WO1997043989A1 - Compositions, methods and devices for the transdermal delivery of drugs - Google Patents
Compositions, methods and devices for the transdermal delivery of drugs Download PDFInfo
- Publication number
- WO1997043989A1 WO1997043989A1 PCT/US1997/008636 US9708636W WO9743989A1 WO 1997043989 A1 WO1997043989 A1 WO 1997043989A1 US 9708636 W US9708636 W US 9708636W WO 9743989 A1 WO9743989 A1 WO 9743989A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- progesterone
- composition
- drug
- weight
- wafer
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F6/00—Contraceptive devices; Pessaries; Applicators therefor
- A61F6/06—Contraceptive devices; Pessaries; Applicators therefor for use by females
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
- A61K47/38—Cellulose; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/70—Web, sheet or filament bases ; Films; Fibres of the matrix type containing drug
- A61K9/7023—Transdermal patches and similar drug-containing composite devices, e.g. cataplasms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/02—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/14—Esters of carboxylic acids, e.g. fatty acid monoglycerides, medium-chain triglycerides, parabens or PEG fatty acid esters
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
Definitions
- the present invention relates to compositions for the transdermal delivery of hormones comprising a hydrogel-forming base mixture and a skin permeation enhancer. Methods for the treatment of disorders responsive to the administration of hormones are also provided. The present invention further relates to devices for the transdermal delivery of drugs.
- Conventional dosage types include sublingual (under the tongue), oral (capsules, tablets, liquids), injectable, nasal and parenteral (suppository and non-oral) forms. While oral dosage forms comprise a substantial majority of all present dosage forms and offer ease of administration and low cost-per-use, they can suffer from inconvenient dosing intervals, side effects and reduced efficacy. Conventional dosage forms have disadvantages in certain patients, including unpredictable blood levels, difficult or uncomfortable administration and poor compliance. In order to maintain optimum blood levels, some conventional forms of drug delivery require frequent doses which can be difficult to remember or understand, pa ⁇ icularly for the elderly patient. Failure to comply with a recommended drug regimen can endanger a patient's health.
- Controlled drug delivery systems have been introduced within the last decade to eliminate or reduce the limitations of conventional dosage forms.
- One type of controlled delivery is transdermal delivery, which involves delivery of a therapeutic agent through the skin for distribution within the body by the circulation of the blood.
- Transdermal delivery can be compared to continuous, controlled intravenous delivery of a drug using the skin as a port of entry instead of an intravenous needle.
- the therapeutic agent passes through the outer layers of the skin, diffuses into the capillaries, or tiny blood vessels in the skin, and then is transported into the main circulatory system.
- drugs which have successfully been delivered transdermally include scopolamine for the treatment of motion sickness, nitroglycerin for the treatment of angina, estrogen and combined estrogen/progestogen for menopausal symptoms and osteoporosis, isosorbide dinitrate for angina, clonidine for hypertension, nicotine for smoking cessation, fentanyl for pain management and testosterone for male hypogonadism.
- progesterone is used in the treatment of premenstrual syndrome, menopausal hormone replacement therapy (in combination with estrogen), infertility and a variety of gynecological conditions.
- progesterone is chiefly produced by the corpus luteum of the ovary with smaller amounts made in the adrenal cortex in both sexes, and in the testes of males.
- mitochondria Within the cytoplasm of cells are minute organelles called the mitochondria, which convert cholesterol to pregnenolone.
- Pregnenolone on being transferred to the cytoplasm, is converted to progesterone or DHEA depending on cell type and body needs.
- progesterone With the development of the corpus luteum at ovulation, the ovarian production of progesterone rapidly rises from 2-3 mg per day to an average of 22 mg per day, peak production being as high as 30 mg per day, a week or so after ovulation. After 10 to 12 days, if fertilization does not occur, ovarian production of progesterone falls dramatically. It is the sudden decline in progesterone levels which triggers the shedding of the secretory endometrium leading to a renewal of the entire menstrual cycle. During pregnancy, production of progesterone is taken over by the placenta which secretes an ever increasing supply, reaching 300- 400 mg per day during the third trimester.
- Progesterone as it is secreted into the blood stream is bound within a water soluble protein (termed cortisol binding globulin; CBG), being the same globulin used by cortisol for passage through the plasma. Only a small portion of progesterone (2- 10%) circulates unbound through the plasma.
- CBG cortisol binding globulin
- a molecular framework of progesterone, having been derived from cholesterol, is very similar to the cholesterol molecule, and like cholesterol is fat soluble. As such, it would not be soluble in the watery plasma were it not for the CBG protein carrier.
- progesterone In close proximity to a cell, progesterone is freed from the CBG and passes easily through cell membranes into the cell cytoplasm where, if it encounters and binds to an accessible receptor, forms an activated complex which migrates into the cell nucleus for binding with an accessible DNA segment (genome) resulting in the formation of a specific RNA by which the cellular effects of progesterone are brought into being. If the progesterone passes into a cell lacking an appropriate progesterone receptor, it will simply pass on through and out the cell again. Eventually, progesterone molecules are carried by the blood through the liver where they are inactivated and disposed of by the bile and urine. Progesterone, in addition to its own intrinsic hormonal effect is an important precursor in the biosynthesis of various hormones.
- progesterone Specialized cells in key organs throughout the body use progesterone to synthesize other hormones as needed, specifically the adrenal corticosteroids, estrogen and testosterone. This aspect of progesterone distinguishes it from most other hormones which are at a metabolic endpoint. This means they are unable to be used in further metabolic functions, except to be metabolized for excretion. More specifically, the various synthetic analogues of progesterone now being promoted heavily have undergone molecular alterations at unusual positions that inhibit further metabolism, and thus are not subject to feedback control by the body to prevent excessive or improperly prolonged activity. Unfortunately, these molecular alterations carry a heavy burden of potential undesirable side effects.
- progesterone is known to be metabolized within the skin by the 5- ⁇ -reductase enzyme which converts it to inactive 5- ⁇ -dihydroxyprogesterone (R. Sitruk-Ware, 1995, “Transdermal Application of Steroid Hormones for Contraception,” J. Steroid Biochem. Molec. Biol. 53 (1- 6):247-251).
- 5- ⁇ -reductase enzyme which converts it to inactive 5- ⁇ -dihydroxyprogesterone
- the desired goal of transdermal delivery of progesterone is to be able to maintain consistent serum levels of progesterone at relatively low dosage levels without requiring multiple dosing.
- Low rates of transdermal delivery of progesterone have been reported by various researchers. For example, Guy et al. (1987, "Kinetics of Drug Absorption Across Human Skin In Vivo," Pharmacol. Skin 1:70-76), disclose that about 1.2 ⁇ g/c ⁇ r penetrated in a 24-hour period, when the drug was applied as a thin film on the skin in vivo.
- Barry and Bennett (1987, "Effect of Penetration Enhancers on the Permeation of Mannitol, Hydrocortisone, and Progesterone Through Human Skin, " J. Pharm. Pharmacol.
- permeation enhancers Compounds that act as permeation enhancers have been added to transdermal drug delivery systems for a number of drugs, including progesterone.
- Pfister and Hsieh (1990, in "Permeation Enhancers with Transdermal Drug Delivery Systems: Part II: System Design Considerations," Pharmaceutical Technology, October 1990: 55-60), disclose a wide variety of permeation enhancers.
- isopropyl palmitate and isopropyl myristate are disclosed as cosolvents to enhance the solubility of nitroglycerin in a polymer matrix-type transdermal system, which in turn optimizes the release of the drug from the system.
- ethanol is disclosed as enhancing the solubility of 17- /3-estradiol in the reservoir compartment of a transdermal drug delivery device.
- Other skin penetration enhancers are disclosed, including stearyl alcohol, glycerol, 2- pyrrolidone, urea, propylene glycol, oleic acid, and palmitic acid. D.R.
- Butylurea has also been disclosed as a permeation enhancer.
- U.S. Patent No. 5, 128,376 discloses a method for percutaneous administration of a drug from a mixture of an adjuvant, a solvent and a diol/triol moderator, wherein the solvent, which enhances permeation, may be a substituted urea such as butylurea.
- U.S. Patent No. 4,863,952 discloses an improved method of drug administration using a mixture comprising pyrrolidone carboxylic acid esters as percutaneous promoters, and optionally, substituted ureas such as butylurea. S.K. Han et al.
- Patent Nos. 5,344,655 and 5,254,338 disclose that hydrogel bases containing water soluble polymers such as cellulose derivatives are known in the art for delivery of drugs through the skin.
- U.S. Patent No. 4,693,887 discloses hydrogel compositions for the controlled release of contraceptives such as progesterone.
- the hydrogels are blends of either N-vinyl lactam or a copolymer of N-vinyl lactam and may further comprise spermicides such as urea.
- 5,405,366 discloses an adhesive hydrogel comprising an aqueous mixture of a radiation crosslinkable water- soluble polymer such as a polymer of N-vinyl-2-pyrrolidone and ethylene oxide and a humectant such as propylene glycol which may be used in a transdermal drug delivery system.
- the hydrogel may also contain preservatives such as propyl paraben and methyl paraben.
- U.S. Patent No. 4,593,053 discloses a skin-compatible pressure- sensitive adhesive hydrogel comprising polyvinyl pyrrolidone and polyvinyl alcohol, a polar plasticizer or humectant such as propylene glycol, water and a drug.
- the composition may also contain cellulose derivatives to increase strength and guar gum to increase tackiness.
- the transdermal delivery of progesterone, progestins, estrogens and testosterone from hydrogel matrices comprising permeation enhancers is also known.
- U.S. Patent No. 5,030,629 discloses transdermal formulations containing progesterone, ethanol, saline and an imidazoline penetration enhancer.
- Dosage forms of the formulations for application to the skin include gels, which may comprise inert carriers such as propylene glycol, urea and methylcellulose.
- androgens such as testosterone and estrogens such as estradiol comprising water- and fat-soluble abso ⁇ tion enhancers
- a water-absorbent resin such as a vinyl acetate-acrylic acid ester copolymer that swells to form a hydrogel upon contact with water.
- U.S. Patent No. 5,064,654 discloses a transdermal drug formulation comprising a drug such as progesterone or estradiol, water and ethanol. The formulation may also contain an adhesive or gelling agent such as pectin, guar gum or methyl cellulose.
- 4,942, 158 discloses a composition comprising a combination of isopropyl alcohol and isobutyl alcohol to enhance the transdermal penetration of steroids such as estradiol, or a combination of estradiol with a progestogen.
- the composition may also include water and a gelling agent such as methyl cellulose.
- U.S. Patent No. 4,865,848 discloses compositions for enhancing the transdermal delivery of drugs, including progesterone, comprising sucrose esters as penetration enhancers.
- the permeation enhancer and the drug are dispersed in a matrix which may be a gel or a hydrophilic polymer.
- Natural progesterone which is the form of progesterone that is produced by the body, has been administered in oral, injectable and suppository forms.
- injectable and suppository forms are obvious: they are burdensome to administer.
- oral forms are that they are short acting, and to maintain adequate blood levels, they have to be dosed throughout the day.
- the bulk of orally administered progesterone is metabolized by the digestive system and excreted before it can be used by the body.
- progesterone whether administered orally, vaginally or rectally, has a half life in the body of only about 2.2 hours. Therefore, much larger amounts than the body actually requires must be dosed to maintain effective blood levels.
- progestins fall primarily into two categories. The first group, pregnanes, is derived from 17- ⁇ -acetoxy progesterone. A classic example is medroxy progesterone acetate (Provera). With an increased affinity for progesterone receptors, these compounds have marked progestational activity. They possess anti-estrogenic anti- gonadotropic, and no significant androgenic properties. A second group, estranes, derived from 17- ⁇ -ethinyl-l-nortestosterone, includes norethindrone acetate (aygestin).
- progestins can cause many negative side effects, such as sudden or partial loss of vision, thrombophlebitis, pulmonary embolism, cerebral thrombosis, salt and fluid retention, epilepsy, migraine, asthma, cardiac or renal dysfunction, weight gain, rise in blood pressure, headaches, depression, decreased glucose tolerance leading to diabetes in predisposed individuals, acne, alopecia, hirsutism, decrease in T3 uptake and thyroid regulation.
- a disadvantage of conventional patch systems is that many of them either inco ⁇ orate drugs in an adhesive or require an adhesive to affix the patch to patient's skin. These adhesives can irritate the skin, causing patient non-compliance.
- a non-adhesive transdermal delivery system that can deliver therapeutically effective amounts of hormones such as natural progesterone, progestins, estrogens and testosterone that will reduce or eliminate the skin irritation experienced by many patients using current adhesive patch transdermal delivery systems. This need is satisfied by a particular embodiment of the invention.
- the present invention is directed to compositions for the transdermal delivery of a hormone selected from the group consisting of progesterone, progestin, estrogen, and testosterone, or a combination thereof, said compositions comprising or alternatively consisting or consisting essentially of: (a) a hydrogel, or a base mixture that when combined with water forms a hydrogel; (b) a permeation enhancer selected from the group consisting of urea, hydroxyurea, or an alkylurea; and (c) a hormone selected from the group consisting of progesterone, progestin, estrogen, and testosterone, or a combination thereof.
- the hormone is natural progesterone.
- Natural progesterone is superior to synthetic forms of progesterone, known as progestins, in treating gynecological conditions.
- the invention also provides apparatuses for transdermal hormone delivery containing the compositions of the invention. Methods of transdermal drug delivery, and of treatment of disorders responsive to the administration of hormones, are also provided.
- Transdermal delivery according to the invention offers significant advantages over conventional means of drug administration. It is a comfortable, convenient and noninvasive means of drug delivery. The variable rates of abso ⁇ tion and metabolism encountered in oral treatment are avoided, and side effects such as gastrointestinal irritation and the like are eliminated. Transdermal drug delivery also makes possible a high degree of control over blood concentrations of particular drugs.
- Transdermal drug delivery according to the invention also helps provide patients with a drug's maximum therapeutic effect and decreases the risk of adverse side effects or diminished therapeutic effect due to excessive or insufficient blood concentrations.
- the therapeutic effect of a drug is typically achieved only when the drug is within a specific concentration range in the bloodstream. This blood concentration range is often called the drug's "therapeutic window. " Below this range the drug may be ineffective, and above it the drug may cause unwanted side effects.
- Many conventional forms of drug delivery administer higher concentrations than are required in order to maintain effective blood levels between doses. However, blood levels often fall below effective concentrations prior to administration of the next dose.
- Transdermal drug delivery systems using the compositions of the present invention are designed to maintain a precise and continuous flow of drug into the bloodstream.
- transdermal drug delivery can frequently maximize a drug's therapeutic effect by avoiding the gastrointestinal ("GI") tract and "first pass" liver metabolism.
- Oral drug delivery is often unreliable because the achievement of therapeutic blood levels depends on several factors, including the drug's chemical composition, the patient's physical condition, chemical and physical reactions between the drug and substances in the GI tract and the timing of drug administration.
- GI tract abso ⁇ tion a drug must pass through the liver before entering the bloodstream.
- the liver metabolizes a large portion of the drug.
- orally dosed drugs must generally be administered at levels which exceed optimal therapeutic levels, potentially resulting in adverse side effects.
- Transdermal drug delivery systems avoid many of the problems associated with conventional drug delivery, and are capable of conveniently and consistently delivering drugs over a number of hours or days.
- the transdermal drug delivery systems of the invention may also improve the safety of drug administration, since they can be removed quickly and easily. If a patient has an adverse reaction to a drug, rapid removal of the transdermal drug delivery device can minimize the extent of such an adverse reaction.
- compositions comprising (i) a gelling agent consisting of methylcellulose or at least one natural gum, or a mixture thereof; (ii) at least one natural gum; (iii) glucose; (iv) propylparaben; (v) methyl paraben; and (vi) sodium chloride.
- the compositions of the present invention may further comprise a glycolic, alcoholic or oil-based additive such as propylene glycol.
- the compositions may further comprise coloring, fragrance or other pharmaceutically acceptable additives.
- compositions of the present invention may also comprise pectin.
- compositions of the present invention consist essentially of methyl cellulose, a natural gum selected from the xanthan and guar gums, glucose, propyl paraben, methyl paraben, sodium chloride and pectin.
- compositions of the present invention comprise 50-80% (by weight) methyl cellulose, 15-25% of a natural gum selected from the xanthan and guar gums, 3-
- compositions comprise about 63 % methylcellulose, about 21 % guar gum, about 5% glucose, about 3.5% propylparaben, about 3% methyl paraben, about 3% pectin and about 1.5% sodium chloride.
- compositions of the present invention further comprise a drug selected from the group consisting of nicotine, nitroglycerin, albuterol, VERAPAMIL ® , scopolamine, n-butylurea, fentanyl, mo ⁇ hine, butaconazole, acetylsalicylic acid, MINOXIDIL ® , lidocaine, racemic menthol, methyl salicylate, benzalkonium chloride, DEET ® , phenobarbital, iodine, insulin, salicylic acid, nonoxynol-9, erythromycin, tetracycline, cephalosporins, and acetaminophen.
- a drug selected from the group consisting of nicotine, nitroglycerin, albuterol, VERAPAMIL ® , scopolamine, n-butylurea, fentanyl, mo ⁇ hine, butaconazole, acetylsalicylic acid, MI
- compositions of the present invention further comprise a substituted urea of the formula R-NH-CO-NH 2 wherein R is " hydrogen, hydroxyl, or a lower alkyl having from 1 to 8 carbon atoms selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert-butyl, pentyl, hexyl, heptyl and octyl.
- R is " hydrogen, hydroxyl, or a lower alkyl having from 1 to 8 carbon atoms selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert-butyl, pentyl, hexyl, heptyl and octyl.
- the substituted urea is butylurea.
- the compositions of the present invention may be provided in the form of a hydrogel comprising water and a base mixture, said base mixture comprising: (i) a gelling agent consisting of methylcellulose or at least one natural gum, or a mixture thereof; (ii) at least one natural gum; (iii) glucose; (iv) propylparaben; (v) methyl paraben; and (vi) sodium chloride.
- the compositions of the present invention may be provided in the form of a dry powder comprising (a) a drug; and
- a base mixture comprising (i) a gelling agent consisting of methylcellulose or at least one natural gum, or a mixture thereof; (ii) at least one natural gum; (iii) glucose; (iv) propylparaben; (v) methyl paraben; and (vi) sodium chloride.
- compositions of the present invention may also be provided in the form of a paste comprising
- compositions comprising:
- a hydrogel comprising water and a base mixture, said base mixture comprising or consisting essentially of: (i) a gelling agent consisting of methylcellulose or at least one natural gum, or a mixture thereof; (ii) at least one natural gum; (iii) glucose; (iv) propylparaben; (v) methyl paraben; and (vi) sodium chloride;
- a hormone selected from the group consisting of progesterone, progestin, estrogen, and testosterone, or a mixture of any two or more of the foregoing.
- compositions of the present invention may further comprise a glycolic, alcoholic or oil-based additive such as propylene glycol.
- the compositions may further comprise coloring, fragrance, or other pharmaceutically acceptable additives.
- the compositions of the present invention may also comprise pectin.
- the hydrogel-forming base mixture of the present invention consists essentially of methyl cellulose, a natural gum selected from the xanthan and guar gums, glucose, propyl paraben, methyl paraben, pectin and sodium chloride. More preferably, the hydrogel-forming base mixture of the present invention consists essentially of 50-80% (by weight) methyl cellulose, 15-25% of a natural gum selected from the xanthan and guar gums, 3-7% glucose, 2-3.5% propylparaben, 1.5- 3% methylparaben, 0.75-3.5% pectin, and 1-3% sodium chloride.
- the base mixture consists essentially of about 63% methylcellulose, about 21 % guar gum, about 5% glucose, about 3.5% propylparaben, about 3 % methyl paraben, about 3% pectin and about 1.5% sodium chloride.
- compositions of the present invention consist essentially of:
- compositions of the present invention consist essentially of: ( a ) about 9% by weight of a base mixture consisting essentially of: about
- methyl cellulose about 21 % guar gum, about 5% glucose, about 3.5% propylparaben, about 3% methylparaben, about 1.5% sodium chloride and about 3% pectin;
- compositions of the present invention may be used as vehicles or carriers for the delivery of a wide variety of drugs to a subject.
- Drugs that can be delivered using the compositions of the present invention include, but are not limited to nicotine, nitroglycerin, albuterol, VERAPAMIL ® , scopolamine, n-butylurea, fentanyl, mo ⁇ hine, butaconazole, acetylsalicylic acid, MINOXIDIL ® , lidocaine, racemic menthol, methyl salicylate, benzalkonium chloride, DEET ® , phenobarbital, iodine, insulin, salicylic acid, nonoxynol-9, erythromycin, tetracycline, cephalosporins, and acetaminophen.
- compositions of the present invention are particularly useful as pharmaceutically acceptable bases for the delivery of drugs such as hormones selected from the group consisting of progesterone, progestin, estrogen, testosterone, and mixtures of any two or more of the foregoing.
- the compositions of the present invention are particularly useful for the transdermal delivery of these hormones.
- compositions of the present invention are particularly useful for the transdermal delivery of progesterone.
- the compositions of the present invention are useful in a variety of vaginal applications.
- compositions of the present invention are useful as vaginal lubricants, spermicides, and to treat vaginal yeast infections.
- the present invention provides devices for transdermal delivery of drugs that do not require the use of an adhesive that has the potential to irritate the skin.
- a watch or watch-like device of the invention may comprise a watch, or are in the form of a watch.
- a watch or watch-like device of the invention has a watch-case with a recessed chamber on the back face of the watch-case in contact with the skin.
- the recessed chamber contains a wafer having a top face and a bottom face containing a drug and a pharmaceutically acceptable base.
- the recessed chamber of the watch-case comprises a fluid-filled or hydrogel cushion later situated against the top face of the drug-containing wafer.
- the recessed chamber of the watch-case comprises a heating layer having a top face and a bottom face in which the bottom face of the heating layer is situated against the top face of the drug-containing wafer.
- the recessed chamber of the watch-case comprises permanent, closed cell having a top face and a bottom face, the bottom face of which is situated against the top face of the heating layer.
- a device for the transdermal delivery of a drug to a subject which comprises a disc-shaped drug reservoir.
- the reservoir has a recessed chamber which can contain a wafer having a top face and a bottom face comprising a drug and a pharmaceutically acceptable base.
- the reservoir clips on to the back face of a watch or watch-like device such that drug- containing wafer is maintained in contact with the subject's skin.
- the reservoir slides onto a band capable of being attached to the limb of a subject such that the drug-containing wafer is maintained in contact with the skin of the subject's limb.
- the recessed chamber of the clip-on or slide-on drug reservoir comprises a fluid-filled or hydrogel cushion later situated against the top face of the drug-containing wafer.
- the recessed chamber of the clip-on or slide-on drug reservoir comprises a heating layer having a top face and a bottom face in which the bottom face of the heating layer is situated against the top face of the drug-containing wafer.
- the recessed chamber of the clip-on or slide-on drug reservoir comprises permanent, closed cell having a top face and a bottom face, the bottom face of which is situated against the top face of the heating layer.
- a device for the transdermal delivery of a drug to a subject in accordance with the present invention comprises a glove wherein the inside of the glove is lined with a layer containing a composition of the invention comprising a pharmaceutically acceptable hydrogel base and a drug.
- a device for the transdermal delivery of a drug to a subject in accordance with the present invention comprises a band or a strap having a surface coated with a drug-containing composition of the present invention, wherein the band or strap is capable of being attached to the limb of a subject such that the drug- containing composition is maintained in contact with the skin of the subject's limb.
- kits comprising the recessed chamber- type, the clip-on drug reservoir-type, the slide-on drug reservoir-type, the glove-type or the band-type of transdermal drug delivery devices.
- the present invention also provides methods for the treatment of conditions responsive to hormone replacement therapy such as premenstrual syndrome, menopause, infertility, dysfunctional bleeding, co ⁇ us luteum failure, senile vulvo-vaginitis, hypogonadism and osteoporosis.
- the invention also provides methods of contraception in males and females.
- the invention further provides a method of delivering a hormone or mixture of hormones to a subject.
- These methods of the invention involve placing a composition of the invention in contact with the skin of a subject such that an effective amount of the hormone or mixture of hormones is delivered transdermally to said subject.
- the present invention also provides methods for the treatment of vaginal disorders such as yeast infections.
- the present invention also provides methods of providing contraception using spermicides.
- the present invention further provides methods for providing vaginal lubrication. These methods involve placing a composition of the present invention inside the vagina of a subject.
- FIG. 1 Cross-sectional view of a Membrane-Moderated Transdermal Drug Delivery System.
- the system comprises: a drug reservoir 1 that may contain a composition of the present invention; a drug-impermeable backing layer 2, which can be plastic, metal, metallic laminate or any other pharmaceutically acceptable material; a rate-controlling polymeric membrane 3; and an adhesive layer 4, which can be any adhesive known in the art.
- Arrows 5 show an exemplary route of passage of the drug out the device into the skin.
- Figure 1 is adapted from R. Sitruk-Ware, 1989, "Transdermal Delivery of Steroids, " Contraception 39 (l): l-20, at page 9, Figure 5.
- Figure 2. Cross-sectional view of an Adhesive Diffusion-Controlled Transdermal
- the system comprises: a drug reservoir layer 1 which may comprise a composition of the present invention; a drug-impermeable backing layer 2, which can be plastic, metal, metallic laminate or any other pharmaceutically acceptable material; a rate-controlling adhesive layer 3; and an adhesive layer 4.
- the adhesive layers may comprise any adhesive known in the art.
- Arrows 5 show an exemplary route of passage of the drug out the device into the skin.
- Figure 2 is adapted from R. Sitruk-Ware, 1989, "Transdermal Delivery of Steroids," Contraception 39 (l): l-20, at page 10, Figure 6.
- FIG. 3 Cross-sectional view of a Microreservoir-Type Transdermal Drug Delivery System.
- the system comprises: an adhesive foam pad 1 made of flexible polyurethane; an occlusive baseplate 2 comprising an aluminum foil disc; an adhesive rim 3; microscopic drug reservoirs 4; and a polymer matrix 5 which may comprise a composition of the present invention.
- Arrows 6 show an exemplary route of passage of the drug out the device into the skin.
- Figure 3 is adapted from R. Sitruk-Ware, 1989, "Transdermal Delivery of Steroids," Contraception 39 (l):l-20, at page 12, Figure 8.
- Figure 4. Cross-sectional view of a Matrix Dispersion-Type Transdermal Drug
- the system comprises: a drug-impermeable backing layer 1 , which may be plastic, metal, metallic laminate or any other pharmaceutically acceptable material; an absorbent pad 2; an occlusive baseplate 3 comprising an aluminum foil disc; a drug reservoir 4 which may contain a composition of the present invention; and an adhesive rim 5.
- Arrows 6 depict an exemplary route of passage of the drug out the device into the skin.
- Figure 4 is adapted from R. Sitruk-Ware, 1989, "Transdermal Delivery of Steroids, " Contraception 39 (l): l-20, at page 11 , Figure 7.
- FIG. 5 Perspective view of the top face of a recessed-chamber type of transdermal drug delivery device of the present invention, comprising: a watch case 1 and a band or strap 2.
- FIG. 6 Perspective view of the back face of one embodiment of the recessed- chamber type of transdermal drug delivery device of the present invention, comprising: a watch case 1 ; a recessed chamber 2 capable of containing a wafer comprising a drug and a pharmaceutically acceptable base, which may comprise a composition of the present invention; and a band or strap 3.
- FIG. 7 Cross-sectional view of one embodiment of the recessed-chamber type of transdermal drug delivery device of the present invention, comprising: a watch case 1 ; a recessed chamber 2; a band or strap 3; and a wafer 4 comprising a drug and a pharmaceutically acceptable base which may comprise a composition of the present invention.
- Figure 8. Cross-sectional view of another embodiment of the recessed-chamber type of transdermal drug delivery device of the present invention, comprising: a watch case 1 ; a recessed chamber 2; a band or strap 3; a fluid-filled cushion 4; a wafer 5 comprising a drug and a pharmaceutically acceptable base which may comprise a composition of the present invention; and a non-slip material 6 on the rim of the recessed chamber.
- FIG. 9 Perspective view of one embodiment of the top face of a watch or watch-like device having the clip-on drug reservoir type of transdermal drug delivery device of the present invention attached to its back face comprising: a watch case 1 ; a band or strap 2; and a clip-on drug reservoir 3.
- FIG. 10 Cross-sectional view of one embodiment of the clip-on or slide-on drug reservoir type of transdermal drug delivery device of the present invention, comprising: a plurality of clips 1 capable of attaching the clip-on drug reservoir to the back face of a watch or watch-like device; a recessed chamber 2; a wafer 3 comprising a drug and a pharmaceutically acceptable base which may comprise a composition of the present invention; and a soft flair skin seal 4.
- FIG 11. Cross-sectional view of another embodiment of the clip-on or slide- on drug reservoir type of transdermal drug delivery device of the present invention, comprising: a plurality of clips 1 capable of attaching the drug reservoir to the back face of a watch or watch-like device; a recessed chamber 2; a permanent closed cell 3 comprising dense polyethylene capable of providing slight downward pressure on the drug-containing wafer; a heating layer 4 comprising a means for heating the drug- containing wafer; a wafer 5 comprising a drug and a pharmaceutically acceptable base which may comprise a composition of the present invention; and a soft flair skin seal 6.
- FIG. 13 Perspective view of the bottom face of one embodiment of the clip- on or slide-on drug reservoir type of transdermal drug delivery device of the present invention, comprising: a plurality of clips 1 capable of attaching the clip-on drug reservoir to the back face of a watch or watch-like device; a recessed chamber 2; a soft flair skin seal 3; and a plurality of slots 4 that are capable of having a band or strap threaded through them.
- FIG. 14 Cross-sectional view of one embodiment of the slide-on drug reservoir type of transdermal drug delivery device of the present invention attached to a band, comprising: a slide-on drug reservoir 1; a band or strap 2; a recessed chamber 3; a wafer 4 comprising a drug and a pharmaceutically acceptable base which may comprise a composition of the present invention; and a soft flair skin seal 5.
- FIG. 15 Perspective view of the bottom face of one embodiment of the slide- on drug reservoir type of transdermal drug delivery device of the present invention, comprising: a plurality of slots 1 that are capable of having a band or strap threaded through them; a wafer 2 comprising a drug and a pharmaceutically acceptable base which may comprise a composition of the present invention; a soft flair skin seal 3; and a band or strap 4.
- FIG. 16 Cross-sectional view of the Single-Chamber Diffusion Cell used in the skin permeation measurements described in Section 6.2.
- the cell comprises: a retaining nut 1; a flanged PVC washer 2 with aluminum foil coating 3; an o-ring 4; a skin disc 5, which is cemented to a nylon washer 6; a teflon-coated magnetic stirring button 7; receptor fluid 8; and a stopper 9 comprising a foil-coated flanged washer.
- Figure 17 A graph depicting the cumulative amount of progesterone penetrated, ( ⁇ g/cm 2 , y-axis) against time, (hours x-axis), from sample A-l through a dialysis membrane, as described in Section 6.2.2. The graph shows a penetration rate of 30.8 ⁇ g/cm 2 of progesterone through the dialysis membrane.
- Figure 18 A graph depicting the cumulative amount of progesterone penetrated, ( ⁇ g/cm 2 , y-axis) against time, (hours, x-axis), from covered and not-covered samples of A-l through human skin, as described in Section 6.2.3. Curve 1 corresponds to the covered sample of A-l and Curve 2 corresponds to the not-covered sample of A-l .
- Figure 19 A graph depicting the cumulative amount of progesterone penetrated, ( ⁇ g/cm 2 , y-axis) against time, (hours, x-axis), from samples A-l , A-2, and A-3 through human skin, as described in Section 6.2.4.
- Curve 1 corresponding to sample A-l, shows a rate of 3.1 ⁇ g/cm 2 /hr.
- Curve 2 corresponding to sample A-2, shows a rate of 1.9 ⁇ g/cm 2 /hr.
- Curve 3 corresponding to sample A-3, shows a rate of 9.8 ⁇ g/cm 2 /hr.
- FIG. 20 Cross-sectional view of the redesigned Diffusion Cell Assembly used in the skin permeation measurements described in Section 6.4.
- the cell comprises: a glass receptor 1 ; a magnetic stirrer 2; saline 3; a teflon washer 4; a notch which eliminates air bubbles 5; an assembled donor 6; a donor chamber 7; test material on epidermal membrane cemented to nylon washer 8; a foil cover 9; an O-ring 10; a flanged washer 11; and a nut 12.
- FIG. 21 Progesterone penetration of human skin in vitro.
- compositions comprising (i) a gelling agent consisting of methylcellulose or at least one natural gum, or a mixture thereof; (ii) at least one natural gum; (iii) glucose; (iv) propylparaben; (v) methyl paraben; and (vi) sodium chloride.
- compositions of the present invention may further comprise a glycolic, alcoholic or oil-based additive.
- glycolic, alcoholic, or oil-based additives that may be used in the compositions of the present invention include, but are not limited to propylene glycol, glycerin, mineral oil, corn oil, bran oil, rice oil, soy oil, ethylene glycol, xylene and alcohols such as ethyl alcohol.
- a preferred additive is propylene glycol.
- the compositions may further comprise coloring, fragrance or other pharmaceutically acceptable additives.
- the compositions of the present invention may also comprise pectin.
- compositions of the present invention consist essentially of methyl cellulose, a natural gum selected from the xanthan and guar gums, glucose, propyl paraben, methyl paraben, sodium chloride and pectin.
- compositions of the present invention comprise 50-80% (by weight) methyl cellulose, 15-25% of a natural gum selected from the xanthan and guar gums, 3- 7% glucose, 2-3.5% propylparaben, 1.5-3% methylparaben, 1-3 % sodium chloride and 0.75-3.5 % pectin. Even more preferably, the compositions comprise about 63% methylcellulose, about 21 % guar gum, about 5% glucose, about 3.5% propylparaben, about 3% methyl paraben, about 3 % pectin and about 1.5% sodium chloride.
- drugs are included in the compositions of the present invention.
- compositions of the present invention further comprise a substituted urea of the formula R-NH-CO-NH 2 wherein R is hydrogen, hydroxyl, or a lower alkyl having from 1 to 8 carbon atoms selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert-butyl, pentyl, hexyl, heptyl and octyl.
- the substituted urea is butylurea.
- compositions of the present invention may be provided in the form of a dry powder comprising
- compositions of the present invention may also be provided in the form of a paste comprising
- a base mixture comprising (i) a gelling agent consisting of methylcellulose or at least one natural gum, or a mixture thereof; (ii) at least one natural gum; (iii) glucose; (iv) propylparaben; (v) methyl paraben; and (vi) sodium chloride.
- Dry powder and paste compositions of the present invention can form hydrogels upon the addition of water.
- compositions for the transdermal delivery or hormones such as progesterone, progestin, estrogen, testosterone, or combinations thereof, said compositions alternatively consisting of or consisting essentially of: (a) a hydrogel, or a base mixture that when combined with water forms a hydrogel; (b) a permeation enhancer selected from the group consisting of urea, hydroxyurea, or an alkylurea; and (c) a hormone selected from the group consisting of progesterone, progestin, estrogen, testosterone, or combinations thereof.
- the hormone is natural progesterone. Natural progesterone is superior to synthetic forms of progesterone, known as progestins, in treating gynecological conditions.
- the invention also provides apparatuses for transdermal hormone delivery containing the compositions of the invention. Methods of transdermal delivery, and of treatment of disorders responsive to the administration of hormones are also provided.
- the compositions of the invention provide more effective and efficient means for delivery of a therapeutically effective amount of the hormone(s) contained therein to the bloodstream of a patient.
- compositions comprising:
- a hydrogel comprising water and a base mixture, said base mixture comprising or consisting essentially of: (i) a gelling agent consisting of methylcellulose or at least one natural gum, or a mixture thereof; (ii) at least one natural gum; (iii) glucose; (iv) propylparaben; (v) methyl paraben; and (vi) sodium chloride;
- a hormone selected from the group consisting of progesterone, progestin, estrogen, and testosterone, or a mixture of any two or more of the foregoing.
- the composition may also optionally contain a glycolic, alcoholic or oil-based additive.
- glycolic, alcoholic or oil-based additives examples include propylene glycol, glycerin, mineral oil, corn oil, bran oil, rice oil, soy oil, ethylene glycol, xylene, and alcohols such as ethyl alcohol.
- a preferred additive is propylene glycol.
- the compositions may also contain colorings, fragrances or other pharmaceutically acceptable additives.
- the water used in the inventive compositions is distilled water.
- the compositions of the present invention may also comprise pectin.
- the hydrogel-forming base mixture of the present invention consists essentially of methyl cellulose, a natural gum selected from the xanthan and guar gums, glucose, propyl paraben, methyl paraben, pectin and sodium chloride.
- the hydrogel-forming base mixture of the present invention consists essentially of 50-80% (by weight) methyl cellulose, 15-25 % of a natural gum selected from the xanthan and guar gums, 3-7% glucose, 2-3.5% propylparaben, 1.5- 3 % methylparaben, 0.75-3.5 % pectin, and 1-3 % sodium chloride. Even more preferably, the base mixture consists essentially of about 63% methylcellulose, about 21 % guar gum, about 5% glucose, about 3.5% propylparaben, about 3% methyl paraben, about 3% pectin and about 1.5% sodium chloride.
- compositions of the present invention consist essentially of:
- compositions of the present invention consist essentially of:
- a base mixture consisting essentially of: about 63% (by weight) methyl cellulose, about 21 % guar gum, about 5% glucose, about 3.5% propylparaben, about 3% methylparaben, about
- the hydrogel-forming base mixture may optionally include pectin.
- Natural gums which may be used in the hydrogel-forming base mixture include guar gum and xanthin gum.
- the hydrogel-forming base mixture preferably comprises a methylcellulose gelling agent; however inclusion of methylcellulose is not required. In place of the methylcellulose gelling agent, additional amounts of natural gums such as guar or xanthin gums, or mixtures thereof, may be substituted.
- the hydrogel-forming base mixture of the present invention is preferably made by way of example as follows: A dry powder form is mixed together of each of the natural gum, glucose, propylparaben, methylparaben, and sodium chloride ingredients. Methyl cellulose and pectin dry powders are optionally included. The dry powder mixture is then micronized to a size of approximately one micron. Upon the addition of water, the mixture forms a hydrogel, which varies from a semi-fluid to a solid rubber consistency, depending on the amount of water added.
- the hydrogel-forming base mixture of the present invention consists essentially of 50-80% (percentages are by weight) methyl cellulose, 15-25% of a natural gum, 3-7% glucose, 2-3.5% propylparaben, 1.5-3% methylparaben, and 1-3% sodium chloride.
- the mixture may include 0.75-3.5% pectin.
- the base mixture consists essentially of 63% methylcellulose, 21 % guar gum, 5% glucose, 3.5% propylparaben, 3% methyl paraben, 3% pectin and 1.5% sodium chloride.
- the present invention provides a composition for the transdermal delivery of natural progesterone comprising, or alternatively, consisting essentially of:
- a base mixture comprising: (i) a gelling agent consisting of methylcellulose or at least one natural gum, or mixtures thereof; (ii) at least one natural gum; (iii) glucose; (iv) propylparaben; (v) methyl paraben; and (vi) sodium chloride; which may optionally include pectin; (b) 0.5-15% by weight of a substituted urea permeation enhancer of the formula R-NH-CO-NH 2 , wherein R is hydrogen, hydroxyl or lower alkyl having from 1 to 8 carbon atoms; (c) 5-20% by weight of a hormone selected from the group consisting of progesterone, progestins, estrogens, and testosterone, or mixtures thereof; (d) 0-20% by weight propylene glycol; and
- a particularly preferred embodiment of the present invention is directed to a composition for the transdermal delivery of natural progesterone consisting of: (a) 9% by weight of a base mixture consisting essentially of (1) methylcellulose; (2) guar gum; (3) glucose; (4) propylparaben; (5) methyl paraben; (6) pectin; and (7) sodium chloride;
- the above composition of the present invention is formed by first mixing the base mixture powder, substituted alkyl urea and progesterone in propylene glycol to form a paste. The paste is then heated without boiling until the progesterone and substituted alkyl urea are dissolved and the paste becomes a liquid. This hot glycol mixture is then added to tepid water while stirring with a blade type stirring unit at a speed sufficient to from a vortex at the surface. The hot glycol mixture is added to the stirring water in this manner for 5 to 15 minutes, or until the proper or desired viscosity is achieved. The mixture is then removed from the stirred container and placed in a barrier container for storage to prevent evaporation. Shelf life of the finished product should be several years.
- compositions enhancers of the present invention can comprise monosubstituted lower-alky 1 ureas of the formula:
- R-NH-CO-NH 2 wherein R is hydrogen, hydroxyl or lower alkyl having from 1 to 8 carbon atoms.
- the lower alkyl group has from 1 to 6 carbon atoms; more preferably from 1 to 4 carbon atoms; and most preferably from 3 to 4 carbon atoms.
- Specific examples of the lower alkyl group R include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert- butyl, pentyl, hexyl, heptyl and octyl.
- a particularly preferred monosubstituted lower- alky 1 urea useful in the present invention is butylurea.
- the monosubstituted lower alkyl ureas are known compounds and are readily available from commercial "sources such as the Aldrich Chemical Co. (Milwaukee, WI).
- the permeation enhancer is butylurea.
- the urea derivatives may function as transdermal penetration enhancers.
- Urea derivatives such as butylurea can also function as spermicides when the compositions of the present invention are used in vaginal applications.
- Drugs which may be used in the compositions of the present invention and the disorders which such compositions may be used to treat include, but are not limited to nicotine for smoking cessation, nitroglycerin for angina pectoris, albuterol as an antiasthmatic, VERAPAMIL ® for hypertension, scopolamine for motion sickness, n-butylurea for he ⁇ es sores, fentanyl for acute pain, mo ⁇ hine for pain, steroid hormones for osteoporosis, estrogen and progestin for hormonal replacement, butaconazole for vaginal yeast infection, acetylsalicylic acid as aspirin for pain, MINOXIDIL ® for hair growth, lidocaine for pain, racemic menthol for pain, methyl salicylate for pain, benzalkonium chloride as a germicide, DEET ® for insect control, phenobarbital as a sedative, iodine as an antiseptic, insulin as an antidiabe
- hormones examples include progesterone, progestin, estrogen and testosterone, or a mixture of any two or more of the foregoing. Depending on the application, the present invention may be used to deliver each of these hormones alone, or in various combinations. In a preferred embodiment, the hormones are delivered transdermally using the compositions of the present invention.
- progestins useful in the present invention include but are not limited to are medroxy progesterone acetate, norethindrone, norethindrone acetate, norgestrel, and ethynodiol diacetate.
- Specific examples of estrogens useful in the present invention include but are not limited to 17-/3- estradiol, diethylstilbestrol, estropipate (formerly known as piperazine estrone sulfate), estrone and estriol.
- a most preferred hormone is pregn-4-ene-3,20-dione, which has a molecular weight of 314.47, and is also known as "natural progesterone", or simply,
- progesterone Natural progesterone is produced in the human body, but it can be also be isolated from certain plants, and is available commercially.
- micronized powder forms of natural progesterone useful in the present invention are available from both the Upjohn Chemical Co. (Kalamazoo, MI) and the Berlex Chemical Co. (North Surburban, IL).
- the compositions of the present invention are also suitable for delivery, transdermal and otherwise, of synthetic progestins, estrogens such as 17-/3- estradiol and testosterone. Synthetic progestins, estrogens and testosterone which may be used in the present invention are readily available from a variety of commercial sources well known to those skilled in the art.
- compositions of the present invention are administered vaginally.
- the compositions are delivered transdermal ly, which but necessarily involves application of the composition to a selected intact surface of the skin for a period of time sufficient to provide the desired blood level of the drug.
- the composition is administered to a patient via use of a transdermal delivery device.
- transdermal delivery devices are known and may be used to administer therapeutically effective amounts of hormones using the compositions of the invention.
- Four general types of transdermal drug delivery devices are taught, for example, by R. Sitruk-Ware (1989, "Transdermal Delivery of Steroids," Contraception 39 (l): l-20.
- Transdermal delivery devices are also disclosed by Pfister and Hsieh (1990, "Permeation Enhancers Compatible with Transdermal Drug Delivery Systems: Part II: System Design Considerations," Pharmaceutical Technology (October 1990): 55-60.
- the composition of the present invention may be administered using a membrane-moderated transdermal drug delivery system, as shown in Figure 1.
- the inventive composition is contained within the drug reservoir 1.
- the rate of delivery of the hormone through the skin is simultaneously enhanced by use of the hydrogel and the monosubstituted alkyl urea penetration enhancer of the invention and moderated by a rate -controlling polymeric membrane 3.
- the inventive composition may also be administered to a patient via an adhesive diffusion-controlled transdermal drug delivery system, (see Figure 2) in which the rate of delivery of the hormone through the skin is simultaneously enhanced by use of the hydrogel and the monosubstituted alkyl urea penetration enhancer of the invention and moderated by a rate-controlling adhesive layer 3.
- transdermal drug delivery device suitable for use with the present invention is the microreservoir-type system (see Figure 3).
- this type of system consists of a suspension of the solid hormone in the inventive hydrogel-forming base mixture.
- the hormone suspension is dispersed homogeneously in a lipophilic polymer to form numerous microscopic spheres of drug reservoir 4.
- This dispersion is thermodynamically unstable and is stabilized by cross- linking the polymer chains in situ.
- the mixture is formed into a disc and is then covered by an occlusive baseplate 2 and optionally surrounded by an adhesive rim 3.
- a preferred transdermal delivery device useful for application of the compositions of the present invention is a matrix dispersion type system ( Figure 4).
- the composition of the present invention is molded into a disc of a certain thickness and is applied onto an occlusive baseplate 3 in a compartment fabricated from a drug-impermeable plastic backing 1.
- An adhesive may be applied along the circumference of the device to form an adhesive rim 5 around the disc.
- the present invention provides transdermal delivery devices in form of a watch or watch-like device which have the advantage that an adhesive is not necessary to maintain contact of a drug- containing transdermal delivery composition with the patient's skin.
- a watch or watch-like device of the invention has a watch- case with a recessed chamber on the back face of the watch-case in contact with the skin.
- the recessed chamber contains a wafer having a top face and a bottom face containing a drug and a pharmaceutically acceptable base.
- the recessed chamber of the watch-case comprises a fluid-filled or hydrogel cushion later situated against the top face of the drug-containing wafer.
- the recessed chamber of the watch-case comprises a heating layer having a top face and a bottom face in which the bottom face of the heating layer is situated against the top face of the drug-containing wafer.
- the recessed chamber of the watch-case comprises permanent, closed cell having a top face and a bottom face, the bottom face of which is situated against the top face of the heating layer.
- the heating layer of the inventive devices can comprise a printed circuit board or a plastic or metallic layer which contains a plurality of metal electrical wires or lines comprising circuitry which provide heating.
- the heating layer of the inventive devices can be battery-powered or solar-powered. In one embodiment the heating layer is powered via the battery of the watch. In another embodiment, the heating layer is powered by a solar cell.
- the permanent, closed cell layer of the inventive devices can comprise a dense polymeric material.
- the permanent closed cell layer provides sight downward pressure on the drug filled wafer, to aid in maintaining contact of the wafer with the subject's skin.
- a device for the transdermal delivery of a drug to a subject which comprises a disc-shaped drug reservoir.
- the reservoir has a recessed chamber which can contain a wafer having a top face and a bottom face comprising a drug and a pharmaceutically acceptable base.
- the drug reservoir may comprise clear plastic or vinyl.
- the reservoir clips on to the back face of a watch or watch ⁇ like device such that drug-containing wafer is maintained in contact with the subject's skin.
- the reservoir clips over the posts and band of the wafer.
- the reservoir slides onto a band capable of being attached to the limb of a subject such that the drug-containing wafer is maintained in contact with the skin of the subject's limb.
- the recessed chamber of the clip-on or slide-on drug reservoir comprises a fluid-filled or hydrogel cushion later situated against the top face of the drug-containing wafer.
- the recessed chamber of the clip-on or slide-on drug reservoir comprises a heating layer as described above having a top face and a bottom face in which the bottom face of the heating layer is situated against the top face of the drug-containing wafer.
- the recessed chamber of the clip-on or slide-on drug reservoir comprises permanent, closed cell as described above having a top face and a bottom face, the bottom face of which is situated against the top face of the heating layer.
- a device for the transdermal delivery of a drug to a subject in accordance with the present invention comprises a glove wherein the inside of the glove is lined with a layer containing a composition of the invention comprising a pharmaceutically acceptable hydrogel base and a drug.
- a device for the transdermal delivery of a drug to a subject in accordance with the present invention comprises a band or a strap having a surface coated with a drug-containing composition of the present invention, wherein the band or strap is capable of being attached to the limb of a subject such that the drug- containing composition is maintained in contact with the skin of the subject's limb.
- kits comprising the recessed chamber-type, the clip-on drug reservoir-type, the slide-on drug reservoir-type, the glove-type or the band-type of transdermal drug delivery devices.
- the kit may comprise a watch or watch-like device with a recessed chamber and comprising a band or strap, and a plurality of drug-containing wafers. The wafers may be provided in sealed packages for long shelf life.
- the kit may comprise at least one drug reservoir, which may be clear plastic, vinyl or some other acceptable material, and a plurality of drug-containing wafers. Again, the wafers may be provided in sealed packages for long shelf life.
- the watch-like device of the present invention provides a recessed-chamber type of transdermal drug delivery device.
- a perspective view of the front face of this device is shown in Figure 5.
- the device comprises a watch case (1) and a band or strap (2) to attach the device to a limb (e.g. , wrist, arm or leg) of the user.
- the back face ( Figure 6) of the device has a recessed-chamber capable (2) of containing a drug-containing wafer, which may comprise a composition of the present invention.
- the recessed chamber of the watch is approximately 3.6 cm in diameter, corresponding to an area of about 10 cm 2 .
- the chamber is at least deep enough to accommodate a drug-containing wafer approximately 1.5 mm thick. When the wafer loses its ability to deliver the drug, the patient can simply replace it with another "refill" wafer.
- the watch case may comprise stainless steel or plastic, and the recessed chamber may be lined with teflon or some other inert material.
- the rim of the recessed chamber is coated with a non-slip or a soft flair skin seal material to aid in maintaining contact of the drug-containing wafer with the user's skin, and to prevent the drug-containing wafer from drying out.
- the front face of the watch need not actually contain a functional watch dial, although such can be the case.
- recessed-chamber type of transdermal drug delivery device comprises a recessed chamber 2 deep enough to contain both a drug-containing wafer 2 and a fluid- filled cushion 4.
- the fluid-filled cushion may comprise the hydrogel alone, without the hormone.
- the recessed chamber is deep enough to accommodate both a 1.5 mm thick hormone- containing wafer and a fluid-filled cushion on the roof of the chamber. This fluid cushion prevents the wafer from buckling and minimizes air bubbles.
- the device of this embodiment may also comprise a soft, non-slip material 6 on the rim of the recessed chamber.
- FIG. 9 A perspective view of the top face of the clip-on drug reservoir type transdermal drug delivery device of the present invention is shown in Figure 9, comprising a watch case 1, a band or strap 2 and a clip-on drug reservoir 3.
- the clip-on drug reservoir transdermal drug delivery device of the present invention ( Figure 10) comprises a plurality of clips 1 capable of attaching the clip-on drug reservoir to the back face of a watch; a recessed chamber 2; a drug-containing wafer 3 which may comprise a composition of the present invention; and a soft flair skin seal 4.
- the front face of the watch need not actually contain a functional watch dial, although such can be the case.
- Figure 13 shows a cross-sectional view of a watch having the clip-on drug reservoir type of transdermal drug delivery device of the present invention attached to its back face, comprising: a watch case 1; a band or strap 2; the clip-on drug reservoir 3 of the present invention; a recessed chamber 4, a drug-containing wafer 5; and a soft flair skin seal 6.
- Another preferred embodiment of the present invention which also eliminates the need for an adhesive is a glove lined with the hormone-containing composition of the present invention.
- the glove may be latex or any material that is compatible with the compositions of the present invention.
- This embodiment of the invention is suitable when 24 hour contact of patient's skin with the hormone-containing composition of the invention is not necessary to achieve therapeutically effective blood levels of the hormone. The patient simply wears the lined gloves to bed and then removes them in the morning and washes their hands.
- a means is provided for heating the hormone-containing composition while it is in contact with the patient's skin.
- Such heating increases the rate of penetration of the drug into the patient's bloodstream.
- a battery-powered heater capable of warming the system approximately 10° F above ambient temperature can be built into the watch-like transdermal device of the present invention to increase the delivery rate of the hormone through the patient's skin.
- compositions of the invention does not involve topical pre-administration of acetone or similar solvents in order to achieve transdermal delivery of therapeutically effective amounts to the bloodstream.
- compositions of the present invention may contain a wide variety of drugs, and may be used to treat a wide variety of disorders.
- the present invention provides methods for treatment of disorders responsive to the administration of hormones comprising administering to the patient transdermally a composition of the invention comprising a therapeutically effective amount of a hormone selected from the group consisting of progesterone, progestin, estrogen, and testosterone, and a mixture of any two or more of the foregoing.
- the compositions of the present invention are useful for treating a variety of disorders which benefit from hormone replacement therapy, including premenstrual syndrome, menopausal symptoms, infertility, and osteoporosis.
- compositions of the present invention may also be treated with the compositions of the present invention, as well as male disorders such as hypogonadism.
- the compositions of the present invention may also be used to provide contraception to male and female subjects.
- the subject is preferably an animal, including but not limited to animals such as cows, pigs, chickens, etc. , and is preferably a mammal, and most preferably human.
- Human subjects may be male or female.
- the condition being treated is one specific to women, such as premenstrual syndrome, menopausal symptoms, and gynecological problems, the subject is a woman.
- compositions useful for providing contraception to female subjects comprising progesterone, progestins, estrogens and mixtures thereof.
- compositions of the present invention comprising progesterone, mixtures of progesterone and one or more estrogens, or mixtures of one or more estrogens and one or more progestins may be used to provide contraception to females.
- Compositions of the present invention comprising testosterone are useful for providing contraception to male subjects.
- compositions of the present invention comprising estrogen are particularly useful in treatment of menopause.
- women lose their ability to produce estrogen.
- a diminished estrogen supply in post-menopausal women can cause such symptoms as hot flashes, insomnia, vaginal atrophy, irritability, anxiety, moodiness and excessive sweating.
- Natural or synthetic estrogens may be formulated into the compositions of the present invention for the treatment of these menopausal symptoms.
- Estrogen alone may be used to treat menopause in some women.
- women who use estrogen and who have an intact uterus preferably also take progesterone to prevent uterine cancer.
- Compositions of the present invention comprising estrogen, progesterone or mixtures thereof may be used to treat or prevent osteoporosis.
- Estrogen is known to prevent bone loss, while progesterone is known to stimulate bone growth.
- Osteoporosis is a progressive deterioration of the skeletal system through the loss of bone mass, and is related to the inability to produce estrogen.
- Osteoporosis Foundation According to the National Osteoporosis Foundation, osteoporosis currently affects approximately 25 million people in the United States. It is estimated that 80% of all hip fractures in elderly patients are associated with osteoporosis.
- Co ⁇ us luteum failure is another condition that the compositions of the present invention may be used to treat, and can result in infertility and/or irregular bleeding.
- compositions of the present invention comprising low dosages of testosterone can be used to treat senile vulvo-vaginitis, a post-menopausal condition.
- compositions of the present invention comprising natural progesterone, synthetic progestins, estrogens and mixtures thereof may be used to provide contraception to a subject.
- the present invention provides methods for the treatment of premenstrual syndrome, menopausal symptoms, infertility, osteoporosis, dysfunctional bleeding, co ⁇ us luteum failure, post-menopausal senile vulvo-vaginitis and hypogonadism, and methods for providing contraception, by using compositions of the present invention containing one or more hormones.
- the methods entail providing a composition of the present invention comprising a hydrogel-forming base mixture, a skin permeation enhancer, a hormone selected from the group consisting of natural progesterone, progestins, estrogens, testosterone and mixtures thereof, an optional glycolic, alcoholic or oil-based additive, and water, and contacting said composition to the skin of a patient to permit the hormone in the composition to be absorbed into the skin.
- the present invention provides a method for the treatment of premenstrual syndrome in which a composition of the present invention comprising natural progesterone is contacted with the skin of a patient suffering from premenstrual syndrome.
- the present invention provides a method for the treatment of menopausal symptoms such as hot flashes, insomnia, vaginal atrophy, irritability, anxiety, moodiness and excessive sweating in which a composition of the present invention comprising a hormone selected from the group consisting of natural progesterone, progestins, estrogens, testosterone, and mixtures thereof, is contacted with the skin of a patient suffering from any of such symptoms.
- a composition of the present invention comprising natural progesterone is contacted with the skin of a patient suffering from any of such symptoms.
- the present invention provides a method for the treatment of osteoporosis in which a composition of the present invention comprising natural progesterone, estrogen, or a mixture thereof is contacted with the skin of a patient suffering from osteoporosis.
- the present invention provides a method for the prevention of osteoporosis in which a composition of the present invention comprising natural progesterone, estrogen, or a mixture thereof is contacted with the skin of a patient who has the potential to develop osteoporosis.
- the present invention provides a method for the treatment of dysfunctional bleeding in which a composition of the present invention comprising natural progesterone or a mixture of natural progesterone and an estrogen is contacted with the skin of a patient suffering from dysfunctional bleeding.
- the present invention provides a method for the treatment of co ⁇ us luteum failure in which a composition of the present invention comprising natural progesterone or a mixture of natural progesterone and an estrogen is contacted with the skin of a patient suffering from co ⁇ us luteum failure.
- the present invention provides a method for the treatment of post-menopausal senile vulvo-vaginitis in which a composition of the present invention comprising testosterone is contacted with the skin of a patient suffering from senile vulvo-vaginitis.
- the present invention provides a method for the treatment of hypogonadism in which a composition of the present invention comprising testosterone is contacted with the skin of a patient suffering from hypogonadism.
- the present invention provides a method for providing contraception in which a composition of the present invention comprising natural progesterone, synthetic progestins, estrogens or mixtures thereof is contacted with the skin of a subject in need of contraception.
- the compositions of the invention may be sued to provide contraception to both males and females.
- the inventive composition comprising the appropriate hormone or hormone mixture is contacted with the subject's skin for a period of time sufficient to permit a therapeutically effective amount of the hormone or mixture of hormones to be absorbed through the skin into the subject's bloodstream.
- progesterone in the blood which will be considered therapeutically effective will, of course, vary from patient to patient, and with the type of condition being treated. As a general rule, a therapeutically effective amount can be considered the level of progesterone one would anticipate during the second half of the luteal phase of the menstrual cycle. However, other levels produced by the normal functions of the body may also be effective, depending on the circumstances. For example, a mean serum level of 2 ng/ml progesterone gives an optimal effect in the treatment of menopause using a combination of progesterone and estrogen. According to W.S. Maxon (1987, "Use of Progesterone in the Treatment of
- progesterone production by the ovarian co ⁇ us luteum dramatically increases and may reach 50 mg per day.
- serum concentrations of progesterone average 5 to 25 ng/ml.
- Progesterone is released in a pulsatile fashion, which correlates with LH pulsatility.
- placenta contributes an additional 25 to 40 mg/day of progesterone. Concentrations during gestation increase progressively to a mean of 175 ng/ml at term.
- blood level of progesterone can fluctuate widely during the luteal phase of the cycle, i.e. at least from 0-50 ng/ml. Generally, however, the fluctuation is on the order of 0-18 ng/ml.
- the preferred method of assaying the level of progesterone in the blood is by standard radioimmunoassay, which is well known to those skilled in the art.
- compositions of the present invention are also useful for treating vaginal conditions such as yeast infections and vaginal dryness.
- Compositions of the present invention are also useful as spermicides.
- compositions of the present invention comprising a drug selected from butylurea and butaconazole can be used to treat vaginal yeast infections.
- Compositions of the invention comprising butylurea can also be used as spermicides.
- Compositions of the present invention, with or without drug(s) may be used to treat vaginal dryness.
- compositions of the present invention comprising progesterone, estrogen, progestin, testosterone and a mixture of any two or more of the foregoing may also be administered vaginally to treat a variety of vaginal or gynecological conditions.
- the present invention provides methods for treating vaginal yeast infections.
- the present invention also provides methods for treating vaginal dryness.
- the invention also provides methods for contraception which involve the vaginal administration of compositions of the present invention comprising urea or urea derivatives such as butylurea. These methods involve the placement of a composition of the present invention inside the vagina of a subject.
- a hydrogel-forming base mixture of the invention is prepared a by mixing together the following seven dry powder ingredients:
- the mixture is micronized to a size of approximately one micron.
- composition for the transdermal delivery of natural progesterone containing the ingredients: 2 % butylurea
- Excised full-thickness human skin was obtained from elective surgery, (e.g. , breast reductions) or occasionally, from amputated legs.
- the tissue was stored in plastic containers at refrigerator temperature until use, at which time the subcutaneous fat and connective tissue were carefully trimmed off.
- Skin specimen thickness was in the range 5 1.5 - 2.0 mm, with characteristic histological features preserved.
- Tritium-labeled progesterone was obtained from NEN-DuPont (Wilmington, DE), with the label at ring positions 1, 2, 6, and 7. It was inco ⁇ orated in the test compositions in the range 100 to 500 DPM (Disintegration Per Minute) per microgram of progesterone.
- the labeled progesterone was inco ⁇ orated at the pre-polymerization Q stage, i.e. , just as water is added to form the hydrogel polymer. Aliquots of the gelling mixtures were added to small amounts of tritiated progesterone in scintillation vials (after the solvent had been evaporated), and stirred briskly with a small spatula.
- Diffusion Cell held vertically and inverted ( Figure 13).
- Half-inch diameter discs of skin to which nylon washers had been cemented with cyanoacrylate were used.
- the available diffusional area of each skin disc enclosed by the nylon washer was 0.495 cm 2 .
- the cell was assembled from parts of a commercial PVC coupling manufactured by Genova Products, Inc. , Davison, MI.
- the body of the coupling serves as the receptor chamber.
- the skin disc (5) kept flat by the nylon washer (6) cemented to its surface, is seated in the bottom end of the chamber, dermal side in contact with the receptor fluid (8).
- an O-ring (4) and a flanged PVC washer (2) coated with aluminum foil (3) are positioned under the nylon washer on the skin, and a retaining nut (1) is carefully tightened under all.
- a small, teflon-covered magnetic stirring button (7) and the receptor fluid are introduced, after which the top end of the coupling is stoppered with another foil-coated flanged washer (9).
- the cell is held in a rack on a magnetic stirrer, in a 32° C incubator.
- the magnetic stirring button is used to stir the receptor fluid in the inverted cell, and this button spins in direct contact with the dermal side of the skin.
- the 1 ml of fluid in the receptor chamber is mixed instantaneously as stirring is begun, and continuously during the course of the experiment.
- the receptor fluid was physiological saline, and 0.5 ml samples were removed (with replacement) at one-hour intervals.
- Intradermal Penetration Intradermal penetration was assessed with 0.5-inch diameter discs of skin, to which nylon washers were cemented, as described above. Discs with attached washers were placed in wells drilled in a plastic block. The block was then covered with a sheet of clear plastic to minimize dehydration of the skin.
- the epidermis was to be separated from the dermis for measurement of radioactivity, this separation was done by exposing the disc to an infrared heat lamp, held at 4 inches from the surface for 30 seconds, and peeling off the epidermis with fine forceps. Scintillation cocktail was added to the dissolved tissue samples, for determination of radioactivity. Thirteen formulations were used in the Examples described hereinbelow. The formulations were prepared as described above by addition of radioactive progesterone during polymerization, before the final viscosity had developed. The thirteen formulations are give below.
- the hydrogel-forming base mixture used in all thirteen formulations consisted of: 63% methyl cellulose, 21 % guar guam, 5% glucose, 3% pectin, 1.5% sodium chloride, 3.5% propyl paraben, and 3% methyl paraben.
- the dialysis membrane used was Spectra/Por R No. 1 dialysis tubing, purchased from Thomas Scientific, Swedesboro, NJ.
- the dialysis membrane was positioned in the diffusion cell as described above.
- Figure 14 is a plot of the cumulative amount of progesterone penetrated ( ⁇ g/cm 2 ) over a period of about 1.5 hours.
- Figure 14 shows that penetration occurred at a rate of about 30 ⁇ g/cm 2 /Hr. Note that there appeared to be no lag time, i.e. , the rate of penetration appeared to be linear from the time of contact. Such a time course for penetration is consistent with diffusion through pores (i.e. , the solvent-filled pores of the dialysis membrane).
- FIG. 15 shows a plot of the cumulative amount of progesterone penetration ( ⁇ g/cm 2 ) over a period of about 8 hours for uncovered and covered samples of A-l . Clearly, penetration proceeds more rapidly through the skin when the application site is covered.
- Rates of transdermal penetration of progesterone were determined for three samples: A-l , A-2, and A-3. Each of these samples was of the same viscosity, but each contained a different kind of penetration enhancer. (The compositions of these samples are given above in Section 6.2.1). The penetration data for samples A-l , A-2 and A-3 are given in Table 1. Table 1. Transdermal Penetration of Progesterone ⁇ g/cm 2
- n-butylurea was clearly the most efficacious of the three in promoting the penetration of progesterone through the skin.
- the highest rate observed, (18.1 ⁇ g/cm 2 /Hr, sample A-3, skin #1) was for the composition of the present invention containing n-butylurea. Note that the rate of 18.1 ⁇ g/cm 2 /Hr was substantially lower than that measured through dialysis membrane (Section 6.3.2).
- the test system does not impose an upper limit on the rate of penetration, and therefore, the measured rates are a true indication of how fast the drug can be transferred through the skin from these formulations.
- samples A-l, A-2, A-3, B-l , B-2, B-3, C-1 , C-2, and C-3 were compared by measuring intradermal penetration rather than transdermal penetration.
- the observed dilution-dependent decrease in transdermal penetration was contrary to expectation, since normally, lowering drug concentration the receptor increases penetration, as it increases the concentration gradient across the skin. The decrease was especially unexpected because in this model, the receptor volume is at a minimum (only the fluid in the dermal compartment of the skin). All measurements were performed on skin from a single donor. Results are given in Table 3.
- Transdermal penetration using samples A-3 and C-3 was measured using skin from the same donor as was used in Section 6.3.5. Measurements were performed in two ways: with the sampling from the diffusion cells during the first eight hours, and without such sampling (Table 4). In this experiment, the amounts that passed through the skin were much higher when the diffusion cells were not sampled, however the best hourly average for transdermal penetration rate (Sample C-3, 4.8 ⁇ g/cm 2 /Hr), was still only about one-half the lowest rate measured with n-butylurea in Section 6.2.4 (Table 1).
- the transdermal penetration of the progesterone through full-thickness skin was compared to penetration through skin from which the epidermis had been removed.
- the rate increased only about 2-fold after this maneuver, indicating that the dermis is an important factor in limiting the rate of penetration through the skin.
- Discs of skin were prepared as described in "Methods", for measurement of intradermal penetration, but using much larger washers to provide test sites of about 10 cm, in area.
- a test formula containing 20% progesterone (Sample Z) was applied, and uptake into the skin was allowed to proceed for one hour, and for 20 hours. Surface residue was removed at the indicated times, using the rinse/ wipe procedure described.
- Skin surface biopsies were taken from 1 & 20-Hr skin discs, and coated with photographic emulsion (Illford K-2 emulsion, Illford, London), then allowed to develop for 14 days. Rinsing and fixing were done as prescribed by the manufacturer.
- a high magnification study of the sections was undertaken, to establish that the pattern of darkness, or opacity, seen in these low-magnification images was really due to silver grains.
- a view of the underside of a control biopsy showed an uprooted vellus hair enveloped by a mixture sebum and compacted corneocytes (keratinized stratum corneum cells). It resembled a horn with a collar, emerging from the background undersurface of the stratum corneum.
- the horn is the coated hair, and the collar a portion of the infundibulum, or funnel-shaped opening of the pilosebaceous duct, that emerges on the surface of the skin.
- the polygonal outlines of individual corneocytes are sometimes discernible, a convenient metric (about 40 microns) for the scale of the images.
- the underside of a section from skin which had been in contact with a tritiated 20% progesterone formula for 20 hours appeared to be stained a deep blue color, most intensely at follicular structures. At higher magnification, the blue stain could be resolved into individual silver grains, seen as a stippling of blue dots about 500 nanometers in diameter.
- compositions of the present invention demonstrate that progesterone penetration rates using compositions of the present invention are in the range 1-20 ⁇ g/cm 2 /hr, and appear to be sustainable for at least 48 hours (Table 6).
- the rate at the higher end of this range is in excess of two orders of magnitude greater the 1.2 ⁇ g/cm 2 /day rate reported by Guy et al. , (1987, "Kinetics of Drug Abso ⁇ tion Across Human Skin In Vivo, " Pharmacol. Skin 1: 70-76). (discussed supra).
- Barry and Bennett (1987, "Effect of Penetration Enhancers on the Permeation of Mannitol, Hydrocortisone, and Progesterone Through Human Skin," J. Pharm. Pharmacol.
- compositions of the present invention provide unexpectedly superior delivery to the blood of the hormone contained within them and a very promising aspect of a sustained-released transdermal delivery system for progesterone.
- the radioactive species in the receptor of the diffusion cell was extracted from the saline receptor fluid, and shown by thin layer chromatography to be indistinguishable from the progesterone which had been applied to the surface of the skin.
- Two progesterone formulations were tested, one with 2% enhancer (Test Formulation 5 A-4) and one with 4% enhancer (Test Formulation B-4).
- Mean penetration rates of 3.2 and 6.1 micrograms per centimeter squared per hour were determined, for the 2% and the 4% enhancer formulations, respectively (see Figure 21).
- Test Formulation A-4 which is the same formula used in the in vivo study described in Section 6.5. Impressive (clinically significant) serum and saliva levels were achieved when Formulation A-4 was applied to human test subjects during a single 8-hour application using a 10 x 10 cm 2 patch. It is likely that concentrations observed in vitro could rise substantially, with continuous in vivo application of a patch, using an optimized formula (e.g. , with higher enhancer concentration). In such instance, the patch could be reduced in size to something which could be worn around the wrist, or applied at some other convenient body site.
- Test formulations A-4 and B-4 had the following compositions:
- the polymer (hydrogel-forming base mixture) was composed of the following ingredients: 63 % methyl cellulose, 21 % guar gum, 5% glucose, 3% pectin, 1.5% sodium chloride, 3.5% propyl paraben and 3% methyl paraben.
- Diffusion Cell Design and Assembly Transepidermal penetration was measured using single chamber diffusion cells.
- the diffusion cell described in Section 6.2 was redesigned to work with epidermal membranes, which are much thinner and more fragile than the full-thickness skin from which they are isolated.
- the same polyvinyl chloride ("PVC") couplings described in Section 6.2 were used to make a donor assembly ( Figure 20) which could be partially immersed, so that the underside of the epidermal membrane was in contact with saline held in a glass receptor.
- a half-inch diameter epidermal membrance disc was positioned such that its dermal surface was in contact with normal saline (2.5 ml) held in a glass receptor.
- the diffusion area was 0.71 cm 2 .
- the saline was stirred continuously using a small stirring bar.
- Diffusion cells were incubated at 32 °C. The cells were covered with a plastic sheet to prevent evaporation from the receptor.
- Progesterone penetration was estimated by measuring the amount of tritiated progesterone contained in each aliquot, using standard techniques. Plots of the cumulative amount collected in the receptor with time were used to determine progesterone penetration rates, expressed in ⁇ g/cm 2 /hour (see Figure 21). Four separate determinations were made for each of the two formulations tested. The extracted labeled material was shown by thin layer chromatography to be indistinguishable from fresh natural progesterone. The membranes were examined visually with a hand lens before each experiment, to confirm that no visible defects were present.
- Tritiated progesterone was purchased from NEN-Dupont (96 Ci/mmole), and was labeled in ring positions 1 , 2, 6, and 7. Compounding was as follows:
- Enhancer was dissolved in an appropriate volume of water, in a heavy 2-ounce "shot glass", stirred with a straight steel rod at 1500 RPM. A weighed amount of polymer was then dusted onto the vortex of stirring solution, to ensure rapid, thorough mixing. As soon as thickening was evident, the progesterone-glycol solution from No. 1 above was added, and the stirring rate increased to 3000 RPM. Stirring was continued for 2-3 minutes, until the mixture was the consistency of dough, and adhered to the rod; then the stirrer was turned off. With the help of a small spatula, the labeled polymer mixture was transferred to a clean scintillation vial, which was then tightly capped. The final mixtures had 5-600 dpm/microgram of hormone.
- TLC Thin layer chromatography
- Test Formulation A-4 Fifty mg of Test Formulation A-4 was extracted with 1 ml of ethyl acetate and 5- ⁇ l aliquots spotted for analysis. These aliquots also yielded a single spot on visualization, with an R f that was indistinguishable from that obtained with the "cold" stock solution.
- spots were scraped from the TLC plate and suspended in scintillation cocktail, spuriously high counts were observed because of the fluor bound to the silica gel. These counts were found to decrease as the silica settled, but as this procedure was time-consuming, the practice was adopted of centrifuging the suspension at lOOOxg for 5 minutes and counting the supernatant.
- Receptor fluid (2.5 ml) was removed from an assembled diffusion cell about 5 hours after Test Formulation A-4 had been applied. This fluid was extracted with 5 ml of ethyl acetate with vigorous shaking for 10 minutes. Five- ⁇ l aliquots were counted or applied to a TLC plate to assess their identity and purity. On visualization with UV after the plates were developed, three UV-absorbing spots could be detected, one with R f 0.68. As may be seen in Table 10, about 75% of the applied counts were recovered in this "progesterone spot" . No radioactivity could be detected in the other UV-absorbing spots, or elsewhere on the TLC plate.
- TLC Thin-layer chromatography
- receptor fluid was extracted with ethyl acetate after five hours of transepidermal penetration. This ethyl acetate extract was concentrated and then spotted on a TLC plate. Visual examination of the TLC plate then showed three spots, one of which was at R f 0.68. All of the radioactivity recoverable from the TLC plate was (Table 10) again confined to this "progesterone spot. " It thus appears that little chemical or enzymatic changes occur when progesterone passed through the epidermis, and that tritium in the receptor is a true indicator of progesterone penetrating.
- DMP refers to "Disintegrations Per Minute", which is a measure of radioactivity.
- the receptor fluid (2.5 ml) from a diffusion cells was extracted with 5 ml of ethyl acetate, and concentrated to about 100 ⁇ l.
- Five- ⁇ l aliquots were added directly to scintillation cocktail and counted, or spotted on a TLC plate.
- DPM in the second line of the Table represent the amount of progesterone recovered from the "Progesterone Spot" (R f 0.68). UV- absorbing material appeared at two other spots on the TLC plate, but these were found to be non-radioactive.
- transdermal progesterone formulation used in this study was the same as Test Formulation A-4, described in Section 6.4, supra, except that the formulation used in this study did not contain radioactive progesterone.
- Baseline progesterone ranges in both saliva and serum in men normally range from 0.02 to 0.05 nanograms per ml.
- salivary levels of progesterone were chosen for measurements is that salivary levels reflect the free plasma fraction. Many factors may alter the level of binding proteins in the blood or affect the binding of steroids to them. This greatly complicates the inte ⁇ retation of the total plasma steroid levels. Serum measurement of total progesterone must therefore be inte ⁇ reted with caution. These considerations are not a significant problem in the current study, however, since the concentration of progesterone binding proteins would not be expected to change during the brief duration of the of the experiment. Saliva samples avoid these problems by giving an index of the free plasma level.
- the lipid, soluble, unconjugated steroids enter saliva predominantly via the intracellular route. Their salivary concentrations are not dependent on saliva flow rate, and their salivary concentrations closely approximate their unbound concentration in plasma. Unconjugated steroids enter saliva by diffusing through the cells of the saliva glands and their concentration in saliva does not depend on the rate of saliva production. Both the saliva progesterone assay and serum assays for progesterone were performed by radioimmunoassay which uses the competition between radioactive and non-radioactive progesterone for a fixed number of antibody binding sites to determine the progesterone concentration present in the specimen.
- Transdermal delivery of natural progesterone was assessed in three male volunteers by measurement of serum and salivary progesterone levels, at time points ranging from zero to 48 hours following patch application.
- the serum progesterone data is shown in Table 11 and the saliva progesterone data is shown in Table 12. All three men showed baseline serum levels of 0.4 ng/ml and saliva values of 0.04, 0.04 and 0.02 ng/ml, consistent with established normal values in men.
- Subject #3's salivary progesterone level measured 25 ng/ml at around 8 hours in a single determination, with a lower value of 0.13 at 24 hours.
- Subjects #1 and #2 showed decreases in serum progesterone at about 24 hours, around 9 hours following patch removal, the same time that their saliva hormone concentration reached its highest value during the experiment. Both serum and salivary concentrations had decreased, but were still supernormal, at about 48 hours, 33 hours after discontinuing administration of the hormone.
- Subject #3 showed a decline in both serum and salivary concentrations at 24 hours; he was not tested at 48 hours. 6.5.4 DISCUSSION
- the pu ⁇ ose of this study was to determine whether the transdermal formulation of natural progesterone of the present invention could provide a safe and effective method for the transdermal administration of natural progesterone to humans.
- the formulation delivered significant amount of hormone without skin irritation or any other detectable adverse effects.
- the results suggest that the formulation has utility as a safe and effective device to deliver natural progesterone.
Abstract
Description

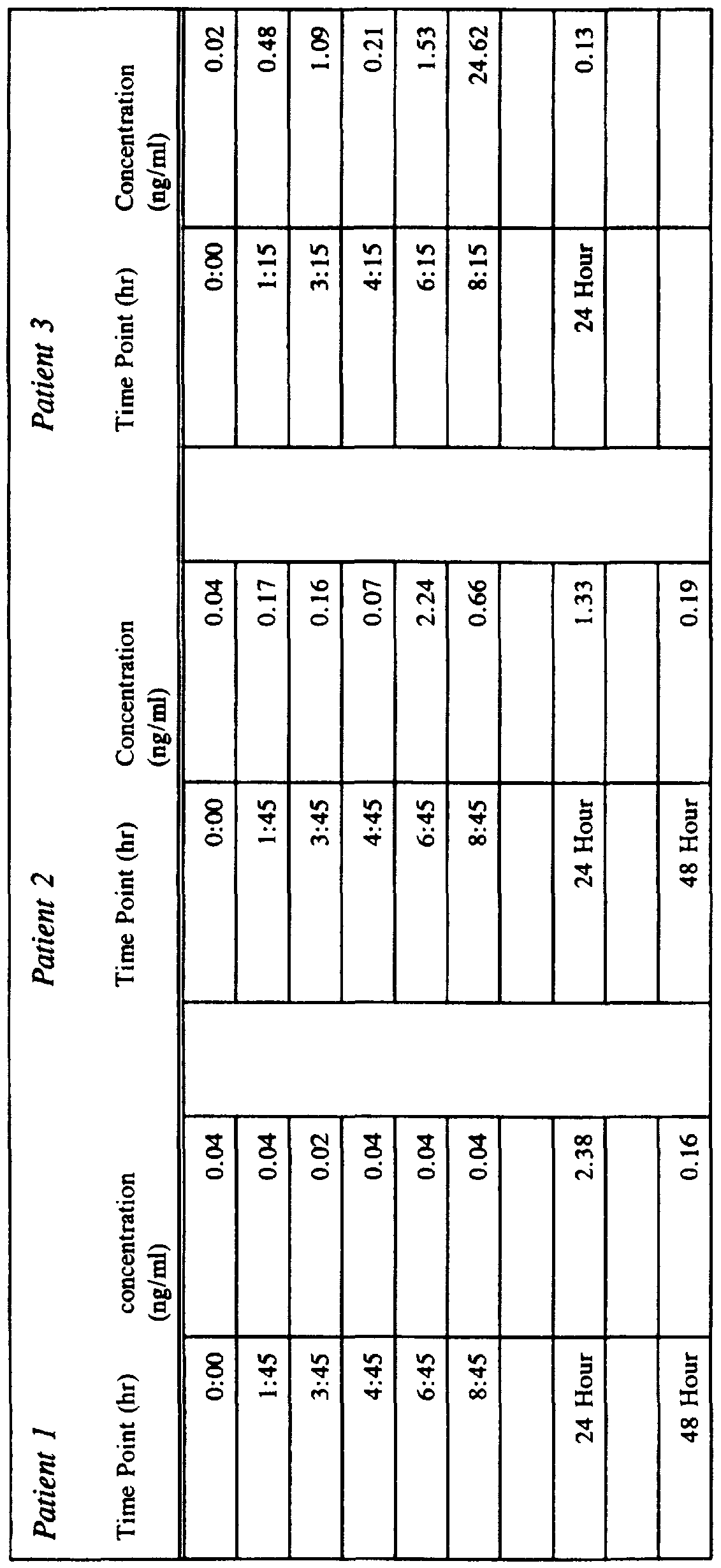
Claims
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU31368/97A AU3136897A (en) | 1996-05-22 | 1997-05-22 | Compositions, methods and devices for the transdermal delivery of drugs |
| JP09542696A JP2000513336A (en) | 1996-05-22 | 1997-05-22 | Compositions, methods and devices for transdermal delivery of drugs |
| EP97926655A EP0954260A1 (en) | 1996-05-22 | 1997-05-22 | Compositions, methods and devices for the transdermal delivery of drugs |
| IL12718997A IL127189A0 (en) | 1996-05-22 | 1997-05-22 | Compositions methods and devices for the transdermal delivery of drugs |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US65228096A | 1996-05-22 | 1996-05-22 | |
| US08/652,280 | 1996-05-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1997043989A1 true WO1997043989A1 (en) | 1997-11-27 |
Family
ID=24616242
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1997/008636 WO1997043989A1 (en) | 1996-05-22 | 1997-05-22 | Compositions, methods and devices for the transdermal delivery of drugs |
Country Status (6)
| Country | Link |
|---|---|
| EP (1) | EP0954260A1 (en) |
| JP (1) | JP2000513336A (en) |
| AU (1) | AU3136897A (en) |
| CA (1) | CA2256365A1 (en) |
| IL (1) | IL127189A0 (en) |
| WO (1) | WO1997043989A1 (en) |
Cited By (71)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1117356A1 (en) * | 1998-09-29 | 2001-07-25 | Zars, Inc. | Methods and apparatus for improved administration of fentanyl and sufentanil |
| WO2001068138A2 (en) * | 2000-03-10 | 2001-09-20 | Epicept, Inc. | Intradermal-penetration agents for topical local anesthetic administration |
| WO2002051421A2 (en) * | 2000-12-22 | 2002-07-04 | Dr. August Wolff Gmbh & Co. | Gel composition and trans-scrotal application of a composition for the treatment of hypogonadism |
| JP2002535347A (en) * | 1999-01-28 | 2002-10-22 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフトング | Crude drug preparation containing biotin |
| EP1309301A1 (en) * | 2000-04-27 | 2003-05-14 | Morton I. Hyson | Medicated wrap |
| EP1401408A1 (en) * | 2001-06-11 | 2004-03-31 | Zars, Inc. | Controlled heat induced rapid delivery of pharmaceuticals from skin depot |
| JP2004513079A (en) * | 2000-08-10 | 2004-04-30 | エス・アイ・エス・シユロブ・インステイテユート・フオー・サイエンス・リミテツド | Pharmaceutical composition comprising an analgesic peptide |
| EP1875905A2 (en) | 2003-04-28 | 2008-01-09 | Bayer Schering Pharma Aktiengesellschaft | Pharmaceutical compound in the form of a hydrogel for transdermal application of active substances |
| EP2124938A1 (en) * | 2007-01-24 | 2009-12-02 | Bomac Research Limited | Topical formulation |
| US7915244B2 (en) * | 2005-03-24 | 2011-03-29 | Emory University | Methods for the treatment of a traumatic central nervous injury |
| WO2011063775A2 (en) | 2009-11-25 | 2011-06-03 | Zentiva, K.S. | Pectin complexes of sartans and pharmaceutical compositions based thereon |
| US20120201904A1 (en) * | 2009-10-19 | 2012-08-09 | Won Seog Choi | Skin external composition comprising a salt and sugar as active ingredients for preventing and treating vaginosis and the use thereof |
| US8388546B2 (en) | 2006-10-23 | 2013-03-05 | Bard Access Systems, Inc. | Method of locating the tip of a central venous catheter |
| US8388541B2 (en) | 2007-11-26 | 2013-03-05 | C. R. Bard, Inc. | Integrated system for intravascular placement of a catheter |
| US8437833B2 (en) | 2008-10-07 | 2013-05-07 | Bard Access Systems, Inc. | Percutaneous magnetic gastrostomy |
| US8478382B2 (en) | 2008-02-11 | 2013-07-02 | C. R. Bard, Inc. | Systems and methods for positioning a catheter |
| US8512256B2 (en) | 2006-10-23 | 2013-08-20 | Bard Access Systems, Inc. | Method of locating the tip of a central venous catheter |
| US8614203B2 (en) | 2005-03-24 | 2013-12-24 | Emory University | Methods for the treatment of a central nervous system injury via a tapered administration protocol |
| US8633178B2 (en) | 2011-11-23 | 2014-01-21 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| USD699359S1 (en) | 2011-08-09 | 2014-02-11 | C. R. Bard, Inc. | Ultrasound probe head |
| US8781555B2 (en) | 2007-11-26 | 2014-07-15 | C. R. Bard, Inc. | System for placement of a catheter including a signal-generating stylet |
| US8784336B2 (en) | 2005-08-24 | 2014-07-22 | C. R. Bard, Inc. | Stylet apparatuses and methods of manufacture |
| US8801693B2 (en) | 2010-10-29 | 2014-08-12 | C. R. Bard, Inc. | Bioimpedance-assisted placement of a medical device |
| US8933059B2 (en) | 2012-06-18 | 2015-01-13 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| USD724745S1 (en) | 2011-08-09 | 2015-03-17 | C. R. Bard, Inc. | Cap for an ultrasound probe |
| EP2806742A4 (en) * | 2012-01-26 | 2015-07-29 | Therapeuticsmd Inc | Transdermal hormone replacement therapies |
| US9125816B2 (en) | 2000-08-30 | 2015-09-08 | Besins Healthcare Inc. | Pharmaceutical composition and method for treating hypogonadism |
| US9125578B2 (en) | 2009-06-12 | 2015-09-08 | Bard Access Systems, Inc. | Apparatus and method for catheter navigation and tip location |
| EP2861233A4 (en) * | 2012-06-18 | 2015-10-07 | Therapeuticsmd Inc | Transdermal hormone replacement therapies |
| US9180091B2 (en) | 2012-12-21 | 2015-11-10 | Therapeuticsmd, Inc. | Soluble estradiol capsule for vaginal insertion |
| US9211107B2 (en) | 2011-11-07 | 2015-12-15 | C. R. Bard, Inc. | Ruggedized ultrasound hydrogel insert |
| US9289382B2 (en) | 2012-06-18 | 2016-03-22 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US9339206B2 (en) | 2009-06-12 | 2016-05-17 | Bard Access Systems, Inc. | Adaptor for endovascular electrocardiography |
| US9433641B2 (en) | 2009-10-19 | 2016-09-06 | Won Seog Choi | Skin external composition comprising a salt and sugar as active ingredients for preventing and treating vaginosis and the use thereof |
| US9445734B2 (en) | 2009-06-12 | 2016-09-20 | Bard Access Systems, Inc. | Devices and methods for endovascular electrography |
| US9456766B2 (en) | 2007-11-26 | 2016-10-04 | C. R. Bard, Inc. | Apparatus for use with needle insertion guidance system |
| US9492097B2 (en) | 2007-11-26 | 2016-11-15 | C. R. Bard, Inc. | Needle length determination and calibration for insertion guidance system |
| US9521961B2 (en) | 2007-11-26 | 2016-12-20 | C. R. Bard, Inc. | Systems and methods for guiding a medical instrument |
| US9532724B2 (en) | 2009-06-12 | 2017-01-03 | Bard Access Systems, Inc. | Apparatus and method for catheter navigation using endovascular energy mapping |
| US9549685B2 (en) | 2007-11-26 | 2017-01-24 | C. R. Bard, Inc. | Apparatus and display methods relating to intravascular placement of a catheter |
| US9554716B2 (en) | 2007-11-26 | 2017-01-31 | C. R. Bard, Inc. | Insertion guidance system for needles and medical components |
| US9636031B2 (en) | 2007-11-26 | 2017-05-02 | C.R. Bard, Inc. | Stylets for use with apparatus for intravascular placement of a catheter |
| US9649048B2 (en) | 2007-11-26 | 2017-05-16 | C. R. Bard, Inc. | Systems and methods for breaching a sterile field for intravascular placement of a catheter |
| US9827267B2 (en) * | 2013-10-01 | 2017-11-28 | Won Seog Choi | Mixture of sodium chloride and sugar in the manufacture of a medicament employed for treating lax vagina syndrome or colpoxerosis disease in a mammal |
| US9839372B2 (en) | 2014-02-06 | 2017-12-12 | C. R. Bard, Inc. | Systems and methods for guidance and placement of an intravascular device |
| US9901714B2 (en) | 2008-08-22 | 2018-02-27 | C. R. Bard, Inc. | Catheter assembly including ECG sensor and magnetic assemblies |
| US9931349B2 (en) | 2016-04-01 | 2018-04-03 | Therapeuticsmd, Inc. | Steroid hormone pharmaceutical composition |
| US10046139B2 (en) | 2010-08-20 | 2018-08-14 | C. R. Bard, Inc. | Reconfirmation of ECG-assisted catheter tip placement |
| US20180264019A1 (en) * | 2011-07-14 | 2018-09-20 | Able Cerebral, Llc | Composition, device and method for delayed and sustained release of brain energy molecules |
| US10098894B2 (en) | 2014-07-29 | 2018-10-16 | Therapeuticsmd, Inc. | Transdermal cream |
| US10206932B2 (en) | 2014-05-22 | 2019-02-19 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US10258630B2 (en) | 2014-10-22 | 2019-04-16 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10286077B2 (en) | 2016-04-01 | 2019-05-14 | Therapeuticsmd, Inc. | Steroid hormone compositions in medium chain oils |
| US10328087B2 (en) | 2015-07-23 | 2019-06-25 | Therapeuticsmd, Inc. | Formulations for solubilizing hormones |
| US10349890B2 (en) | 2015-06-26 | 2019-07-16 | C. R. Bard, Inc. | Connector interface for ECG-based catheter positioning system |
| US10449330B2 (en) | 2007-11-26 | 2019-10-22 | C. R. Bard, Inc. | Magnetic element-equipped needle assemblies |
| US10471072B2 (en) | 2012-12-21 | 2019-11-12 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10471148B2 (en) | 2012-06-18 | 2019-11-12 | Therapeuticsmd, Inc. | Progesterone formulations having a desirable PK profile |
| US10524691B2 (en) | 2007-11-26 | 2020-01-07 | C. R. Bard, Inc. | Needle assembly including an aligned magnetic element |
| US10537581B2 (en) | 2012-12-21 | 2020-01-21 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10639008B2 (en) | 2009-10-08 | 2020-05-05 | C. R. Bard, Inc. | Support and cover structures for an ultrasound probe head |
| US10751509B2 (en) | 2007-11-26 | 2020-08-25 | C. R. Bard, Inc. | Iconic representations for guidance of an indwelling medical device |
| US10806740B2 (en) | 2012-06-18 | 2020-10-20 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US10820885B2 (en) | 2012-06-15 | 2020-11-03 | C. R. Bard, Inc. | Apparatus and methods for detection of a removable cap on an ultrasound probe |
| WO2020101590A3 (en) * | 2018-06-12 | 2020-12-24 | Kemikoglu Yahya Yagiz | Medication carrier watch |
| US10973584B2 (en) | 2015-01-19 | 2021-04-13 | Bard Access Systems, Inc. | Device and method for vascular access |
| US10992079B2 (en) | 2018-10-16 | 2021-04-27 | Bard Access Systems, Inc. | Safety-equipped connection systems and methods thereof for establishing electrical connections |
| US11000207B2 (en) | 2016-01-29 | 2021-05-11 | C. R. Bard, Inc. | Multiple coil system for tracking a medical device |
| US11103213B2 (en) | 2009-10-08 | 2021-08-31 | C. R. Bard, Inc. | Spacers for use with an ultrasound probe |
| US11246875B2 (en) | 2012-12-21 | 2022-02-15 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11266661B2 (en) | 2012-12-21 | 2022-03-08 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5021124B2 (en) * | 2001-08-29 | 2012-09-05 | 日東電工株式会社 | Medical adhesive composition, medical adhesive tape and transdermal absorption tape formulation using the same |
| EP2343963B1 (en) | 2008-10-08 | 2019-04-10 | Agile Therapeutics, Inc. | Transdermal delivery |
| ES2795455T3 (en) | 2008-10-08 | 2020-11-23 | Agile Therapeutics Inc | Transdermal delivery |
| US9192614B2 (en) | 2008-10-08 | 2015-11-24 | Agile Therapeutics, Inc. | Contraceptive transdermal delivery of hormones |
| WO2010111488A1 (en) | 2009-03-27 | 2010-09-30 | Agile Therapeutics, Inc. | Transdermal delivery |
| KR102109188B1 (en) * | 2013-04-01 | 2020-05-11 | 삼성전자주식회사 | Temperature sensitive liposome comprising cationic lipid and use thereof |
| KR101887088B1 (en) * | 2017-02-28 | 2018-08-09 | (주)강앤박메디컬 | Needle free solution injecting apparatus and method of needle free injecting solution using the same |
| KR101871886B1 (en) * | 2017-11-20 | 2018-06-27 | 김선수 | Non-smoking bracelet with pill receptacle |
| KR101995868B1 (en) * | 2018-07-10 | 2019-07-03 | 김선수 | Bracelet with pill storage structure |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5232705A (en) * | 1990-08-31 | 1993-08-03 | Alza Corporation | Dosage form for time-varying patterns of drug delivery |
-
1997
- 1997-05-22 AU AU31368/97A patent/AU3136897A/en not_active Abandoned
- 1997-05-22 JP JP09542696A patent/JP2000513336A/en active Pending
- 1997-05-22 CA CA002256365A patent/CA2256365A1/en not_active Abandoned
- 1997-05-22 IL IL12718997A patent/IL127189A0/en unknown
- 1997-05-22 WO PCT/US1997/008636 patent/WO1997043989A1/en not_active Application Discontinuation
- 1997-05-22 EP EP97926655A patent/EP0954260A1/en not_active Withdrawn
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5232705A (en) * | 1990-08-31 | 1993-08-03 | Alza Corporation | Dosage form for time-varying patterns of drug delivery |
Cited By (158)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1117356A1 (en) * | 1998-09-29 | 2001-07-25 | Zars, Inc. | Methods and apparatus for improved administration of fentanyl and sufentanil |
| EP1117356B1 (en) * | 1998-09-29 | 2006-03-15 | Zars, Inc. | Apparatus for improved administration of fentanyl and sufentanil |
| JP2002535347A (en) * | 1999-01-28 | 2002-10-22 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフトング | Crude drug preparation containing biotin |
| AU2001245449B2 (en) * | 2000-03-10 | 2007-03-22 | Epicept, Inc. | Intradermal-penetration agents for topical local anesthetic administration |
| WO2001068138A2 (en) * | 2000-03-10 | 2001-09-20 | Epicept, Inc. | Intradermal-penetration agents for topical local anesthetic administration |
| WO2001068138A3 (en) * | 2000-03-10 | 2002-01-17 | Epicept Inc | Intradermal-penetration agents for topical local anesthetic administration |
| US6455066B1 (en) | 2000-03-10 | 2002-09-24 | Epicept Corporation | Intradermal-penetration agents for topical local anesthetic administration |
| US6746689B2 (en) | 2000-03-10 | 2004-06-08 | Epicept Corporation | Intradermal-penetration agents for topical local anesthetic administration |
| EP1309301A4 (en) * | 2000-04-27 | 2007-04-04 | Morton I Hyson | Medicated wrap |
| EP1309301A1 (en) * | 2000-04-27 | 2003-05-14 | Morton I. Hyson | Medicated wrap |
| JP4860096B2 (en) * | 2000-08-10 | 2012-01-25 | エス・アイ・エス・シユロブ・インステイテユート・フオー・サイエンス・リミテツド | Pharmaceutical composition comprising an analgesic peptide |
| JP2004513079A (en) * | 2000-08-10 | 2004-04-30 | エス・アイ・エス・シユロブ・インステイテユート・フオー・サイエンス・リミテツド | Pharmaceutical composition comprising an analgesic peptide |
| US9125816B2 (en) | 2000-08-30 | 2015-09-08 | Besins Healthcare Inc. | Pharmaceutical composition and method for treating hypogonadism |
| US9132089B2 (en) | 2000-08-30 | 2015-09-15 | Besins Healthcare Inc. | Pharmaceutical composition and method for treating hypogonadism |
| WO2002051421A3 (en) * | 2000-12-22 | 2002-11-07 | August Wolff Gmbh & Co Dr | Gel composition and trans-scrotal application of a composition for the treatment of hypogonadism |
| WO2002051421A2 (en) * | 2000-12-22 | 2002-07-04 | Dr. August Wolff Gmbh & Co. | Gel composition and trans-scrotal application of a composition for the treatment of hypogonadism |
| EP1401408A4 (en) * | 2001-06-11 | 2007-11-07 | Zars Inc | Controlled heat induced rapid delivery of pharmaceuticals from skin depot |
| EP1401408A1 (en) * | 2001-06-11 | 2004-03-31 | Zars, Inc. | Controlled heat induced rapid delivery of pharmaceuticals from skin depot |
| EP1875905A2 (en) | 2003-04-28 | 2008-01-09 | Bayer Schering Pharma Aktiengesellschaft | Pharmaceutical compound in the form of a hydrogel for transdermal application of active substances |
| US7915244B2 (en) * | 2005-03-24 | 2011-03-29 | Emory University | Methods for the treatment of a traumatic central nervous injury |
| US8614203B2 (en) | 2005-03-24 | 2013-12-24 | Emory University | Methods for the treatment of a central nervous system injury via a tapered administration protocol |
| US8455468B2 (en) | 2005-03-24 | 2013-06-04 | Emory University | Methods for the treatment of a traumatic central nervous system injury |
| US11207496B2 (en) | 2005-08-24 | 2021-12-28 | C. R. Bard, Inc. | Stylet apparatuses and methods of manufacture |
| US10004875B2 (en) | 2005-08-24 | 2018-06-26 | C. R. Bard, Inc. | Stylet apparatuses and methods of manufacture |
| US8784336B2 (en) | 2005-08-24 | 2014-07-22 | C. R. Bard, Inc. | Stylet apparatuses and methods of manufacture |
| US9265443B2 (en) | 2006-10-23 | 2016-02-23 | Bard Access Systems, Inc. | Method of locating the tip of a central venous catheter |
| US8388546B2 (en) | 2006-10-23 | 2013-03-05 | Bard Access Systems, Inc. | Method of locating the tip of a central venous catheter |
| US9345422B2 (en) | 2006-10-23 | 2016-05-24 | Bard Acess Systems, Inc. | Method of locating the tip of a central venous catheter |
| US8512256B2 (en) | 2006-10-23 | 2013-08-20 | Bard Access Systems, Inc. | Method of locating the tip of a central venous catheter |
| US9833169B2 (en) | 2006-10-23 | 2017-12-05 | Bard Access Systems, Inc. | Method of locating the tip of a central venous catheter |
| US8858455B2 (en) | 2006-10-23 | 2014-10-14 | Bard Access Systems, Inc. | Method of locating the tip of a central venous catheter |
| US8774907B2 (en) | 2006-10-23 | 2014-07-08 | Bard Access Systems, Inc. | Method of locating the tip of a central venous catheter |
| EP2124938A4 (en) * | 2007-01-24 | 2010-04-14 | Bomac Research Ltd | Topical formulation |
| US8748467B2 (en) | 2007-01-24 | 2014-06-10 | Bayer Intellectual Property Gmbh | Topical formulation |
| EP2124938A1 (en) * | 2007-01-24 | 2009-12-02 | Bomac Research Limited | Topical formulation |
| US11123099B2 (en) | 2007-11-26 | 2021-09-21 | C. R. Bard, Inc. | Apparatus for use with needle insertion guidance system |
| US9999371B2 (en) | 2007-11-26 | 2018-06-19 | C. R. Bard, Inc. | Integrated system for intravascular placement of a catheter |
| US11707205B2 (en) | 2007-11-26 | 2023-07-25 | C. R. Bard, Inc. | Integrated system for intravascular placement of a catheter |
| US8781555B2 (en) | 2007-11-26 | 2014-07-15 | C. R. Bard, Inc. | System for placement of a catheter including a signal-generating stylet |
| US10231753B2 (en) | 2007-11-26 | 2019-03-19 | C. R. Bard, Inc. | Insertion guidance system for needles and medical components |
| US11529070B2 (en) | 2007-11-26 | 2022-12-20 | C. R. Bard, Inc. | System and methods for guiding a medical instrument |
| US9492097B2 (en) | 2007-11-26 | 2016-11-15 | C. R. Bard, Inc. | Needle length determination and calibration for insertion guidance system |
| US10165962B2 (en) | 2007-11-26 | 2019-01-01 | C. R. Bard, Inc. | Integrated systems for intravascular placement of a catheter |
| US10105121B2 (en) | 2007-11-26 | 2018-10-23 | C. R. Bard, Inc. | System for placement of a catheter including a signal-generating stylet |
| US11779240B2 (en) | 2007-11-26 | 2023-10-10 | C. R. Bard, Inc. | Systems and methods for breaching a sterile field for intravascular placement of a catheter |
| US11134915B2 (en) | 2007-11-26 | 2021-10-05 | C. R. Bard, Inc. | System for placement of a catheter including a signal-generating stylet |
| US10238418B2 (en) | 2007-11-26 | 2019-03-26 | C. R. Bard, Inc. | Apparatus for use with needle insertion guidance system |
| US10342575B2 (en) | 2007-11-26 | 2019-07-09 | C. R. Bard, Inc. | Apparatus for use with needle insertion guidance system |
| US9456766B2 (en) | 2007-11-26 | 2016-10-04 | C. R. Bard, Inc. | Apparatus for use with needle insertion guidance system |
| US10449330B2 (en) | 2007-11-26 | 2019-10-22 | C. R. Bard, Inc. | Magnetic element-equipped needle assemblies |
| US9521961B2 (en) | 2007-11-26 | 2016-12-20 | C. R. Bard, Inc. | Systems and methods for guiding a medical instrument |
| US9526440B2 (en) | 2007-11-26 | 2016-12-27 | C.R. Bard, Inc. | System for placement of a catheter including a signal-generating stylet |
| US10966630B2 (en) | 2007-11-26 | 2021-04-06 | C. R. Bard, Inc. | Integrated system for intravascular placement of a catheter |
| US10524691B2 (en) | 2007-11-26 | 2020-01-07 | C. R. Bard, Inc. | Needle assembly including an aligned magnetic element |
| US9549685B2 (en) | 2007-11-26 | 2017-01-24 | C. R. Bard, Inc. | Apparatus and display methods relating to intravascular placement of a catheter |
| US8388541B2 (en) | 2007-11-26 | 2013-03-05 | C. R. Bard, Inc. | Integrated system for intravascular placement of a catheter |
| US10602958B2 (en) | 2007-11-26 | 2020-03-31 | C. R. Bard, Inc. | Systems and methods for guiding a medical instrument |
| US10849695B2 (en) | 2007-11-26 | 2020-12-01 | C. R. Bard, Inc. | Systems and methods for breaching a sterile field for intravascular placement of a catheter |
| US9681823B2 (en) | 2007-11-26 | 2017-06-20 | C. R. Bard, Inc. | Integrated system for intravascular placement of a catheter |
| US9649048B2 (en) | 2007-11-26 | 2017-05-16 | C. R. Bard, Inc. | Systems and methods for breaching a sterile field for intravascular placement of a catheter |
| US9636031B2 (en) | 2007-11-26 | 2017-05-02 | C.R. Bard, Inc. | Stylets for use with apparatus for intravascular placement of a catheter |
| US10751509B2 (en) | 2007-11-26 | 2020-08-25 | C. R. Bard, Inc. | Iconic representations for guidance of an indwelling medical device |
| US9554716B2 (en) | 2007-11-26 | 2017-01-31 | C. R. Bard, Inc. | Insertion guidance system for needles and medical components |
| US8478382B2 (en) | 2008-02-11 | 2013-07-02 | C. R. Bard, Inc. | Systems and methods for positioning a catheter |
| US8971994B2 (en) | 2008-02-11 | 2015-03-03 | C. R. Bard, Inc. | Systems and methods for positioning a catheter |
| US9901714B2 (en) | 2008-08-22 | 2018-02-27 | C. R. Bard, Inc. | Catheter assembly including ECG sensor and magnetic assemblies |
| US11027101B2 (en) | 2008-08-22 | 2021-06-08 | C. R. Bard, Inc. | Catheter assembly including ECG sensor and magnetic assemblies |
| US8437833B2 (en) | 2008-10-07 | 2013-05-07 | Bard Access Systems, Inc. | Percutaneous magnetic gastrostomy |
| US9907513B2 (en) | 2008-10-07 | 2018-03-06 | Bard Access Systems, Inc. | Percutaneous magnetic gastrostomy |
| US9339206B2 (en) | 2009-06-12 | 2016-05-17 | Bard Access Systems, Inc. | Adaptor for endovascular electrocardiography |
| US9125578B2 (en) | 2009-06-12 | 2015-09-08 | Bard Access Systems, Inc. | Apparatus and method for catheter navigation and tip location |
| US9532724B2 (en) | 2009-06-12 | 2017-01-03 | Bard Access Systems, Inc. | Apparatus and method for catheter navigation using endovascular energy mapping |
| US11419517B2 (en) | 2009-06-12 | 2022-08-23 | Bard Access Systems, Inc. | Apparatus and method for catheter navigation using endovascular energy mapping |
| US10271762B2 (en) | 2009-06-12 | 2019-04-30 | Bard Access Systems, Inc. | Apparatus and method for catheter navigation using endovascular energy mapping |
| US9445734B2 (en) | 2009-06-12 | 2016-09-20 | Bard Access Systems, Inc. | Devices and methods for endovascular electrography |
| US10231643B2 (en) | 2009-06-12 | 2019-03-19 | Bard Access Systems, Inc. | Apparatus and method for catheter navigation and tip location |
| US10912488B2 (en) | 2009-06-12 | 2021-02-09 | Bard Access Systems, Inc. | Apparatus and method for catheter navigation and tip location |
| US10639008B2 (en) | 2009-10-08 | 2020-05-05 | C. R. Bard, Inc. | Support and cover structures for an ultrasound probe head |
| US11103213B2 (en) | 2009-10-08 | 2021-08-31 | C. R. Bard, Inc. | Spacers for use with an ultrasound probe |
| US9433641B2 (en) | 2009-10-19 | 2016-09-06 | Won Seog Choi | Skin external composition comprising a salt and sugar as active ingredients for preventing and treating vaginosis and the use thereof |
| US9446069B2 (en) | 2009-10-19 | 2016-09-20 | Won Seog Choi | Skin external composition comprising a sea salt and sugar as active ingredients for preventing and treating vaginosis and the use thereof |
| US20120201904A1 (en) * | 2009-10-19 | 2012-08-09 | Won Seog Choi | Skin external composition comprising a salt and sugar as active ingredients for preventing and treating vaginosis and the use thereof |
| WO2011063775A2 (en) | 2009-11-25 | 2011-06-03 | Zentiva, K.S. | Pectin complexes of sartans and pharmaceutical compositions based thereon |
| WO2011063774A2 (en) | 2009-11-25 | 2011-06-03 | Zentiva, K.S. | Pectin complexes of steroids and pharmaceutical compositions based thereon |
| US10046139B2 (en) | 2010-08-20 | 2018-08-14 | C. R. Bard, Inc. | Reconfirmation of ECG-assisted catheter tip placement |
| US8801693B2 (en) | 2010-10-29 | 2014-08-12 | C. R. Bard, Inc. | Bioimpedance-assisted placement of a medical device |
| US9415188B2 (en) | 2010-10-29 | 2016-08-16 | C. R. Bard, Inc. | Bioimpedance-assisted placement of a medical device |
| US20180264019A1 (en) * | 2011-07-14 | 2018-09-20 | Able Cerebral, Llc | Composition, device and method for delayed and sustained release of brain energy molecules |
| US10953025B2 (en) * | 2011-07-14 | 2021-03-23 | Able Cerebral, Llc | Composition, device and method for delayed and sustained release of brain energy molecules |
| USD754357S1 (en) | 2011-08-09 | 2016-04-19 | C. R. Bard, Inc. | Ultrasound probe head |
| USD724745S1 (en) | 2011-08-09 | 2015-03-17 | C. R. Bard, Inc. | Cap for an ultrasound probe |
| USD699359S1 (en) | 2011-08-09 | 2014-02-11 | C. R. Bard, Inc. | Ultrasound probe head |
| US9211107B2 (en) | 2011-11-07 | 2015-12-15 | C. R. Bard, Inc. | Ruggedized ultrasound hydrogel insert |
| US8846649B2 (en) | 2011-11-23 | 2014-09-30 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US10675288B2 (en) | 2011-11-23 | 2020-06-09 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US11103516B2 (en) | 2011-11-23 | 2021-08-31 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US11793819B2 (en) | 2011-11-23 | 2023-10-24 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US8633178B2 (en) | 2011-11-23 | 2014-01-21 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US8846648B2 (en) | 2011-11-23 | 2014-09-30 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US8987237B2 (en) | 2011-11-23 | 2015-03-24 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US9248136B2 (en) | 2011-11-23 | 2016-02-02 | Therapeuticsmd, Inc. | Transdermal hormone replacement therapies |
| EP2806742A4 (en) * | 2012-01-26 | 2015-07-29 | Therapeuticsmd Inc | Transdermal hormone replacement therapies |
| AU2013211876B2 (en) * | 2012-01-26 | 2017-02-09 | Therapeuticsmd, Inc. | Transdermal hormone replacement therapies |
| US10820885B2 (en) | 2012-06-15 | 2020-11-03 | C. R. Bard, Inc. | Apparatus and methods for detection of a removable cap on an ultrasound probe |
| US11529360B2 (en) | 2012-06-18 | 2022-12-20 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US9012434B2 (en) | 2012-06-18 | 2015-04-21 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US11110099B2 (en) | 2012-06-18 | 2021-09-07 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US11865179B2 (en) | 2012-06-18 | 2024-01-09 | Therapeuticsmd, Inc. | Progesterone formulations having a desirable PK profile |
| US8987238B2 (en) | 2012-06-18 | 2015-03-24 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US10639375B2 (en) | 2012-06-18 | 2020-05-05 | Therapeuticsmd, Inc. | Progesterone formulations |
| US11033626B2 (en) | 2012-06-18 | 2021-06-15 | Therapeuticsmd, Inc. | Progesterone formulations having a desirable pk profile |
| EP2861233A4 (en) * | 2012-06-18 | 2015-10-07 | Therapeuticsmd Inc | Transdermal hormone replacement therapies |
| US9006222B2 (en) | 2012-06-18 | 2015-04-14 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US10471148B2 (en) | 2012-06-18 | 2019-11-12 | Therapeuticsmd, Inc. | Progesterone formulations having a desirable PK profile |
| US11166963B2 (en) | 2012-06-18 | 2021-11-09 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US10806740B2 (en) | 2012-06-18 | 2020-10-20 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US8933059B2 (en) | 2012-06-18 | 2015-01-13 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US10052386B2 (en) | 2012-06-18 | 2018-08-21 | Therapeuticsmd, Inc. | Progesterone formulations |
| US9301920B2 (en) | 2012-06-18 | 2016-04-05 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US9289382B2 (en) | 2012-06-18 | 2016-03-22 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11116717B2 (en) | 2012-12-21 | 2021-09-14 | Therapeuticsmd, Inc. | Soluble estradiol capsule for vaginal insertion |
| US10835487B2 (en) | 2012-12-21 | 2020-11-17 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11246875B2 (en) | 2012-12-21 | 2022-02-15 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11241445B2 (en) | 2012-12-21 | 2022-02-08 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11497709B2 (en) | 2012-12-21 | 2022-11-15 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US9180091B2 (en) | 2012-12-21 | 2015-11-10 | Therapeuticsmd, Inc. | Soluble estradiol capsule for vaginal insertion |
| US10471072B2 (en) | 2012-12-21 | 2019-11-12 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11304959B2 (en) | 2012-12-21 | 2022-04-19 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11266661B2 (en) | 2012-12-21 | 2022-03-08 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10888516B2 (en) | 2012-12-21 | 2021-01-12 | Therapeuticsmd, Inc. | Soluble estradiol capsule for vaginal insertion |
| US10806697B2 (en) | 2012-12-21 | 2020-10-20 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11351182B2 (en) | 2012-12-21 | 2022-06-07 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11065197B2 (en) | 2012-12-21 | 2021-07-20 | Therapeuticsmd, Inc. | Soluble estradiol capsule for vaginal insertion |
| US11622933B2 (en) | 2012-12-21 | 2023-04-11 | Therapeuticsmd, Inc. | Soluble estradiol capsule for vaginal insertion |
| US10568891B2 (en) | 2012-12-21 | 2020-02-25 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11123283B2 (en) | 2012-12-21 | 2021-09-21 | Therapeuticsmd, Inc. | Soluble estradiol capsule for vaginal insertion |
| US10537581B2 (en) | 2012-12-21 | 2020-01-21 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US9827267B2 (en) * | 2013-10-01 | 2017-11-28 | Won Seog Choi | Mixture of sodium chloride and sugar in the manufacture of a medicament employed for treating lax vagina syndrome or colpoxerosis disease in a mammal |
| US9839372B2 (en) | 2014-02-06 | 2017-12-12 | C. R. Bard, Inc. | Systems and methods for guidance and placement of an intravascular device |
| US10863920B2 (en) | 2014-02-06 | 2020-12-15 | C. R. Bard, Inc. | Systems and methods for guidance and placement of an intravascular device |
| US10206932B2 (en) | 2014-05-22 | 2019-02-19 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US11103513B2 (en) | 2014-05-22 | 2021-08-31 | TherapeuticsMD | Natural combination hormone replacement formulations and therapies |
| US10098894B2 (en) | 2014-07-29 | 2018-10-16 | Therapeuticsmd, Inc. | Transdermal cream |
| US10258630B2 (en) | 2014-10-22 | 2019-04-16 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10398708B2 (en) | 2014-10-22 | 2019-09-03 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10668082B2 (en) | 2014-10-22 | 2020-06-02 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10973584B2 (en) | 2015-01-19 | 2021-04-13 | Bard Access Systems, Inc. | Device and method for vascular access |
| US10349890B2 (en) | 2015-06-26 | 2019-07-16 | C. R. Bard, Inc. | Connector interface for ECG-based catheter positioning system |
| US11026630B2 (en) | 2015-06-26 | 2021-06-08 | C. R. Bard, Inc. | Connector interface for ECG-based catheter positioning system |
| US10912783B2 (en) | 2015-07-23 | 2021-02-09 | Therapeuticsmd, Inc. | Formulations for solubilizing hormones |
| US10328087B2 (en) | 2015-07-23 | 2019-06-25 | Therapeuticsmd, Inc. | Formulations for solubilizing hormones |
| US11000207B2 (en) | 2016-01-29 | 2021-05-11 | C. R. Bard, Inc. | Multiple coil system for tracking a medical device |
| US10532059B2 (en) | 2016-04-01 | 2020-01-14 | Therapeuticsmd, Inc. | Steroid hormone pharmaceutical composition |
| US9931349B2 (en) | 2016-04-01 | 2018-04-03 | Therapeuticsmd, Inc. | Steroid hormone pharmaceutical composition |
| US10286077B2 (en) | 2016-04-01 | 2019-05-14 | Therapeuticsmd, Inc. | Steroid hormone compositions in medium chain oils |
| WO2020101590A3 (en) * | 2018-06-12 | 2020-12-24 | Kemikoglu Yahya Yagiz | Medication carrier watch |
| US11621518B2 (en) | 2018-10-16 | 2023-04-04 | Bard Access Systems, Inc. | Safety-equipped connection systems and methods thereof for establishing electrical connections |
| US10992079B2 (en) | 2018-10-16 | 2021-04-27 | Bard Access Systems, Inc. | Safety-equipped connection systems and methods thereof for establishing electrical connections |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0954260A1 (en) | 1999-11-10 |
| IL127189A0 (en) | 1999-09-22 |
| CA2256365A1 (en) | 1997-11-27 |
| JP2000513336A (en) | 2000-10-10 |
| AU3136897A (en) | 1997-12-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1997043989A1 (en) | Compositions, methods and devices for the transdermal delivery of drugs | |
| TW565462B (en) | A novel composition for transdermal administration of estrogen, progestin or a mixture thereof | |
| JP5727494B2 (en) | Transdermal pharmaceutical composition comprising an active agent | |
| AU718811B2 (en) | Fatty acid esters of glycolic acid and its salts as permeation enhancers | |
| AU2006201918B2 (en) | Improved transdermal contraceptive delivery system and process | |
| US5762956A (en) | Transdermal contraceptive delivery system and process | |
| US7045145B1 (en) | Transdermal contraceptive delivery system and process | |
| US5651973A (en) | Therapeutically effective topical application of ST1435 | |
| Fraser et al. | An initial pharmacokinetic study with a Metered Dose Transdermal System® for delivery of the progestogen Nestorone® as a possible future contraceptive | |
| US3787571A (en) | Trichloroethanol and trifluoroethanol topical and percutaneous steroid adsorption promoters | |
| Henzl | Optimizing delivery of therapeutics: percutaneous technologies | |
| KR100481459B1 (en) | Composition for transdermal administration of an estrogen, a progestin or a mixture thereof | |
| James et al. | Percutaneous absorption of dexamethasone 3 H in rats and mice: Comparative Study with oral administration | |
| Bierman et al. | Transdermal Controlled Delivery of Drugs With Special Reference to Development and Evaluation of Transdermal Contraceptive Devices YW Chien, Ph. D., TY Chien, Ph. D., YC Huang, Ph. D. | |
| IL95508A (en) | Pharmaceutical compositions for transdermal administration |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AU AZ BA BB BG BR BY CA CN CU CZ EE GE GH HU IL IS JP KG KP KR KZ LC LK LR LT LV MD MG MK MN MX NO NZ PL RO RU SG SI SK TJ TM TR TT UA US UZ VN YU AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH KE LS MW SD SZ UG AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2256365 Country of ref document: CA Ref country code: CA Ref document number: 2256365 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1997926655 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 333481 Country of ref document: NZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 1997926655 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1997926655 Country of ref document: EP |