WO1999017809A2 - Contrast-enhanced diagnostic imaging method for monitoring interventional therapies - Google Patents
Contrast-enhanced diagnostic imaging method for monitoring interventional therapies Download PDFInfo
- Publication number
- WO1999017809A2 WO1999017809A2 PCT/US1998/020182 US9820182W WO9917809A2 WO 1999017809 A2 WO1999017809 A2 WO 1999017809A2 US 9820182 W US9820182 W US 9820182W WO 9917809 A2 WO9917809 A2 WO 9917809A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- tissue
- contrast agent
- state
- relaxivity
- contrast
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 90
- 238000002560 therapeutic procedure Methods 0.000 title claims abstract description 60
- 238000012544 monitoring process Methods 0.000 title claims abstract description 30
- 238000002059 diagnostic imaging Methods 0.000 title claims abstract description 14
- 239000002872 contrast media Substances 0.000 claims abstract description 135
- 230000027455 binding Effects 0.000 claims abstract description 102
- 239000003795 chemical substances by application Substances 0.000 claims abstract description 79
- 230000001419 dependent effect Effects 0.000 claims abstract description 34
- 210000001519 tissue Anatomy 0.000 claims description 197
- 102000008100 Human Serum Albumin Human genes 0.000 claims description 82
- 108091006905 Human Serum Albumin Proteins 0.000 claims description 82
- -1 clusters Substances 0.000 claims description 39
- 102000004169 proteins and genes Human genes 0.000 claims description 31
- 108090000623 proteins and genes Proteins 0.000 claims description 31
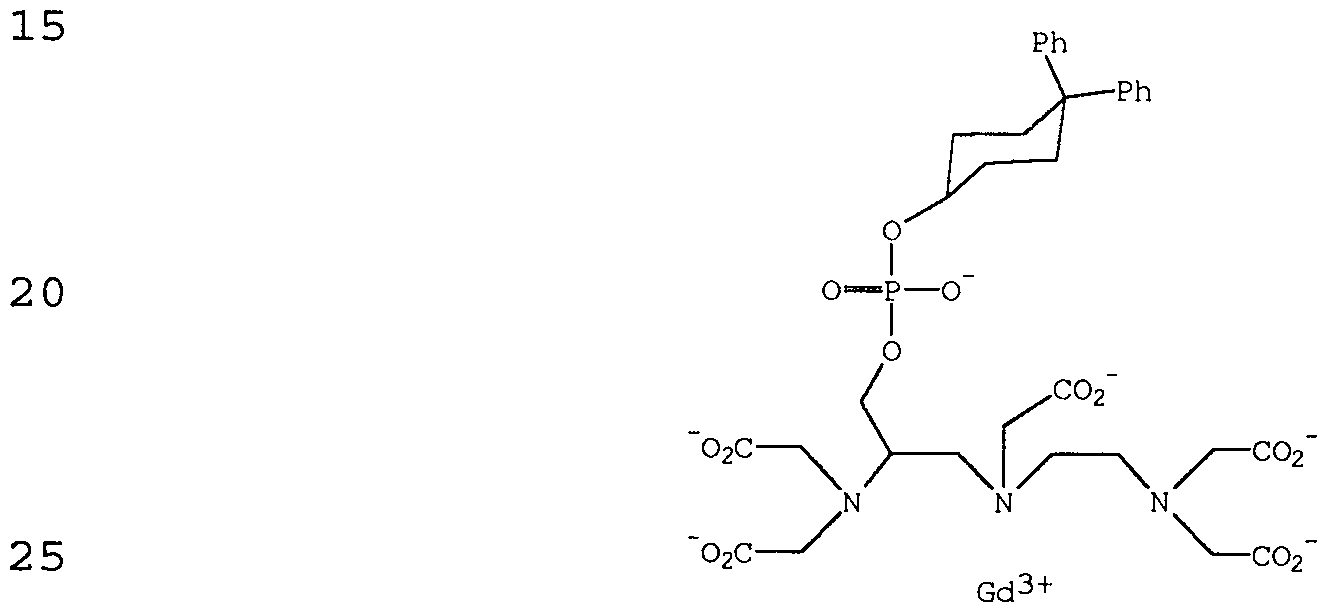
- ZSDLYSDDELEVDD-UFTMZEDQSA-K 2-[[(2r)-2-[bis(carboxylatomethyl)amino]-3-[(4,4-diphenylcyclohexyl)oxy-oxidophosphoryl]oxypropyl]-[2-[bis(carboxylatomethyl)amino]ethyl]amino]acetate;gadolinium(3+);hydron Chemical compound [H+].[H+].[H+].[Gd+3].C1CC(OP([O-])(=O)OC[C@@H](CN(CCN(CC([O-])=O)CC([O-])=O)CC(=O)[O-])N(CC([O-])=O)CC([O-])=O)CCC1(C=1C=CC=CC=1)C1=CC=CC=C1 ZSDLYSDDELEVDD-UFTMZEDQSA-K 0.000 claims description 27
- 239000013522 chelant Substances 0.000 claims description 26
- 238000003384 imaging method Methods 0.000 claims description 25
- 229910052751 metal Inorganic materials 0.000 claims description 25
- 239000002184 metal Substances 0.000 claims description 25
- 229910021645 metal ion Inorganic materials 0.000 claims description 21
- 230000005298 paramagnetic effect Effects 0.000 claims description 19
- 210000004369 blood Anatomy 0.000 claims description 15
- 239000008280 blood Substances 0.000 claims description 15
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 14
- 150000003839 salts Chemical class 0.000 claims description 14
- 125000003118 aryl group Chemical group 0.000 claims description 13
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 13
- 229910019142 PO4 Inorganic materials 0.000 claims description 10
- 230000002209 hydrophobic effect Effects 0.000 claims description 10
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 9
- 229910052688 Gadolinium Inorganic materials 0.000 claims description 9
- 239000002245 particle Substances 0.000 claims description 9
- 239000010452 phosphate Substances 0.000 claims description 9
- 150000003384 small molecules Chemical class 0.000 claims description 9
- 125000001931 aliphatic group Chemical group 0.000 claims description 8
- 125000001424 substituent group Chemical group 0.000 claims description 8
- 239000002738 chelating agent Substances 0.000 claims description 7
- 239000000975 dye Substances 0.000 claims description 7
- 229910052742 iron Inorganic materials 0.000 claims description 7
- 229910052757 nitrogen Inorganic materials 0.000 claims description 7
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 claims description 6
- 230000004962 physiological condition Effects 0.000 claims description 6
- 229940124530 sulfonamide Drugs 0.000 claims description 6
- 239000007864 aqueous solution Substances 0.000 claims description 5
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 5
- 229910052760 oxygen Inorganic materials 0.000 claims description 5
- 239000001301 oxygen Substances 0.000 claims description 5
- 230000036961 partial effect Effects 0.000 claims description 5
- 102000004895 Lipoproteins Human genes 0.000 claims description 4
- QPCDCPDFJACHGM-UHFFFAOYSA-N N,N-bis{2-[bis(carboxymethyl)amino]ethyl}glycine Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(=O)O)CCN(CC(O)=O)CC(O)=O QPCDCPDFJACHGM-UHFFFAOYSA-N 0.000 claims description 4
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 4
- 239000012530 fluid Substances 0.000 claims description 4
- 229910052736 halogen Inorganic materials 0.000 claims description 4
- 150000002367 halogens Chemical class 0.000 claims description 4
- 125000000623 heterocyclic group Chemical group 0.000 claims description 4
- 229920000642 polymer Polymers 0.000 claims description 4
- 229910052717 sulfur Inorganic materials 0.000 claims description 4
- 239000011593 sulfur Substances 0.000 claims description 4
- VTLYFUHAOXGGBS-UHFFFAOYSA-N Fe3+ Chemical compound [Fe+3] VTLYFUHAOXGGBS-UHFFFAOYSA-N 0.000 claims description 3
- 102000005720 Glutathione transferase Human genes 0.000 claims description 3
- 125000002015 acyclic group Chemical group 0.000 claims description 3
- 125000002252 acyl group Chemical group 0.000 claims description 3
- 125000000539 amino acid group Chemical group 0.000 claims description 3
- 125000004122 cyclic group Chemical group 0.000 claims description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 3
- 230000002757 inflammatory effect Effects 0.000 claims description 3
- 210000003093 intracellular space Anatomy 0.000 claims description 3
- 239000002502 liposome Substances 0.000 claims description 3
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 3
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 claims description 3
- 210000001179 synovial fluid Anatomy 0.000 claims description 3
- DGJPRBXSVZULQO-PFEQFJNWSA-N (2r)-1-(3-phenyl-1,2-oxazol-5-yl)-2-(pyrrolidin-1-ylmethyl)butan-1-one;hydrochloride Chemical compound Cl.C([C@@H](CC)C(=O)C=1ON=C(C=1)C=1C=CC=CC=1)N1CCCC1 DGJPRBXSVZULQO-PFEQFJNWSA-N 0.000 claims description 2
- JPVYNHNXODAKFH-UHFFFAOYSA-N Cu2+ Chemical compound [Cu+2] JPVYNHNXODAKFH-UHFFFAOYSA-N 0.000 claims description 2
- 229910052692 Dysprosium Inorganic materials 0.000 claims description 2
- 102000030914 Fatty Acid-Binding Human genes 0.000 claims description 2
- WDLRUFUQRNWCPK-UHFFFAOYSA-N Tetraxetan Chemical compound OC(=O)CN1CCN(CC(O)=O)CCN(CC(O)=O)CCN(CC(O)=O)CC1 WDLRUFUQRNWCPK-UHFFFAOYSA-N 0.000 claims description 2
- 150000008431 aliphatic amides Chemical class 0.000 claims description 2
- 125000003545 alkoxy group Chemical group 0.000 claims description 2
- 125000004448 alkyl carbonyl group Chemical group 0.000 claims description 2
- 125000005196 alkyl carbonyloxy group Chemical group 0.000 claims description 2
- 125000004414 alkyl thio group Chemical group 0.000 claims description 2
- 125000004432 carbon atom Chemical group C* 0.000 claims description 2
- 210000001175 cerebrospinal fluid Anatomy 0.000 claims description 2
- BFGKITSFLPAWGI-UHFFFAOYSA-N chromium(3+) Chemical compound [Cr+3] BFGKITSFLPAWGI-UHFFFAOYSA-N 0.000 claims description 2
- RZESKRXOCXWCFX-UHFFFAOYSA-N 2-[bis[2-[carboxymethyl-[2-(methylamino)-2-oxoethyl]amino]ethyl]amino]acetic acid Chemical compound CNC(=O)CN(CC(O)=O)CCN(CC(O)=O)CCN(CC(O)=O)CC(=O)NC RZESKRXOCXWCFX-UHFFFAOYSA-N 0.000 claims 1
- 101000889990 Homo sapiens Apolipoprotein(a) Proteins 0.000 claims 1
- 101000799549 Homo sapiens Aspartate aminotransferase, mitochondrial Proteins 0.000 claims 1
- 101000988802 Homo sapiens Hematopoietic prostaglandin D synthase Proteins 0.000 claims 1
- WAEMQWOKJMHJLA-UHFFFAOYSA-N Manganese(2+) Chemical compound [Mn+2] WAEMQWOKJMHJLA-UHFFFAOYSA-N 0.000 claims 1
- 150000004696 coordination complex Chemical class 0.000 claims 1
- MMIPFLVOWGHZQD-UHFFFAOYSA-N manganese(3+) Chemical compound [Mn+3] MMIPFLVOWGHZQD-UHFFFAOYSA-N 0.000 claims 1
- 230000008859 change Effects 0.000 abstract description 29
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 55
- 238000002595 magnetic resonance imaging Methods 0.000 description 41
- 239000000243 solution Substances 0.000 description 30
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 28
- 239000000126 substance Substances 0.000 description 25
- 235000002639 sodium chloride Nutrition 0.000 description 21
- 230000036425 denaturation Effects 0.000 description 20
- 238000004925 denaturation Methods 0.000 description 20
- 239000000523 sample Substances 0.000 description 20
- 239000002904 solvent Substances 0.000 description 18
- 150000002148 esters Chemical class 0.000 description 17
- 239000003446 ligand Substances 0.000 description 16
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical group NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 15
- 230000005291 magnetic effect Effects 0.000 description 15
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical class C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 14
- 150000001875 compounds Chemical class 0.000 description 14
- 239000000203 mixture Substances 0.000 description 14
- 238000012634 optical imaging Methods 0.000 description 14
- 230000001965 increasing effect Effects 0.000 description 13
- 239000008194 pharmaceutical composition Substances 0.000 description 13
- 230000000694 effects Effects 0.000 description 12
- 230000001338 necrotic effect Effects 0.000 description 11
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical class CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 10
- 206010028851 Necrosis Diseases 0.000 description 10
- 102000012404 Orosomucoid Human genes 0.000 description 10
- 108010061952 Orosomucoid Proteins 0.000 description 10
- 230000017074 necrotic cell death Effects 0.000 description 10
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical group NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 10
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 9
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 9
- 230000007423 decrease Effects 0.000 description 9
- 235000021317 phosphate Nutrition 0.000 description 9
- 230000035945 sensitivity Effects 0.000 description 9
- 231100000419 toxicity Toxicity 0.000 description 9
- 230000001988 toxicity Effects 0.000 description 9
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 8
- 239000002585 base Substances 0.000 description 8
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 8
- 238000001727 in vivo Methods 0.000 description 8
- 238000002679 ablation Methods 0.000 description 7
- 125000000217 alkyl group Chemical group 0.000 description 7
- 150000001412 amines Chemical class 0.000 description 7
- 238000002347 injection Methods 0.000 description 7
- 239000007924 injection Substances 0.000 description 7
- 125000005647 linker group Chemical group 0.000 description 7
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 7
- 230000003287 optical effect Effects 0.000 description 7
- 230000001575 pathological effect Effects 0.000 description 7
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 6
- 239000004215 Carbon black (E152) Substances 0.000 description 6
- 238000005481 NMR spectroscopy Methods 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 238000010317 ablation therapy Methods 0.000 description 6
- 238000002835 absorbance Methods 0.000 description 6
- 239000007795 chemical reaction product Substances 0.000 description 6
- 229930195733 hydrocarbon Natural products 0.000 description 6
- 150000002430 hydrocarbons Chemical class 0.000 description 6
- 239000011780 sodium chloride Substances 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 6
- 238000004679 31P NMR spectroscopy Methods 0.000 description 5
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical group NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 5
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical group [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 5
- 206010028980 Neoplasm Diseases 0.000 description 5
- 241000700159 Rattus Species 0.000 description 5
- 150000001241 acetals Chemical group 0.000 description 5
- 239000004202 carbamide Chemical group 0.000 description 5
- 239000013068 control sample Substances 0.000 description 5
- UIWYJDYFSGRHKR-UHFFFAOYSA-N gadolinium atom Chemical compound [Gd] UIWYJDYFSGRHKR-UHFFFAOYSA-N 0.000 description 5
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 150000003457 sulfones Chemical group 0.000 description 5
- 229910052721 tungsten Inorganic materials 0.000 description 5
- 229920001817 Agar Polymers 0.000 description 4
- 102000004506 Blood Proteins Human genes 0.000 description 4
- 108010017384 Blood Proteins Proteins 0.000 description 4
- 239000008272 agar Substances 0.000 description 4
- 229940024606 amino acid Drugs 0.000 description 4
- 150000001413 amino acids Chemical class 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- 210000004027 cell Anatomy 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 230000000052 comparative effect Effects 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 238000009792 diffusion process Methods 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 239000003814 drug Substances 0.000 description 4
- 239000003937 drug carrier Substances 0.000 description 4
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 4
- 150000002500 ions Chemical class 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 210000004185 liver Anatomy 0.000 description 4
- 230000033001 locomotion Effects 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- 229910052750 molybdenum Inorganic materials 0.000 description 4
- 239000007800 oxidant agent Substances 0.000 description 4
- 230000001590 oxidative effect Effects 0.000 description 4
- 230000005414 paramagnetic center Effects 0.000 description 4
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 4
- 229910052698 phosphorus Inorganic materials 0.000 description 4
- 230000036470 plasma concentration Effects 0.000 description 4
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical group [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- 101100226596 Gallus gallus FABP gene Proteins 0.000 description 3
- 108090001030 Lipoproteins Proteins 0.000 description 3
- 239000002616 MRI contrast agent Substances 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 241000283973 Oryctolagus cuniculus Species 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 239000002671 adjuvant Substances 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 230000015271 coagulation Effects 0.000 description 3
- 238000005345 coagulation Methods 0.000 description 3
- 239000010941 cobalt Substances 0.000 description 3
- 229910017052 cobalt Inorganic materials 0.000 description 3
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 3
- 230000000875 corresponding effect Effects 0.000 description 3
- LWNLXVXSCCLRRZ-UHFFFAOYSA-N dichlorophosphane Chemical compound ClPCl LWNLXVXSCCLRRZ-UHFFFAOYSA-N 0.000 description 3
- 235000014113 dietary fatty acids Nutrition 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 239000002270 dispersing agent Substances 0.000 description 3
- 238000010494 dissociation reaction Methods 0.000 description 3
- 230000005593 dissociations Effects 0.000 description 3
- 230000002708 enhancing effect Effects 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 230000029142 excretion Effects 0.000 description 3
- 239000000194 fatty acid Substances 0.000 description 3
- 229930195729 fatty acid Natural products 0.000 description 3
- 150000004665 fatty acids Chemical class 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 229910052739 hydrogen Inorganic materials 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- 230000014759 maintenance of location Effects 0.000 description 3
- 230000004060 metabolic process Effects 0.000 description 3
- 230000007935 neutral effect Effects 0.000 description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- 239000012044 organic layer Substances 0.000 description 3
- 235000019271 petrolatum Nutrition 0.000 description 3
- 239000002953 phosphate buffered saline Substances 0.000 description 3
- 230000000704 physical effect Effects 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 238000012552 review Methods 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 239000011877 solvent mixture Substances 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 150000003456 sulfonamides Chemical class 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- 238000002604 ultrasonography Methods 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- KBPLFHHGFOOTCA-UHFFFAOYSA-N 1-Octanol Chemical compound CCCCCCCCO KBPLFHHGFOOTCA-UHFFFAOYSA-N 0.000 description 2
- LLPKQRMDOFYSGZ-UHFFFAOYSA-N 2,5-dimethyl-1h-imidazole Chemical compound CC1=CN=C(C)N1 LLPKQRMDOFYSGZ-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical group CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- 208000037260 Atherosclerotic Plaque Diseases 0.000 description 2
- 0 CCCCOc(cc1S(O)(=O)=O)ccc1OCC(CN(CCN(CC(O)=O)CC(O)=O)CC(O)=O)N(C[*-])C* Chemical compound CCCCOc(cc1S(O)(=O)=O)ccc1OCC(CN(CCN(CC(O)=O)CC(O)=O)CC(O)=O)N(C[*-])C* 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical compound NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- 108010070675 Glutathione transferase Proteins 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 102000003886 Glycoproteins Human genes 0.000 description 2
- 108090000288 Glycoproteins Proteins 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical class COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 2
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 2
- 108010043958 Peptoids Proteins 0.000 description 2
- 239000004264 Petrolatum Substances 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 239000013011 aqueous formulation Substances 0.000 description 2
- 239000007900 aqueous suspension Substances 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- 238000009529 body temperature measurement Methods 0.000 description 2
- 210000000988 bone and bone Anatomy 0.000 description 2
- 150000001649 bromium compounds Chemical class 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 150000001720 carbohydrates Chemical class 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 230000007248 cellular mechanism Effects 0.000 description 2
- 125000003636 chemical group Chemical group 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 150000002012 dioxanes Chemical class 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 235000019197 fats Nutrition 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- LGMLJQFQKXPRGA-VPVMAENOSA-K gadopentetate dimeglumine Chemical compound [Gd+3].CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO.CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO.OC(=O)CN(CC([O-])=O)CCN(CC([O-])=O)CCN(CC(O)=O)CC([O-])=O LGMLJQFQKXPRGA-VPVMAENOSA-K 0.000 description 2
- 210000001035 gastrointestinal tract Anatomy 0.000 description 2
- 125000005456 glyceride group Chemical group 0.000 description 2
- 210000002216 heart Anatomy 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 230000002440 hepatic effect Effects 0.000 description 2
- 210000003494 hepatocyte Anatomy 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 229960004657 indocyanine green Drugs 0.000 description 2
- MOFVSTNWEDAEEK-UHFFFAOYSA-M indocyanine green Chemical compound [Na+].[O-]S(=O)(=O)CCCCN1C2=CC=C3C=CC=CC3=C2C(C)(C)C1=CC=CC=CC=CC1=[N+](CCCCS([O-])(=O)=O)C2=CC=C(C=CC=C3)C3=C2C1(C)C MOFVSTNWEDAEEK-UHFFFAOYSA-M 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 150000004694 iodide salts Chemical class 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 229920002521 macromolecule Polymers 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 238000013507 mapping Methods 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 230000001613 neoplastic effect Effects 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- 239000000346 nonvolatile oil Substances 0.000 description 2
- 108020004707 nucleic acids Proteins 0.000 description 2
- 102000039446 nucleic acids Human genes 0.000 description 2
- 150000007523 nucleic acids Chemical class 0.000 description 2
- 239000002773 nucleotide Substances 0.000 description 2
- 125000003729 nucleotide group Chemical group 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 238000005192 partition Methods 0.000 description 2
- 229940066842 petrolatum Drugs 0.000 description 2
- ACVYVLVWPXVTIT-UHFFFAOYSA-M phosphinate Chemical compound [O-][PH2]=O ACVYVLVWPXVTIT-UHFFFAOYSA-M 0.000 description 2
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 2
- 238000002428 photodynamic therapy Methods 0.000 description 2
- 230000035790 physiological processes and functions Effects 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 2
- 239000012217 radiopharmaceutical Substances 0.000 description 2
- 229940121896 radiopharmaceutical Drugs 0.000 description 2
- 230000002799 radiopharmaceutical effect Effects 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 230000003595 spectral effect Effects 0.000 description 2
- 238000001356 surgical procedure Methods 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000007704 transition Effects 0.000 description 2
- 238000012285 ultrasound imaging Methods 0.000 description 2
- 150000003751 zinc Chemical class 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- VMSLCPKYRPDHLN-UHFFFAOYSA-N (R)-Humulone Chemical compound CC(C)CC(=O)C1=C(O)C(CC=C(C)C)=C(O)C(O)(CC=C(C)C)C1=O VMSLCPKYRPDHLN-UHFFFAOYSA-N 0.000 description 1
- 125000006091 1,3-dioxolane group Chemical class 0.000 description 1
- VFWCMGCRMGJXDK-UHFFFAOYSA-N 1-chlorobutane Chemical compound CCCCCl VFWCMGCRMGJXDK-UHFFFAOYSA-N 0.000 description 1
- VUQPJRPDRDVQMN-UHFFFAOYSA-N 1-chlorooctadecane Chemical class CCCCCCCCCCCCCCCCCCCl VUQPJRPDRDVQMN-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- QWENRTYMTSOGBR-UHFFFAOYSA-N 1H-1,2,3-Triazole Chemical compound C=1C=NNN=1 QWENRTYMTSOGBR-UHFFFAOYSA-N 0.000 description 1
- KJUGUADJHNHALS-UHFFFAOYSA-N 1H-tetrazole Chemical compound C=1N=NNN=1 KJUGUADJHNHALS-UHFFFAOYSA-N 0.000 description 1
- LDGWQMRUWMSZIU-LQDDAWAPSA-M 2,3-bis[(z)-octadec-9-enoxy]propyl-trimethylazanium;chloride Chemical compound [Cl-].CCCCCCCC\C=C/CCCCCCCCOCC(C[N+](C)(C)C)OCCCCCCCC\C=C/CCCCCCCC LDGWQMRUWMSZIU-LQDDAWAPSA-M 0.000 description 1
- CHHHXKFHOYLYRE-UHFFFAOYSA-M 2,4-Hexadienoic acid, potassium salt (1:1), (2E,4E)- Chemical compound [K+].CC=CC=CC([O-])=O CHHHXKFHOYLYRE-UHFFFAOYSA-M 0.000 description 1
- CVOFKRWYWCSDMA-UHFFFAOYSA-N 2-chloro-n-(2,6-diethylphenyl)-n-(methoxymethyl)acetamide;2,6-dinitro-n,n-dipropyl-4-(trifluoromethyl)aniline Chemical compound CCC1=CC=CC(CC)=C1N(COC)C(=O)CCl.CCCN(CCC)C1=C([N+]([O-])=O)C=C(C(F)(F)F)C=C1[N+]([O-])=O CVOFKRWYWCSDMA-UHFFFAOYSA-N 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- LEACJMVNYZDSKR-UHFFFAOYSA-N 2-octyldodecan-1-ol Chemical compound CCCCCCCCCCC(CO)CCCCCCCC LEACJMVNYZDSKR-UHFFFAOYSA-N 0.000 description 1
- WMPPDTMATNBGJN-UHFFFAOYSA-N 2-phenylethylbromide Chemical class BrCCC1=CC=CC=C1 WMPPDTMATNBGJN-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-M 3-phenylpropionate Chemical compound [O-]C(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-M 0.000 description 1
- UUQQLWRHPNEFIG-UHFFFAOYSA-N 4,4-diphenylcyclohexan-1-ol Chemical compound C1CC(O)CCC1(C=1C=CC=CC=1)C1=CC=CC=C1 UUQQLWRHPNEFIG-UHFFFAOYSA-N 0.000 description 1
- NSPMIYGKQJPBQR-UHFFFAOYSA-N 4H-1,2,4-triazole Chemical compound C=1N=CNN=1 NSPMIYGKQJPBQR-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 102000011767 Acute-Phase Proteins Human genes 0.000 description 1
- 108010062271 Acute-Phase Proteins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- 102000009091 Amyloidogenic Proteins Human genes 0.000 description 1
- 108010048112 Amyloidogenic Proteins Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 1
- 201000009030 Carcinoma Diseases 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 208000000094 Chronic Pain Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical class C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 102000016942 Elastin Human genes 0.000 description 1
- 108010014258 Elastin Proteins 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical group C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 1
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 1
- 108010067306 Fibronectins Proteins 0.000 description 1
- 102000016359 Fibronectins Human genes 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 208000032612 Glial tumor Diseases 0.000 description 1
- 206010018338 Glioma Diseases 0.000 description 1
- 102000006395 Globulins Human genes 0.000 description 1
- 108010044091 Globulins Proteins 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 229920002683 Glycosaminoglycan Polymers 0.000 description 1
- 206010073069 Hepatic cancer Diseases 0.000 description 1
- 101000958041 Homo sapiens Musculin Proteins 0.000 description 1
- 101000617823 Homo sapiens Solute carrier organic anion transporter family member 6A1 Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 102000007547 Laminin Human genes 0.000 description 1
- 108010085895 Laminin Proteins 0.000 description 1
- NNJVILVZKWQKPM-UHFFFAOYSA-N Lidocaine Chemical compound CCN(CC)CC(=O)NC1=C(C)C=CC=C1C NNJVILVZKWQKPM-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 1
- 208000001894 Nasopharyngeal Neoplasms Diseases 0.000 description 1
- 206010061308 Neonatal infection Diseases 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- 102100037369 Nidogen-1 Human genes 0.000 description 1
- 206010030113 Oedema Diseases 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 102000009890 Osteonectin Human genes 0.000 description 1
- 108010077077 Osteonectin Proteins 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 108010038988 Peptide Hormones Proteins 0.000 description 1
- 102000015731 Peptide Hormones Human genes 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000004721 Polyphenylene oxide Substances 0.000 description 1
- 229920001214 Polysorbate 60 Polymers 0.000 description 1
- 206010067268 Post procedural infection Diseases 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 102000007327 Protamines Human genes 0.000 description 1
- 108010007568 Protamines Proteins 0.000 description 1
- 101710197567 Protein LIFEGUARD 4 Proteins 0.000 description 1
- 102100024094 Protein lifeguard 4 Human genes 0.000 description 1
- 102000016611 Proteoglycans Human genes 0.000 description 1
- 108010067787 Proteoglycans Proteins 0.000 description 1
- HVUMOYIDDBPOLL-XWVZOOPGSA-N Sorbitan monostearate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O HVUMOYIDDBPOLL-XWVZOOPGSA-N 0.000 description 1
- 101710172711 Structural protein Proteins 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 102000007000 Tenascin Human genes 0.000 description 1
- 108010008125 Tenascin Proteins 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- 108060008245 Thrombospondin Proteins 0.000 description 1
- 102000002938 Thrombospondin Human genes 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 108010031318 Vitronectin Proteins 0.000 description 1
- 102100035140 Vitronectin Human genes 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 206010000269 abscess Diseases 0.000 description 1
- 229940124532 absorption promoter Drugs 0.000 description 1
- FMIZLDOLVIXTAG-NXHHSFFQSA-N acetic acid (2R)-2-amino-3-(2-aminoethylamino)propan-1-ol Chemical compound CC(O)=O.CC(O)=O.CC(O)=O.CC(O)=O.CC(O)=O.NCCNC[C@@H](N)CO FMIZLDOLVIXTAG-NXHHSFFQSA-N 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- MPRWIVBRDAXEAF-UHFFFAOYSA-N acetyloxy ethaneperoxoate Chemical group CC(=O)OOOC(C)=O MPRWIVBRDAXEAF-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 206010002022 amyloidosis Diseases 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 210000003423 ankle Anatomy 0.000 description 1
- 230000001640 apoptogenic effect Effects 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 210000001367 artery Anatomy 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 230000003143 atherosclerotic effect Effects 0.000 description 1
- JXLHNMVSKXFWAO-UHFFFAOYSA-N azane;7-fluoro-2,1,3-benzoxadiazole-4-sulfonic acid Chemical compound N.OS(=O)(=O)C1=CC=C(F)C2=NON=C12 JXLHNMVSKXFWAO-UHFFFAOYSA-N 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- SQVRNKJHWKZAKO-UHFFFAOYSA-N beta-N-Acetyl-D-neuraminic acid Natural products CC(=O)NC1C(O)CC(O)(C(O)=O)OC1C(O)C(O)CO SQVRNKJHWKZAKO-UHFFFAOYSA-N 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-N beta-phenylpropanoic acid Natural products OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 1
- 238000006065 biodegradation reaction Methods 0.000 description 1
- 239000013060 biological fluid Substances 0.000 description 1
- 238000012984 biological imaging Methods 0.000 description 1
- 239000000090 biomarker Substances 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 210000000133 brain stem Anatomy 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 239000008364 bulk solution Substances 0.000 description 1
- 235000019437 butane-1,3-diol Nutrition 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000011088 calibration curve Methods 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229940081733 cetearyl alcohol Drugs 0.000 description 1
- 230000003196 chaotropic effect Effects 0.000 description 1
- 230000005591 charge neutralization Effects 0.000 description 1
- 150000004697 chelate complex Chemical class 0.000 description 1
- 229920001429 chelating resin Polymers 0.000 description 1
- 230000010109 chemoembolization Effects 0.000 description 1
- 230000000973 chemotherapeutic effect Effects 0.000 description 1
- ZPEIMTDSQAKGNT-UHFFFAOYSA-N chlorpromazine Chemical compound C1=C(Cl)C=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 ZPEIMTDSQAKGNT-UHFFFAOYSA-N 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 229940039231 contrast media Drugs 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 239000002254 cytotoxic agent Substances 0.000 description 1
- 231100000599 cytotoxic agent Toxicity 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 238000013480 data collection Methods 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 150000008050 dialkyl sulfates Chemical class 0.000 description 1
- 125000005265 dialkylamine group Chemical group 0.000 description 1
- 150000001983 dialkylethers Chemical class 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- GXGAKHNRMVGRPK-UHFFFAOYSA-N dimagnesium;dioxido-bis[[oxido(oxo)silyl]oxy]silane Chemical compound [Mg+2].[Mg+2].[O-][Si](=O)O[Si]([O-])([O-])O[Si]([O-])=O GXGAKHNRMVGRPK-UHFFFAOYSA-N 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- GAFRWLVTHPVQGK-UHFFFAOYSA-N dipentyl sulfate Chemical class CCCCCOS(=O)(=O)OCCCCC GAFRWLVTHPVQGK-UHFFFAOYSA-N 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 1
- 235000019797 dipotassium phosphate Nutrition 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 229920002549 elastin Polymers 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 230000005670 electromagnetic radiation Effects 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000008387 emulsifying waxe Substances 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940095399 enema Drugs 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 210000003722 extracellular fluid Anatomy 0.000 description 1
- 210000002744 extracellular matrix Anatomy 0.000 description 1
- 108091022862 fatty acid binding Proteins 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 229920002313 fluoropolymer Polymers 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 229940044350 gadopentetate dimeglumine Drugs 0.000 description 1
- 229940016115 gadoterate meglumine Drugs 0.000 description 1
- RYHQMKVRYNEBNJ-BMWGJIJESA-K gadoterate meglumine Chemical compound [Gd+3].CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO.OC(=O)CN1CCN(CC([O-])=O)CCN(CC([O-])=O)CCN(CC([O-])=O)CC1 RYHQMKVRYNEBNJ-BMWGJIJESA-K 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 229960002449 glycine Drugs 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 235000015220 hamburgers Nutrition 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 206010019692 hepatic necrosis Diseases 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 102000046949 human MSC Human genes 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical compound [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- BCGWQEUPMDMJNV-UHFFFAOYSA-N imipramine Chemical compound C1CC2=CC=CC=C2N(CCCN(C)C)C2=CC=CC=C21 BCGWQEUPMDMJNV-UHFFFAOYSA-N 0.000 description 1
- 229960004801 imipramine Drugs 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- 238000007917 intracranial administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007919 intrasynovial administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 229910021644 lanthanide ion Inorganic materials 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 231100001231 less toxic Toxicity 0.000 description 1
- 229960004194 lidocaine Drugs 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 231100000149 liver necrosis Toxicity 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 210000005228 liver tissue Anatomy 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004324 lymphatic system Anatomy 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229940099273 magnesium trisilicate Drugs 0.000 description 1
- 229910000386 magnesium trisilicate Inorganic materials 0.000 description 1
- 235000019793 magnesium trisilicate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 229940042472 mineral oil Drugs 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000011733 molybdenum Substances 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- 125000001421 myristyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 201000008383 nephritis Diseases 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 108010008217 nidogen Proteins 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- GLDOVTGHNKAZLK-UHFFFAOYSA-N octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCCCO GLDOVTGHNKAZLK-UHFFFAOYSA-N 0.000 description 1
- HGASFNYMVGEKTF-UHFFFAOYSA-N octan-1-ol;hydrate Chemical compound O.CCCCCCCCO HGASFNYMVGEKTF-UHFFFAOYSA-N 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 229960003330 pentetic acid Drugs 0.000 description 1
- 230000010412 perfusion Effects 0.000 description 1
- KHIWWQKSHDUIBK-UHFFFAOYSA-N periodic acid Chemical class OI(=O)(=O)=O KHIWWQKSHDUIBK-UHFFFAOYSA-N 0.000 description 1
- 150000002978 peroxides Chemical class 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 230000010363 phase shift Effects 0.000 description 1
- 150000004713 phosphodiesters Chemical class 0.000 description 1
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 1
- FAIAAWCVCHQXDN-UHFFFAOYSA-N phosphorus trichloride Chemical compound ClP(Cl)Cl FAIAAWCVCHQXDN-UHFFFAOYSA-N 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- 229950010765 pivalate Drugs 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 239000001818 polyoxyethylene sorbitan monostearate Substances 0.000 description 1
- 235000010989 polyoxyethylene sorbitan monostearate Nutrition 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229940113124 polysorbate 60 Drugs 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 150000004032 porphyrins Chemical class 0.000 description 1
- 230000008092 positive effect Effects 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 239000004302 potassium sorbate Substances 0.000 description 1
- 235000010241 potassium sorbate Nutrition 0.000 description 1
- 229940069338 potassium sorbate Drugs 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 229960003712 propranolol Drugs 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 208000023958 prostate neoplasm Diseases 0.000 description 1
- 229950008679 protamine sulfate Drugs 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 229940100618 rectal suppository Drugs 0.000 description 1
- 239000006215 rectal suppository Substances 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 230000029058 respiratory gaseous exchange Effects 0.000 description 1
- SQVRNKJHWKZAKO-OQPLDHBCSA-N sialic acid Chemical compound CC(=O)N[C@@H]1[C@@H](O)C[C@@](O)(C(O)=O)OC1[C@H](O)[C@H](O)CO SQVRNKJHWKZAKO-OQPLDHBCSA-N 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 230000003381 solubilizing effect Effects 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000001587 sorbitan monostearate Substances 0.000 description 1
- 235000011076 sorbitan monostearate Nutrition 0.000 description 1
- 229940035048 sorbitan monostearate Drugs 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 230000003637 steroidlike Effects 0.000 description 1
- 238000005556 structure-activity relationship Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 229910021653 sulphate ion Inorganic materials 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- MHXBHWLGRWOABW-UHFFFAOYSA-N tetradecyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCCCCCCCCCCCCCC MHXBHWLGRWOABW-UHFFFAOYSA-N 0.000 description 1
- QBVXKDJEZKEASM-UHFFFAOYSA-M tetraoctylammonium bromide Chemical compound [Br-].CCCCCCCC[N+](CCCCCCCC)(CCCCCCCC)CCCCCCCC QBVXKDJEZKEASM-UHFFFAOYSA-M 0.000 description 1
- 238000004861 thermometry Methods 0.000 description 1
- 238000000015 thermotherapy Methods 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 description 1
- 239000010937 tungsten Substances 0.000 description 1
- 238000000108 ultra-filtration Methods 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 239000003871 white petrolatum Substances 0.000 description 1
- 210000002268 wool Anatomy 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/06—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations
- A61K49/08—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations characterised by the carrier
- A61K49/10—Organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0013—Luminescence
- A61K49/0017—Fluorescence in vivo
- A61K49/005—Fluorescence in vivo characterised by the carrier molecule carrying the fluorescent agent
- A61K49/0052—Small organic molecules
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/06—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations
- A61K49/08—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations characterised by the carrier
- A61K49/085—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations characterised by the carrier conjugated systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/06—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations
- A61K49/08—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations characterised by the carrier
- A61K49/10—Organic compounds
- A61K49/101—Organic compounds the carrier being a complex-forming compound able to form MRI-active complexes with paramagnetic metals
- A61K49/103—Organic compounds the carrier being a complex-forming compound able to form MRI-active complexes with paramagnetic metals the complex-forming compound being acyclic, e.g. DTPA
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/06—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations
- A61K49/08—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations characterised by the carrier
- A61K49/10—Organic compounds
- A61K49/101—Organic compounds the carrier being a complex-forming compound able to form MRI-active complexes with paramagnetic metals
- A61K49/106—Organic compounds the carrier being a complex-forming compound able to form MRI-active complexes with paramagnetic metals the complex-forming compound being cyclic, e.g. DOTA
Landscapes
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Radiology & Medical Imaging (AREA)
- Medicinal Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Magnetic Resonance Imaging Apparatus (AREA)
- Apparatus For Radiation Diagnosis (AREA)
- Ultra Sonic Daignosis Equipment (AREA)
- Analysing Materials By The Use Of Radiation (AREA)
- Transforming Light Signals Into Electric Signals (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Investigating Or Analysing Materials By Optical Means (AREA)
Abstract
Description


















Claims



















Priority Applications (16)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU96686/98A AU742438C (en) | 1997-10-02 | 1998-09-24 | Contrast-enhanced diagnostic imaging method for monitoring interventional therapies |
| NZ503402A NZ503402A (en) | 1997-10-02 | 1998-09-24 | Contrast-enhanced diagnostic imaging method for monitoring interventional therapies |
| SK484-2000A SK4842000A3 (en) | 1997-10-02 | 1998-09-24 | Contrast-enhanced diagnostic imaging method for monitoring interventional therapies |
| KR1020007003527A KR20010030854A (en) | 1997-10-02 | 1998-09-24 | Contrast-enhanced diagnostic imaging method for monitoring interventional therapies |
| DE69819925T DE69819925T2 (en) | 1997-10-02 | 1998-09-24 | Contrast-enhanced diagnostic image generation method for monitoring therapeutic interventions |
| AT98950704T ATE254479T1 (en) | 1997-10-02 | 1998-09-24 | METHOD FOR IMPROVED CONTRAST IMAGING FOR THERAPY MONITORING |
| JP2000514677A JP2001518523A (en) | 1997-10-02 | 1998-09-24 | Contrast-enhanced diagnostic imaging method for monitoring interventional therapy |
| IL13498598A IL134985A0 (en) | 1997-10-02 | 1998-09-24 | A method for contrast enhanced diagnostic imaging |
| DK98950704T DK1019094T3 (en) | 1997-10-02 | 1998-09-24 | Contrast-enhanced diagnostic imaging method for monitoring interventional therapies |
| HU0101245A HUP0101245A3 (en) | 1997-10-02 | 1998-09-24 | Contrast-enhanced diagnostic imaging method for monitoring interventional therapies |
| EP98950704A EP1019094B1 (en) | 1997-10-02 | 1998-09-24 | Contrast-enhanced diagnostic imaging method for monitoring interventional therapies |
| CA002303426A CA2303426C (en) | 1997-10-02 | 1998-09-24 | Contrast-enhanced diagnostic imaging method for monitoring interventional therapies |
| BR9812716-0A BR9812716A (en) | 1997-10-02 | 1998-09-24 | Imaging methods for enhanced contrast diagnosis to monitor interventional therapies |
| IS5399A IS2041B (en) | 1997-10-02 | 2000-03-10 | Shadow-enhancing imaging method to monitor the intervention |
| NO20001707A NO321966B1 (en) | 1997-10-02 | 2000-04-03 | Use of a contrast agent for the preparation of a diagnostic agent for monitoring image contrast during intervention therapy. |
| HK01100360A HK1030744A1 (en) | 1997-10-02 | 2001-01-12 | Contrast-enhanced diagnostic imaging method for monitoring interventional therapies. |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US94298997A | 1997-10-02 | 1997-10-02 | |
| US08/942,989 | 1997-10-02 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO1999017809A2 true WO1999017809A2 (en) | 1999-04-15 |
| WO1999017809A3 WO1999017809A3 (en) | 1999-05-20 |
Family
ID=25478927
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1998/020182 WO1999017809A2 (en) | 1997-10-02 | 1998-09-24 | Contrast-enhanced diagnostic imaging method for monitoring interventional therapies |
Country Status (20)
| Country | Link |
|---|---|
| US (2) | US6861045B1 (en) |
| EP (1) | EP1019094B1 (en) |
| JP (2) | JP2001518523A (en) |
| KR (1) | KR20010030854A (en) |
| AT (1) | ATE254479T1 (en) |
| AU (1) | AU742438C (en) |
| BR (1) | BR9812716A (en) |
| CA (1) | CA2303426C (en) |
| DE (1) | DE69819925T2 (en) |
| DK (1) | DK1019094T3 (en) |
| ES (1) | ES2206996T3 (en) |
| HK (1) | HK1030744A1 (en) |
| HU (1) | HUP0101245A3 (en) |
| IL (1) | IL134985A0 (en) |
| IS (1) | IS2041B (en) |
| NO (1) | NO321966B1 (en) |
| NZ (1) | NZ503402A (en) |
| PT (1) | PT1019094E (en) |
| SK (1) | SK4842000A3 (en) |
| WO (1) | WO1999017809A2 (en) |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001052906A2 (en) * | 2000-01-22 | 2001-07-26 | Epix Medical, Inc. | Magnetic resonance imaging using contrast agents prodrugs bioactivated by enzymatic cleavage |
| US6495118B1 (en) | 1997-09-26 | 2002-12-17 | Schering Aktiengesellschaft | Lipophilic metal complexes for necrosis and infarction imaging |
| US6548044B1 (en) | 1999-11-22 | 2003-04-15 | Epix Medical, Inc. | Imaging sexual response |
| EP1423136A1 (en) * | 2001-08-10 | 2004-06-02 | Epix Medical, Inc. | Polypeptide conjugates with extended circulating half-lives |
| EP1747026A1 (en) * | 2004-05-18 | 2007-01-31 | Siemens Aktiengesellschaft | Biomolecular contrast agents for therapy control in radiation therapy with proton or ion beams |
| EP1944312A1 (en) | 2003-03-03 | 2008-07-16 | Dyax Corporation | Peptides that specifically bind HGF receptor (CMET) and uses thereof |
| US7412279B2 (en) | 2001-07-30 | 2008-08-12 | Epix Pharmaceuticals, Inc. | Systems and methods for targeted magnetic resonance imaging of the vascular system |
| EP2014310A2 (en) | 2002-03-01 | 2009-01-14 | Dyax Corporation | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| EP2048504A1 (en) * | 1999-05-05 | 2009-04-15 | Spectros Corporation | Detecting, localizing, and targeting internal sites in vivo using optical contrast agents |
| US7585492B2 (en) | 2004-05-18 | 2009-09-08 | Siemens Aktiengesellschaft | Biomolecular contrast agents for therapy success and dose monitoring in radiation therapy with proton or ion beams |
| US8263040B2 (en) | 2006-10-18 | 2012-09-11 | Bayer Schering Pharma Ag | Metal chelates having a perfluorinated PEG radical, processes for their preparation, and their use |
| WO2014160039A1 (en) * | 2013-03-13 | 2014-10-02 | Lantheus Medical Imaging, Inc. | Process for manufacture of gadofosveset trisodium monohydrate |
| US9056138B2 (en) | 2002-03-01 | 2015-06-16 | Bracco Suisse Sa | Multivalent constructs for therapeutic and diagnostic applications |
| US9200017B2 (en) | 2009-03-19 | 2015-12-01 | The General Hospital Corporation | Multimodal imaging of fibrin |
| US9629934B2 (en) | 2002-03-01 | 2017-04-25 | Dyax Corp. | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| US9889213B2 (en) | 2009-04-17 | 2018-02-13 | The General Hospital Corporation | Multimodal imaging of fibrin |
| US9913875B2 (en) | 2015-03-16 | 2018-03-13 | California Institute Of Technology | Botulinum neurotoxin-specific capture agents, compositions, and methods of using and making |
| US10383590B2 (en) | 2015-09-28 | 2019-08-20 | General Electric Company | Methods and systems for adaptive scan control |
| US10471162B2 (en) | 2014-06-20 | 2019-11-12 | The General Hospital Corporation | Collagen targeted imaging probes |
| US10598671B2 (en) | 2015-07-15 | 2020-03-24 | Indi Molecular, Inc. | IL-17F-specific capture agents, compositions, and methods of using and making |
| US10975123B2 (en) | 2014-05-05 | 2021-04-13 | California Institute Of Technology | Mutant Akt-specific capture agents, compositions, and methods of using and making |
| US11358982B2 (en) | 2016-11-01 | 2022-06-14 | Ohio State Innovation Foundation | Methods for the iodination of biomolecules |
| US11414460B2 (en) | 2019-07-19 | 2022-08-16 | Institute For Systems Biology | KRAS-specific capture agents, compositions, and methods of making and using |
| US11638764B2 (en) | 2018-11-08 | 2023-05-02 | Indi Molecular, Inc. | Theranostic capture agents, compositions, and methods of using and making |
| US11719705B2 (en) | 2017-06-15 | 2023-08-08 | Indi Molecular, Inc. | IL-17F and IL-17A-specific capture agents, compositions, and methods of using and making |
| US11733246B2 (en) | 2020-11-03 | 2023-08-22 | Indi Molecular, Inc. | Compositions, imaging, and therapeutic methods targeting folate receptor 1 (FOLR1) |
| US11884707B2 (en) | 2016-09-29 | 2024-01-30 | Regeneron Pharmaceuticals, Inc. | Compositions for detection, inhibition and imaging of indoleamine 2, 3-dioxygenase 1 (IDO1) and methods of making and using same |
| US11919972B2 (en) | 2018-11-02 | 2024-03-05 | Regeneron Pharmaceuticals, Inc. | Peptide libraries with non-canonical amino acids |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL134985A0 (en) * | 1997-10-02 | 2001-05-20 | Epix Medical Inc | A method for contrast enhanced diagnostic imaging |
| US7794693B2 (en) | 2002-03-01 | 2010-09-14 | Bracco International B.V. | Targeting vector-phospholipid conjugates |
| US8623822B2 (en) | 2002-03-01 | 2014-01-07 | Bracco Suisse Sa | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| US20050064045A1 (en) * | 2003-09-18 | 2005-03-24 | Sheng-Ping Zhong | Injectable therapeutic formulations |
| JP2006248978A (en) * | 2005-03-10 | 2006-09-21 | Mebiopharm Co Ltd | New liposome preparation |
| EP1957996A1 (en) * | 2005-11-29 | 2008-08-20 | Koninklijke Philips Electronics N.V. | Distinguishing bound and unbound contrast agents using magnetic resonance |
| WO2007073792A1 (en) * | 2005-12-19 | 2007-07-05 | Bayer Schering Pharma Aktiengesellschaft | Process for preparing 4,4- diphenylcyclohexanol |
| CA2660717A1 (en) | 2006-08-17 | 2008-02-21 | Epix Pharmaceuticals, Inc. | Methods for lymph system imaging |
| US20100092389A1 (en) * | 2008-10-10 | 2010-04-15 | The General Hospital Corporation | Detection of atherosclerosis using indocyanine green |
| US9423480B2 (en) * | 2008-10-27 | 2016-08-23 | The University Of Western Ontario | System and method for magnetic resonance imaging |
| US8891842B2 (en) | 2010-02-02 | 2014-11-18 | Koninklijke Philips N.V. | Functional imaging |
| US11160454B2 (en) | 2013-04-08 | 2021-11-02 | Sunnybrook Research Institute | System and method for imaging biomarkers indicative of cardiac thermal ablation lesions |
| JP2015087167A (en) * | 2013-10-29 | 2015-05-07 | キヤノン株式会社 | Image processing method and image processing system |
| WO2015089505A2 (en) | 2013-12-13 | 2015-06-18 | The Trustees Of The University Of Pennsylvania | Coaxial ablation probe and method and system for real-time monitoring of ablation therapy |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996023526A2 (en) * | 1995-02-01 | 1996-08-08 | Epix Medical, Inc. | Diagnostic imaging contrast agents with extended blood retention |
| WO1997036619A2 (en) * | 1996-04-01 | 1997-10-09 | Epix Medical, Inc. | Bioactivated diagnostic imaging contrast agents |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5250285A (en) | 1985-05-08 | 1993-10-05 | The General Hospital Corporation | Hydroxy-aryl metal chelates for diagnostic NMR imaging |
| US5094848A (en) | 1989-06-30 | 1992-03-10 | Neorx Corporation | Cleavable diphosphate and amidated diphosphate linkers |
| IT1269839B (en) | 1994-05-26 | 1997-04-15 | Bracco Spa | CONJUGATES OF BILIARY ACIDS, THEIR DERIVATIVES WITH METALLIC COMPLEXES AND RELATED USES |
| CA2539215A1 (en) | 1995-02-01 | 1996-08-08 | Epix Pharmaceuticals, Inc. | Diagnostic imaging contrast agents with extended blood retention |
| DE19529512C2 (en) | 1995-08-10 | 2000-11-23 | Siemens Ag | Phase contrast MR angiography method and arrangement for carrying out the method |
| DE19543785A1 (en) | 1995-11-24 | 1997-05-28 | Philips Patentverwaltung | MR method and arrangement for carrying out the method |
| IT1283650B1 (en) | 1996-08-02 | 1998-04-23 | Bracco Spa | HIGH RELAXATION PARAMAGNETIC CHELATES IN SERUM |
| US5919967A (en) | 1997-04-11 | 1999-07-06 | Epix Medical, Inc. | Process for synthesizing phosphodiesters |
| IL134985A0 (en) * | 1997-10-02 | 2001-05-20 | Epix Medical Inc | A method for contrast enhanced diagnostic imaging |
| IT1304501B1 (en) | 1998-12-23 | 2001-03-19 | Bracco Spa | USE OF BILIARY ACID DERIVATIVES CONJUGATED WITH METALLIC COMPLEXES LIKE "BLOOD POOL AGENTS" FOR THE DIAGNOSTIC INVESTIGATION THROUGH RESONANCE |
-
1998
- 1998-09-24 IL IL13498598A patent/IL134985A0/en not_active IP Right Cessation
- 1998-09-24 HU HU0101245A patent/HUP0101245A3/en unknown
- 1998-09-24 SK SK484-2000A patent/SK4842000A3/en unknown
- 1998-09-24 AT AT98950704T patent/ATE254479T1/en not_active IP Right Cessation
- 1998-09-24 DK DK98950704T patent/DK1019094T3/en active
- 1998-09-24 JP JP2000514677A patent/JP2001518523A/en active Pending
- 1998-09-24 WO PCT/US1998/020182 patent/WO1999017809A2/en not_active Application Discontinuation
- 1998-09-24 NZ NZ503402A patent/NZ503402A/en unknown
- 1998-09-24 AU AU96686/98A patent/AU742438C/en not_active Ceased
- 1998-09-24 ES ES98950704T patent/ES2206996T3/en not_active Expired - Lifetime
- 1998-09-24 EP EP98950704A patent/EP1019094B1/en not_active Expired - Lifetime
- 1998-09-24 CA CA002303426A patent/CA2303426C/en not_active Expired - Fee Related
- 1998-09-24 KR KR1020007003527A patent/KR20010030854A/en not_active Application Discontinuation
- 1998-09-24 PT PT98950704T patent/PT1019094E/en unknown
- 1998-09-24 BR BR9812716-0A patent/BR9812716A/en not_active Application Discontinuation
- 1998-09-24 DE DE69819925T patent/DE69819925T2/en not_active Expired - Lifetime
-
2000
- 2000-03-10 IS IS5399A patent/IS2041B/en unknown
- 2000-04-03 NO NO20001707A patent/NO321966B1/en not_active IP Right Cessation
- 2000-09-08 US US09/887,706 patent/US6861045B1/en not_active Expired - Fee Related
-
2001
- 2001-01-12 HK HK01100360A patent/HK1030744A1/en not_active IP Right Cessation
-
2004
- 2004-10-08 US US10/961,872 patent/US7175829B2/en not_active Expired - Fee Related
-
2005
- 2005-09-22 JP JP2005276942A patent/JP2006077020A/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996023526A2 (en) * | 1995-02-01 | 1996-08-08 | Epix Medical, Inc. | Diagnostic imaging contrast agents with extended blood retention |
| WO1997036619A2 (en) * | 1996-04-01 | 1997-10-09 | Epix Medical, Inc. | Bioactivated diagnostic imaging contrast agents |
Non-Patent Citations (6)
| Title |
|---|
| BRASCH R. ET AL.: "Assessing Tumor Angiogenisis Using Macromolecular MR Imaging Contrast Media" JOURNAL OF MAGNETIC RESONANCE IMAGING, vol. 7, no. 1, February 1997, pages 68-74, XP002094297 * |
| DUPAS BENOIT ET AL.: "Delineation of Liver Necrosis Using Double Contrast-Enhanced MRI" JOURNAL OF MAGNETIC RESONANCE IMAGING , vol. 7, no. 3, 1997, pages 472-477, XP002094271 cited in the application * |
| HYNYNEN ET AL.: "The feasibility of using MRI to monitor and guide noninvasive ultrasound surgery." ULTRASOUND IN MED. AND BIOL., vol. 19, no. 1, 1993, pages 91-92, XP002095678 * |
| HYNYNEN K. ET AL.: "The Usefulness of a Contrast Agent and Gradient-Recalled Acquisition in a Steady-State Imaging Sequence for Magnetic Resonance Imaging-Guided Noninvasive Ultrasound Surgery" INVESTIGATIVE RADIOLOGY, vol. 29, no. 10, 1994, pages 897-903, XP002094270 * |
| LAUFFER R B ET AL: "MS-325: A SMALL-MOLECULE VASCULAR IMAGING AGENT FOR MAGNETIC RESONANCE IMAGING" ACADEMIC RADIOLOGY, vol. 3, no. SUPPL. 02, August 1996, pages S356-S358, XP000197827 * |
| R.M.M. SEIBEL ET AL.: "Image-guided minimally invasive therapy" SURGICAL ENDOSCOPY, vol. 11, no. 2, 1997, pages 154-162, XP002095677 * |
Cited By (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6495118B1 (en) | 1997-09-26 | 2002-12-17 | Schering Aktiengesellschaft | Lipophilic metal complexes for necrosis and infarction imaging |
| EP2048504A1 (en) * | 1999-05-05 | 2009-04-15 | Spectros Corporation | Detecting, localizing, and targeting internal sites in vivo using optical contrast agents |
| US6548044B1 (en) | 1999-11-22 | 2003-04-15 | Epix Medical, Inc. | Imaging sexual response |
| US6969507B2 (en) | 1999-11-22 | 2005-11-29 | Epix Pharmaceuticals, Inc. | Imaging sexual response |
| WO2001052906A3 (en) * | 2000-01-22 | 2002-01-24 | Epix Medical Inc | Magnetic resonance imaging using contrast agents prodrugs bioactivated by enzymatic cleavage |
| WO2001052906A2 (en) * | 2000-01-22 | 2001-07-26 | Epix Medical, Inc. | Magnetic resonance imaging using contrast agents prodrugs bioactivated by enzymatic cleavage |
| US7412279B2 (en) | 2001-07-30 | 2008-08-12 | Epix Pharmaceuticals, Inc. | Systems and methods for targeted magnetic resonance imaging of the vascular system |
| EP1423136A1 (en) * | 2001-08-10 | 2004-06-02 | Epix Medical, Inc. | Polypeptide conjugates with extended circulating half-lives |
| EP1423136A4 (en) * | 2001-08-10 | 2007-07-25 | Epix Pharm Inc | Polypeptide conjugates with extended circulating half-lives |
| US9629934B2 (en) | 2002-03-01 | 2017-04-25 | Dyax Corp. | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| US9056138B2 (en) | 2002-03-01 | 2015-06-16 | Bracco Suisse Sa | Multivalent constructs for therapeutic and diagnostic applications |
| EP2014310A2 (en) | 2002-03-01 | 2009-01-14 | Dyax Corporation | KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy |
| EP1944312A1 (en) | 2003-03-03 | 2008-07-16 | Dyax Corporation | Peptides that specifically bind HGF receptor (CMET) and uses thereof |
| US7585492B2 (en) | 2004-05-18 | 2009-09-08 | Siemens Aktiengesellschaft | Biomolecular contrast agents for therapy success and dose monitoring in radiation therapy with proton or ion beams |
| EP1747026A1 (en) * | 2004-05-18 | 2007-01-31 | Siemens Aktiengesellschaft | Biomolecular contrast agents for therapy control in radiation therapy with proton or ion beams |
| US8263040B2 (en) | 2006-10-18 | 2012-09-11 | Bayer Schering Pharma Ag | Metal chelates having a perfluorinated PEG radical, processes for their preparation, and their use |
| US9200017B2 (en) | 2009-03-19 | 2015-12-01 | The General Hospital Corporation | Multimodal imaging of fibrin |
| US9889213B2 (en) | 2009-04-17 | 2018-02-13 | The General Hospital Corporation | Multimodal imaging of fibrin |
| US10106562B2 (en) | 2013-03-13 | 2018-10-23 | Lantheus Medical Imaging, Inc. | Process for manufacture of gadofosveset trisodium monohydrate |
| WO2014160039A1 (en) * | 2013-03-13 | 2014-10-02 | Lantheus Medical Imaging, Inc. | Process for manufacture of gadofosveset trisodium monohydrate |
| CN105209439A (en) * | 2013-03-13 | 2015-12-30 | 蓝瑟尔斯医学影像有限公司 | Process for manufacture of gadofosveset trisodium monohydrate |
| US10975123B2 (en) | 2014-05-05 | 2021-04-13 | California Institute Of Technology | Mutant Akt-specific capture agents, compositions, and methods of using and making |
| US10471162B2 (en) | 2014-06-20 | 2019-11-12 | The General Hospital Corporation | Collagen targeted imaging probes |
| US11723944B2 (en) | 2015-03-16 | 2023-08-15 | Indi Molecular, Inc. | Botulinum neurotoxin-specific capture agents, compositions, and methods of using and making |
| US9913875B2 (en) | 2015-03-16 | 2018-03-13 | California Institute Of Technology | Botulinum neurotoxin-specific capture agents, compositions, and methods of using and making |
| US11007245B2 (en) | 2015-03-16 | 2021-05-18 | Indi Molecular, Inc. | Botulinum neurotoxin-specific capture agents, compositions, and methods of using and making |
| US10598671B2 (en) | 2015-07-15 | 2020-03-24 | Indi Molecular, Inc. | IL-17F-specific capture agents, compositions, and methods of using and making |
| US10383590B2 (en) | 2015-09-28 | 2019-08-20 | General Electric Company | Methods and systems for adaptive scan control |
| US11884707B2 (en) | 2016-09-29 | 2024-01-30 | Regeneron Pharmaceuticals, Inc. | Compositions for detection, inhibition and imaging of indoleamine 2, 3-dioxygenase 1 (IDO1) and methods of making and using same |
| US11358982B2 (en) | 2016-11-01 | 2022-06-14 | Ohio State Innovation Foundation | Methods for the iodination of biomolecules |
| US11719705B2 (en) | 2017-06-15 | 2023-08-08 | Indi Molecular, Inc. | IL-17F and IL-17A-specific capture agents, compositions, and methods of using and making |
| US11919972B2 (en) | 2018-11-02 | 2024-03-05 | Regeneron Pharmaceuticals, Inc. | Peptide libraries with non-canonical amino acids |
| US11638764B2 (en) | 2018-11-08 | 2023-05-02 | Indi Molecular, Inc. | Theranostic capture agents, compositions, and methods of using and making |
| US11414460B2 (en) | 2019-07-19 | 2022-08-16 | Institute For Systems Biology | KRAS-specific capture agents, compositions, and methods of making and using |
| US11733246B2 (en) | 2020-11-03 | 2023-08-22 | Indi Molecular, Inc. | Compositions, imaging, and therapeutic methods targeting folate receptor 1 (FOLR1) |
Also Published As
| Publication number | Publication date |
|---|---|
| SK4842000A3 (en) | 2000-11-07 |
| ATE254479T1 (en) | 2003-12-15 |
| IL134985A0 (en) | 2001-05-20 |
| JP2001518523A (en) | 2001-10-16 |
| EP1019094B1 (en) | 2003-11-19 |
| US7175829B2 (en) | 2007-02-13 |
| IS5399A (en) | 2000-03-10 |
| AU742438B2 (en) | 2002-01-03 |
| CA2303426A1 (en) | 1999-04-15 |
| US6861045B1 (en) | 2005-03-01 |
| PT1019094E (en) | 2004-03-31 |
| NO20001707L (en) | 2000-05-31 |
| ES2206996T3 (en) | 2004-05-16 |
| NO20001707D0 (en) | 2000-04-03 |
| KR20010030854A (en) | 2001-04-16 |
| CA2303426C (en) | 2008-09-23 |
| DE69819925T2 (en) | 2004-09-02 |
| US20050118103A1 (en) | 2005-06-02 |
| DK1019094T3 (en) | 2004-02-16 |
| NO321966B1 (en) | 2006-07-31 |
| HK1030744A1 (en) | 2001-05-18 |
| WO1999017809A3 (en) | 1999-05-20 |
| DE69819925D1 (en) | 2003-12-24 |
| HUP0101245A2 (en) | 2001-08-28 |
| EP1019094A2 (en) | 2000-07-19 |
| BR9812716A (en) | 2000-08-22 |
| HUP0101245A3 (en) | 2003-10-28 |
| IS2041B (en) | 2005-09-15 |
| AU9668698A (en) | 1999-04-27 |
| AU742438C (en) | 2003-05-22 |
| NZ503402A (en) | 2002-03-01 |
| JP2006077020A (en) | 2006-03-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU742438C (en) | Contrast-enhanced diagnostic imaging method for monitoring interventional therapies | |
| AU689700B2 (en) | Diagnostic imaging contrast agents with extended blood reention | |
| WO2008022263A2 (en) | Methods for lymph system imaging | |
| AU3138702A (en) | Contrast-enhanced diagnostic imaging method for monitoring interventional therapies | |
| MXPA00002869A (en) | Contrast-enhanced diagnostic imaging method for monitoring interventional therapies | |
| CZ20001189A3 (en) | Diagnostic agent for monitoring intervention therapies | |
| CA2539215A1 (en) | Diagnostic imaging contrast agents with extended blood retention |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 134985 Country of ref document: IL |
|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GD GE GH GM HR HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GD GE GH GM HR HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 96686/98 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2303426 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 503402 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2000/002869 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4842000 Country of ref document: SK Ref document number: PV2000-1189 Country of ref document: CZ Ref document number: 1020007003527 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2000 514677 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998950704 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998950704 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2000-1189 Country of ref document: CZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020007003527 Country of ref document: KR |
|
| WWR | Wipo information: refused in national office |
Ref document number: PV2000-1189 Country of ref document: CZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 96686/98 Country of ref document: AU |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1998950704 Country of ref document: EP |
|
| WWR | Wipo information: refused in national office |
Ref document number: 1020007003527 Country of ref document: KR |