dispersion in oil, 9.32 mmol) in DMF (30 mL). After workup, the crude product was chromatographed on silica gel with 30% ethyl acetate in hexane to obtain the title compound.
Step E: 1 -tert-Butoxycarbonyl-4-(2,3-dimethylphenyl)-2-S)-(2- hydroxyethyPpiperazin-5-one
The product from Step D was dissolved in methanol (40 mL) and 10% Pd/C was added (0.160 g). The reaction was shaken under 60 psi hydrogen overnight. The catalyst was removed by filtration, and the solvent evaporated to give the title compound.
Step F: 1 -tert-Butoxycarbonyl-4-(2,3-dimethylphenyI)-2(S)-(2- methoxyethyl)piperazin-5-one
The product from Step E (0.241 g, 0.688 mmol) was dissolved in DMF (10 mL) containing methyl iodide (0.21 mL, 3.44 mmol) and the stirred solution cooled to 0°C under nitrogen. Sodium hydride (0.070 g, 60% dispersion in oil, 1.72 mmol) was added and the reaction stirred for 1h. The reaction was quenched with water, and the
DMF removed under vacuum. The residue was partitioned between ethyl acetate and water, and the organic phase washed with saturated brine and dried over magnesium sulfate. The crude product was chromatographed on silica gel with 40% ethyl acetate in hexane to give the title compound.
Step G: 4-(2,3-Dimethylphenyl)-2(S)-(2-methoxyethyl)-1 -[4-(1 - triphenylmethylimidazolyl)methyl]piperazin-5-one
The product from Step F (0.1 13 g, 0.312 mmol) was converted to the title compound according to the procedure described in Example 24, Step E, except using 30% trifluoroacetic acid in dichloromethane (10 mL) for 1 h for the initial deprotection. The volatiles were removed in vacua, and the residue dissolved in
dichloroethane. Triethylamine was added to obtain pH 5. Sodium triacetoxyborohydride (0.100 g, 0.468 mmol) and 1 -triphenylmethyl- 4-imidazolylcarboxaldehyde (0.1 164 g, 0.343 mmol) was added. The reaction was stirred overnight at 20°C then poured into saturated sodium bicarbonate solution. The organic phase was washed with saturated brine and dried over magnesium sulfate. Silica gel
chromatography using 5% methanol in chloroform as eluant yielded the title compound.
Step H: 1 -[ 1-(4-Cyanobenzyl)imidazol-5-ylmethyl]-4-(2,3- dimethylphenyl)-2(S)-(2-methoxyethyl)piperazin-5-one ditrifluoroacetic acid salt
The product from Step G (0.182 g, 0.312 mmol) was converted to the title compound according to the procedure described in Example 25, using 4-cyanobenzylbromide (0.061 g, 0.312 mmol) in acetonitrile (10 mL), followed by reaction of the crude imidazolium salt with triethylsilane (0.13 mL) and trifluoroacetic acid (2 mL) in dichloromethane (6 mL). Purification was accomplished by reverse phase preparative HPLC with a mixed gradient of 0%-70%
acetonitrile/0.1 % TFA; 100%-30% 0.1 % aqueous TFA over 60 min.
The title compound was isolated after lyophilization from water. FAB ms (m+ 1 ) 458.
Anal. Calc. for C27H31N5O2• 0.35 H2O• 2.0 TFA:
C, 53.81 ; H, 4.91; N, 10.21.
Found: C, 53.83; H, 4.95; N, 10.29.
EXAMPLE 6
N-[2(S)-N'-(1 -(4-Cyanophenyl-methyl)-1H-imidazol-5-ylacetyl)amino- 3(S)-methylpentyl]-N-1 -naahthylmethyl-glycyl-methionine methyl ester
Preparation of N-[2(S)-N'-(1 -(4-Cyanophenylmethyl)-1 H-imidazol-5- ylacetyl)amino-3(S)-methylpentyl]-N- 1 -naphthylmethyl-glycyl- methionine bis trifluoroacetate
Step A: Preparation of 1-(Triphenvlmethyl)- 1 H-imidazol-4-ylacetic acid methyl ester (23)
To a suspension of 1H-imidazole-4-acetic acid methyl ester hydrochloride (1, 7.48, 42.4 mmol) in methylene chloride (200 ml) was added triethylamine (17.7 ml, 127 mmol) and tripheny Imethyl bromide (16.4 g, 50.8 mmol) and stirred for 72 h. After this time, reaction mixture was washed with sat. aq. sodium bicarbonate ( 100 ml) and water (100 ml). The organic layer was evaporated in vacua and purified by flash chromatography (30-100% ethyl acetate/hexanes gradient elution) to provide 23 as a white solid.
1H NMR (CDCI3, 400 MHz) δ 7.35 (1 H, s), 7.31 (9H, m), 7.22 (6H, m), 6.76 (1H, s), 3.68 (3H, s) and 3.60 (2H, s) ppm.
Step B: Preparation of l -(4-NitrophenylmethyI)-1H-imidazol-5- ylacetic acid methyl ester (16)
To a solution of l -(triphenylmethyl)-1H-imidazol-4-ylacetic acid methyl ester (23, 274 mg, 0.736 mmol) in acetonitrile (10 ml) was added 4-nitrobenzylbromide (159 mg, 0.736 mmol) and heated to 55°C for 16 h. After this time, the reaction was cooled to room temperature, treated with ethyl acetate (20 ml) and the resulting precipitate was filtered. The filtrate was concentrated to dryness in vacua and the residue was redissolved in acetonitrile (4 ml) and heated to 65°C for 3 h. After this time, the reaction mixture was evaporated to dryness and combined with initial precipitate. This residue was dissolved in methanol (5 ml ) and heated to reflux for 30 min. The resulting solution was evaporated in vacuo and the residue was purified by flash chromatography (2-5% methanol/methylene chloride gradient elution ) to provide 16.
1H NMR (CDCI3, 400 MHz) δ 8.20 (2H, d, J=8.8 Hz), 7.53 (1H, s), 7.19 (2H, d, J=8.8 Hz), 7.03 (1H, s), 5.28 (2H, s), 3.61 (3H, s) and 3.44 (2H, s) ppm.
Step C: 1 -(4-Nitrophenylmethyl)-1H-imidazol-5-ylacetic acid
hydrochloride
1 -(4-Nitrophenylmethyl)-1H-imidazol-5-ylacetic acid methyl ester (0.1 15 g, 0.42 mmol ) was dissolved in 1.ON hydrochloric acid (10 ml ) and heated at 55°C for 3 h. The solution was evaporated in vacuo to give the compound as a white solid.
1 H NMR (CD3OD, 400 MHz) δ 9.06 ( 1H, s), 8.27 (2H, d, J=8.8 Hz), 7.61 (1 H, s), 7.55 (2H, d, J=8.8 Hz), 5.63 (2H, s) and 3.81 (2H, s) ppm.
Step D: N-[2(S)-N'-(1 -(4-Nitrophenylmethyl)-1 H-imidazol-5- ylacetyl)amino-3(S)-methylpentyl]-N- 1 -naphthylmethyl- glvcyl-methionine methyl ester bis trifluoroacetate
To a solution of 1 -(4-nitrophenyImethyI)-1H-imidazol- 5-ylacetic acid hydrochloride, N-[2(S)-amino-3(S)-methylpentyl]-N- naphthylmethyl-glycyl-methionine methyl ester bis hydrochloride (209 mg, 0.392 mmol) and 3-hydroxy- l ,2,3-benzotriazin-4(3H)-one (HOOBT, 64 mg, 0.39 mmol) in methylene chloride ( 10 ml) was added 1 -(3-dimethylaminopropyl)-3-ethyIcarbodiimide hydrochloride (EDC, 75.2 mg, 0.392 mmol) and triethylamine (219 μl, 1.57 mmol) and the mixture stirred overnight at room temperature. After this time, satd. aq. sodium bicarbonate (10 ml) was added and the mixture was extracted with methylene chloride. The combined extracts were washed with satd. aq. sodium bicarbonate ( 10 ml) and the solvent evaporated in vacuo.
EXAMPLE 7
Preparation of N-(2(R)-amino-3-mercaptopropyl)-valyl-isoleucyl- leucine methyl ester (Compound 1)
Step A. Preparation of N-(2(R)-t-butoxycarbonyI-amino-3- triphenyl-methylmercaptopropyl)-valyl-isoleucyl-leucine methyl ester
The tripeptide ester valyl-isoleucyl-leucine methyl ester was synthesized using conventional solution phase peptide synthesis methods. The trifluoroacetate salt of this tripeptide (360 mg, 0.77 mmol) was dissolved in 5 mL of methanol with 147 mg (1.5 mmol) of potassium acetate and 670 mg (1.5 mmol) of N-Boc-S-tritylcysteinal (prepared using the procedure of Goel, Krolls, Stier, and Kesten Org. Syn. 67: 69-74 (1988) for the preparation of N-Boc-leucinal) was added. Sodium cyanoborohydride (47 mg, 0.75 mmol) was added and the mixture was stirred overnight. The mixture was diluted with ether and washed with water, 5% ammonium hydroxide and brine. The solution was dried
(sodium sulfate) and evaporated to give a white foam which was purified by chromatography (1 -15% acetone in methylene chloride). The title compound was obtained as an oily material. Step B. Preparation of N-(2(R)-amino-3-mercaptopropyl)-valyl- isoleucyl-leucine methyl ester
A sample of the protected pseudopeptide prepared as described in Step A (728 mg, 0.92 mmol) was dissolved in 100 mL of methylene chloride, 50 mL of TFA was added and the resulting yellow solution was treated immediately with 0.80 mL (5 mmol) of triethylsilane. After 45 min, the solvents were evaporated and the residue was partitioned between hexane and 0.1 % aqueous TFA. The aqueous solution was lyophilized. This material was further purified by reverse phase HPLC (5-95% acetonitrile/0.1 % TFA/water) to afford the title compound. 1H NMR (CD3OD) δ 8.65 (1 H, d), 4.45 ( 1 H, m), 4.3 ( 1 H, d), 3.7 (3H, s), 3.4 (1 H, m), 3.15 (1 H, d), 2.75-2.95 (m), 0.8- 1.05 ( 18 H, m). FAB mass spectrum, m/z = 447 (M + 1).
Anal. Calcd for C2 1H42N4O4S• 1.8 TFA:
C, 45.24; H, 6.75; N, 8.56.
Found: C, 45.26; H, 6.77; N. 8.50.
EXAMPLE 8
N-(2(R)-amino-3-mercaptopropyl)-valyl-isoleucyl-leucine (Compound 2)
Step A. Preparation of N-(2(R)-t-butoxycarbonylamino-3- triphenylmethylmercaptopropyl)-valyl-isoleucyl-leucine The product of Example 7, Step A (60 mg, 0.076 mmol) was dissolved in 1 mL of methanol and 150 μL of I N NaOH was added. After stirring overnight, the solution was acidified with 150 μL of 10% citric acid and the product was extracted with ether. The ether solution was washed with water and brine and dried (sodium sulfate).
Evaporation provided the title compound as a solid.
Step B. Preparation of N-(2(R)-amino-3-mercaptopropyl)-valyl- isoleucyl-leucine
Using the method of Example 7, Step B, the protecting groups were removed with TFA and triethylsilane to provide the title compound. FAB mass spectrum, m/z = 433 (M+l).
Anal. Calcd for C20H40N4O4S• 2 TFA:
C, 43.63; H, 6.41 ; N, 8.48.
Found: C, 43.26; H, 6.60; N. 8.49. EXAMPLE 9
Preparation of 2(S)-[2(S)-[2(R)-Amino-3-mercapto]-propylamino-3(S)- methyl]pentyloxy-3-phenylpropionyl-homoserine lactone (Compound 3) ann 2(S)-[2(S)-[2(R)-Amino-3-mercapto]-propylamino-3(S)- methyllpentyloxy-3-phenyl-propionyl-homoserine (Compound 44
Step A: Preparation of N-(α-chloroacetyl)-L-isoleucinol
To a stirred solution of L-isoleucinoI (20 g, 0.17 mol) and triethylamine (28.56 ml, 0.204 mol) in CH2CI2 (500 ml) at -78°C was added chloroacetyl chloride (16.3 ml, 0.204 mol) over 5 minutes. The cooling bath was removed and the solution allowed to warm to -20°C.
The mixture was diluted with EtOAc and washed sequentially with 1 M
HCl, and brine and dried (Na2SO4). Evaporation in vacuo afforded the amide title compound (35 g, 100%).
Rf = 0.3 CH2Cl2: MeOH (95:5);
1H NMR (CDCI3) δ 6.80 ( 1 H, brd, J = 5 Hz), 4.10 (2H, s), 3.84 ( 1 H, m), 3.79 (2H, m), 2.65 (1H, brs), 1.72 (1H, m), 1.55 (1 H, m), 1.17
(1H, m), 0.96 (3H, d, J = 6Hz) 0.90 (3H,t, J=6 Hz). Step B: Preparation of 5(S)-[ 1 (S)-methyI]propyl-2,3,5,6-tetra- hydro-4H-1 ,4-oxazin-3-one
To a stirred solution of N-(a-chloroacetyl)-L-isoleucinol (7.4 g, 0.038 mol) in THF (125 ml) under argon at 0°C was slowly added sodium hydride (2.2 g of a 60% dispersion in mineral oil, 0.055
mol) with concomitant gas evolution. After completing the addition, the mixture was warmed to room temperature (R.T.) and stirred for 16 hr. Water (2.8 ml) was added and the solvents evaporated in vacuo. The residue was dissolved in CHCl3 (70 ml) and washed with water saturated NaCl solution. The organic layer was dried (Na2SO4) and evaporated in vacuo. The residue was chromatographed using silica gel eluting with CH2Cl2:MeOH (96:4) to afford the lactam title compound (4.35 g, 72%) as a white solid.
Rf = 0.35 CH2Cl2:MeOH (95:5);
1H NMR δ (CDCI3) 6.72 (1H, brs), 4.20 (1H, d, J = 14.5 Hz), 4.10 ( 1H, d, J = 14.5 Hz), 3.88 (1H, dd, J = 9 and 3.5 Hz), 3.58 (1H, dd, J = 9 and 6.5 Hz), 3.45 (1H, brqt, J = 3.5 Hz), 1.70-1.45 (2H, m), 1.34 - 1.15 ( 1 H, m), 0.96 (3H, t, J = 6.5 Hz), 0.94 (3H, d, J = 6.5 Hz). Step C: Preparation of N-(tert-butoxycarbonyl)-5(S)-[ 1 (S)- methynpropyl-2,3,5,6-tetrahydro-4H-1 ,4-oxazin-3-one
5(S)-[1(S)-Methyl]propyl-2,3,5,6-tetrahydro 4H-1 ,4- oxazin-3-one (12.2 g, 0.0776 mol) and DMAP (18.9 g, 0.155 mol) were dissolved in methylene chloride ( 120 ml) under argon at R.T. Boc anhydride (33.9 g, 0.155 mol) was added to the stirred solution in one portion, with concomitant gas evolution and the mixture was stirred at R.T. for 16 hr. The solvent was evaporated in vacuo and the residue was taken up in ethyl acetate and washed sequentially with 10% citric acid, 50% NaHCO3 and finally brine. The organic extract was dried (Na2SO4) and evaporated in vacuo. Chromatography of the residue over silica gel eluting with 20% EtOAc in hexanes afforded the title compound ( 14.1 g, 71 %) as a white solid.
Rf = 0.75 EtOAcmexanes (20:80); mp 59-60°C
Anal. Calc'd. for C1 3H23O4N :
C, 60.68; H,9.01 ; N, 5.44.
Found: C, 60.75; H, 9.01 ; N, 5.58.
1 H NMR (CDCI3) δ 4.25 (1H, d, J = 15 Hz), 4.15 ( 1 H, d, J = 15 Hz), 4.15 - 4.00 (2H, m), 3.73 ( 1H, dd, J = 10 and 2 Hz), 1.88 (1 H, qt, J = 6
Hz), 1.55 (9H, s), 1.50 - 1.36 (1 H, m), 1.35 - 1.19 (1 H, m) 1.00 (3H, d, J = 6 Hz) 0.95 (3H, d, J = 6.5 Hz).
Step D: Preparation of N-(tert-Butoxycarbonyl)-2(S)-benzyl-5(S)- [1 (S)-methyl]propyl-2,3,5,6-tetrahydro-4H-1 ,4-oxazin-3- one
A solution of N-(tert-butoxycarbonyl)-5(S)-[ 1(S)- methyl]propyl-2,3,5,6-tetrahydro-4H-1 ,4-oxazin-3-one (5.75 g, 22.34 mmol) in DME (100 ml) under argon was cooled to -60°C. The cold solution was transferred via canula to a second flask containing sodium bis(trimethylsilyl)amide (24.58 ml of a 1 M solution in THF, 24.58 mmol) at -78°C under argon. After stirring for 10 minutes, benzyl bromide (2.25 ml, 18.99 mmol) was added over 5 minutes and the resulting mixture was stirred at -78°C for 3 hours. After this time, the reaction mixture was transferred via cannula to another flask containing sodium bis(trimethylsilyl)amide (24.58 ml of a 1 M solution in THF, 24.58 mmol) at -78°C, under argon. After stirring for a further 5 minutes, the reaction was quenched by the addition of saturated aqueous ammonium chloride solution (24.6 ml) and allowed to warm to room temperature. This mixture was diluted with brine (50 ml) and water (20 ml) and then extracted with ethyl acetate (2 x 100 ml). The organic extracts were washed with brine (50 ml) and evaporated in vacuo to afford an oil. Chromatography of the residue over silica gel (230-400 mesh, 300 g) eluting with 10-20% ethyl acetate in hexanes afforded the title compound (5.12 g, 67%) as a clear oil.
Rf = 0.25 EtOAc.Ηexanes (20:80);
1H NMR (CDCI3) δ 7.35 - 7.15 (5H, m), 4.31 (1 H, dd, J = 6 and 2 Hz), 4.03 (1 H, d, J = 12 Hz), 3.88 (1H, dd, J = 6 and 1 Hz), 3.66 (1H, dd, J = 12 and 2 Hz), 3.29 (1H, dd, J = 12 and 3 Hz), 1.54 (9H, s), 3.12 (1H, dd, J = 12 and 7 Hz), 1.47 (1 H, m), 1.25 (1 H, m), 1.10 ( 1H, m), 0.83 (3H, d, J = 6 Hz), 0.80 (3H, t, J = 6 Hz).
Step E: Preparation of N-(tert-butoxycarbonyl)-2(S)-[2(S)-amino-
3(S)-methyllpentyloxy-3-phenyl-propionic acid
To a stirred solution of N-(tert-butoxycarbonyl)-2(S)- benzyl-5(S)-[1 (S)-methyl]-propyl-2,3,5,6-tetrahydro-4H-1 ,4-oxazin- 3-one (5.1 g, 14.7 mmol) in THF (150 ml) and water (50 ml) at 0°C was added hydrogen peroxide (15 ml of a 30% aqueous solution, 132 mmol) and lithium hydroxide (3.0 g, 63.9 mmol). After stirring for 30 minutes, the reaction was quenched with a solution of sodium sulfite (28.25 g, 0.224 mol) in water (70 ml). The THF was evaporated in vacuo and the aqueous phase was acidified to pH 3-4 by addition of 10% citric acid solution and extracted with EtOAc. The organic extracts were dried (Na2SO4), evaporated in vacuo and the residue purified by chromatography over silica gel eluting with 4% MeOH in CH2CI2 to give the lactam 2(S)-benzyl-5(S)-[1 (S)-methyl]propyl-2,3,5,6- tetrahydro-4H-1 ,4-oxazin-3-one (0.82 g 22%) and then with 20% MeOH in CH2CI2 to afford the title compound (4.03 g, 75%) as a viscous oil. Rf = 0.4 MeOH:CH2Cl2 (5:95) + 0.3% AcOH;
1H NMR (d6 DMSO) δ 7.35 - 7.10 (5H, m), 6.68 (1H, br, s), 3.75 ( 1 H, dd, J = 7.5 and 2.5 Hz) 3.54 (1 H, m), 3.5 - 3.2 (2H, m) 2.99 (1 H, dd, J = 12.5 and 2.5 Hz), 2.75 (1H, dd, J = 12.5 and 7.5 Hz), 1.50 - 1.35
( 1 1H, m), 0.98 ( 1 H, sept, J = 6 Hz), 0.78 (3H, t, J = 6 Hz), 0.65 (3H, d,
J = 6 Hz);
FAB MS 366 (MH+) 266 (MH2+ - CO2tBu). Step F: Preparation of N-(tert-butoxycarbonyl)-2(S)-[2(S)-amino-
3(S)-methyl]-pentyloxy-3-phenyl-propionyl-homoserine lactone
To a stirred solution of N-(tert-butoxycarbonyl)-2(S)-[2(S)- amino-3(S)-methyl]-pentyloxy-3-phenylpropionic acid (0.53 g, 1.45 mmol) and 3-hydroxy-1 ,2,3,-benzotriazin-4(3H)-one (HOOBT) (0.26 g, 1.6 mmol) in DMF (15 ml) at room temperature was added EDC (0.307 g, 1.6 mmol) and L-homoserine lactone hydrochloride (0.219 g, 6.0 mmol). The pH was adjusted to pH= 6.5 by addition of NEt3 (the pH was monitored by application of an aliquot of the reaction mixture to a
moist strip of pH paper). After stirring at room temperature for 16 hr, the reaction was diluted with EtOAc and washed with saturated NaHCO3 and then brine and dried (NaSO4). Evaporation in vacuo (sufficient to remove DMF) and chromatography over silica gel eluting with 5% acetone in CH2CI2 afforded the title compound (520 mg, 80%) as a white solid, mp 1 15-1 17°C.
Rf = 0.3 Acetone: CH2CI2 (5:95).
1H NMR (CDCI3 ) δ 7.73 (1 H, brd, J=5 Hz), 7.40-7.15 (5H, m), 4.68 (1H, dt, J=9 and 7.5 Hz), 4.65-4.35 (2H, m), 4.33-4.18 (1 H, m), 4.20 (1H, dd, J=7 and 3 Hz), 3.78 (1H, m), 3.49 (1H, dd, J=7.5 and 4.0 Hz), 3.37 (1H, dd, J=7.5 and 6.5 Hz), 3.15 (1H, dd, .1=11.5 and 2 Hz), 2.86 (1H, dd, J=11.5 and 7.5 Hz), 2.68 (1H, m) 2.11 (1H, q, J=9 Hz), 1.55- 1.30 (11H, m), 1.07 (1H, m), 0.87 (3H, t, J=6.3 Hz), 0.79 (3H, d, J=6 Hz).
Step G: Preparation of 2(S)-[ 2(S)-amino-3(S)-methyl]-pentyloxy-3- phenylpropionyl-homoserine lactone hydrochloride
Anhydrous HCl gas was bubbled through a cold (0°C) solution of N-(tert-butoxycarbonyl)-2(S)-[2(S)-amino-3(S)-methyl] pentyloxy-3-phenylpropionyI-homoserine lactone (3.0 g, 6.7 mmol) in ethyl acetate (120 ml) until a saturated solution was obtained. The resulting mixture was stirred at 0°C for 1 hr. The solution was purged with nitrogen and the mixture concentrated in vacuo to afford the title compound as a sticky foam which was used without further purification.
1H NMR (d6 DMSO) δ 8.60 (1 H, d, J=7 Hz), 8.08 (3H, brs), 7.35-7.15 (5H, m), 4.60 (1H, qt, J=8 Hz), 4.36 (1H, t J=7.5 Hz), 4.22 (1H, q, J=7.5 Hz), 4.15-3.95 (2H, m), 3.64 (1 H, dd, J=9 and 2.5 Hz), 3.15-3.00 (2H, m), 2.92 (1H, dd, J=12.5 and 5.0 Hz), 2.40-2.15 (2H, m), 1.65 ( 1 H, m), 1.43 (1H, m), 1.07 (1H, m), 0.82 (3H, t, J=6 Hz), 0.72 (3H, d, J=6.0 Hz).
Step H: Preparation of 2(S)-[2(S)-[2(R)-(tert-butoxycarbonyl)- amino-3-triphenylmethylmercap-to]propylamino-3(S)- methyl]-pentyloxy-3-phenylpropionyl-homoserine lactone 2(S)-[2(S)-Amino-3(S)-methyl]pentyloxy-3-phenyl- propionyl-homoserine hydrochloride (6.7 mmol) and N-(tert-butoxy- carbonyl)-S-triphenylmethylcysteine aldehyde (0.74 g, 7.5 mmol) (prepared from N-(tert-butoxycarbonyl)-S-triphenyImethylcysteine by the procedure of Goel, O.P.; Krolls, U.; Stier, M.; Keston, S. Ore. Syn. 1988, 67, 69.) and potassium acetate (3.66 g, 8.2 mmol) were dissolved in methanol (48 ml). Activated 4A molecular sieves (6g) and then Na(CN)BH3 (0.70 g, 10.7 mmol) were added and the resulting slurry was stirred under argon at room temperature for 16 hr. The solids were removed by filtration and the filtrate evaporated in vacuo. The residue was dissolved in EtOAc and washed sequentially with saturated aqueous NaHCO3 and brine and then dried (Na2SO4). Evaporation in vacuo afforded an oil which was purified by chromatography over silica gel eluting with a gradient of 30-50% EtOAc in hexane to afford the title compound (2.34 g, 45%) contaminated with a small amount of the corresponding methyl ester.
1 H NMR (CD3OD) δ 7.60-7.05(20H, m), 4.64 (1 H, d, J=9.0Hz), 4.39 ( 1 H, br t, J=9Hz), 4.25(1 H, m), 3.93 (1H, m), 3.75-3.60( 1H, m), 3.55 (1H, dd, J=9.0 and 2Hz), 3.20 ( 1H, dd, J=9.0 and 6.0 Hz), 3.04 ( 1 H, dd, J=15.0 and 5.0 Hz), 2.85 (1 H, dd, J=15.0 and 9.0 Hz), 2.60 (1H, dd, J=12.0 and 5.0 Hz), 2.50-2.15 (7H, m), 1.45 (9H, s), 1.40-1.20 (1 H, m), 1.07 ( 1 H, m), 0.87 (3H, t, J=6 Hz), 0.67 (3H, d, J=6.0 Hz).
Step I: Preparation of 2(S)-[2(S)-[2(R)-Amino-3-mercapto]- propylamino-3(S)-methyl]pentyloxy-3-phenylpropionyI- homoserine lactone
To a stirred solution of 2(S)-[2(S)-[2(R)-(tert-butoxy- carbonyl)amino-3-triphenylmethylmercapto]-propylamino-3(S)- methyl]pentyloxy-3-phenylpropionyl-homoserine lactone (2.72 g, 3.49 mmol) in CH2CI2 (90 ml) was added HSiEt3 (2.16 ml, 13.5 mmol) and TFA (43.2 ml, 0.56 mol) and the solution was stirred at R.T. under
argon for 2 hrs. The solvent was evaporated in vacuo and the residue partitioned between 0.1 % aqueous TFA (200 ml) and hexanes (100 ml). The aqueous layer was separated and washed with hexanes (20 ml) and then lyophilised. The resulting white lyophilate was chromatographed in 5 equal portions over a Waters Prepak cartridge (C-18, 15-20 mM 100 A) eluting with a gradient of 95:5 to 5:95 0.1 % TFA in H20 : 0.1 % TFA in CH3CN at 100 ml/min over 60 min. The desired compound eluted after 19 min. The CH3CN was evaporated in vacuo and the aqueous solution lyophilised to afford the title compound (1.95 g, 77%) as the TFA salt.
The salt is hygroscopic and is prone to disulphide formation if left in solution and exposed to air.
1H NMR δ (CD3OD) 7.40-7.15 (5H,m), 4.55-5.40 (2H, m), 4.33 (1H, m), 4.18 (1 H, m), 3.90-3.62 (3H, m), 3.53 ( 1H, dd, J= 10.5 and 4.0 Hz), 3.37 (1H, dd, J=10.5 and 6.0 Hz), 3.23 (1 H, m), 3.15-2.95 (2H, m),
2.88 (1H, dd, J=12.5 and 5.0 Hz), 2.55-2.25 (2H, m), 1.92 ( 1 H, m), 1.49 (1H, m), 1.23 (1H, m), 0.94 (3H, t, J=6 Hz), 0.90 (3H, d, J=6Hz).
FAB MS 873 (2M-H+) 438 (MH+) 361 (MH±Ph)
Anal, calc'd for C22H36O4N3S 2.6 TFA:
C, 43.58; H, 5.25; N, 5,82.
Found: C, 43.62; H, 5.07; N, 5.80.
Step J: Preparation of 2(S)-[2(S)-[2(R)-Amino-3-mercapto]- propylamino-3(S)-methyl]pentyloxy-3-phenylpropionyl- homoserine
2(S)-[2(S)-[2(R)-Amino-3-mercapto]propyl-amino-3(S)- methyl]pentyloxy-3-phenylpropionyl-homoserine lactone (0.00326 mmol) was dissolved in methanol (0.0506 ml) and 1N sodium hydroxide (0.0134 ml) was added followed by methanol (0.262 ml). The
conversion of the lactone to the hydroxy-acid was confirmed by HPLC analysis and NMR.
EXAMPLE 10
Preparation of 2(S)-[2(S)-[2(R)-Amino-3-mercapto]-propylamino-3(S)- methy1]pentyloxy-3-phenylpropionyl-methionine
Step A: Preparation of 2(S)-[2(S)-{2(R)-(tert-butoxy-carbonyl)- amino-3-triphenyl-methylmercaptoj-propylamino-3(S)- methyl ]pentyIoxy-3-phenyl-propionyl-methionine
To a solution of 2(S)-[2(S)-[2(R)-(tert-butoxycarbonyl)- amino-3-triphenylmethylmercapto]-propylamino-3(S)-methyl]- pentyloxy-3-phenylpropionyl-methionine methyl ester (120 mg, 0.143 mmol) in methanol (4 ml) was added sodium hydroxide ( 1N, 0.57 ml, 0.57 mmol) and the resulting mixture was stirred at room temperature for 3 hours. Another portion of sodium hydroxide (1 N, 0.25 ml) was added and stirring continued for 0.5 hours. The reaction mixture was concentrated and the residue was dissolved in a minimum amount of water and neutralized with hydrochloric acid (1 N, 0.87 ml). The aqueous solution was extracted with ethyl acetate three times. The combined extracts were dried (Na2SO4) and concentrated to yield the title compound (1 10 mg, 0.133 mmol, 93%). NMR (CD3OD) δ 0.70 (3H, d, J=6Hz), 0.80 (3H, t, J=6Hz), 1.05 (H, m), 1.34 (9H, s), 1.60 (H, m), 1.95 (3H, S), 2.7-2.9 (3H, m), 2.95-3.1 (2H, m), 3.95 (H, d of d, J=8, 4Hz), 4.27 (H, d of d, J=8.6Hz), 7.1-7.4 (20H, m). Step B: Preparation of 2(S)-[2(S)-[2(R)-Amino-3-mercapto]- propylamino-3(S)-methyl]pentyloxy-3-phenylpropionyl- methionine
The title compound was prepared in the same manner as that described in Example 9, Step I, but using 2(S)-[2(S)-[2(R)-(tert- butoxycarbonyl)-amino-3-triphenylmethylmercapto|-propylamino-3(S)- methyl]-pentyloxy-3-phenylpropionyl-methionine in place of 2(S)-[2(S)- [2(R)-(tert-butoxycarbonyl)-amino-3-triphenylmethylmercapto]- propylamino-3(S)-methyl]-pentyloxy-3-phenylpropionyl-homoserine lactone. NMR (CD3OD) δ 0.82 (3H, d, J=6Hz), 0.95 (3H, t, J=6Hz),
1.20 (H, m), 1.40 (H, m), 1.85 (H, m), 2.10 (3H, s), 2.4-2.6 (2H, m), 3.1 -3.2 (2H, m), 3.35 (H, d of d, J=14, 6Hz), 3.55 (H, d of d, J=14, 5Hz), 4.20 (H, d of d, J=10, 5Hz), 4.63 (H, d of d, J=10.6Hz), 7.27 (5H, m).
Anal. Calcd for C23H39N3O4S2•2CF3CO2H•2H2O:
C, 43.25; H, 6.05; N, 5.60.
Found: C, 43.09; H, 6.01 ; N, 5.46.
EXAMPLE 11
Preparation of 2(S)-[2(S)-[2(R)-Amino-3-mercapto]-propylamino-3(S)- methyl]pentyloxy-3-phenylpropionyl-methionine sulfone methyl__ester (Compound 5) Step A: Preparation of Methionine sulfone methyl ester
Thionyl chloride (2.63 ml, 36 mmol) was added dropwise to a stirred solution of N-Boc-Met sulfone (5 g, 18 mmol) in methanol (40 ml) cooled at 0°C. After the completion of the addition, the resulting mixture was warmed to room temperature and stirred overnight. The reaction mixture was recooled to 0°C and slowly treated with solid sodium bicarbonate to adjust the pH to 7. The mixture was concentrated in vacuo to remove methanol and the residue was dissolved in a minimum amount of water (solution pH ca. 10) and extracted with ethyl acetate four times. The combined extracts were dried (Na2SO4) and concentrated to give the title compound (1.5 g). NMR (CD3OD) δ 2.04 (H, m), 2.21 (H, m), 2.98 (3H, s), 3.23 (2H, t, J=7Hz), 3.63 (H, d of d, J=8.6Hz), 3.77 (3H, s).
Step B: Preparation of N-(tert-Butoxycarbonyl)-2(S)-[2(S)-amino- 3(S)-methyl]-pentyloxy-3-phenyl-propionyl-methionine sulfone methyl ester
The title compound was prepared in the same fashion as that described in Example 9, Step F, but using methionine sulfone methyl ester in place of homoserine lactone hydrochloride. NMR
(CD3OD) δ 0.80 (3H, d, J=6Hz), 0.88 (3H, t, J=6Hz), 1.12 (H, m), 1.47 (9H, s), 2.10 (H, m), 2.32 (H, m), 2.93 (3H, s), 3.5-3.7 (2H, m), 3.74 (3H, s), 4.01 (H, d of d, J=7.4Hz), 4.60 (H, d of d, J=9.5Hz), 6.60 (H, d, J=8Hz), 7.25 (5H, m).
Step C: Preparation of 2(S)-[ 2(S)-Amino-3(S)-methyl]-pentyloxy-3- phenylpropionyl-methionine sulfone methyl ester hydrochloride
The title compound was prepared in the same fashion as that described in Example 9, Step G, but using N-(tert-butoxycarbonyl)- 2(S)-[2(S)-amino-3(S)-methyl lpentyloxy-3-phenylpropionyl-methionine sulfone methyl ester in place of N-(tert-butoxycarbonyl)-2(S)-[2(S)- amino-3(S)-methyl ]pentyloxy-3-phenylpropionyl-homoserine lactone. NMR (CD3OD) δ 0.85 (3H, d, J=6Hz), 0.94 (3H, t, J=6Hz), 1.20 (H, m), 1.52 (H, m), 1.72 (H, m), 2.14 (H, m), 2.38 (H, m), 2.98 (3H, s), 3.57 (H, d of d, J=12, 6Hz), 3.73 (H, d of d, .1= 12, 9Hz), 3.78 (3H, s), 4.15 (H, d of d, J=8.6Hz), 4.63 (H, d of d, J=8.5Hz), 7.30 (5H, m).
Step D: Preparation of 2(S)-[2(S)-[2(R)-(tert-Butoxy-carbonyI)- amino-3-triphenylmethylmercapto]-propylamino-3(S)- methyl]pentyloxy-3-phenyl-propionyl-methionine sulfone methyl ester
The title compound was prepared in a similar fashion as that described in Example 9, Step H, but using 2(S)-[2(S)-amino-3(S)- methyl]pentyloxy-3-phenyl-propionyl-methionine sulfone methyl ester hydrochloride in place of 2(S)-[2(S)-amino-3(S)-methyl]pentyloxy-3- phenylpropionyl-homoserine lactone hydrochloride. NMR (CD3OD) δ 0.70 (3H, d, J=6Hz), 0.88 (3H, t, J=6Hz), 1.10 (H, m), 1.47 (9H, s), 2.15 (H, m), 2.67 (H, m), 2.92 (3H, s), 3.67 (H, m), 4.68 (H, d of d, J= 10 , 6Hz), 7.15~7.45 (20H, m).
Step E: Preparation of 2(S)-[2(S)-[2(R)-Amino-3-mercaρto]- propylamino-3(S)-methyl]pentyloxy-3-phenylpropionyl- methionine sulfone methyl ester
The title compound was prepared in a similar fashion as that described in Example 9, Step I, but using 2(S)-[2(S)-[2(R)-(tert- butoxycarbonyl)amino-3-triphenylmethylmercapto)propylamino-3(S)- methyl]-pentyloxy-3-phenylpropionyl-methionine sulfone methyl ester in place of 2(S)-[2(S)-[2(R)-(tert-butoxy-carbonyl)-amino-3-triphenyl- methylmercapto]propylamino-3(S)-methyl]pentyloxy-3-phenyl- propionyl-homoserine lactone. NMR (CD3OD) δ 0.83 (3H, d, J=6Hz), 0.93 (3H, t, J=6Hz), 1.20 (H, m), 1.51 (H, m), 1.80 (H, m), 2.22 (H, m), 2.43 (H, m), 3.00 (3H, s), 3.78 (3H, s), 4.20 (H, d of d, J=8.4Hz), 4.72 (H, d of d, J=10, 6Hz), 7.30 (5H, m).
FABMS m/z 532 (MH+).
EXAMPLE 12
Preparation of 2(S)-[2(S)-[2(R)- Amino-3-mercapto]-propylamino-3(S)- methyl]-pentyloxy-3-phenylpropionyl-methionine sulfone (Compound 6)
Step A: Preparation of 2(S)-[2(S)-[2(R)-(tert-Butoxy-carbonyl)- amino-3-triphenylmethylmercapto]-propylamino-3(S)- methyllpentyloxy-3-phenyl-propionyl-methionine sulfone The title compound was prepared in a similar fashion as that described in Example 10, Step A, but using 2(S)-[2(S)-[2(R)-(tert- butoxycarbonyl)amino-3-triphenylmethylmercapto)-propylamino-3(S)- methyl]-pentyloxy-3-phenylpropionyl-methionine sulfone methyl ester in place of 2(S)-[2(S)-[2(R)-(tert-butoxycarbonyl)amino-3-triphenyl- methylmercapto]propylamino-3(S)-methyl]pentyloxy-methionine methyl ester. NMR (CD3OD) δ 0.79 (3H, d, J=6Hz), 0.90 (3H, t, J=6Hz), 1.47 (9H, s), 2.92 (3H, s), 4.08 (H, m), 4.32 (H, m), 7.15-7.35 (20H, m).
Step B: Preparation of 2(S)-[2(S)-[2(R)-Amino-3-mercapto]- propylamino-3(S)-methyl]-pentyloxy-3-phenylpropionyI- methionine sulfone
The title compound was prepared in a similar fashion as that described in Example 9, Step I, but using 2(S)-[2(S)-[2(R)-(tert- butoxycarbonyl)amino-triphenylmethylmercapto]propylamino-3(S)- methyl]-pentyloxy-3-phenylpropionyl-methionine sulfone in place of 2(S)-[2(S)-[2(R)-(tert-butoxycarbonyl)amino-3-triphenylmethyI- mercapto]propylamino-3(S)-methyl]-pentyloxy-3-phenylpropionyl- 3(S)-methyl]pentyloxy-3-phenylpropionyl-homoserine lactone. NMR (CD3OD) δ 0.84 (3H, d, J=6Hz), 0.94 (3H, t, J=6Hz), 1.21 (H, m), 1.50 (H, m), 1.82 (H, m), 2.24 (H, m), 2.47 (H, m), 2.98 (3H, s), 3.6-3.75 (3H, m), 4.20 (H, d of d, J=9.5Hz), 4.64 (H, d of d, J=9.6Hz), 7.30 (5H, m).
Anal. Calcd for C23H39N3O6S2•3CF3CO2H:
C, 40.51 ; H, 4.92; N, 4.89.
Found: C, 40.47; H, 5.1 1 ; N, 4.56.
EXAMPLE 13
Preparation of 2(S)-[2(S)-[2(R)-Amino-3-mercapto]-propylamino-3(S)- methyl]-pentyloxy-3-phenylpropionyl-methionine sulfone isopropyl ester
The title compound was prepared using methods A-E from
Example 1 1 , except for Method A. Methionine sulfone isopropyl ester
was prepared by coupling t-butyloxycarbonyl-methionine sulfone with isopropyl alcohol using dicyclohexylcarbodiimide (DCC) and 4- dimethylaminopyridine (DMAP) followed by deprotection with HCl in EtOAc. NMR (CD3OD) δ 0.83 (3H, d, J = 6 Hz), 0.94 (3H, t, J = 6 Hz), 1.1 1 -1.56 (2H, m), 1.28 (6H, d, J = 6 Hz), 1.8- 1.96 ( 1 H, m), 2.12- 2.27 (1H, m), 2.89-3.0 (2H, m), 3.01 (3H, s), 3.06-3.3 (4H, m), 3.42 (1H, dd, J = 6, 13 Hz), 3.65 (1H, dd, J = 6,13 Hz), 3.68-3.91 (3H, m), 4.2-4.27 (1 H, m), 4.61 -4.7 (1H, m), 4.96-5.12 (2H, m), 7.19-7.44 (5H, m).
Anal. Calc d. for C26H45N3O6S2• 2 CF3CO2H:
C, 44.07; H, 5.67; N, 4.97;
Found: C, 44.35; H, 5.68; N, 5.23
EXAMPLE 14
N-(2(R)-amino-3-mercaptopropyl)-valyl-isoleucyl-methionine methyl ester (Compound 7):
The title compound is prepared in accordance with U.S. Pat. No. 5,238,922, incorporated by reference.
EXAMPLE 15
N-(2(R)-amino-3-mercaptopropyl)-valyl-isoleucyl-methionine
(Compound 8):
The title compound is prepared in accordance with U.S. Pat. No. 5,238,922, incorporated by reference.
BIOLOGICAL ASSAYS.
The ability of compounds of the present invention to inhibit cancer can be demonstrated using the following assays.
EXAMPLE 16
In vitro inhibition
Transferase Assays. Isoprenyl-protein transferase activity assays were carried out at 30 °C unless noted otherwise. A typical reaction contained (in a final volume of 50 μL): [3H]farnesyl
diphosphate or [3H]geranylgeranyl diphosphate, Ras protein , 50 mM HEPES, pH 7.5, 5 mM MgCl2, 5 mM dithiothreitol and isoprenyl- protein transferase. The FPTase employed in the assay was prepared by recombinant expression as described in Omer, C.A., Krai, A.M., Diehl, R.E., Prendergast, G.C., Powers, S., Allen, CM., Gibbs, J.B. and Kohl, N.E. (1993) Biochemistry 32:5167-5176. The geranylgeranyl-protein transferase-type I employed in the assay was prepared as described in U.S. Pat. No. 5,470,832, incorporated by reference. After thermally pre-equilibrating the assay mixture in the absence of enzyme, reactions were initiated by the addition of isoprenyl-protein transferase and stopped at timed intervals (typically 15 min) by the addition of 1 M HCl in ethanol (1 mL). The quenched reactions were allowed to stand for 15
m (to complete the precipitation process). After adding 2 mL of 100% ethanol, the reactions were vacuum-filtered through Whatman GF/C filters. Filters were washed four times with 2 mL aliquots of 100% ethanol, mixed with scintillation fluid (10 mL) and then counted in a Beckman LS3801 scintillation counter .
For inhibition studies, assays were run as described above, except inhibitors were prepared as concentrated solutions in 100% dimethyl sulfoxide and then diluted 20-fold into the enzyme assay mixture. IC50 values were determined with both transferase substrates near KM concentrations. Nonsaturating substrate conditions for inhibitor IC50 determinations were as follows: FTase, 650 nM Ras- CVLS, 100 nM farnesyl diphosphate; GGPTase-I, 500 nM Ras-CAIL, 100 nM geranylgeranyl diphosphate.
*(IC50 is the concentration of the test compound which gives 50% inhibition of FTase or GGTase-type I under the described assay conditions.
EXAMPLE 17
In vivo ras prenylation assay
The cell lines used in this assay consist of either Ratl or NIH3T3 cells transformed by either viral Ha-ras; an N-ras chimeric gene in which the C-terminal hypervariable region of v -Ha-ras was substituted with the corresponding region from the N-ras gene; or ras-CVLL, a v-Ha-ras mutant in which the C-terminal exon encodes leucine instead of serine, making the encoded protein a substrate for geranylgeranylation by GGPTase I. The assay can also be performed using cell lines transformed with human Ha-ras, N-ras or Ki4B-ras. The assay is performed essentially as described in DeClue, J.E. et al., Cancer Research 51 :712-717, (1991 ). Cells in 10 cm dishes at 50-75% confluency are treated with the test compound(s) (final concentration of solvent, methanol or dimethyl sulfoxide, is 0.1 %). After 4 hours at 37°C, the cells are labelled in 3 ml methionine -free DMEM supplemented with 10% regular DMEM, 2% fetal bovine serum, 400
μCi[35S]methionine (1000 Ci/mmol) and test compound(s). Cells treated with lovastatin, a compound that blocks Ras processing in cells by inhibiting the rate-limiting step in the isoprenoid biosynthetic pathway (Hancock, J.F. et al. Cell, 57: 1 167 ( 1989); DeClue, J.E.
et al. Cancer Res., 51 :712 (1991 ); Sinensky, M. et al. /. Biol. Chem., 265:19937 (1990)), serve as a positive control in this assay. After an additional 20 hours, the cells are lysed in 1 ml lysis buffer (1 % NP40/20 mM HEPES, pH 7.5/5 mM MgCl2/lmM DTT/10 mg/ml aprotinen/2 mg/ml leupeptin/2 mg/ml antipain/0.5 mM PMSF) and the lysates cleared by centrifugation al 100,000 x g for 45 min. Alternatively, four hours after the additon of the labelling media, the media is removed, the cells washed, and 3 ml of media containing the same or a different test compound added. Following an additional 16 hour incubation, the lysis is carried out as above. Aliquots of lysates containing equal numbers of acid-precipitable counts are bought to 1 ml with IP buffer (lysis buffer lacking DTT) and immunoprecipitated with the ras-specific monoclonal antibody Y 13-259 (Furth, M.E. et al., J. Virol. 43:294-304, ( 1982)). Following a 2 hour antibody incubation at 4°C, 200 μl of a 25%
suspension of protein A-Sepharose coated with rabbit anti rat IgG is added for 45 min. The immunoprecipitates are washed four times with IP buffer (20 nM HEPES, pH 7.5/1 mM EDTA/1 % Triton X-100.0.5% deoxycholate/0.1 %/SDS/0.1 M NaCl) boiled in SDS-PAGE sample buffer and loaded on 13% acrylamide gels. When the dye front reached the bottom, the gel is fixed, soaked in Enlightening, dried and
autoradiographed. The intensities of the bands corresponding to prenylated and nonprenylated Ras proteins are compared to determine the percent inhibition of prenyl transfer to protein.
Groups of in viva ras prenylation experiments are separated in Table 2 according to direct comparison of assay results on acrylamide gel chromatography plates. Legend: +++, > 90% of Ras protein
unprocessed; ++, 60 - 90% unprocessed; +, 30 - 60% unprocessed; +/-, 10 - 30% unprocessed; -, < 10% unprocessed.
EXAMPLE 18
In vivo growth inhibition assay
To determine the biological consequences of FPTase inhibition, the effect of the compounds of the instant invention on the anchorage-independent growth of Ratl cells transformed with either a v-ras, v-raf, or v-mos oncogene is tested. Cell lines transformed with human Ha-ras, N-ras or Ki4B-ras can also be utilized. Cells transformed by v-Raf and v-Mos may be included in the analysis to evaluate the specificity of instant compounds for Ras-induced cell transformation.
Rat 1 cells transformed with either v-ras, v-raf, or v-mos are seeded at a density of 1 x 104 cells per plate (35 mm in diameter) in a 0.3% top agarose layer in medium A (Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum) over a bottom agarose layer (0.6%). Both layers contain 0.1 % methanol or an appropriate concentration of the instant compound (dissolved in methanol at 1000 times the final concentration used in the assay).
The cells are fed twice weekly with 0.5 ml of medium A containing 0.1 % methanol or the concentration of the instant compound. Photomicrographs are taken 16 days after the cultures are seeded and comparisons are made.