WO1999041383A1 - Antigen library immunization - Google Patents
Antigen library immunization Download PDFInfo
- Publication number
- WO1999041383A1 WO1999041383A1 PCT/US1999/002944 US9902944W WO9941383A1 WO 1999041383 A1 WO1999041383 A1 WO 1999041383A1 US 9902944 W US9902944 W US 9902944W WO 9941383 A1 WO9941383 A1 WO 9941383A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polypeptide
- antigens
- antigen
- recombinant
- cells
- Prior art date
Links
- 108091007433 antigens Proteins 0.000 title claims abstract description 430
- 239000000427 antigen Substances 0.000 title claims abstract description 428
- 102000036639 antigens Human genes 0.000 title claims abstract description 417
- 230000003053 immunization Effects 0.000 title abstract description 34
- 238000002649 immunization Methods 0.000 title abstract description 30
- 238000000034 method Methods 0.000 claims abstract description 218
- 230000028993 immune response Effects 0.000 claims abstract description 97
- 230000001976 improved effect Effects 0.000 claims abstract description 93
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 60
- 244000052769 pathogen Species 0.000 claims abstract description 58
- 206010020751 Hypersensitivity Diseases 0.000 claims abstract description 49
- 201000011510 cancer Diseases 0.000 claims abstract description 47
- 208000023275 Autoimmune disease Diseases 0.000 claims abstract description 18
- 208000027866 inflammatory disease Diseases 0.000 claims abstract description 3
- 230000002757 inflammatory effect Effects 0.000 claims abstract description 3
- 108090000623 proteins and genes Proteins 0.000 claims description 259
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 203
- 210000004027 cell Anatomy 0.000 claims description 197
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 193
- 229920001184 polypeptide Polymers 0.000 claims description 184
- 150000007523 nucleic acids Chemical class 0.000 claims description 156
- 102000039446 nucleic acids Human genes 0.000 claims description 148
- 108020004707 nucleic acids Proteins 0.000 claims description 148
- 229960005486 vaccine Drugs 0.000 claims description 129
- 102000004169 proteins and genes Human genes 0.000 claims description 103
- 239000013566 allergen Substances 0.000 claims description 84
- 230000000890 antigenic effect Effects 0.000 claims description 74
- 238000012216 screening Methods 0.000 claims description 74
- 230000001717 pathogenic effect Effects 0.000 claims description 72
- 241001465754 Metazoa Species 0.000 claims description 64
- 230000002068 genetic effect Effects 0.000 claims description 61
- 238000012360 testing method Methods 0.000 claims description 58
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 55
- 241000700605 Viruses Species 0.000 claims description 53
- 201000010099 disease Diseases 0.000 claims description 51
- 229960004784 allergens Drugs 0.000 claims description 47
- 210000001744 T-lymphocyte Anatomy 0.000 claims description 46
- 230000014509 gene expression Effects 0.000 claims description 46
- 102000040430 polynucleotide Human genes 0.000 claims description 46
- 108091033319 polynucleotide Proteins 0.000 claims description 46
- 239000002157 polynucleotide Substances 0.000 claims description 46
- 239000013598 vector Substances 0.000 claims description 46
- 239000003795 chemical substances by application Substances 0.000 claims description 39
- 230000001681 protective effect Effects 0.000 claims description 37
- 239000003053 toxin Substances 0.000 claims description 34
- 231100000765 toxin Toxicity 0.000 claims description 34
- 108700012359 toxins Proteins 0.000 claims description 34
- 238000001727 in vivo Methods 0.000 claims description 33
- 230000005847 immunogenicity Effects 0.000 claims description 32
- 230000002163 immunogen Effects 0.000 claims description 29
- 208000015181 infectious disease Diseases 0.000 claims description 29
- 230000004044 response Effects 0.000 claims description 29
- 241000588724 Escherichia coli Species 0.000 claims description 22
- 101710091045 Envelope protein Proteins 0.000 claims description 21
- 101710188315 Protein X Proteins 0.000 claims description 21
- 241000607734 Yersinia <bacteria> Species 0.000 claims description 19
- 239000002773 nucleotide Substances 0.000 claims description 19
- 125000003729 nucleotide group Chemical group 0.000 claims description 19
- 238000002823 phage display Methods 0.000 claims description 19
- 239000013603 viral vector Substances 0.000 claims description 17
- 238000004519 manufacturing process Methods 0.000 claims description 16
- 108020004705 Codon Proteins 0.000 claims description 15
- 241000710842 Japanese encephalitis virus Species 0.000 claims description 15
- 230000000172 allergic effect Effects 0.000 claims description 15
- 208000010668 atopic eczema Diseases 0.000 claims description 15
- 231100000655 enterotoxin Toxicity 0.000 claims description 13
- 102000037865 fusion proteins Human genes 0.000 claims description 13
- 108020001507 fusion proteins Proteins 0.000 claims description 13
- 230000001939 inductive effect Effects 0.000 claims description 13
- 230000002829 reductive effect Effects 0.000 claims description 13
- 241000894007 species Species 0.000 claims description 13
- 241000124008 Mammalia Species 0.000 claims description 12
- 238000000684 flow cytometry Methods 0.000 claims description 12
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 claims description 12
- 230000001988 toxicity Effects 0.000 claims description 12
- 231100000419 toxicity Toxicity 0.000 claims description 12
- 208000001490 Dengue Diseases 0.000 claims description 11
- 206010012310 Dengue fever Diseases 0.000 claims description 11
- 241000223960 Plasmodium falciparum Species 0.000 claims description 11
- 208000025729 dengue disease Diseases 0.000 claims description 11
- 230000000694 effects Effects 0.000 claims description 11
- 239000000147 enterotoxin Substances 0.000 claims description 11
- 230000001404 mediated effect Effects 0.000 claims description 11
- 239000002245 particle Substances 0.000 claims description 10
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 10
- 230000003612 virological effect Effects 0.000 claims description 10
- 208000004006 Tick-borne encephalitis Diseases 0.000 claims description 9
- 241000607479 Yersinia pestis Species 0.000 claims description 9
- 244000045947 parasite Species 0.000 claims description 9
- 241000894006 Bacteria Species 0.000 claims description 8
- 102000009016 Cholera Toxin Human genes 0.000 claims description 8
- 108010049048 Cholera Toxin Proteins 0.000 claims description 8
- 230000006044 T cell activation Effects 0.000 claims description 8
- 241000607626 Vibrio cholerae Species 0.000 claims description 8
- 206010003645 Atopy Diseases 0.000 claims description 7
- 239000002671 adjuvant Substances 0.000 claims description 7
- 230000009260 cross reactivity Effects 0.000 claims description 7
- 239000013604 expression vector Substances 0.000 claims description 7
- 238000010186 staining Methods 0.000 claims description 7
- 101710146739 Enterotoxin Proteins 0.000 claims description 6
- 241000700584 Simplexvirus Species 0.000 claims description 6
- 230000005867 T cell response Effects 0.000 claims description 6
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 claims description 6
- 208000002672 hepatitis B Diseases 0.000 claims description 6
- 210000004962 mammalian cell Anatomy 0.000 claims description 6
- 201000006417 multiple sclerosis Diseases 0.000 claims description 6
- 231100000699 Bacterial toxin Toxicity 0.000 claims description 5
- 208000035473 Communicable disease Diseases 0.000 claims description 5
- 206010039710 Scleroderma Diseases 0.000 claims description 5
- 230000005784 autoimmunity Effects 0.000 claims description 5
- 239000000688 bacterial toxin Substances 0.000 claims description 5
- 230000016396 cytokine production Effects 0.000 claims description 5
- 230000035755 proliferation Effects 0.000 claims description 5
- 241001674044 Blattodea Species 0.000 claims description 4
- 241000590002 Helicobacter pylori Species 0.000 claims description 4
- 101100476480 Mus musculus S100a8 gene Proteins 0.000 claims description 4
- 241000191967 Staphylococcus aureus Species 0.000 claims description 4
- 230000006052 T cell proliferation Effects 0.000 claims description 4
- 230000005860 defense response to virus Effects 0.000 claims description 4
- 208000035475 disorder Diseases 0.000 claims description 4
- 239000000428 dust Substances 0.000 claims description 4
- 230000004968 inflammatory condition Effects 0.000 claims description 4
- 210000001685 thyroid gland Anatomy 0.000 claims description 4
- 244000056139 Brassica cretica Species 0.000 claims description 3
- 235000003351 Brassica cretica Nutrition 0.000 claims description 3
- 235000003343 Brassica rupestris Nutrition 0.000 claims description 3
- 240000007817 Olea europaea Species 0.000 claims description 3
- 240000007594 Oryza sativa Species 0.000 claims description 3
- 235000007164 Oryza sativa Nutrition 0.000 claims description 3
- 241000607447 Yersinia enterocolitica Species 0.000 claims description 3
- 241000607477 Yersinia pseudotuberculosis Species 0.000 claims description 3
- 239000003659 bee venom Substances 0.000 claims description 3
- QKSKPIVNLNLAAV-UHFFFAOYSA-N bis(2-chloroethyl) sulfide Chemical compound ClCCSCCCl QKSKPIVNLNLAAV-UHFFFAOYSA-N 0.000 claims description 3
- 230000024245 cell differentiation Effects 0.000 claims description 3
- 210000004443 dendritic cell Anatomy 0.000 claims description 3
- 229940037467 helicobacter pylori Drugs 0.000 claims description 3
- 239000012678 infectious agent Substances 0.000 claims description 3
- 210000002540 macrophage Anatomy 0.000 claims description 3
- 235000010460 mustard Nutrition 0.000 claims description 3
- 235000009566 rice Nutrition 0.000 claims description 3
- 235000001543 Corylus americana Nutrition 0.000 claims description 2
- 240000007582 Corylus avellana Species 0.000 claims description 2
- 235000007466 Corylus avellana Nutrition 0.000 claims description 2
- 208000027761 Hepatic autoimmune disease Diseases 0.000 claims description 2
- 241000186359 Mycobacterium Species 0.000 claims description 2
- 235000002725 Olea europaea Nutrition 0.000 claims description 2
- 108700006640 OspA Proteins 0.000 claims description 2
- 108700023315 OspC Proteins 0.000 claims description 2
- 201000009594 Systemic Scleroderma Diseases 0.000 claims description 2
- 206010042953 Systemic sclerosis Diseases 0.000 claims description 2
- 230000034994 death Effects 0.000 claims description 2
- 230000000688 enterotoxigenic effect Effects 0.000 claims description 2
- 239000013569 fungal allergen Substances 0.000 claims description 2
- 230000028709 inflammatory response Effects 0.000 claims description 2
- 210000001616 monocyte Anatomy 0.000 claims description 2
- 239000009342 ragweed pollen Substances 0.000 claims description 2
- 201000000596 systemic lupus erythematosus Diseases 0.000 claims description 2
- 229940046536 tree pollen allergenic extract Drugs 0.000 claims description 2
- 208000035408 type 1 diabetes mellitus 1 Diseases 0.000 claims description 2
- 108091060545 Nonsense suppressor Proteins 0.000 claims 2
- 239000013573 pollen allergen Substances 0.000 claims 2
- 241000589968 Borrelia Species 0.000 claims 1
- 206010014596 Encephalitis Japanese B Diseases 0.000 claims 1
- 206010014611 Encephalitis venezuelan equine Diseases 0.000 claims 1
- 208000005176 Hepatitis C Diseases 0.000 claims 1
- 201000005807 Japanese encephalitis Diseases 0.000 claims 1
- 208000012658 Skin autoimmune disease Diseases 0.000 claims 1
- 241000193996 Streptococcus pyogenes Species 0.000 claims 1
- 102100021696 Syncytin-1 Human genes 0.000 claims 1
- 102100032467 Transmembrane protease serine 13 Human genes 0.000 claims 1
- 208000002687 Venezuelan Equine Encephalomyelitis Diseases 0.000 claims 1
- 201000009145 Venezuelan equine encephalitis Diseases 0.000 claims 1
- 230000006472 autoimmune response Effects 0.000 claims 1
- 239000013575 birch pollen allergen Substances 0.000 claims 1
- 239000013574 grass pollen allergen Substances 0.000 claims 1
- 206010022000 influenza Diseases 0.000 claims 1
- 230000002401 inhibitory effect Effects 0.000 claims 1
- 210000001179 synovial fluid Anatomy 0.000 claims 1
- 208000026935 allergic disease Diseases 0.000 abstract description 47
- 230000007815 allergy Effects 0.000 abstract description 46
- 230000001225 therapeutic effect Effects 0.000 abstract description 8
- 235000018102 proteins Nutrition 0.000 description 98
- 108020004414 DNA Proteins 0.000 description 89
- 230000006798 recombination Effects 0.000 description 38
- 238000005215 recombination Methods 0.000 description 38
- 241000282414 Homo sapiens Species 0.000 description 34
- 150000001413 amino acids Chemical group 0.000 description 31
- 238000003556 assay Methods 0.000 description 31
- 238000000338 in vitro Methods 0.000 description 31
- 230000001965 increasing effect Effects 0.000 description 26
- 239000000203 mixture Substances 0.000 description 26
- 241000699670 Mus sp. Species 0.000 description 25
- 230000006698 induction Effects 0.000 description 25
- 238000011282 treatment Methods 0.000 description 24
- 235000001014 amino acid Nutrition 0.000 description 23
- 229940024606 amino acid Drugs 0.000 description 23
- 102000003886 Glycoproteins Human genes 0.000 description 22
- 108090000288 Glycoproteins Proteins 0.000 description 22
- 238000013459 approach Methods 0.000 description 22
- 230000003472 neutralizing effect Effects 0.000 description 21
- 241000711549 Hepacivirus C Species 0.000 description 20
- 102100034349 Integrase Human genes 0.000 description 19
- 210000000612 antigen-presenting cell Anatomy 0.000 description 19
- 238000009739 binding Methods 0.000 description 19
- 238000003752 polymerase chain reaction Methods 0.000 description 19
- 239000000758 substrate Substances 0.000 description 19
- 230000027455 binding Effects 0.000 description 18
- 210000003936 merozoite Anatomy 0.000 description 18
- 238000004458 analytical method Methods 0.000 description 16
- 230000015572 biosynthetic process Effects 0.000 description 16
- 239000012634 fragment Substances 0.000 description 16
- 238000009396 hybridization Methods 0.000 description 16
- 102000004127 Cytokines Human genes 0.000 description 15
- 108090000695 Cytokines Proteins 0.000 description 15
- 238000002347 injection Methods 0.000 description 15
- 239000007924 injection Substances 0.000 description 15
- 239000000523 sample Substances 0.000 description 15
- 239000002253 acid Substances 0.000 description 14
- 150000007513 acids Chemical class 0.000 description 14
- 210000003719 b-lymphocyte Anatomy 0.000 description 14
- 230000000875 corresponding effect Effects 0.000 description 14
- 230000006872 improvement Effects 0.000 description 14
- 238000000746 purification Methods 0.000 description 14
- 238000003786 synthesis reaction Methods 0.000 description 14
- 101710194807 Protective antigen Proteins 0.000 description 13
- 230000001580 bacterial effect Effects 0.000 description 13
- 230000035772 mutation Effects 0.000 description 13
- 239000000047 product Substances 0.000 description 13
- 241000282693 Cercopithecidae Species 0.000 description 12
- 238000012286 ELISA Assay Methods 0.000 description 12
- 241000150562 Hantaan orthohantavirus Species 0.000 description 12
- 241000700721 Hepatitis B virus Species 0.000 description 12
- 108091028043 Nucleic acid sequence Proteins 0.000 description 12
- 210000003743 erythrocyte Anatomy 0.000 description 12
- 210000001519 tissue Anatomy 0.000 description 12
- 108010041986 DNA Vaccines Proteins 0.000 description 11
- 229940021995 DNA vaccine Drugs 0.000 description 11
- 241000282412 Homo Species 0.000 description 11
- 108700018351 Major Histocompatibility Complex Proteins 0.000 description 11
- 241000710771 Tick-borne encephalitis virus Species 0.000 description 11
- 241000710959 Venezuelan equine encephalitis virus Species 0.000 description 11
- 230000004913 activation Effects 0.000 description 11
- 238000009472 formulation Methods 0.000 description 11
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 description 11
- 238000002255 vaccination Methods 0.000 description 11
- 238000002965 ELISA Methods 0.000 description 10
- 230000036039 immunity Effects 0.000 description 10
- 239000013612 plasmid Substances 0.000 description 10
- 238000006467 substitution reaction Methods 0.000 description 10
- 241000700588 Human alphaherpesvirus 1 Species 0.000 description 9
- 241000701074 Human alphaherpesvirus 2 Species 0.000 description 9
- 108010052285 Membrane Proteins Proteins 0.000 description 9
- 238000006243 chemical reaction Methods 0.000 description 9
- 238000011161 development Methods 0.000 description 9
- 230000018109 developmental process Effects 0.000 description 9
- 239000007788 liquid Substances 0.000 description 9
- 238000002703 mutagenesis Methods 0.000 description 9
- 231100000350 mutagenesis Toxicity 0.000 description 9
- 238000004091 panning Methods 0.000 description 9
- 241000725619 Dengue virus Species 0.000 description 8
- 108010002350 Interleukin-2 Proteins 0.000 description 8
- 102000000588 Interleukin-2 Human genes 0.000 description 8
- 241000219061 Rheum Species 0.000 description 8
- 208000003455 anaphylaxis Diseases 0.000 description 8
- 206010003246 arthritis Diseases 0.000 description 8
- 210000004369 blood Anatomy 0.000 description 8
- 239000008280 blood Substances 0.000 description 8
- 239000003153 chemical reaction reagent Substances 0.000 description 8
- 206010012601 diabetes mellitus Diseases 0.000 description 8
- 238000005516 engineering process Methods 0.000 description 8
- 230000006870 function Effects 0.000 description 8
- 244000052613 viral pathogen Species 0.000 description 8
- 206010008631 Cholera Diseases 0.000 description 7
- 241000725303 Human immunodeficiency virus Species 0.000 description 7
- 102000004388 Interleukin-4 Human genes 0.000 description 7
- 108090000978 Interleukin-4 Proteins 0.000 description 7
- 108010002616 Interleukin-5 Proteins 0.000 description 7
- 102000000743 Interleukin-5 Human genes 0.000 description 7
- 102000018697 Membrane Proteins Human genes 0.000 description 7
- 101710105714 Outer surface protein A Proteins 0.000 description 7
- 238000010171 animal model Methods 0.000 description 7
- 238000010367 cloning Methods 0.000 description 7
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 7
- 238000001514 detection method Methods 0.000 description 7
- 239000003814 drug Substances 0.000 description 7
- 230000009545 invasion Effects 0.000 description 7
- 102000005962 receptors Human genes 0.000 description 7
- 108020003175 receptors Proteins 0.000 description 7
- 239000000243 solution Substances 0.000 description 7
- 241000193738 Bacillus anthracis Species 0.000 description 6
- 241000589969 Borreliella burgdorferi Species 0.000 description 6
- 108091026890 Coding region Proteins 0.000 description 6
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 6
- 108091054438 MHC class II family Proteins 0.000 description 6
- 108010057081 Merozoite Surface Protein 1 Proteins 0.000 description 6
- 230000003321 amplification Effects 0.000 description 6
- 239000000872 buffer Substances 0.000 description 6
- 229940079593 drug Drugs 0.000 description 6
- 238000009169 immunotherapy Methods 0.000 description 6
- 238000011534 incubation Methods 0.000 description 6
- 238000001990 intravenous administration Methods 0.000 description 6
- 210000004153 islets of langerhan Anatomy 0.000 description 6
- 230000004048 modification Effects 0.000 description 6
- 238000012986 modification Methods 0.000 description 6
- 238000003199 nucleic acid amplification method Methods 0.000 description 6
- 230000002265 prevention Effects 0.000 description 6
- 230000008569 process Effects 0.000 description 6
- 210000002966 serum Anatomy 0.000 description 6
- 210000003491 skin Anatomy 0.000 description 6
- 235000002639 sodium chloride Nutrition 0.000 description 6
- 208000024891 symptom Diseases 0.000 description 6
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 5
- 206010006187 Breast cancer Diseases 0.000 description 5
- 208000026310 Breast neoplasm Diseases 0.000 description 5
- 241000710831 Flavivirus Species 0.000 description 5
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 5
- 101000591210 Homo sapiens Receptor-type tyrosine-protein phosphatase-like N Proteins 0.000 description 5
- 102100037850 Interferon gamma Human genes 0.000 description 5
- 108010074328 Interferon-gamma Proteins 0.000 description 5
- 102000003816 Interleukin-13 Human genes 0.000 description 5
- 108090000176 Interleukin-13 Proteins 0.000 description 5
- 208000016604 Lyme disease Diseases 0.000 description 5
- 241000699666 Mus <mouse, genus> Species 0.000 description 5
- 241000187479 Mycobacterium tuberculosis Species 0.000 description 5
- 102100034091 Receptor-type tyrosine-protein phosphatase-like N Human genes 0.000 description 5
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 5
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 description 5
- 239000004480 active ingredient Substances 0.000 description 5
- 238000007792 addition Methods 0.000 description 5
- 239000000443 aerosol Substances 0.000 description 5
- 230000005875 antibody response Effects 0.000 description 5
- 208000006673 asthma Diseases 0.000 description 5
- 244000052616 bacterial pathogen Species 0.000 description 5
- 230000001413 cellular effect Effects 0.000 description 5
- 239000002299 complementary DNA Substances 0.000 description 5
- 230000001186 cumulative effect Effects 0.000 description 5
- 238000002474 experimental method Methods 0.000 description 5
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 5
- 231100000518 lethal Toxicity 0.000 description 5
- 230000001665 lethal effect Effects 0.000 description 5
- 210000004698 lymphocyte Anatomy 0.000 description 5
- 230000003211 malignant effect Effects 0.000 description 5
- 239000012528 membrane Substances 0.000 description 5
- 239000003471 mutagenic agent Substances 0.000 description 5
- 238000006386 neutralization reaction Methods 0.000 description 5
- 231100000252 nontoxic Toxicity 0.000 description 5
- 230000003000 nontoxic effect Effects 0.000 description 5
- 230000001105 regulatory effect Effects 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 210000004881 tumor cell Anatomy 0.000 description 5
- 101710132601 Capsid protein Proteins 0.000 description 4
- 101710204837 Envelope small membrane protein Proteins 0.000 description 4
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 4
- 108010010803 Gelatin Proteins 0.000 description 4
- 108010004889 Heat-Shock Proteins Proteins 0.000 description 4
- 102000002812 Heat-Shock Proteins Human genes 0.000 description 4
- 241000589989 Helicobacter Species 0.000 description 4
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 4
- 108010073816 IgE Receptors Proteins 0.000 description 4
- 102000009438 IgE Receptors Human genes 0.000 description 4
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 4
- 101710145006 Lysis protein Proteins 0.000 description 4
- 108091054437 MHC class I family Proteins 0.000 description 4
- 241000186362 Mycobacterium leprae Species 0.000 description 4
- 108091005461 Nucleic proteins Proteins 0.000 description 4
- 108091034117 Oligonucleotide Proteins 0.000 description 4
- 108700005078 Synthetic Genes Proteins 0.000 description 4
- 108091008874 T cell receptors Proteins 0.000 description 4
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 4
- 101800000970 Vacuolating cytotoxin Proteins 0.000 description 4
- 230000001093 anti-cancer Effects 0.000 description 4
- 230000001363 autoimmune Effects 0.000 description 4
- 238000004113 cell culture Methods 0.000 description 4
- 230000000295 complement effect Effects 0.000 description 4
- 150000001875 compounds Chemical class 0.000 description 4
- 238000012217 deletion Methods 0.000 description 4
- 230000037430 deletion Effects 0.000 description 4
- 238000010586 diagram Methods 0.000 description 4
- 230000004069 differentiation Effects 0.000 description 4
- 206010014599 encephalitis Diseases 0.000 description 4
- 230000004927 fusion Effects 0.000 description 4
- 239000008273 gelatin Substances 0.000 description 4
- 229920000159 gelatin Polymers 0.000 description 4
- 235000019322 gelatine Nutrition 0.000 description 4
- 235000011852 gelatine desserts Nutrition 0.000 description 4
- 238000001415 gene therapy Methods 0.000 description 4
- 210000002443 helper t lymphocyte Anatomy 0.000 description 4
- 208000006454 hepatitis Diseases 0.000 description 4
- 231100000283 hepatitis Toxicity 0.000 description 4
- 210000003630 histaminocyte Anatomy 0.000 description 4
- 210000000987 immune system Anatomy 0.000 description 4
- 238000003018 immunoassay Methods 0.000 description 4
- 238000002955 isolation Methods 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- 239000002502 liposome Substances 0.000 description 4
- 201000004792 malaria Diseases 0.000 description 4
- 239000003550 marker Substances 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- 230000007246 mechanism Effects 0.000 description 4
- 201000001441 melanoma Diseases 0.000 description 4
- 230000003505 mutagenic effect Effects 0.000 description 4
- 230000003287 optical effect Effects 0.000 description 4
- 238000005457 optimization Methods 0.000 description 4
- 239000008194 pharmaceutical composition Substances 0.000 description 4
- 238000011176 pooling Methods 0.000 description 4
- 230000003389 potentiating effect Effects 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 238000012545 processing Methods 0.000 description 4
- 230000010076 replication Effects 0.000 description 4
- 210000004215 spore Anatomy 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 238000002560 therapeutic procedure Methods 0.000 description 4
- 230000020192 tolerance induction in gut-associated lymphoid tissue Effects 0.000 description 4
- 238000013518 transcription Methods 0.000 description 4
- 230000035897 transcription Effects 0.000 description 4
- 238000001890 transfection Methods 0.000 description 4
- 201000008827 tuberculosis Diseases 0.000 description 4
- KISWVXRQTGLFGD-UHFFFAOYSA-N 2-[[2-[[6-amino-2-[[2-[[2-[[5-amino-2-[[2-[[1-[2-[[6-amino-2-[(2,5-diamino-5-oxopentanoyl)amino]hexanoyl]amino]-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]-5-(diaminomethylideneamino)p Chemical compound C1CCN(C(=O)C(CCCN=C(N)N)NC(=O)C(CCCCN)NC(=O)C(N)CCC(N)=O)C1C(=O)NC(CO)C(=O)NC(CCC(N)=O)C(=O)NC(CCCN=C(N)N)C(=O)NC(CO)C(=O)NC(CCCCN)C(=O)NC(C(=O)NC(CC(C)C)C(O)=O)CC1=CC=C(O)C=C1 KISWVXRQTGLFGD-UHFFFAOYSA-N 0.000 description 3
- 239000004475 Arginine Substances 0.000 description 3
- 206010003267 Arthritis reactive Diseases 0.000 description 3
- 241000283690 Bos taurus Species 0.000 description 3
- 241000282472 Canis lupus familiaris Species 0.000 description 3
- 101710098119 Chaperonin GroEL 2 Proteins 0.000 description 3
- 241000710829 Dengue virus group Species 0.000 description 3
- 241000196324 Embryophyta Species 0.000 description 3
- 102100031940 Epithelial cell adhesion molecule Human genes 0.000 description 3
- 241000283073 Equus caballus Species 0.000 description 3
- 241000282326 Felis catus Species 0.000 description 3
- 241000233866 Fungi Species 0.000 description 3
- 102100021181 Golgi phosphoprotein 3 Human genes 0.000 description 3
- 102000012153 HLA-B27 Antigen Human genes 0.000 description 3
- 108010061486 HLA-B27 Antigen Proteins 0.000 description 3
- 102000008949 Histocompatibility Antigens Class I Human genes 0.000 description 3
- 102000018713 Histocompatibility Antigens Class II Human genes 0.000 description 3
- 101000920667 Homo sapiens Epithelial cell adhesion molecule Proteins 0.000 description 3
- 241000282620 Hylobates sp. Species 0.000 description 3
- 206010061218 Inflammation Diseases 0.000 description 3
- 241000712431 Influenza A virus Species 0.000 description 3
- 102000004877 Insulin Human genes 0.000 description 3
- 108090001061 Insulin Proteins 0.000 description 3
- 241000588747 Klebsiella pneumoniae Species 0.000 description 3
- 240000007472 Leucaena leucocephala Species 0.000 description 3
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 3
- 102000043131 MHC class II family Human genes 0.000 description 3
- 101710125418 Major capsid protein Proteins 0.000 description 3
- 241000283923 Marmota monax Species 0.000 description 3
- 102000047918 Myelin Basic Human genes 0.000 description 3
- 101710107068 Myelin basic protein Proteins 0.000 description 3
- 101710105711 Outer surface protein C Proteins 0.000 description 3
- 241000282577 Pan troglodytes Species 0.000 description 3
- 102000002727 Protein Tyrosine Phosphatase Human genes 0.000 description 3
- 241000242680 Schistosoma mansoni Species 0.000 description 3
- 241001505901 Streptococcus sp. 'group A' Species 0.000 description 3
- 229930006000 Sucrose Natural products 0.000 description 3
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 102000003425 Tyrosinase Human genes 0.000 description 3
- 108060008724 Tyrosinase Proteins 0.000 description 3
- 208000036142 Viral infection Diseases 0.000 description 3
- 230000010530 Virus Neutralization Effects 0.000 description 3
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 3
- 238000002835 absorbance Methods 0.000 description 3
- 238000009825 accumulation Methods 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 230000003213 activating effect Effects 0.000 description 3
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 238000002619 cancer immunotherapy Methods 0.000 description 3
- 230000022534 cell killing Effects 0.000 description 3
- 210000000170 cell membrane Anatomy 0.000 description 3
- 230000000139 costimulatory effect Effects 0.000 description 3
- 238000004132 cross linking Methods 0.000 description 3
- 238000002784 cytotoxicity assay Methods 0.000 description 3
- 231100000263 cytotoxicity test Toxicity 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- UQLDLKMNUJERMK-UHFFFAOYSA-L di(octadecanoyloxy)lead Chemical compound [Pb+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O UQLDLKMNUJERMK-UHFFFAOYSA-L 0.000 description 3
- 230000029087 digestion Effects 0.000 description 3
- 239000003623 enhancer Substances 0.000 description 3
- 210000003979 eosinophil Anatomy 0.000 description 3
- 238000011156 evaluation Methods 0.000 description 3
- 101150086609 groEL2 gene Proteins 0.000 description 3
- 230000004054 inflammatory process Effects 0.000 description 3
- 230000000977 initiatory effect Effects 0.000 description 3
- 229940125396 insulin Drugs 0.000 description 3
- 238000007918 intramuscular administration Methods 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 108020004999 messenger RNA Proteins 0.000 description 3
- 238000010369 molecular cloning Methods 0.000 description 3
- 238000010172 mouse model Methods 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- 238000004806 packaging method and process Methods 0.000 description 3
- -1 phosphoramidite triester Chemical class 0.000 description 3
- 108010040473 pneumococcal surface protein A Proteins 0.000 description 3
- 230000000069 prophylactic effect Effects 0.000 description 3
- 108020000494 protein-tyrosine phosphatase Proteins 0.000 description 3
- 230000002285 radioactive effect Effects 0.000 description 3
- 208000002574 reactive arthritis Diseases 0.000 description 3
- 230000008439 repair process Effects 0.000 description 3
- 238000012552 review Methods 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- 230000003248 secreting effect Effects 0.000 description 3
- 230000028327 secretion Effects 0.000 description 3
- 238000002741 site-directed mutagenesis Methods 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 230000009870 specific binding Effects 0.000 description 3
- 210000004989 spleen cell Anatomy 0.000 description 3
- 239000005720 sucrose Substances 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- 241000712461 unidentified influenza virus Species 0.000 description 3
- 241001515965 unidentified phage Species 0.000 description 3
- 230000009385 viral infection Effects 0.000 description 3
- 238000011179 visual inspection Methods 0.000 description 3
- 101710149790 64 kDa protein Proteins 0.000 description 2
- 102000005369 Aldehyde Dehydrogenase Human genes 0.000 description 2
- 108020002663 Aldehyde Dehydrogenase Proteins 0.000 description 2
- 241000710929 Alphavirus Species 0.000 description 2
- 206010002556 Ankylosing Spondylitis Diseases 0.000 description 2
- 101710145634 Antigen 1 Proteins 0.000 description 2
- 101710203310 Apical membrane antigen 1 Proteins 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 102100022717 Atypical chemokine receptor 1 Human genes 0.000 description 2
- 235000014469 Bacillus subtilis Nutrition 0.000 description 2
- 208000035143 Bacterial infection Diseases 0.000 description 2
- 235000018185 Betula X alpestris Nutrition 0.000 description 2
- 235000018212 Betula X uliginosa Nutrition 0.000 description 2
- 241000123966 Blomia tropicalis Species 0.000 description 2
- 241000589567 Brucella abortus Species 0.000 description 2
- 101100459439 Caenorhabditis elegans nac-2 gene Proteins 0.000 description 2
- 241000700198 Cavia Species 0.000 description 2
- 101710104159 Chaperonin GroEL Proteins 0.000 description 2
- 108010009685 Cholinergic Receptors Proteins 0.000 description 2
- 102000011022 Chorionic Gonadotropin Human genes 0.000 description 2
- 108010062540 Chorionic Gonadotropin Proteins 0.000 description 2
- 101710190612 Circulating cathodic antigen Proteins 0.000 description 2
- 241001149956 Cladosporium herbarum Species 0.000 description 2
- 101710094648 Coat protein Proteins 0.000 description 2
- 108020004635 Complementary DNA Proteins 0.000 description 2
- 102000018832 Cytochromes Human genes 0.000 description 2
- 108010052832 Cytochromes Proteins 0.000 description 2
- 241000710815 Dengue virus 2 Species 0.000 description 2
- 108010045579 Desmoglein 1 Proteins 0.000 description 2
- 102100034579 Desmoglein-1 Human genes 0.000 description 2
- 102100034577 Desmoglein-3 Human genes 0.000 description 2
- 108091029865 Exogenous DNA Proteins 0.000 description 2
- 241000724791 Filamentous phage Species 0.000 description 2
- 206010016952 Food poisoning Diseases 0.000 description 2
- 208000019331 Foodborne disease Diseases 0.000 description 2
- 102000030902 Galactosyltransferase Human genes 0.000 description 2
- 108060003306 Galactosyltransferase Proteins 0.000 description 2
- 108091022930 Glutamate decarboxylase Proteins 0.000 description 2
- 102000008214 Glutamate decarboxylase Human genes 0.000 description 2
- 102100035857 Glutamate decarboxylase 2 Human genes 0.000 description 2
- 108010070675 Glutathione transferase Proteins 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- 239000000579 Gonadotropin-Releasing Hormone Substances 0.000 description 2
- 102100029100 Hematopoietic prostaglandin D synthase Human genes 0.000 description 2
- 208000009889 Herpes Simplex Diseases 0.000 description 2
- 101000678879 Homo sapiens Atypical chemokine receptor 1 Proteins 0.000 description 2
- 101000873786 Homo sapiens Glutamate decarboxylase 2 Proteins 0.000 description 2
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 2
- 241000701024 Human betaherpesvirus 5 Species 0.000 description 2
- 206010061598 Immunodeficiency Diseases 0.000 description 2
- 208000029462 Immunodeficiency disease Diseases 0.000 description 2
- 206010022004 Influenza like illness Diseases 0.000 description 2
- 108010036012 Iodide peroxidase Proteins 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- 241000710770 Langat virus Species 0.000 description 2
- 102000043129 MHC class I family Human genes 0.000 description 2
- 101710164702 Major outer membrane protein Proteins 0.000 description 2
- 206010027476 Metastases Diseases 0.000 description 2
- 241000588622 Moraxella bovis Species 0.000 description 2
- 101710159910 Movement protein Proteins 0.000 description 2
- 241000710908 Murray Valley encephalitis virus Species 0.000 description 2
- 241000204045 Mycoplasma hyopneumoniae Species 0.000 description 2
- 241000588652 Neisseria gonorrhoeae Species 0.000 description 2
- 102000005348 Neuraminidase Human genes 0.000 description 2
- 108010006232 Neuraminidase Proteins 0.000 description 2
- 108010051791 Nuclear Antigens Proteins 0.000 description 2
- 102000019040 Nuclear Antigens Human genes 0.000 description 2
- 102000007999 Nuclear Proteins Human genes 0.000 description 2
- 108010089610 Nuclear Proteins Proteins 0.000 description 2
- 108020004711 Nucleic Acid Probes Proteins 0.000 description 2
- 108090001074 Nucleocapsid Proteins Proteins 0.000 description 2
- 101710141454 Nucleoprotein Proteins 0.000 description 2
- 108700026244 Open Reading Frames Proteins 0.000 description 2
- 241000713112 Orthobunyavirus Species 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- 102100039867 Outer mitochondrial transmembrane helix translocase Human genes 0.000 description 2
- 206010034277 Pemphigoid Diseases 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- 208000037581 Persistent Infection Diseases 0.000 description 2
- 241000224017 Plasmodium berghei Species 0.000 description 2
- 241000223830 Plasmodium yoelii Species 0.000 description 2
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 2
- 241000288906 Primates Species 0.000 description 2
- 101710083689 Probable capsid protein Proteins 0.000 description 2
- 108050001408 Profilin Proteins 0.000 description 2
- 102000011195 Profilin Human genes 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- 102000007066 Prostate-Specific Antigen Human genes 0.000 description 2
- 108010072866 Prostate-Specific Antigen Proteins 0.000 description 2
- 239000004365 Protease Substances 0.000 description 2
- 108010076504 Protein Sorting Signals Proteins 0.000 description 2
- 102100024147 Protein phosphatase 1 regulatory subunit 14A Human genes 0.000 description 2
- 241000589517 Pseudomonas aeruginosa Species 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 108020004511 Recombinant DNA Proteins 0.000 description 2
- 208000035415 Reinfection Diseases 0.000 description 2
- 241000702670 Rotavirus Species 0.000 description 2
- 241001354013 Salmonella enterica subsp. enterica serovar Enteritidis Species 0.000 description 2
- 241000293869 Salmonella enterica subsp. enterica serovar Typhimurium Species 0.000 description 2
- 229920002684 Sepharose Polymers 0.000 description 2
- 206010040070 Septic Shock Diseases 0.000 description 2
- 108010017898 Shiga Toxins Proteins 0.000 description 2
- 241000607768 Shigella Species 0.000 description 2
- 208000021386 Sjogren Syndrome Diseases 0.000 description 2
- 102000004598 Small Nuclear Ribonucleoproteins Human genes 0.000 description 2
- 108010003165 Small Nuclear Ribonucleoproteins Proteins 0.000 description 2
- 241000589970 Spirochaetales Species 0.000 description 2
- 101000857870 Squalus acanthias Gonadoliberin Proteins 0.000 description 2
- 241000710888 St. Louis encephalitis virus Species 0.000 description 2
- 241000194019 Streptococcus mutans Species 0.000 description 2
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 2
- 102000014267 Thyroid peroxidases Human genes 0.000 description 2
- 102000003911 Thyrotropin Receptors Human genes 0.000 description 2
- 108090000253 Thyrotropin Receptors Proteins 0.000 description 2
- 206010044248 Toxic shock syndrome Diseases 0.000 description 2
- 231100000650 Toxic shock syndrome Toxicity 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 2
- 241000711975 Vesicular stomatitis virus Species 0.000 description 2
- 108010003533 Viral Envelope Proteins Proteins 0.000 description 2
- 241001492404 Woodchuck hepatitis virus Species 0.000 description 2
- 102000034337 acetylcholine receptors Human genes 0.000 description 2
- 208000009956 adenocarcinoma Diseases 0.000 description 2
- 230000002009 allergenic effect Effects 0.000 description 2
- 230000004075 alteration Effects 0.000 description 2
- 125000000539 amino acid group Chemical group 0.000 description 2
- 230000005975 antitumor immune response Effects 0.000 description 2
- 230000003190 augmentative effect Effects 0.000 description 2
- 208000022362 bacterial infectious disease Diseases 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 238000009395 breeding Methods 0.000 description 2
- 230000001488 breeding effect Effects 0.000 description 2
- 229940056450 brucella abortus Drugs 0.000 description 2
- 239000006172 buffering agent Substances 0.000 description 2
- 210000004899 c-terminal region Anatomy 0.000 description 2
- 238000009566 cancer vaccine Methods 0.000 description 2
- 229940022399 cancer vaccine Drugs 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 230000002860 competitive effect Effects 0.000 description 2
- 238000010276 construction Methods 0.000 description 2
- 230000001461 cytolytic effect Effects 0.000 description 2
- 230000001086 cytosolic effect Effects 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- 230000006735 deficit Effects 0.000 description 2
- 239000003599 detergent Substances 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 231100000676 disease causative agent Toxicity 0.000 description 2
- 235000021186 dishes Nutrition 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 230000003325 follicular Effects 0.000 description 2
- 108010055409 ganglioside receptor Proteins 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 238000011239 genetic vaccination Methods 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 210000002288 golgi apparatus Anatomy 0.000 description 2
- XLXSAKCOAKORKW-AQJXLSMYSA-N gonadorelin Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 XLXSAKCOAKORKW-AQJXLSMYSA-N 0.000 description 2
- 229940035638 gonadotropin-releasing hormone Drugs 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 238000013537 high throughput screening Methods 0.000 description 2
- 229960001340 histamine Drugs 0.000 description 2
- 210000005260 human cell Anatomy 0.000 description 2
- 229940084986 human chorionic gonadotropin Drugs 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- 230000002519 immonomodulatory effect Effects 0.000 description 2
- 230000001900 immune effect Effects 0.000 description 2
- 230000007813 immunodeficiency Effects 0.000 description 2
- 230000028802 immunoglobulin-mediated neutralization Effects 0.000 description 2
- 238000012750 in vivo screening Methods 0.000 description 2
- 230000002458 infectious effect Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 238000003780 insertion Methods 0.000 description 2
- 230000037431 insertion Effects 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 210000002490 intestinal epithelial cell Anatomy 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 150000002617 leukotrienes Chemical class 0.000 description 2
- 238000007834 ligase chain reaction Methods 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 201000011475 meningoencephalitis Diseases 0.000 description 2
- 230000009401 metastasis Effects 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- 230000003278 mimic effect Effects 0.000 description 2
- 230000033607 mismatch repair Effects 0.000 description 2
- 231100000219 mutagenic Toxicity 0.000 description 2
- 231100000707 mutagenic chemical Toxicity 0.000 description 2
- 239000002853 nucleic acid probe Substances 0.000 description 2
- 229940094443 oxytocics prostaglandins Drugs 0.000 description 2
- 239000012188 paraffin wax Substances 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 210000004180 plasmocyte Anatomy 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 230000003449 preventive effect Effects 0.000 description 2
- 150000003180 prostaglandins Chemical class 0.000 description 2
- 238000001742 protein purification Methods 0.000 description 2
- 238000003259 recombinant expression Methods 0.000 description 2
- 210000001563 schizont Anatomy 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 238000010187 selection method Methods 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 230000001568 sexual effect Effects 0.000 description 2
- 230000011664 signaling Effects 0.000 description 2
- 229910001415 sodium ion Inorganic materials 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000007790 solid phase Substances 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 230000002269 spontaneous effect Effects 0.000 description 2
- 108010088201 squamous cell carcinoma-related antigen Proteins 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 239000002511 suppository base Substances 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 230000008685 targeting Effects 0.000 description 2
- 210000001550 testis Anatomy 0.000 description 2
- 229940031351 tetravalent vaccine Drugs 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 239000000196 tragacanth Substances 0.000 description 2
- 235000010487 tragacanth Nutrition 0.000 description 2
- 229940116362 tragacanth Drugs 0.000 description 2
- 230000005030 transcription termination Effects 0.000 description 2
- 230000014616 translation Effects 0.000 description 2
- 150000003626 triacylglycerols Chemical class 0.000 description 2
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 2
- 229940118696 vibrio cholerae Drugs 0.000 description 2
- 230000001018 virulence Effects 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- DQJCDTNMLBYVAY-ZXXIYAEKSA-N (2S,5R,10R,13R)-16-{[(2R,3S,4R,5R)-3-{[(2S,3R,4R,5S,6R)-3-acetamido-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy}-5-(ethylamino)-6-hydroxy-2-(hydroxymethyl)oxan-4-yl]oxy}-5-(4-aminobutyl)-10-carbamoyl-2,13-dimethyl-4,7,12,15-tetraoxo-3,6,11,14-tetraazaheptadecan-1-oic acid Chemical compound NCCCC[C@H](C(=O)N[C@@H](C)C(O)=O)NC(=O)CC[C@H](C(N)=O)NC(=O)[C@@H](C)NC(=O)C(C)O[C@@H]1[C@@H](NCC)C(O)O[C@H](CO)[C@H]1O[C@H]1[C@H](NC(C)=O)[C@@H](O)[C@H](O)[C@@H](CO)O1 DQJCDTNMLBYVAY-ZXXIYAEKSA-N 0.000 description 1
- GEYOCULIXLDCMW-UHFFFAOYSA-N 1,2-phenylenediamine Chemical compound NC1=CC=CC=C1N GEYOCULIXLDCMW-UHFFFAOYSA-N 0.000 description 1
- 102100024341 10 kDa heat shock protein, mitochondrial Human genes 0.000 description 1
- 108010041801 2',3'-Cyclic Nucleotide 3'-Phosphodiesterase Proteins 0.000 description 1
- 102100040458 2',3'-cyclic-nucleotide 3'-phosphodiesterase Human genes 0.000 description 1
- VGONTNSXDCQUGY-RRKCRQDMSA-N 2'-deoxyinosine Chemical group C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC2=O)=C2N=C1 VGONTNSXDCQUGY-RRKCRQDMSA-N 0.000 description 1
- RNAMYOYQYRYFQY-UHFFFAOYSA-N 2-(4,4-difluoropiperidin-1-yl)-6-methoxy-n-(1-propan-2-ylpiperidin-4-yl)-7-(3-pyrrolidin-1-ylpropoxy)quinazolin-4-amine Chemical compound N1=C(N2CCC(F)(F)CC2)N=C2C=C(OCCCN3CCCC3)C(OC)=CC2=C1NC1CCN(C(C)C)CC1 RNAMYOYQYRYFQY-UHFFFAOYSA-N 0.000 description 1
- JZCAMRDTJYUXNN-ASTDGNLGSA-N 2-ethyl-6-methylpiperidine;2-methyl-6-[(e)-undec-2-enyl]piperidine Chemical compound CCC1CCCC(C)N1.CCCCCCCC\C=C\CC1CCCC(C)N1 JZCAMRDTJYUXNN-ASTDGNLGSA-N 0.000 description 1
- CYDQOEWLBCCFJZ-UHFFFAOYSA-N 4-(4-fluorophenyl)oxane-4-carboxylic acid Chemical compound C=1C=C(F)C=CC=1C1(C(=O)O)CCOCC1 CYDQOEWLBCCFJZ-UHFFFAOYSA-N 0.000 description 1
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 1
- 101710166488 6 kDa early secretory antigenic target Proteins 0.000 description 1
- 102100035841 60S ribosomal protein L7 Human genes 0.000 description 1
- 102000040352 A family Human genes 0.000 description 1
- 108091072132 A family Proteins 0.000 description 1
- 239000013607 AAV vector Substances 0.000 description 1
- 241000238876 Acari Species 0.000 description 1
- 241000606750 Actinobacillus Species 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 101710102499 Alanine and proline-rich secreted protein Apa Proteins 0.000 description 1
- 102100027211 Albumin Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 101710172128 Alpha-amylase/trypsin inhibitor Proteins 0.000 description 1
- 101710095306 Alpha-crystallin Proteins 0.000 description 1
- 108091093088 Amplicon Proteins 0.000 description 1
- 206010002198 Anaphylactic reaction Diseases 0.000 description 1
- 108020005544 Antisense RNA Proteins 0.000 description 1
- 101100272788 Arabidopsis thaliana BSL3 gene Proteins 0.000 description 1
- 101100178203 Arabidopsis thaliana HMGB3 gene Proteins 0.000 description 1
- 101001126433 Arabidopsis thaliana Pyrophosphate-fructose 6-phosphate 1-phosphotransferase subunit alpha 2 Proteins 0.000 description 1
- 101000999895 Arabidopsis thaliana Pyrophosphate-fructose 6-phosphate 1-phosphotransferase subunit beta 2 Proteins 0.000 description 1
- 235000017060 Arachis glabrata Nutrition 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- 235000010777 Arachis hypogaea Nutrition 0.000 description 1
- 235000018262 Arachis monticola Nutrition 0.000 description 1
- 206010003399 Arthropod bite Diseases 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 1
- 241000972773 Aulopiformes Species 0.000 description 1
- 208000032116 Autoimmune Experimental Encephalomyelitis Diseases 0.000 description 1
- 101000977023 Azospirillum brasilense Uncharacterized 17.8 kDa protein in nodG 5'region Proteins 0.000 description 1
- 102000019260 B-Cell Antigen Receptors Human genes 0.000 description 1
- 108010012919 B-Cell Antigen Receptors Proteins 0.000 description 1
- 208000003950 B-cell lymphoma Diseases 0.000 description 1
- 101000585552 Bacillus anthracis Protective antigen Proteins 0.000 description 1
- 244000063299 Bacillus subtilis Species 0.000 description 1
- 101000961984 Bacillus thuringiensis Uncharacterized 30.3 kDa protein Proteins 0.000 description 1
- 208000009299 Benign Mucous Membrane Pemphigoid Diseases 0.000 description 1
- 102100026189 Beta-galactosidase Human genes 0.000 description 1
- 241000589978 Borrelia hermsii Species 0.000 description 1
- 241001148604 Borreliella afzelii Species 0.000 description 1
- 241001148605 Borreliella garinii Species 0.000 description 1
- 241000565673 Borreliella tanukii Species 0.000 description 1
- 241000582024 Borreliella turdi Species 0.000 description 1
- 241001148106 Brucella melitensis Species 0.000 description 1
- 125000001433 C-terminal amino-acid group Chemical group 0.000 description 1
- 101100038180 Caenorhabditis briggsae rpb-1 gene Proteins 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 101100507655 Canis lupus familiaris HSPA1 gene Proteins 0.000 description 1
- 244000025254 Cannabis sativa Species 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 108090000565 Capsid Proteins Proteins 0.000 description 1
- 102100032378 Carboxypeptidase E Human genes 0.000 description 1
- 108010058255 Carboxypeptidase H Proteins 0.000 description 1
- 108010022366 Carcinoembryonic Antigen Proteins 0.000 description 1
- 102100025475 Carcinoembryonic antigen-related cell adhesion molecule 5 Human genes 0.000 description 1
- 102000014914 Carrier Proteins Human genes 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 102000004178 Cathepsin E Human genes 0.000 description 1
- 108090000611 Cathepsin E Proteins 0.000 description 1
- 208000003163 Cavernous Hemangioma Diseases 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 description 1
- 102000011682 Centromere Protein A Human genes 0.000 description 1
- 108010076303 Centromere Protein A Proteins 0.000 description 1
- 102100023321 Ceruloplasmin Human genes 0.000 description 1
- 241000282994 Cervidae Species 0.000 description 1
- CXRFDZFCGOPDTD-UHFFFAOYSA-M Cetrimide Chemical compound [Br-].CCCCCCCCCCCCCC[N+](C)(C)C CXRFDZFCGOPDTD-UHFFFAOYSA-M 0.000 description 1
- 101710163595 Chaperone protein DnaK Proteins 0.000 description 1
- 108010059013 Chaperonin 10 Proteins 0.000 description 1
- 102000019034 Chemokines Human genes 0.000 description 1
- 108010012236 Chemokines Proteins 0.000 description 1
- 241000256135 Chironomus thummi Species 0.000 description 1
- 241001647372 Chlamydia pneumoniae Species 0.000 description 1
- 241001647378 Chlamydia psittaci Species 0.000 description 1
- 241000498849 Chlamydiales Species 0.000 description 1
- 101710117490 Circumsporozoite protein Proteins 0.000 description 1
- 241000193468 Clostridium perfringens Species 0.000 description 1
- 108010065152 Coagulase Proteins 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 102000000503 Collagen Type II Human genes 0.000 description 1
- 108010041390 Collagen Type II Proteins 0.000 description 1
- 102000004510 Collagen Type VII Human genes 0.000 description 1
- 108010017377 Collagen Type VII Proteins 0.000 description 1
- 102100028256 Collagen alpha-1(XVII) chain Human genes 0.000 description 1
- 101710082950 Collagen alpha-1(XVII) chain Proteins 0.000 description 1
- 208000002330 Congenital Heart Defects Diseases 0.000 description 1
- 241000557626 Corvus corax Species 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- 108050006400 Cyclin Proteins 0.000 description 1
- 102000001493 Cyclophilins Human genes 0.000 description 1
- 108010068682 Cyclophilins Proteins 0.000 description 1
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 1
- 229930105110 Cyclosporin A Natural products 0.000 description 1
- 108010036949 Cyclosporine Proteins 0.000 description 1
- 241001137307 Cyprinodon variegatus Species 0.000 description 1
- 241000701022 Cytomegalovirus Species 0.000 description 1
- 101710112752 Cytotoxin Proteins 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- 238000012270 DNA recombination Methods 0.000 description 1
- 102000052510 DNA-Binding Proteins Human genes 0.000 description 1
- 108700020911 DNA-Binding Proteins Proteins 0.000 description 1
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 1
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 1
- 201000004624 Dermatitis Diseases 0.000 description 1
- 241000238713 Dermatophagoides farinae Species 0.000 description 1
- 241000238740 Dermatophagoides pteronyssinus Species 0.000 description 1
- 108010061573 Dermatophagoides pteronyssinus antigen p 5 Proteins 0.000 description 1
- 108010061638 Dermatophagoides pteronyssinus antigen p 7 Proteins 0.000 description 1
- 108010032035 Desmoglein 3 Proteins 0.000 description 1
- 206010012735 Diarrhoea Diseases 0.000 description 1
- 241000605721 Dichelobacter nodosus Species 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- 241000255925 Diptera Species 0.000 description 1
- 101000644901 Drosophila melanogaster Putative 115 kDa protein in type-1 retrotransposable element R1DM Proteins 0.000 description 1
- 102100021238 Dynamin-2 Human genes 0.000 description 1
- 108010031111 EBV-encoded nuclear antigen 1 Proteins 0.000 description 1
- 108010043290 Echinococcus granulosus antigen 5 Proteins 0.000 description 1
- 102100038132 Endogenous retrovirus group K member 6 Pro protein Human genes 0.000 description 1
- 101000747702 Enterobacteria phage N4 Uncharacterized protein Gp2 Proteins 0.000 description 1
- 101001095863 Enterobacteria phage T4 RNA ligase 1 Proteins 0.000 description 1
- 241000702374 Enterobacteria phage fd Species 0.000 description 1
- 101800001467 Envelope glycoprotein E2 Proteins 0.000 description 1
- 101710181478 Envelope glycoprotein GP350 Proteins 0.000 description 1
- 206010014950 Eosinophilia Diseases 0.000 description 1
- 239000004593 Epoxy Substances 0.000 description 1
- 108010008655 Epstein-Barr Virus Nuclear Antigens Proteins 0.000 description 1
- 101000867232 Escherichia coli Heat-stable enterotoxin II Proteins 0.000 description 1
- 241001646716 Escherichia coli K-12 Species 0.000 description 1
- 101000758599 Escherichia coli Uncharacterized 14.7 kDa protein Proteins 0.000 description 1
- 241001534152 Escherichia virus FI Species 0.000 description 1
- 241001524679 Escherichia virus M13 Species 0.000 description 1
- 208000010201 Exanthema Diseases 0.000 description 1
- 208000009386 Experimental Arthritis Diseases 0.000 description 1
- 102100020903 Ezrin Human genes 0.000 description 1
- 208000004929 Facial Paralysis Diseases 0.000 description 1
- 108010087819 Fc receptors Proteins 0.000 description 1
- 102000009109 Fc receptors Human genes 0.000 description 1
- 208000015220 Febrile disease Diseases 0.000 description 1
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 1
- 241000589602 Francisella tularensis Species 0.000 description 1
- 108091006027 G proteins Proteins 0.000 description 1
- 102000030782 GTP binding Human genes 0.000 description 1
- 108091000058 GTP-Binding Proteins 0.000 description 1
- 101150014889 Gad1 gene Proteins 0.000 description 1
- 101710177291 Gag polyprotein Proteins 0.000 description 1
- 208000007882 Gastritis Diseases 0.000 description 1
- 102000016354 Glucuronosyltransferase Human genes 0.000 description 1
- 108010092364 Glucuronosyltransferase Proteins 0.000 description 1
- 102100035902 Glutamate decarboxylase 1 Human genes 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 108010015899 Glycopeptides Proteins 0.000 description 1
- 102000002068 Glycopeptides Human genes 0.000 description 1
- 208000003084 Graves Ophthalmopathy Diseases 0.000 description 1
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 1
- 102000004144 Green Fluorescent Proteins Human genes 0.000 description 1
- 208000031886 HIV Infections Diseases 0.000 description 1
- 101150091750 HMG1 gene Proteins 0.000 description 1
- 108700010013 HMGB1 Proteins 0.000 description 1
- 101150021904 HMGB1 gene Proteins 0.000 description 1
- 101710178376 Heat shock 70 kDa protein Proteins 0.000 description 1
- 101710152018 Heat shock cognate 70 kDa protein Proteins 0.000 description 1
- 101710154606 Hemagglutinin Proteins 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 102100037907 High mobility group protein B1 Human genes 0.000 description 1
- 102100022128 High mobility group protein B2 Human genes 0.000 description 1
- 108010093488 His-His-His-His-His-His Proteins 0.000 description 1
- 108010088652 Histocompatibility Antigens Class I Proteins 0.000 description 1
- 101000853617 Homo sapiens 60S ribosomal protein L7 Proteins 0.000 description 1
- 101000721661 Homo sapiens Cellular tumor antigen p53 Proteins 0.000 description 1
- 101000883515 Homo sapiens Chitinase-3-like protein 1 Proteins 0.000 description 1
- 101000924311 Homo sapiens Desmoglein-3 Proteins 0.000 description 1
- 101000817607 Homo sapiens Dynamin-2 Proteins 0.000 description 1
- 101001040734 Homo sapiens Golgi phosphoprotein 3 Proteins 0.000 description 1
- 101001045791 Homo sapiens High mobility group protein B2 Proteins 0.000 description 1
- 101000878605 Homo sapiens Low affinity immunoglobulin epsilon Fc receptor Proteins 0.000 description 1
- 101000972485 Homo sapiens Lupus La protein Proteins 0.000 description 1
- 101000578784 Homo sapiens Melanoma antigen recognized by T-cells 1 Proteins 0.000 description 1
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 description 1
- 101001133056 Homo sapiens Mucin-1 Proteins 0.000 description 1
- 101000623901 Homo sapiens Mucin-16 Proteins 0.000 description 1
- 101001109800 Homo sapiens Pro-neuregulin-1, membrane-bound isoform Proteins 0.000 description 1
- 101001136981 Homo sapiens Proteasome subunit beta type-9 Proteins 0.000 description 1
- 101000687323 Homo sapiens Rabenosyn-5 Proteins 0.000 description 1
- 101001130441 Homo sapiens Ras-related protein Rap-2a Proteins 0.000 description 1
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 1
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 1
- 101000938391 Homo sapiens Transmembrane protein Proteins 0.000 description 1
- 241000598436 Human T-cell lymphotropic virus Species 0.000 description 1
- 241000701806 Human papillomavirus Species 0.000 description 1
- 102100028084 Hyaluronan and proteoglycan link protein 1 Human genes 0.000 description 1
- 101710191341 Hyaluronan and proteoglycan link protein 1 Proteins 0.000 description 1
- 108010003272 Hyaluronate lyase Proteins 0.000 description 1
- 102000001974 Hyaluronidases Human genes 0.000 description 1
- 101150090364 ICP0 gene Proteins 0.000 description 1
- 101150027427 ICP4 gene Proteins 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 206010022941 Iridocyclitis Diseases 0.000 description 1
- 241000710843 Japanese encephalitis virus group Species 0.000 description 1
- 208000003456 Juvenile Arthritis Diseases 0.000 description 1
- 206010059176 Juvenile idiopathic arthritis Diseases 0.000 description 1
- 102100022905 Keratin, type II cytoskeletal 1 Human genes 0.000 description 1
- 101710194922 Keratin, type II cytoskeletal 1 Proteins 0.000 description 1
- 201000008225 Klebsiella pneumonia Diseases 0.000 description 1
- 241000710912 Kunjin virus Species 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 1
- 102000003855 L-lactate dehydrogenase Human genes 0.000 description 1
- 108700023483 L-lactate dehydrogenases Proteins 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 1
- 108010028554 LDL Cholesterol Proteins 0.000 description 1
- 108010054278 Lac Repressors Proteins 0.000 description 1
- 101000768930 Lactococcus lactis subsp. cremoris Uncharacterized protein in pepC 5'region Proteins 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 241000222722 Leishmania <genus> Species 0.000 description 1
- 241000222732 Leishmania major Species 0.000 description 1
- 101710180643 Leishmanolysin Proteins 0.000 description 1
- 108010032926 Lep d I allergen Proteins 0.000 description 1
- 101000976302 Leptospira interrogans Uncharacterized protein in sph 3'region Proteins 0.000 description 1
- 101000778886 Leptospira interrogans serogroup Icterohaemorrhagiae serovar Lai (strain 56601) Uncharacterized protein LA_2151 Proteins 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- 241000186779 Listeria monocytogenes Species 0.000 description 1
- 108700027766 Listeria monocytogenes hlyA Proteins 0.000 description 1
- 241000255640 Loa loa Species 0.000 description 1
- 206010024887 Louping ill Diseases 0.000 description 1
- 102100038007 Low affinity immunoglobulin epsilon Fc receptor Human genes 0.000 description 1
- 108060001084 Luciferase Proteins 0.000 description 1
- 239000005089 Luciferase Substances 0.000 description 1
- 102100022742 Lupus La protein Human genes 0.000 description 1
- 208000030289 Lymphoproliferative disease Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 108010010995 MART-1 Antigen Proteins 0.000 description 1
- 101710175243 Major antigen Proteins 0.000 description 1
- 101710087657 Manganese ABC transporter substrate-binding lipoprotein Proteins 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 108010028253 Measles virus nucleoprotein Proteins 0.000 description 1
- 102100028389 Melanoma antigen recognized by T-cells 1 Human genes 0.000 description 1
- 102000000440 Melanoma-associated antigen Human genes 0.000 description 1
- 108050008953 Melanoma-associated antigen Proteins 0.000 description 1
- 102000012750 Membrane Glycoproteins Human genes 0.000 description 1
- 108010090054 Membrane Glycoproteins Proteins 0.000 description 1
- 201000009906 Meningitis Diseases 0.000 description 1
- 101710162106 Merozoite surface antigen 2 Proteins 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 101000768804 Micromonospora olivasterospora Uncharacterized 10.9 kDa protein in fmrO 5'region Proteins 0.000 description 1
- 102100027869 Moesin Human genes 0.000 description 1
- 241000713869 Moloney murine leukemia virus Species 0.000 description 1
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 description 1
- 241000588621 Moraxella Species 0.000 description 1
- 101000808384 Morganella morganii Urease subunit beta Proteins 0.000 description 1
- 102100034256 Mucin-1 Human genes 0.000 description 1
- 102100023123 Mucin-16 Human genes 0.000 description 1
- 108010063954 Mucins Proteins 0.000 description 1
- 102000015728 Mucins Human genes 0.000 description 1
- 208000012192 Mucous membrane pemphigoid Diseases 0.000 description 1
- 208000034486 Multi-organ failure Diseases 0.000 description 1
- 208000010718 Multiple Organ Failure Diseases 0.000 description 1
- 101000979318 Mus musculus NHP2-like protein 1 Proteins 0.000 description 1
- 108010021466 Mutant Proteins Proteins 0.000 description 1
- 102000008300 Mutant Proteins Human genes 0.000 description 1
- 101100291912 Mycobacterium bovis (strain ATCC BAA-935 / AF2122/97) mpb64 gene Proteins 0.000 description 1
- 101100131080 Mycobacterium bovis (strain ATCC BAA-935 / AF2122/97) mpb70 gene Proteins 0.000 description 1
- 108010048712 Mycobacterium tuberculosis Ag38 antigen Proteins 0.000 description 1
- 102000055324 Myelin Proteolipid Human genes 0.000 description 1
- 101710094913 Myelin proteolipid protein Proteins 0.000 description 1
- 101000761147 Myrmecia pilosula M-myrmeciitoxin-Mp1 Proteins 0.000 description 1
- 108010062010 N-Acetylmuramoyl-L-alanine Amidase Proteins 0.000 description 1
- 101710177157 NHP2-like protein 1 Proteins 0.000 description 1
- 102100023058 NHP2-like protein 1 Human genes 0.000 description 1
- 108091007491 NSP3 Papain-like protease domains Proteins 0.000 description 1
- 241000588650 Neisseria meningitidis Species 0.000 description 1
- 108091092724 Noncoding DNA Proteins 0.000 description 1
- 238000000636 Northern blotting Methods 0.000 description 1
- 102000008297 Nuclear Matrix-Associated Proteins Human genes 0.000 description 1
- 108010035916 Nuclear Matrix-Associated Proteins Proteins 0.000 description 1
- 102000011931 Nucleoproteins Human genes 0.000 description 1
- 108010061100 Nucleoproteins Proteins 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- 241000283283 Orcinus orca Species 0.000 description 1
- 241000150452 Orthohantavirus Species 0.000 description 1
- 101710093908 Outer capsid protein VP4 Proteins 0.000 description 1
- 101710135467 Outer capsid protein sigma-1 Proteins 0.000 description 1
- 101710116435 Outer membrane protein Proteins 0.000 description 1
- 101150012195 PREB gene Proteins 0.000 description 1
- 101150079778 PREP gene Proteins 0.000 description 1
- 206010033645 Pancreatitis Diseases 0.000 description 1
- 206010033701 Papillary thyroid cancer Diseases 0.000 description 1
- 241001631646 Papillomaviridae Species 0.000 description 1
- 241001504519 Papio ursinus Species 0.000 description 1
- 102000001675 Parvalbumin Human genes 0.000 description 1
- 108060005874 Parvalbumin Proteins 0.000 description 1
- 206010034133 Pathogen resistance Diseases 0.000 description 1
- 208000008469 Peptic Ulcer Diseases 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 102100033422 Pericentriolar material 1 protein Human genes 0.000 description 1
- 101710095344 Pericentriolar material 1 protein Proteins 0.000 description 1
- 108010090127 Periplasmic Proteins Proteins 0.000 description 1
- 241000746983 Phleum pratense Species 0.000 description 1
- 101000750404 Phoneutria keyserlingi CRISP-1 Proteins 0.000 description 1
- 102100026918 Phospholipase A2 Human genes 0.000 description 1
- 108010058864 Phospholipases A2 Proteins 0.000 description 1
- 108700019535 Phosphoprotein Phosphatases Proteins 0.000 description 1
- 102000045595 Phosphoprotein Phosphatases Human genes 0.000 description 1
- 102000012288 Phosphopyruvate Hydratase Human genes 0.000 description 1
- 108010022181 Phosphopyruvate Hydratase Proteins 0.000 description 1
- 101710099976 Photosystem I P700 chlorophyll a apoprotein A1 Proteins 0.000 description 1
- 206010035148 Plague Diseases 0.000 description 1
- 101000895911 Plasmodium falciparum (isolate Camp / Malaysia) Erythrocyte-binding antigen 175 Proteins 0.000 description 1
- 101000983333 Plasmodium falciparum (isolate NF54) 25 kDa ookinete surface antigen Proteins 0.000 description 1
- 108010088113 Plasmodium falciparum erythrocyte membrane protein 1 Proteins 0.000 description 1
- 101710183389 Pneumolysin Proteins 0.000 description 1
- 206010035717 Pneumonia klebsiella Diseases 0.000 description 1
- 241000276498 Pollachius virens Species 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 241000605862 Porphyromonas gingivalis Species 0.000 description 1
- 108010076181 Proinsulin Proteins 0.000 description 1
- 102100036691 Proliferating cell nuclear antigen Human genes 0.000 description 1
- 101710201541 Proline-rich antigen Proteins 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 102000004245 Proteasome Endopeptidase Complex Human genes 0.000 description 1
- 108090000708 Proteasome Endopeptidase Complex Proteins 0.000 description 1
- 102100035764 Proteasome subunit beta type-9 Human genes 0.000 description 1
- 101710176177 Protein A56 Proteins 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010001267 Protein Subunits Proteins 0.000 description 1
- 108010010974 Proteolipids Proteins 0.000 description 1
- 102000016202 Proteolipids Human genes 0.000 description 1
- 241000125945 Protoparvovirus Species 0.000 description 1
- 241001062357 Psilocybe cubensis Species 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 241000238711 Pyroglyphidae Species 0.000 description 1
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 1
- 102100022851 Rab5 GDP/GTP exchange factor Human genes 0.000 description 1
- 102100024910 Rabenosyn-5 Human genes 0.000 description 1
- 101900083372 Rabies virus Glycoprotein Proteins 0.000 description 1
- 102100022127 Radixin Human genes 0.000 description 1
- 102100031420 Ras-related protein Rap-2a Human genes 0.000 description 1
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 1
- 102100037404 Receptor-type tyrosine-protein phosphatase N2 Human genes 0.000 description 1
- 101710168689 Receptor-type tyrosine-protein phosphatase N2 Proteins 0.000 description 1
- 101710203837 Replication-associated protein Proteins 0.000 description 1
- 241000725643 Respiratory syncytial virus Species 0.000 description 1
- 241000187562 Rhodococcus sp. Species 0.000 description 1
- 102000004389 Ribonucleoproteins Human genes 0.000 description 1
- 108010081734 Ribonucleoproteins Proteins 0.000 description 1
- 108091028664 Ribonucleotide Proteins 0.000 description 1
- 108010041388 Ribonucleotide Reductases Proteins 0.000 description 1
- 102000000505 Ribonucleotide Reductases Human genes 0.000 description 1
- 102000050114 Ribosomal protein P2 Human genes 0.000 description 1
- 101001121571 Rice tungro bacilliform virus (isolate Philippines) Protein P2 Proteins 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 101100320203 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) YCP4 gene Proteins 0.000 description 1
- 101000999689 Saimiriine herpesvirus 2 (strain 11) Transcriptional regulator ICP22 homolog Proteins 0.000 description 1
- 241000607142 Salmonella Species 0.000 description 1
- 241000607683 Salmonella enterica subsp. enterica serovar Pullorum Species 0.000 description 1
- 241000293871 Salmonella enterica subsp. enterica serovar Typhi Species 0.000 description 1
- 101100503142 Salmonella enteritidis sefA gene Proteins 0.000 description 1
- 206010039587 Scarlet Fever Diseases 0.000 description 1
- 241000242678 Schistosoma Species 0.000 description 1
- 241000242683 Schistosoma haematobium Species 0.000 description 1
- 101000588257 Schistosoma japonicum Paramyosin Proteins 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- 241000446766 Shigella dysenteriae 1 Species 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 241000150288 Sin Nombre orthohantavirus Species 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 244000046101 Sophora japonica Species 0.000 description 1
- 238000002105 Southern blotting Methods 0.000 description 1
- 102100031874 Spectrin alpha chain, non-erythrocytic 1 Human genes 0.000 description 1
- 101000818098 Spirochaeta aurantia Uncharacterized protein in trpE 3'region Proteins 0.000 description 1
- 101710192036 Sporozoite surface protein 2 Proteins 0.000 description 1
- 241000191940 Staphylococcus Species 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 241000194017 Streptococcus Species 0.000 description 1
- 241000193990 Streptococcus sp. 'group B' Species 0.000 description 1
- 101001026590 Streptomyces cinnamonensis Putative polyketide beta-ketoacyl synthase 2 Proteins 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 101800001271 Surface protein Proteins 0.000 description 1
- 101000750896 Synechococcus elongatus (strain PCC 7942 / FACHB-805) Uncharacterized protein Synpcc7942_2318 Proteins 0.000 description 1
- 208000018359 Systemic autoimmune disease Diseases 0.000 description 1
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 1
- 108020005038 Terminator Codon Proteins 0.000 description 1
- 241000223778 Theileria annulata Species 0.000 description 1
- 208000024799 Thyroid disease Diseases 0.000 description 1
- 101710120037 Toxin CcdB Proteins 0.000 description 1
- 241000223997 Toxoplasma gondii Species 0.000 description 1
- 101800001690 Transmembrane protein gp41 Proteins 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 102100031988 Tumor necrosis factor ligand superfamily member 6 Human genes 0.000 description 1
- 108050002568 Tumor necrosis factor ligand superfamily member 6 Proteins 0.000 description 1
- 101150081415 UL55 gene Proteins 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 208000036826 VIIth nerve paralysis Diseases 0.000 description 1
- 241000700618 Vaccinia virus Species 0.000 description 1
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 1
- 241000607598 Vibrio Species 0.000 description 1
- 108010059722 Viral Fusion Proteins Proteins 0.000 description 1
- 108010067390 Viral Proteins Proteins 0.000 description 1
- 108010087302 Viral Structural Proteins Proteins 0.000 description 1
- 206010047642 Vitiligo Diseases 0.000 description 1
- 241000710886 West Nile virus Species 0.000 description 1
- 101000916321 Xenopus laevis Transposon TX1 uncharacterized 149 kDa protein Proteins 0.000 description 1
- 208000003152 Yellow Fever Diseases 0.000 description 1
- 206010048249 Yersinia infections Diseases 0.000 description 1
- 101100398653 Yersinia pestis lamB1 gene Proteins 0.000 description 1
- 101000760088 Zymomonas mobilis subsp. mobilis (strain ATCC 10988 / DSM 424 / LMG 404 / NCIMB 8938 / NRRL B-806 / ZM1) 20.9 kDa protein Proteins 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 238000005903 acid hydrolysis reaction Methods 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 238000005273 aeration Methods 0.000 description 1
- 238000001042 affinity chromatography Methods 0.000 description 1
- 238000001261 affinity purification Methods 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229940074608 allergen extract Drugs 0.000 description 1
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 1
- 230000036783 anaphylactic response Effects 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 201000004612 anterior uveitis Diseases 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 229940124599 anti-inflammatory drug Drugs 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 230000000469 anti-sperm effect Effects 0.000 description 1
- 230000001147 anti-toxic effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 230000030741 antigen processing and presentation Effects 0.000 description 1
- 230000027645 antigenic variation Effects 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 239000008365 aqueous carrier Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 235000009582 asparagine Nutrition 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 238000002820 assay format Methods 0.000 description 1
- 230000002238 attenuated effect Effects 0.000 description 1
- 229940031567 attenuated vaccine Drugs 0.000 description 1
- 229940065181 bacillus anthracis Drugs 0.000 description 1
- 210000003651 basophil Anatomy 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 108010005774 beta-Galactosidase Proteins 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 206010005084 bladder transitional cell carcinoma Diseases 0.000 description 1
- 201000001528 bladder urothelial carcinoma Diseases 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 210000005013 brain tissue Anatomy 0.000 description 1
- 201000008275 breast carcinoma Diseases 0.000 description 1
- 229940038698 brucella melitensis Drugs 0.000 description 1
- 239000007975 buffered saline Substances 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 208000000594 bullous pemphigoid Diseases 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 235000011148 calcium chloride Nutrition 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 210000000234 capsid Anatomy 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 238000012219 cassette mutagenesis Methods 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 230000006037 cell lysis Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 210000003793 centrosome Anatomy 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 238000009614 chemical analysis method Methods 0.000 description 1
- CRQQGFGUEAVUIL-UHFFFAOYSA-N chlorothalonil Chemical compound ClC1=C(Cl)C(C#N)=C(Cl)C(C#N)=C1Cl CRQQGFGUEAVUIL-UHFFFAOYSA-N 0.000 description 1
- 230000002759 chromosomal effect Effects 0.000 description 1
- 208000019069 chronic childhood arthritis Diseases 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 201000010002 cicatricial pemphigoid Diseases 0.000 description 1
- 229960001265 ciclosporin Drugs 0.000 description 1
- 238000003501 co-culture Methods 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 229940001442 combination vaccine Drugs 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 239000003184 complementary RNA Substances 0.000 description 1
- 230000000536 complexating effect Effects 0.000 description 1
- 239000003433 contraceptive agent Substances 0.000 description 1
- 230000002254 contraceptive effect Effects 0.000 description 1
- 229940124462 contraceptive vaccine Drugs 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 229960001681 croscarmellose sodium Drugs 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 239000012228 culture supernatant Substances 0.000 description 1
- 229930182912 cyclosporin Natural products 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 231100000599 cytotoxic agent Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 239000002619 cytotoxin Substances 0.000 description 1
- 238000013500 data storage Methods 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000002716 delivery method Methods 0.000 description 1
- 239000005547 deoxyribonucleotide Substances 0.000 description 1
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 230000000368 destabilizing effect Effects 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 201000002491 encephalomyelitis Diseases 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 230000007071 enzymatic hydrolysis Effects 0.000 description 1
- 238000006047 enzymatic hydrolysis reaction Methods 0.000 description 1
- 238000003114 enzyme-linked immunosorbent spot assay Methods 0.000 description 1
- 230000008029 eradication Effects 0.000 description 1
- 230000003090 exacerbative effect Effects 0.000 description 1
- 201000005884 exanthem Diseases 0.000 description 1
- 208000012997 experimental autoimmune encephalomyelitis Diseases 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 108010055671 ezrin Proteins 0.000 description 1
- 235000013861 fat-free Nutrition 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 108010072250 fimbrillin Proteins 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 1
- 108010006620 fodrin Proteins 0.000 description 1
- 239000013568 food allergen Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 101150029683 gB gene Proteins 0.000 description 1
- 150000002270 gangliosides Chemical class 0.000 description 1
- 238000012817 gel-diffusion technique Methods 0.000 description 1
- 238000001476 gene delivery Methods 0.000 description 1
- 102000054766 genetic haplotypes Human genes 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 244000000058 gram-negative pathogen Species 0.000 description 1
- 229940046528 grass pollen Drugs 0.000 description 1
- 230000005484 gravity Effects 0.000 description 1
- 239000005090 green fluorescent protein Substances 0.000 description 1
- 229960000789 guanidine hydrochloride Drugs 0.000 description 1
- PJJJBBJSCAKJQF-UHFFFAOYSA-N guanidinium chloride Chemical compound [Cl-].NC(N)=[NH2+] PJJJBBJSCAKJQF-UHFFFAOYSA-N 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000005831 heart abnormality Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 239000000185 hemagglutinin Substances 0.000 description 1
- 201000011066 hemangioma Diseases 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 238000012203 high throughput assay Methods 0.000 description 1
- 238000012165 high-throughput sequencing Methods 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 238000002744 homologous recombination Methods 0.000 description 1
- 230000006801 homologous recombination Effects 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 239000003667 hormone antagonist Substances 0.000 description 1
- 229940046533 house dust mites Drugs 0.000 description 1
- 102000054350 human CHI3L1 Human genes 0.000 description 1
- 102000055650 human NRG1 Human genes 0.000 description 1
- 230000028996 humoral immune response Effects 0.000 description 1
- 230000004727 humoral immunity Effects 0.000 description 1
- 229960002773 hyaluronidase Drugs 0.000 description 1
- 238000011574 hybrid mouse model Methods 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 238000001597 immobilized metal affinity chromatography Methods 0.000 description 1
- 230000005934 immune activation Effects 0.000 description 1
- 230000007124 immune defense Effects 0.000 description 1
- 229940127121 immunoconjugate Drugs 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 238000003364 immunohistochemistry Methods 0.000 description 1
- 238000010324 immunological assay Methods 0.000 description 1
- 230000009463 immunological memory response Effects 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 230000003308 immunostimulating effect Effects 0.000 description 1
- 230000001024 immunotherapeutic effect Effects 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 210000004969 inflammatory cell Anatomy 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 239000002054 inoculum Substances 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000007917 intracranial administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 208000002551 irritable bowel syndrome Diseases 0.000 description 1
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 1
- 229960000310 isoleucine Drugs 0.000 description 1
- BPHPUYQFMNQIOC-NXRLNHOXSA-N isopropyl beta-D-thiogalactopyranoside Chemical compound CC(C)S[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O BPHPUYQFMNQIOC-NXRLNHOXSA-N 0.000 description 1
- 201000002215 juvenile rheumatoid arthritis Diseases 0.000 description 1
- 230000002147 killing effect Effects 0.000 description 1
- 238000011005 laboratory method Methods 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 101150012518 lamB gene Proteins 0.000 description 1
- 210000001821 langerhans cell Anatomy 0.000 description 1
- 230000001418 larval effect Effects 0.000 description 1
- 230000002045 lasting effect Effects 0.000 description 1
- 229920000126 latex Polymers 0.000 description 1
- 239000004816 latex Substances 0.000 description 1
- 231100001231 less toxic Toxicity 0.000 description 1
- 150000002632 lipids Chemical group 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 210000003563 lymphoid tissue Anatomy 0.000 description 1
- 230000001589 lymphoproliferative effect Effects 0.000 description 1
- 101710130522 mRNA export factor Proteins 0.000 description 1
- 108010026228 mRNA guanylyltransferase Proteins 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 201000008350 membranous glomerulonephritis Diseases 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 238000012737 microarray-based gene expression Methods 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 230000002438 mitochondrial effect Effects 0.000 description 1
- 108010071525 moesin Proteins 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 230000001459 mortal effect Effects 0.000 description 1
- 101150076681 mpt63 gene Proteins 0.000 description 1
- 101150014428 mpt64 gene Proteins 0.000 description 1
- 230000016379 mucosal immune response Effects 0.000 description 1
- 208000029744 multiple organ dysfunction syndrome Diseases 0.000 description 1
- 238000012243 multiplex automated genomic engineering Methods 0.000 description 1
- 229940031348 multivalent vaccine Drugs 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 210000000663 muscle cell Anatomy 0.000 description 1
- 101150049514 mutL gene Proteins 0.000 description 1
- 230000000869 mutational effect Effects 0.000 description 1
- 206010028417 myasthenia gravis Diseases 0.000 description 1
- QCQYVCMYGCHVMR-AAZUGDAUSA-N n-[(2r,3r,4s,5r)-4,5,6-trihydroxy-1-oxo-3-[(2r,3r,4s,5r,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyhexan-2-yl]acetamide Chemical compound CC(=O)N[C@@H](C=O)[C@H]([C@@H](O)[C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O QCQYVCMYGCHVMR-AAZUGDAUSA-N 0.000 description 1
- 210000000822 natural killer cell Anatomy 0.000 description 1
- 229920006173 natural rubber latex Polymers 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 210000000299 nuclear matrix Anatomy 0.000 description 1
- 238000007899 nucleic acid hybridization Methods 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 210000000287 oocyte Anatomy 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 101150045801 ospA gene Proteins 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 239000003002 pH adjusting agent Substances 0.000 description 1
- 230000002023 papillomaviral effect Effects 0.000 description 1
- 235000010603 pastilles Nutrition 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 235000020232 peanut Nutrition 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 1
- 230000008488 polyadenylation Effects 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 208000005987 polymyositis Diseases 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 239000001103 potassium chloride Substances 0.000 description 1
- 235000011164 potassium chloride Nutrition 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 229940116317 potato starch Drugs 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000001915 proofreading effect Effects 0.000 description 1
- 230000001902 propagating effect Effects 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 230000004952 protein activity Effects 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 230000005180 public health Effects 0.000 description 1
- 108010048484 radixin Proteins 0.000 description 1
- 102000016914 ras Proteins Human genes 0.000 description 1
- 108010014186 ras Proteins Proteins 0.000 description 1
- 206010037844 rash Diseases 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 101150056906 recJ gene Proteins 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000012827 research and development Methods 0.000 description 1
- 208000023504 respiratory system disease Diseases 0.000 description 1
- 230000032537 response to toxin Effects 0.000 description 1
- 230000001177 retroviral effect Effects 0.000 description 1
- 238000003757 reverse transcription PCR Methods 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 239000002336 ribonucleotide Substances 0.000 description 1
- 125000002652 ribonucleotide group Chemical group 0.000 description 1
- 108010055738 ribosomal phosphoprotein P2 Proteins 0.000 description 1
- 238000002702 ribosome display Methods 0.000 description 1
- 108700010850 rotavirus proteins Proteins 0.000 description 1
- 235000019515 salmon Nutrition 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 101150072534 sbcB gene Proteins 0.000 description 1
- 238000010845 search algorithm Methods 0.000 description 1
- 238000004062 sedimentation Methods 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 description 1
- 230000000405 serological effect Effects 0.000 description 1
- 208000026775 severe diarrhea Diseases 0.000 description 1
- 238000007390 skin biopsy Methods 0.000 description 1
- 208000017520 skin disease Diseases 0.000 description 1
- 231100000046 skin rash Toxicity 0.000 description 1
- 231100000430 skin reaction Toxicity 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 239000001540 sodium lactate Substances 0.000 description 1
- 229940005581 sodium lactate Drugs 0.000 description 1
- 235000011088 sodium lactate Nutrition 0.000 description 1
- 239000008279 sol Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 210000003046 sporozoite Anatomy 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 229940032147 starch Drugs 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 229940031626 subunit vaccine Drugs 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 231100000617 superantigen Toxicity 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 229940021747 therapeutic vaccine Drugs 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 208000030045 thyroid gland papillary carcinoma Diseases 0.000 description 1
- 241001147422 tick-borne encephalitis virus group Species 0.000 description 1
- 230000001550 time effect Effects 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 230000001551 toxigenic effect Effects 0.000 description 1
- 230000037317 transdermal delivery Effects 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 206010044412 transitional cell carcinoma Diseases 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 102000035160 transmembrane proteins Human genes 0.000 description 1
- 108091005703 transmembrane proteins Proteins 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical class [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 238000000539 two dimensional gel electrophoresis Methods 0.000 description 1
- 101150115617 umuC gene Proteins 0.000 description 1
- 101150046028 umuD gene Proteins 0.000 description 1
- 241001446247 uncultured actinomycete Species 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241001529453 unidentified herpesvirus Species 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 229940035893 uracil Drugs 0.000 description 1
- 239000004474 valine Substances 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 239000000304 virulence factor Substances 0.000 description 1
- 230000007923 virulence factor Effects 0.000 description 1
- 101150100239 vsr gene Proteins 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- 239000012130 whole-cell lysate Substances 0.000 description 1
- 244000000057 wild-type pathogen Species 0.000 description 1
- 210000005253 yeast cell Anatomy 0.000 description 1
- 210000004340 zona pellucida Anatomy 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1058—Directional evolution of libraries, e.g. evolution of libraries is achieved by mutagenesis and screening or selection of mixed population of organisms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0008—Antigens related to auto-immune diseases; Preparations to induce self-tolerance
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/002—Protozoa antigens
- A61K39/015—Hemosporidia antigens, e.g. Plasmodium antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/02—Bacterial antigens
- A61K39/0225—Spirochetes, e.g. Treponema, Leptospira, Borrelia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/02—Bacterial antigens
- A61K39/025—Enterobacteriales, e.g. Enterobacter
- A61K39/0258—Escherichia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/02—Bacterial antigens
- A61K39/025—Enterobacteriales, e.g. Enterobacter
- A61K39/0291—Yersinia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/02—Bacterial antigens
- A61K39/04—Mycobacterium, e.g. Mycobacterium tuberculosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/02—Bacterial antigens
- A61K39/085—Staphylococcus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/02—Bacterial antigens
- A61K39/09—Lactobacillales, e.g. aerococcus, enterococcus, lactobacillus, lactococcus, streptococcus
- A61K39/092—Streptococcus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/02—Bacterial antigens
- A61K39/105—Delta proteobacteriales, e.g. Lawsonia; Epsilon proteobacteriales, e.g. campylobacter, helicobacter
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/02—Bacterial antigens
- A61K39/107—Vibrio
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/35—Allergens
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/005—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/195—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from bacteria
- C07K14/24—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from bacteria from Enterobacteriaceae (F), e.g. Citrobacter, Serratia, Proteus, Providencia, Morganella, Yersinia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/53—DNA (RNA) vaccination
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/70—Multivalent vaccine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/01—Fusion polypeptide containing a localisation/targetting motif
- C07K2319/02—Fusion polypeptide containing a localisation/targetting motif containing a signal sequence
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/40—Fusion polypeptide containing a tag for immunodetection, or an epitope for immunisation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/70—Fusion polypeptide containing domain for protein-protein interaction
- C07K2319/74—Fusion polypeptide containing domain for protein-protein interaction containing a fusion for binding to a cell surface receptor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/16011—Herpesviridae
- C12N2710/16611—Simplexvirus, e.g. human herpesvirus 1, 2
- C12N2710/16634—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2730/00—Reverse transcribing DNA viruses
- C12N2730/00011—Details
- C12N2730/10011—Hepadnaviridae
- C12N2730/10111—Orthohepadnavirus, e.g. hepatitis B virus
- C12N2730/10122—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2730/00—Reverse transcribing DNA viruses
- C12N2730/00011—Details
- C12N2730/10011—Hepadnaviridae
- C12N2730/10111—Orthohepadnavirus, e.g. hepatitis B virus
- C12N2730/10134—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/16011—Human Immunodeficiency Virus, HIV
- C12N2740/16111—Human Immunodeficiency Virus, HIV concerning HIV env
- C12N2740/16134—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/16011—Human Immunodeficiency Virus, HIV
- C12N2740/16211—Human Immunodeficiency Virus, HIV concerning HIV gagpol
- C12N2740/16222—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2760/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses negative-sense
- C12N2760/00011—Details
- C12N2760/12011—Bunyaviridae
- C12N2760/12111—Hantavirus, e.g. Hantaan virus
- C12N2760/12134—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2770/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses positive-sense
- C12N2770/00011—Details
- C12N2770/24011—Flaviviridae
- C12N2770/24111—Flavivirus, e.g. yellow fever virus, dengue, JEV
- C12N2770/24134—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2770/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses positive-sense
- C12N2770/00011—Details
- C12N2770/24011—Flaviviridae
- C12N2770/24211—Hepacivirus, e.g. hepatitis C virus, hepatitis G virus
- C12N2770/24234—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2770/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses positive-sense
- C12N2770/00011—Details
- C12N2770/36011—Togaviridae
- C12N2770/36111—Alphavirus, e.g. Sindbis virus, VEE, EEE, WEE, Semliki
- C12N2770/36134—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- This invention pertains to the field of methods for developing immunogens that can induce efficient immune responses against a broad range of antigens.
- pathogens and hosts are results of millions of years of evolution, during which the mammalian immune system has evolved sophisticated means to counterattack pathogen invasions.
- bacterial and viral pathogens have simultaneously gained a number of mechanisms to improve their virulence and survival in hosts, providing a major challenge for vaccine research and development despite the powers of modern techniques of molecular and cellular biology.
- pathogen antigens Similar to the evolution of pathogen antigens, several cancer antigens are likely to have gained means to downregulate their immunogenicity as a mechanism to escape the host immune system. Efficient vaccine development is also hampered by the antigenic heterogeneity of different strains of pathogens, driven in part by evolutionary forces as means for the pathogens to escape immune defenses.
- Pathogens also reduce their immunogenicity by selecting antigens that are difficult to express, process and/or transport in host cells, thereby reducing the availability of immunogenic peptides to the molecules initiating and modulating immune responses.
- the mechanisms associated with these challenges are complex, multivariate and rather poorly characterized. Accordingly, a need exists for vaccines that can induce a protective immune response against bacterial and viral pathogens.
- the present invention fulfills this and other needs.
- the present invention provides recombinant multivalent antigenic polypeptides that include a first antigenic determinant from a first disease-associated polypeptide and at least a second antigenic determinant from a second disease-associated polypeptide.
- the disease-associated polypeptides can be selected from the group consisting of cancer antigens, antigens associated with autoimmunity disorders, antigens associated with inflammatory conditions, antigens associated with allergic reactions, antigens associated with infectious agents, and other antigens that are associated with a disease condition.
- the invention provides a recombinant antigen library that contains recombinant nucleic acids that encode antigenic polypeptides.
- the libraries are typically obtained by recombining at least first and second forms of a nucleic acid which includes a polynucleotide sequence that encodes a disease-associated antigenic polypeptide, wherein the first and second forms differ from each other in two or more nucleotides, to produce a library of recombinant nucleic acids.
- Another embodiment of the invention provides methods of obtaining a polynucleotide that encodes a recombinant antigen having improved ability to induce an immune response to a disease condition. These methods involve: (1) recombining at least first and second forms of a nucleic acid which comprises a polynucleotide sequence that encodes an antigenic polypeptide that is associated with the disease condition, wherein the first and second forms differ from each other in two or more nucleotides, to produce a library of recombinant nucleic acids; and (2) screening the library to identify at least one optimized recombinant nucleic acid that encodes an optimized recombinant antigenic polypeptide that has improved ability to induce an immune response to the disease condition.
- These methods optionally further involve: (3) recombining at least one optimized recombinant nucleic acid with a further form of the nucleic acid, which is the same or different from the first and second forms, to produce a further library of recombinant nucleic acids; (4) screening the further library to identify at least one further optimized recombinant nucleic acid that encodes a polypeptide that has improved ability to induce an immune response to the disease condition; and (5) repeating (3) and (4), as necessary, until the further optimized recombinant nucleic acid encodes a polypeptide that has improved ability to induce an immune response to the disease condition.
- the optimized recombinant nucleic acid encodes a multivalent antigenic polypeptide and the screening is accomplished by expressing the library of recombinant nucleic acids in a phage display expression vector such that the recombinant antigen is expressed as a fusion protein with a phage polypeptide that is displayed on a phage particle surface; contacting the phage with a first antibody that is specific for a first serotype of the pathogenic agent and selecting those phage that bind to the first antibody; and contacting those phage that bind to the first antibody with a second antibody that is specific for a second serotype of the pathogenic agent and selecting those phage that bind to the second antibody; wherein those phage that bind to the first antibody and the second antibody express a multivalent antigenic polypeptide.
- the invention also provides methods of obtaining a recombinant viral vector which has an enhanced ability to induce an antiviral response in a cell.
- These methods can include the steps of: (1) recombining at least first and second forms of a nucleic acid which comprise a viral vector, wherein the first and second forms differ from each other in two or more nucleotides, to produce a library of recombinant viral vectors; (2) transfecting the library of recombinant viral vectors into a population of mammalian cells; (3) staining the cells for the presence of Mx protein; and (4) isolating recombinant viral vectors from cells which stain positive for Mx protein, wherein recombinant viral vectors from positive staining cells exhibit enhanced ability to induce an antiviral response.
- Figure 1 shows a schematic representation of a method for generating a chimeric, multivalent antigen that has immunogenic regions from multiple antigens.
- Antibodies to each of the non-chimeric parental immunogenic polypeptides are specific for the respective organisms (A, B, C). After carrying out the recombination and selection methods of the invention, however, a chimeric immunogenic polypeptide is obtained that is recognized by antibodies raised against each of the three parental immunogenic polypeptides.
- Figure 2 shows the principle of family DNA shuffling.
- a family of antigen genes from related pathogens are subjected to shuffling, which results in a library of chimeric and/or mutated antigens. Screening methods are employed to identify those recombinant antigens that are the most immunogenic and/or cross-protective. These can, if desired, be subjected to additional rounds of shuffling and screening.
- Figure 3A- Figure 3B shows a schematic for a method by which one can obtain recombinant polypeptides that can induce a broad-spectrum immune response.
- wild-type immunogenic polypeptides from the pathogens A, B, and C provide protection against the corresponding pathogen from which the polypeptide is derived, but little or no cross-protection against the other pathogens (left panel).
- an A/B/C chimeric polypeptide is obtained that can induce a protective immune response against all three pathogen types (right panel).
- shuffling is used with substrate nucleic acids from two pathogen strains (A, B), which encode polypeptides that are protective only against the corresponding pathogen.
- the resulting chimeric polypeptide can induce an immune response that is effective against not only the two parental pathogen strains, but also against a third strain of pathogen (C).
- Figure 4 diagrams some of the possible factors that can determine whether a particular polynucleotide encodes an immunogenic polypeptide having a desired property, such as enhanced immunogenicity and/or cross-reactivity. Those sequence regions that positively affect a particular property are indicated as plus signs along the antigen gene, while those sequence regions that have a negative effect are shown as minus signs. A pool of related antigen genes are shuffled and screened to obtain those that recombinant nucleic acids that have gained positive sequence regions and lost negative regions. No pre-existing knowledge as to which regions are positive or negative for a particular trait is required.
- Figure 5 shows a schematic representation of the screening strategy for antigen library screening.
- Figure 6 shows a schematic representation of a strategy for pooling and deconvolution as used in antigen library screening.
- Figure 7 is an alignment of the nucleotide sequences of glycoprotein D (gD) from HSV-1 (SEQ ID NO: 1) and HSV-2 (gD-1 (SEQ ID NO: 2) and gD-2 (SEQ ID NO:
- Figure 8 A shows a diagram of a method for expressing HIV gpl20 using genetic vaccine vectors and generation of a library of shuffled gpl20 genes.
- Figure 8B shows PCR primers that are useful for obtaining gpl20 nucleic acid substrates for DNA shuffling reactions.
- Primers suitable for generating substrates include 6025F (SEQ ID NO: 4), 7773R (SEQ ID NO: 5), and primers suitable for amplifying the shuffled nucleic acids include 6196F (SEQ ID NO: 6) and 7746R (SEQ ID NO: 7).
- the primer BssH2-6205F (SEQ ID NO: 8) can be used to clone the resulting fragment into a genetic vaccine vector.
- Figure 9 shows the domain structure of hepadnavirus envelope genes.
- Figure 10 shows a schematic representation of the use of shuffling to obtain hepadnavirus proteins in which the immunogenicity of one antigenic domain is improved.
- Figure 11 shows a strategy in which genes that encode the hepadnavirus proteins having one antigenic domain that has improved immunogenicity are shuffled to obtain recombinant proteins in which all three domains have improved immunogenicity.
- Figure 12 shows the transmembrane organization of the HBsAg polypeptide.
- Figure 13 shows a method for using phage display to obtain recombinant allergens that are not bound by pre-existing IgE.
- Figure 14 shows a strategy for screening of recombinant allergens to identify those that are effective in activating T H cells.
- PBMC or T cell clones from atopic individuals are exposed to antigen-presenting cells that display the antigen variants obtained using the methods of the invention.
- the cultures are tested for induction of T cell proliferation or for a pattern of cytokine synthesis that is indicative of the particular type of T cell activation that is desired. If desired, the allergen variants that test positive in the in vitro assay can be subjected to in vivo testing.
- Figure 15 shows a strategy for screening of recombinant cancer antigens to identify those that are effective in activating T cells of cancer patients.
- Figure 16A and Figure 16B show two different strategies for generating vectors that contain multiple T cell epitopes obtained, for example, by DNA shuffling.
- each individual shuffled epitope-encoding nucleic acid is linked to a single promoter, and multiple promoter-epitope gene constructs can be placed in a single vector.
- the scheme shown in Figure 16B involves linking multiple epitope-encoding nucleic acids to a single promoter.
- Figure 17 shows the sequences of PreS2-S coding regions and corresponding amino acid sequences of different hepatitis B surface antigen (HBsAg) or woodchuck hepatitis B (WHV) proteins. Primers suitable for amplification of this region are also shown.
- HBsAg hepatitis B surface antigen
- HBV woodchuck hepatitis B
- Figure 18 shows primers that are suitable for amplification of large fragments that contain the S2S coding sequences.
- the primers hybridize to regions that are approximately 200 bp outside the desired sequences.
- Figure 19 shows an alignment of the amino acid sequences of surface antigens from different HVB and WHV subtypes.
- Figure 20 shows a diagram of multimeric particles that assemble when an appropriate number of chimeric polypeptides and native HBsAg S monomers are mixed.
- screening describes, in general, a process that identifies optimal antigens.
- properties of the antigen can be used in selection and screening including antigen expression, folding, stability, immunogenicity and presence of epitopes from several related antigens.
- Selection is a form of screening in which identification and physical separation are achieved simultaneously by expression of a selection marker, which, in some genetic circumstances, allows cells expressing the marker to survive while other cells die (or vice versa).
- Screening markers include, for example, luciferase, beta-galactosidase and green fluorescent protein. Selection markers include drug and toxin resistance genes, and the like. Because of limitations in studying primary immune responses in vitro, in vivo studies are particularly useful screening methods.
- the antigens are first introduced to test animals, and the immune responses are subsequently studied by analyzing protective immune responses or by studying the quality or strength of the induced immune response using lymphoid cells derived from the immunized animal.
- spontaneous selection can and does occur in the course of natural evolution, in the present methods selection is performed by man.
- a "exogenous DNA segment”, “heterologous sequence” or a “heterologous nucleic acid”, as used herein, is one that originates from a source foreign to the particular host cell, or, if from the same source, is modified from its original form.
- a heterologous gene in a host cell includes a gene that is endogenous to the particular host cell, but has been modified. Modification of a heterologous sequence in the applications described herein typically occurs through the use of DNA shuffling.
- the terms refer to a DNA segment which is foreign or heterologous to the cell, or homologous to the cell but in a position within the host cell nucleic acid in which the element is not ordinarily found. Exogenous DNA segments are expressed to yield exogenous polypeptides.
- genes are used broadly to refer to any segment of DNA associated with a biological function. Thus, genes include coding sequences and/or the regulatory sequences required for their expression. Genes also include nonexpressed DNA segments that, for example, form recognition sequences for other proteins. Genes can be obtained from a variety of sources, including cloning from a source of interest or synthesizing from known or predicted sequence information, and may include sequences designed to have desired parameters.
- nucleic acid or protein when applied to a nucleic acid or protein, denotes that the nucleic acid or protein is essentially free of other cellular components with which it is associated in the natural state. It is preferably in a homogeneous state although it can be in either a dry or aqueous solution. Purity and homogeneity are typically determined using analytical chemistry techniques such as polyacrylamide gel electrophoresis or high performance liquid chromatography. A protein which is the predominant species present in a preparation is substantially purified. In particular, an isolated gene is separated from open reading frames which flank the gene and encode a protein other than the gene of interest. The term "purified” denotes that a nucleic acid or protein gives rise to essentially one band in an electrophoretic gel.
- nucleic acid or protein is at least about 50% pure, more preferably at least about 85% pure, and most preferably at least about 99% pure.
- naturally-occurring is used to describe an object that can be found in nature as distinct from being artificially produced by man.
- a polypeptide or polynucleotide sequence that is present in an organism (including viruses, bacteria, protozoa, insects, plants or mammalian tissue) that can be isolated from a source in nature and which has not been intentionally modified by man in the laboratory is naturally-occurring.
- nucleic acid refers to deoxyribonucleotides or ribonucleotides and polymers thereof in either single- or double-stranded form.
- nucleic acids containing known analogues of natural nucleotides which have similar binding properties as the reference nucleic acid and are metabolized in a manner similar to naturally occurring nucleotides.
- a particular nucleic acid sequence also implicitly encompasses conservatively modified variants thereof (eg. degenerate codon substitutions) and complementary sequences and as well as the sequence explicitly indicated.
- degenerate codon substitutions may be achieved by generating sequences in which the third position of one or more selected (or all) codons is substituted with mixed-base and/or deoxyinosine residues (Batzer et al. (1991) Nucleic Acid Res.
- nucleic acid is used interchangeably with gene, cDNA, and mRNA encoded by a gene.
- Nucleic acid derived from a gene refers to a nucleic acid for whose synthesis the gene, or a subsequence thereof, has ultimately served as a template.
- an mRNA, a cDNA reverse transcribed from an mRNA, an RNA transcribed from that cDNA, a DNA amplified from the cDNA, an RNA transcribed from the amplified DNA, etc. are all derived from the gene and detection of such derived products is indicative of the presence and/or abundance of the original gene and/or gene transcript in a sample.
- a nucleic acid is "operably linked" when it is placed into a functional relationship with another nucleic acid sequence.
- a promoter or enhancer is operably linked to a coding sequence if it increases the transcription of the coding sequence.
- Operably linked means that the DNA sequences being linked are typically contiguous and, where necessary to join two protein coding regions, contiguous and in reading frame.
- enhancers generally function when separated from the promoter by several kilobases and intronic sequences may be of variable lengths, some polynucleotide elements may be operably linked but not contiguous.
- a specific binding affinity between two molecules means a preferential binding of one molecule for another in a mixture of molecules.
- the binding of the molecules can be considered specific if the binding affinity is about 1 x 10 4 M " ' to about 1 x 10 6 M ' ' or greater.
- the term "recombinant" when used with reference to a cell indicates that the cell replicates a heterologous nucleic acid, or expresses a peptide or protein encoded by a heterologous nucleic acid. Recombinant cells can contain genes that are not found within the native (non-recombinant) form of the cell.
- Recombinant cells can also contain genes found in the native form of the cell wherein the genes are modified and re-introduced into the cell by artificial means.
- the term also encompasses cells that contain a nucleic acid endogenous to the cell that has been modified without removing the nucleic acid from the cell; such modifications include those obtained by gene replacement, site-specific mutation, and related techniques.
- a "recombinant expression cassette” or simply an “expression cassette” is a nucleic acid construct, generated recombinantly or synthetically, with nucleic acid elements that are capable of effecting expression of a structural gene in hosts compatible with such sequences.
- Expression cassettes include at least promoters and optionally, transcription termination signals.
- the recombinant expression cassette includes a nucleic acid to be transcribed (e.g., a nucleic acid encoding a desired polypeptide), and a promoter. Additional factors necessary or helpful in effecting expression may also be used as described herein.
- an expression cassette can also include nucleotide sequences that encode a signal sequence that directs secretion of an expressed protein from the host cell. Transcription termination signals, enhancers, and other nucleic acid sequences that influence gene expression, can also be included in an expression cassette.
- a “multivalent antigenic polypeptide” or a “recombinant multivalent antigenic polypeptide” is a non-naturally occurring polypeptide that includes amino acid sequences from more than one source polypeptide, which source polypeptide is typically a naturally occurring polypeptide. At least some of the regions of different amino acid sequences constitute epitopes that are recognized by antibodies found in a mammal that has been injected with the source polypeptide.
- the source polypeptides from which the different epitopes are derived are usually homologous (i.e., have the same or a similar structure and/or function), and are often from different isolates, serotypes, strains, species, of organism or from different disease states, for example.
- nucleic acid or polypeptide sequences refer to two or more sequences or subsequences that are the same or have a specified percentage of amino acid residues or nucleotides that are the same, when compared and aligned for maximum correspondence, as measured using one of the following sequence comparison algorithms or by visual inspection.
- substantially identical in the context of two nucleic acids or polypeptides, refers to two or more sequences or subsequences that have at least 60%, preferably 80%, most preferably 90-95% nucleotide or amino acid residue identity, when compared and aligned for maximum correspondence, as measured using one of the following sequence comparison algorithms or by visual inspection.
- the substantial identity exists over a region of the sequences that is at least about 50 residues in length, more preferably over a region of at least about 100 residues, and most preferably the sequences are substantially identical over at least about 150 residues.
- the sequences are substantially identical over the entire length of the coding regions.
- sequence comparison typically one sequence acts as a reference sequence to which test sequences are compared.
- test and reference sequences are input into a computer, subsequence coordinates are designated, if necessary, and sequence algorithm program parameters are designated.
- sequence comparison algorithm then calculates the percent sequence identity for the test sequence(s) relative to the reference sequence, based on the designated program parameters.
- Optimal alignment of sequences for comparison can be conducted, e.g., by the local homology algorithm of Smith & Waterman, Adv. Appl. Math. 2:482 (1981), by the homology alignment algorithm of Needleman & Wunsch, J. Mol. Biol. 48:443 (1970), by the search for similarity method of Pearson & Lipman, Proc. Nat 7. Acad. Sci. USA 85:2444 (1988), by computerized implementations of these algorithms (GAP, BESTFIT, FASTA, and TFASTA in the Wisconsin Genetics Software Package, Genetics Computer Group, 575 Science Dr., Madison, WI), or by visual inspection (see generally Ausubel et al, infra).
- HSPs high scoring sequence pairs
- initial neighborhood word hits act as seeds for initiating searches to find longer HSPs containing them.
- the word hits are then extended in both directions along each sequence for as far as the cumulative alignment score can be increased. Cumulative scores are calculated using, for nucleotide sequences, the parameters M (reward score for a pair of matching residues; always > 0) and N (penalty score for mismatching residues; always ⁇ 0). For amino acid sequences, a scoring matrix is used to calculate the cumulative score. Extension of the word hits in each direction are halted when: the cumulative alignment score falls off by the quantity X from its maximum achieved value; the cumulative score goes to zero or below, due to the accumulation of one or more negative-scoring residue alignments; or the end of either sequence is reached.
- the BLAST algorithm parameters W, T, and X determine the sensitivity and speed of the alignment.
- the BLASTP program uses as defaults a wordlength (W) of 3, an expectation (E) of 10, and the BLOSUM62 scoring matrix (see Henikoff & Henikoff (1989) Proc. Natl. Acad. Sci. USA 89:10915).
- the BLAST algorithm In addition to calculating percent sequence identity, the BLAST algorithm also performs a statistical analysis of the similarity between two sequences (see, e.g., Karlin & Altschul (1993) Proc. Nat 7. Acad. Sci. USA 90:5873-5787).
- One measure of similarity provided by the BLAST algorithm is the smallest sum probability (P(N)), which provides an indication of the probability by which a match between two nucleotide or amino acid sequences would occur by chance.
- P(N) the smallest sum probability
- a nucleic acid is considered similar to a reference sequence if the smallest sum probability in a comparison of the test nucleic acid to the reference nucleic acid is less than about 0.1, more preferably less than about 0.01, and most preferably less than about 0.001.
- hybridizing specifically to refers to the binding, duplexing, or hybridizing of a molecule only to a particular nucleotide sequence under stringent conditions when that sequence is present in a complex mixture (e.g., total cellular) DNA or RNA.
- Bod(s) substantially refers to complementary hybridization between a probe nucleic acid and a target nucleic acid and embraces minor mismatches that can be accommodated by reducing the stringency of the hybridization media to achieve the desired detection of the target polynucleotide sequence.
- Stringent hybridization conditions and “stringent hybridization wash conditions” in the context of nucleic acid hybridization experiments such as Southern and northern hybridizations are sequence dependent, and are different under different environmental parameters. Longer sequences hybridize specifically at higher temperatures. An extensive guide to the hybridization of nucleic acids is found in Tijssen (1993) Laboratory Techniques in Biochemistry and Molecular Biology— Hybridization with Nucleic Acid Probes part I chapter 2 “Overview of principles of hybridization and the strategy of nucleic acid probe assays", Elsevier, New York. Generally, highly stringent hybridization and wash conditions are selected to be about 5° C lower than the thermal melting point (T m ) for the specific sequence at a defined ionic strength and pH.
- T m thermal melting point
- a probe will hybridize to its target subsequence, but to no other sequences.
- the T m is the temperature (under defined ionic strength and pH) at which 50% of the target sequence hybridizes to a perfectly matched probe.
- Very stringent conditions are selected to be equal to the T m for a particular probe.
- An example of stringent hybridization conditions for hybridization of complementary nucleic acids which have more than 100 complementary residues on a filter in a Southern or northern blot is 50% formamide with 1 mg of heparin at 42°C, with the hybridization being carried out overnight.
- An example of highly stringent wash conditions is 0.15M NaCl at 72°C for about 15 minutes.
- An example of stringent wash conditions is a 0.2x SSC wash at 65°C for 15 minutes (see, Sambrook, infra., for a description of SSC buffer). Often, a high stringency wash is preceded by a low stringency wash to remove background probe signal.
- An example medium stringency wash for a duplex of, e.g., more than 100 nucleotides, is lx SSC at 45°C for 15 minutes.
- An example low stringency wash for a duplex of, e.g., more than 100 nucleotides, is 4-6x SSC at 40°C for 15 minutes.
- stringent conditions typically involve salt concentrations of less than about 1.0 M Na + ion, typically about 0.01 to 1.0 M Na + ion concentration (or other salts) at pH 7.0 to 8.3, and the temperature is typically at least about 30°C.
- Stringent conditions can also be achieved with the addition of destabilizing agents such as formamide.
- a signal to noise ratio of 2x (or higher) than that observed for an unrelated probe in the particular hybridization assay indicates detection of a specific hybridization.
- Nucleic acids which do not hybridize to each other under stringent conditions are still substantially identical if the polypeptides which they encode are substantially identical. This occurs, e.g., when a copy of a nucleic acid is created using the maximum codon degeneracy permitted by the genetic code.
- a further indication that two nucleic acid sequences or polypeptides are substantially identical is that the polypeptide encoded by the first nucleic acid is immunologically cross reactive with, or specifically binds to, the polypeptide encoded by the second nucleic acid.
- a polypeptide is typically substantially identical to a second polypeptide, for example, where the two peptides differ only by conservative substitutions.
- the specified antibodies bind to a particular protein and do not bind in a significant amount to other proteins present in the sample.
- the antibodies raised against a multivalent antigenic polypeptide will generally bind to the proteins from which one or more of the epitopes were obtained. Specific binding to an antibody under such conditions may require an antibody that is selected for its specificity for a particular protein.
- immunoassay formats may be used to select antibodies specifically immunoreactive with a particular protein.
- solid-phase ELISA immunoassays, Western blots, or immunohistochemistry are routinely used to select monoclonal antibodies specifically immunoreactive with a protein. See Harlow and Lane (1988) Antibodies, A Laboratory Manual, Cold Spring Harbor Publications, New York “Harlow and Lane”), for a description of immunoassay formats and conditions that can be used to determine specific immunoreactivity.
- a specific or selective reaction will be at least twice background signal or noise and more typically more than 10 to 100 times background.
- Consatively modified variations of a particular polynucleotide sequence refers to those polynucleotides that encode identical or essentially identical amino acid sequences, or where the polynucleotide does not encode an amino acid sequence, to essentially identical sequences. Because of the degeneracy of the genetic code, a large number of functionally identical nucleic acids encode any given polypeptide. For instance, the codons CGU, CGC, CGA, CGG, AGA, and AGG all encode the amino acid arginine. Thus, at every position where an arginine is specified by a codon, the codon can be altered to any of the corresponding codons described without altering the encoded polypeptide.
- nucleic acid variations are "silent variations," which are one species of “conservatively modified variations.” Every polynucleotide sequence described herein which encodes a polypeptide also describes every possible silent variation, except where otherwise noted.
- each codon in a nucleic acid except AUG, which is ordinarily the only codon for methionine
- each "silent variation" of a nucleic acid which encodes a polypeptide is implicit in each described sequence.
- G Glycine
- A Alanine
- V Valine
- L Leucine
- I Isoleucine
- Aromatic Phenylalanine
- F Phenylalanine
- Y Tyrosine
- W Tryptophan
- Sulfur-containing Methionine (M), Cysteine (C);
- Acidic Aspartic acid (D), Glutamic acid (E), Asparagine (N), Glutamine (Q). See also, Creighton (1984) Proteins. W.H. Freeman and Company, for additional groupings of amino acids.
- individual substitutions, deletions or additions which alter, add or delete a single amino acid or a small percentage of amino acids in an encoded sequence are also “conservatively modified variations”.
- a "subsequence” refers to a sequence of nucleic acids or amino acids that comprise a part of a longer sequence of nucleic acids or amino acids (e.g., polypeptide) respectively.
- the invention provides a new approach to vaccine development, which is termed "antigen library immunization.”
- antigen library immunization No other technologies are available for generating libraries of related antigens or optimizing known protective antigens.
- the immunization protocols of the invention which use shuffled antigen libraries, provide a means to identify novel antigen sequences.
- Antigen library immunization dramatically expands the diversity of available immunogen sequences, and therefore, these antigen chimera libraries can also provide means to defend against newly emerging pathogen variants of the future.
- the methods of the invention enable the identification of individual chimeric antigens that provide efficient protection against a variety of existing pathogens, providing improved vaccines for troops and civilian populations.
- the methods of the invention provide an evolution-based approach, such as DNA shuffling in particular, that is an optimal approach to improve the immunogenicity of many types of antigens.
- the methods provide means of obtaining optimized cancer antigens useful for preventing and treating malignant diseases.
- an increasing number of self- antigens, causing autoimmune diseases, and allergens, causing atopy, allergy and asthma, have been characterized.
- the immunogenicity and manufacturing of these antigens can likewise be improved with the methods of this invention.
- the antigen library immunization methods of the invention provide a means by which one can obtain a recombinant antigen that has improved ability to induce an immune response to a pathogenic agent.
- a "pathogenic agent” refers to an organism or virus that is capable of infecting a host cell. Pathogenic agents typically include and/or encode a molecule, usually a polypeptide, that is immunogenic in that an immune response is raised against the immunogenic polypeptide. Often, the immune response raised against an immunogenic polypeptide from one serotype of the pathogenic agent is not capable of recognizing, and thus protecting against, a different serotype of the pathogenic agent, or other related pathogenic agents. In other situations, the polypeptide produced by a pathogenic agent is not produced in sufficient amounts, or is not sufficiently immunogenic, for the infected host to raise an effective immune response against the pathogenic agent.
- DNA shuffling use as substrates forms of the nucleic acid that differ from each other in two or more nucleotides, so a library of recombinant nucleic acids results.
- the library is then screened to identify at least one optimized recombinant nucleic acid that encodes an optimized recombinant antigen that has improved ability to induce an immune response to the pathogenic agent or other condition.
- the resulting recombinant antigens often are chimeric in that they are recognized by antibodies (Abs) reacting against multiple pathogen strains, and generally can also elicit broad spectrum immune responses. Specific neutralizing antibodies are known to mediate protection against several pathogens of interest, although additional mechanisms, such as cytotoxic T lymphocytes, are likely to be involved.
- the concept of chimeric, multivalent antigens inducing broadly reacting antibody responses is further illustrated in Figure 1.
- the different forms of the nucleic acids that encode antigenic polypeptides are obtained from members of a family of related pathogenic agents.
- family shuffling This scheme of performing DNA shuffling using nucleic acids from related organisms, known as "family shuffling," is described in Crameri et al. ((1998) Nature 391: 288-291) and is shown schematically in Figure 2.
- Polypeptides of different strains and serotypes of pathogens generally vary between 60-98%, which will allow for efficient family DNA shuffling. Therefore, family DNA shuffling provides an effective approach to generate multivalent, crossprotective antigens. The methods are useful for obtaining individual chimeras that effectively protect against most or all pathogen variants (Figure 3A).
- immunizations using entire libraries or pools of shuffled antigen chimeras can also result in identification of chimeric antigens that protect against pathogen variants that were not included in the starting population of antigens (for example, protection against strain C by shuffled library of chimeras/mutants of strains A and B in Figure 3B).
- the antigen library immunization approach enables the development of immunogenic polypeptides that can induce immune responses against poorly characterized, newly emerging pathogen variants.
- Sequence recombination can be achieved in many different formats and permutations of formats, as described in further detail below. These formats share some common principles.
- the targets for modification vary in different applications, as does the property sought to be acquired or improved.
- candidate targets for acquisition of a property or improvement in a property include genes that encode proteins which have immunogenic and or toxigenic activity when introduced into a host organism.
- the methods use at least two variant forms of a starting target.
- the variant forms of candidate substrates can show substantial sequence or secondary structural similarity with each other, but they should also differ in at least one and preferably at least two positions.
- the initial diversity between forms can be the result of natural variation, e.g., the different variant forms (homologs) are obtained from different individuals or strains of an organism, or constitute related sequences from the same organism (e.g., allelic variations), or constitute homologs from different organisms (interspecific variants).
- initial diversity can be induced, e.g., the variant forms can be generated by error-prone transcription, such as an error-prone PCR or use of a polymerase which lacks proof-reading activity (see, Liao (1990) Gene 88:107-111), of the first variant form, or, by replication of the first form in a mutator strain (mutator host cells are discussed in further detail below, and are generally well known).
- a mutator strain can include any mutants in any organism impaired in the functions of mismatch repair. These include mutant gene products of mutS, mutT, mutH, mutL, ovrD, dcm, vsr, umuC, umuD, sbcB, recJ, etc.
- Impairment can be of the genes noted, or of homologous genes in any organism.
- Other methods of generating initial diversity include methods well known to those of skill in the art, including, for example, treatment of a nucleic acid with a chemical or other mutagen, through spontaneous mutation, and by inducing an error-prone repair system (e.g., SOS) in a cell that contains the nucleic acid.
- SOS error-prone repair system
- Polynucleotide sequences that can positively or negatively affect the immunogenicity of an antigen encoded by the polynucleotide are often scattered throughout the entire antigen gene. Several of these factors are shown diagrammatically in Figure 4.
- Figure 4 By recombining different forms of polynucleotide that encode the antigen using DNA shuffling, followed by selection for those chimeric polynucleotides that encode an antigen that can induce an improved immune response, one can obtain primarily sequences that have a positive influence on antigen immunogenicity. Those sequences that negatively affect antigen immunogenicity are eliminated (Figure 4). One need not know the particular sequences involved.
- the methods of the invention entail performing DNA recombination ("shuffling") and screening or selection to "evolve" individual genes, whole plasmids or viruses, multigene clusters, or even whole genomes (Stemmer (1995) Bio/Technology 13:549-553). Reiterative cycles of recombination and screening/selection can be performed to further evolve the nucleic acids of interest. Such techniques do not require the extensive analysis and computation required by conventional methods for polypeptide engineering. Shuffling allows the recombination of large numbers of mutations in a minimum number of selection cycles, in contrast to natural pair-wise recombination events (e.g., as occur during sexual replication).
- sequence recombination techniques described herein provide particular advantages in that they provide recombination between mutations in any or all of these, thereby providing a very fast way of exploring the manner in which different combinations of mutations can affect a desired result. In some instances, however, structural and/or functional information is available which, although not required for sequence recombination, provides opportunities for modification of the technique.
- DNA shuffling methods of the invention can involve at least one of at least four different approaches to improve immunogenic activity as well as to broaden specificity.
- DNA shuffling can be performed on a single gene.
- several highly homologous genes can be identified by sequence comparison with known homologous genes. These genes can be synthesized and shuffled as a family of homologs, to select recombinants with the desired activity.
- the shuffled genes can be cloned into appropriate host cells, such as E. coli, yeast, plants, fungi, animal cells, and the like, and those that encode antigens having the desired properties can be identified by the methods described below.
- whole genome shuffling can be performed to shuffle genes that encode antigenic polypeptides (along with other genomic nucleic acids). For whole genome shuffling approaches, it is not even necessary to identify which genes are being shuffled. Instead, e.g., bacterial cell or viral genomes are combined and shuffled to acquire recombinant polypeptides that have enhanced ability to induce an immune response, as measured in any of the assays described below.
- antigenic polypep tide-encoding genes can be codon modified to access mutational diversity not present in any naturally occurring gene. Details on each of these procedures can be found below.
- Kits for mutagenesis are commercially available (e.g., Bio-Rad, Amersham International, Yalen Biotechnology).
- the breeding procedure starts with at least two substrates that generally show some degree of sequence identity to each other (i.e., at least about 30%, 50%, 70%, 80% or 90% sequence identity), but differ from each other at certain positions.
- the difference can be any type of mutation, for example, substitutions, insertions and deletions.
- different segments differ from each other in about 5-20 positions.
- the starting materials must differ from each other in at least two nucleotide positions. That is, if there are only two substrates, there should be at least two divergent positions. If there are three substrates, for example, one substrate can differ from the second at a single position, and the second can differ from the third at a different single position.
- the starting DNA segments can be natural variants of each other, for example, allelic or species variants.
- the segments can also be from nonallelic genes showing some degree of structural and usually functional relatedness (e.g., different genes within a superfamily, such as the family of Yersinia V-antigens, for example).
- the starting DNA segments can also be induced variants of each other.
- one DNA segment can be produced by error-prone PCR replication of the other, the nucleic acid can be treated with a chemical or other mutagen, or by substitution of a mutagenic cassette.
- Induced mutants can also be prepared by propagating one (or both) of the segments in a mutagenic strain, or by inducing an error-prone repair system in the cells.
- the second DNA segment is not a single segment but a large family of related segments.
- the different segments forming the starting materials are often the same length or substantially the same length. However, this need not be the case; for example; one segment can be a subsequence of another.
- the segments can be present as part of larger molecules, such as vectors, or can be in isolated form.
- the starting DNA segments are recombined by any of the sequence recombination formats provided herein to generate a diverse library of recombinant DNA segments.
- a library can vary widely in size from having fewer than 10 to more than 10 5 , 10 9 , 10 12 or more members.
- the starting segments and the recombinant libraries generated will include full-length coding sequences and any essential regulatory sequences, such as a promoter and polyadenylation sequence, required for expression.
- the recombinant DNA segments in the library can be inserted into a common vector providing sequences necessary for expression before performing screening/selection.
- the invention provides methods of obtaining recombinant polynucleotides that encode antigens that exhibit improved ability to induce an immune response to a pathogenic agent.
- the methods are applicable to a wide range of pathogenic agents, including potential biological warfare agents and other organisms and polypeptides that can cause disease and toxicity in humans and other animals.
- pathogenic agents including potential biological warfare agents and other organisms and polypeptides that can cause disease and toxicity in humans and other animals.
- the following examples are merely illustrative, and not limiting.
- the methods of the invention are applied to bacterial pathogens, as well as to toxins produced by bacteria and other organisms.
- the polynucleotides of interest encode polypeptides that are present on the surface of the pathogenic organism.
- Yersinia pestis the causative agent of plague
- LD 50 values in mouse of less than 10 bacteria.
- the pneumonic form of the disease is readily spread between humans by aerosol or infectious droplets and can be lethal within days.
- V antigen which is a 37 kDa virulence factor, induces protective immune responses and is currently being evaluated as a subunit vaccine
- the V-antigen alone is not toxic, but Y. pestis isolates that lack the V-antigen are avirulent.
- the Yersinia V- antigen has been successfully produced in E. coli by several groups (Leary et al. (1995) Infect. Immun. 3: 2854).
- V antigen genes from various Yersinia species are subjected to family shuffling.
- the genes encoding the V antigen from Y. pestis, Y. enterocolitica, and Y. pseudotuberculosis, for example, are 92-99% identical at the DNA level, making them ideal for optimization using family shuffling according to the methods of the invention.
- the library of recombinant nucleic acids is screened and/or selected for those that encode recombinant V antigen polypeptides that can induce an improved immune response and/or have greater cross-protectivity.
- Bacillus anthracis the causative agent of anthrax
- the anthrax protective antigen (PA) provides protective immune responses in test animals, and antibodies against PA also provide some protection.
- PA anthrax protective antigen
- LF lethal factor
- the DNA shuffling and antigen library immunization methods of the invention can be used to obtain nontoxic LF.
- Polynucleotides that encode LF from various B. anthracis strains are subjected to family shuffling.
- the resulting library of recombinant LF nucleic acids can then be screened to identify those that encode recombinant LF polypeptides that exhibit reduced toxicity. For example, one can inoculate tissue culture cells with the recombinant LF polypeptides in the presence of PA and select those clones for which the cells survive. If desired, one can then backcross the nontoxic LF polypeptides to retain the immunogenic epitopes of LF. Those that are selected through the first screen can then be subjected to a secondary screen. For example, one can test for the ability of the recombinant nontoxic LF polypeptides to induce an immune response (e.g., CTL or antibody response) in a test animal such as mice. In preferred embodiments, the recombinant nontoxic LF polypeptides are then tested for ability to induce protective immunity in test animals against challenge by different strains of 5. anthracis.
- an immune response e.g., CTL or antibody response
- the protective antigen (PA) of 5. anthracis is also a suitable target for the methods of the invention.
- PA-encoding nucleic acids from various strains of 5. anthracis are subjected to DNA shuffling.
- Screening for ability to induce broad-spectrum antibodies in a test animal is also typically used, either alone or in addition to a preliminary screening method.
- those recombinant polynucleotides that exhibit the desired properties can be backcrossed so that the immunogenic epitopes are maintained.
- the selected recombinants are tested for ability to induce protective immunity against different strains of B. anthracis in a test animal.
- the Staphylococcus aureus and Streptococcus toxins are another example of a target polypeptide that can be altered using the methods of the invention.
- Strains of Staphylococcus aureus and group A Streptococci are involved in a range of diseases, including food poisoning, toxic shock syndrome, scarlet fever and various autoimmune disorders. They secrete a variety of toxins, which include at least five cytolytic toxins, a coagulase, and a variety of enterotoxins.
- the enterotoxins are classified as superantigens in that they crosslink MHC class II molecules with T cell receptors to cause a constitutive T cell activation (Fields et al. (1996) Nature 384: 188).
- enterotoxins have been sequenced and can be phylogenetically grouped into families. Crystal structures have been obtained for several members alone and in complex with MHC class II molecules. Certain mutations in the MHC class II-binding site of the toxins strongly reduce their toxicity and can form the basis of attenuated vaccines (Woody et al. (1997) Vaccine 15: 133). However, a successful immune response to one type of toxin may provide protection against closely related family members, whereas little protection against toxins from the other families is observed.
- family shuffling of enterotoxin genes from various family members can be used to obtain recombinant toxin molecules that have reduced toxicity and can induce a cross-protective immune response.
- Shuffled antigens can also be screened to identify antigens that elicit neutralizing antibodies in an appropriate animal model such as mouse or monkey. Examples of such assays can include ELISA formats in which the elicited antibodies prevent binding of the enterotoxin to the MHC complex and/or T cell receptors on cells or purified forms. These assays can also include formats where the added antibodies would prevent T cells from being cross-linked to appropriate antigen presenting cells.
- Cholera is an ancient, potentially lethal disease caused by the bacterium Vibrio cholerae and an effective vaccine for its prevention is still unavailable. Much of the pathogenesis of this disease is caused by the cholera enterotoxin. Ingestion of microgram quantities of cholera toxin can induce severe diarrhea causing loss of tens of liters of fluid. Cholera toxin is a complex of a single catalytic A subunit with a pentameric ring of identical B subunits. Each subunit is inactive on its own. The B subunits bind to specific ganglioside receptors on the surface of intestinal epithelial cells and trigger the entry of the A subunit into the cell. The A subunit ADP-ribosylates a regulatory G protein initiating a cascade of events causing a massive, sustained flow of electrolytes and water into the intestinal lumen resulting in extreme diarrhea.
- the B subunit of cholera toxin is an attractive vaccine target for a number of reasons. It is a major target of protective antibodies generated during cholera infection and contains the epitopes for antitoxin neutralizing antibodies. It is nontoxic without the A subunit, is orally effective, and stimulates production of a strong IgA-dominated gut mucosal immune response, which is essential in protection against cholera and cholera toxin.
- the B subunit is also being investigated for use as an adjuvant in other vaccine preparations, and therefore, evolved toxins may provide general improvements for a variety of different vaccines.
- the heat-labile enterotoxins (LT) from enterotoxigenic E. coli strains are structurally related to cholera toxin and are 75% identical at the DNA sequence level.
- the genes that encode the related toxins are subjected to DNA shuffling.
- the recombinant toxins are then tested for one or more of a several desirable traits. For example, one can screen for improved cross-reactivity of antibodies raised against the recombinant toxin polypeptides, for lack of toxicity in a cell culture assay, and for ability to induce a protective immune response against the pathogens and/or against the toxins themselves.
- the shuffled clones can be selected by phage display and/or screened by phage ELISA and ELISA assays for the presence of epitopes from the different serotypes.
- Variant proteins with multiple epitopes can then be purified and used to immunize mice or other test animal.
- the animal serum is then assayed for antibodies to the different B chain subtypes and variants that elicit a broad cross-reactive response will be evaluated further in a virulent challenge model.
- the E. coli and V. cholerae toxins can also act as adjuvants that are capable of enhancing mucosal immunity and oral delivery of vaccines and proteins. Accordingly, one can test the library of recombinant toxins for enhancement of the adjuvant activity.
- Shuffled antigens can also be screened for improved expression levels and stability of the B chain pentamer, which may be less stable than when in the presence of the A chain in the hexameric complex. Addition of a heat treatment step or denaturing agents such as salts, urea, and/or guanidine hydrochloride can be included prior to ELISA assays to measure yields of correctly folded molecules by appropriate antibodies. It is sometimes desirable to screen for stable monomeric B chain molecules, in an ELISA format, for example, using antibodies that bind monomeric, but not pentameric B chains. Additionally, the ability of shuffled antigens to elicit neutralizing antibodies in an appropriate animal model such as mouse or monkey can be screened.
- Addition of a heat treatment step or denaturing agents such as salts, urea, and/or guanidine hydrochloride can be included prior to ELISA assays to measure yields of correctly folded molecules by appropriate antibodies. It is sometimes desirable to screen for stable monomeric B chain molecules, in an
- antibodies that bind to the B chain and prevent its binding to its specific ganglioside receptors on the surface of intestinal epithelial cells may prevent disease.
- antibodies that bind to the B chain and prevent its pentamerization or block A chain binding may be useful in preventing disease.
- the bacterial antigens that can be improved by DNA shuffling for use as vaccines also include, but are not limited to, Helicobacter pylori antigens CagA and VacA (Blaser (1996) Aliment. Pharmacol. Ther. 1 : 73-7; Blaser and Crabtree (1996) Am. J. Clin. Pathol. 106: 565-7; Censini et al. (1996) Proc. Nat 'l Acad. Sci.
- H. pylori antigens include, for example, four immunoreactive proteins of 45- 65 kDa as reported by Chatha et al. (1997) Indian J. Med. Res. 105 : 170- 175 and the H. pylori GroES homologue ( ⁇ spA) (Kansau et al. (1996) Mol Microbiol. 22: 1013-1023.
- suitable bacterial antigens include, but are not limited to, the 43-kDa and the fimbrilin (41 kDa) proteins of P. gingivalis (Boutsl et al. (1996) Oral Microbiol. Immunol.
- GC Neisseria gonorrhoeae
- Opa Escherichia coli phase-variable opacity
- Rhodococcus sp. NI86/21 (De Mot et al. (1996) Curr. Microbiol. 33: 26-30); the staphylococcal enterotoxins (SEs) (Wood et al. (1997) FEMS Immunol Med. Microbiol. 17: 1-10), a 42-kDa M. hyopneumoniae NrdF ribonucleotide reductase R2 protein or 15-kDa subunit protein of M. hyopneumoniae (Fagan et al. (1997) Infect. Immun. 65: 2502-2507), the meningococcal antigen PorA protein (Feavers et al. (1997) Clin. Diagn. Lab.
- nocardial antigens of molecular mass approximately 60, 40, 20 and 15-10 kDa Prokesova et al. (1996) Int. J. Immunopharmacol. 18: 661-668
- Staphylococcus aureus antigen ORF-2 Rieneck et al. (1997) Biochim Biophys Ada 1350: 128-132
- GlpQ antigen of Borrelia hermsii Med et al. (1996) J. Clin. Microbiol. 34: 2483-2492
- cholera protective antigen (CPA) (Sciortino (1996; J. Diarrhoeal Dis. Res.
- the methods of the invention are also useful for obtaining recombinant nucleic acids and polypeptides that have enhanced ability to induce an immune response against viral pathogens. While the bacterial recombinants described above are typically administered in polypeptide form, recombinants that confer viral protection are preferably administered in nucleic acid form, as genetic vaccines.
- the methods of the invention solve this problem by performing DNA shuffling on nucleic acids that encode the glycoproteins and identifying those recombinants that exhibit enhanced expression in a host cell, and/or for improved immunogenicity when administered as a genetic vaccine.
- a convenient screening method for these methods is to express the recombinant polynucleotides as fusion proteins to PIG, which results in display of the polypeptides on the surface of the host cell (Whitehorn et al.
- the flaviviruses are another example of a viral pathogen for which the methods of the invention are useful for obtaining a recombinant polypeptide or genetic vaccine that is effective against a viral pathogen.
- the flaviviruses consist of three clusters of antigenically related viruses: Dengue 1-4 (62-77% identity), Japanese, St. Louis and Murray Valley encephalitis viruses (75-82% identity), and the tick-borne encephalitis viruses (77- 96% identity). Dengue virus can induce protective antibodies against SLE and Yellow fever (40-50% identity), but few efficient vaccines are available.
- the polynucleotides that encode envelope proteins of related viruses are subjected to DNA shuffling.
- the resulting recombinant polynucleotides can be tested, either as genetic vaccines or by using the expressed polypeptides, for ability to induce a broadly reacting neutralizing antibody response. Finally, those clones that are favorable in the preliminary screens can be tested for ability to protect a test animal against viral challenge.
- Viral antigens that can be evolved by DNA shuffling for improved activity as vaccines include, but are not limited to, influenza A virus N2 neuraminidase (Kilbourne et al. (1995) Vaccine 13: 1799-1803); Dengue virus envelope (E) and premembrane (prM) antigens (Feighny et al. (1994) Am. J. Trop. Med. Hyg. 50: 322-328; Putnak et al. (1996) Am. J. Trop. Med. Hyg. 55: 504-10); HIV antigens Gag, Pol, Vif and Nef (Vogt et al.
- HIV antigens gpl20 and gpl60 (Achour et al. (1995) Cell. Mol. Biol. 41: 395-400; Hone et al. (1994) £>ev. Biol. Stand. 82: 159-162); gp41 epitope of human immunodeficiency virus (Eckhart et al. (1996) J. Gen. Virol. 77: 2001-2008); rotavirus antigen VP4 (Mattion et al. (1995) J. Virol. 69: 5132-5137); the rotavirus protein VP7 or VP7sc (Emslie et al. (1995) J. Virol.
- HSV herpes simplex virus glycoproteins gB, gC, gD, gE, gG, gH, and gl
- HSV herpes simplex virus glycoproteins
- immediate-early protein ICP47 of herpes simplex virus-type 1 HSV-1 (Banks et al. (1994) Virology 200: 236-245); immediate-early (IE) proteins ICP27, ICP0, and ICP4 of herpes simplex virus (Manickan et al. (1995) J. Virol. 69: 4711-4716); influenza virus nucleoprotein and hemagglutinin (Deck et al. (1997) Vaccine 15: 71-78; Fu et al. (1997) J. Virol. 71: 2715-2721); B19 parvovirus capsid proteins VPl (Kawase et al.
- Virology 111: 359-366) or VP2 (Brown et al. (1994) Virology 198: 477-488); Hepatitis B virus core and e antigen (Schodel et al. (1996) Intervirology 39: 104-106); hepatitis B surface antigen (Shiau and Murray (1997) J. Med. Virol. 51 : 159-166); hepatitis B surface antigen fused to the core antigen of the virus (Id.); Hepatitis B virus core-preS2 particles (Nemeckova et al. (1996) Ada Virol. 40: 273-279); HBV preS2-S protein (Kutinova et al.
- HCV hepatitis C virus
- HBV hepatitis B virus
- Parasites Antigens from parasites can also be optimized by the methods of the invention. These include, but are not limited to, the schistosome gut-associated antigens CAA (circulating anodic antigen) and CCA (circulating cathodic antigen) in Schistosoma mansoni, S. haematobium or S. japonicum (Deelder et al. (1996) Parasitology 112: 21-35); a multiple antigen peptide (MAP) composed of two distinct protective antigens derived from the parasite Schistosoma mansoni (Ferru et al. (1997) Parasite Immunol. 19: 1-11);
- CAA circulating anodic antigen
- CCA circulating cathodic antigen
- MAP multiple antigen peptide
- Leishmania parasite surface molecules Lezama-Davila (1997) Arch. Med. Res. 28: 47-53); third-stage larval (L3) antigens of L. loa (Akue et al. (1997) J Infect. Dis. 175: 158-63); the genes, Tamsl-1 and Tarns 1-2, encoding the 30-and 32-kDa major merozoite surface antigens of Theileria annulata (Ta) (d'Oliveira et al. (1996) Gene 172: 33-39); Plasmodium falciparum merozoite surface antigen 1 or 2 (al-Yaman et al. (1995) Trans. R. Soc. Trop. Med. Hyg. 89: 555-559; Beck et al. (1997) J. Infect. Dis. 175: 921-926; Rzepczyk et al.
- NYVAC-Pf7 encoded Plasmodium falciparum antigens derived from the sporozoite (circumsporozoite protein and sporozoite surface protein 2), liver (liver stage antigen 1), blood (merozoite surface protein 1, serine repeat antigen, and apical membrane antigen 1), and sexual (25-kDa sexual-stage antigen) stages of the parasite life cycle were inserted into a single NYVAC genome to generate NYVAC-Pf7 (Tine et al. (1996) Infect. Immun. 64: 3833-3844); Plasmodium falciparum antigen Pfs230 (Williamson et al. (1996) Mol. Biochem.
- Plasmodium falciparum apical membrane antigen AMA- 1 (Lai et al. (1996) Infect. Immun. 64: 1054-1059); Plasmodium falciparum proteins Pfs28 and Pfs25 (Duffy and Kaslow (1997) Infect. Immun. 65: 1109-1113); Plasmodium falciparum merozoite surface protein, MSP1 (Hui et al. (1996) Infect. Immun. 64: 1502- 1509); the malaria antigen P032 (Ahlborg et al. (1996) Immunology 88: 630-635); Plasmodium falciparum erythrocyte membrane protein 1 (Baruch et al.
- the invention also provides methods of obtaining reagents that are useful for treating allergy.
- the methods involve making a library of recombinant polynucleotides that encode an allergen, and screening the library to identify those recombinant polynucleotides that exhibit improved properties when used as immunotherapeutic reagents for treating allergy.
- specific immunotherapy of allergy using natural antigens carries a risk of inducing anaphylaxis, which can be initiated by cross-linking of high-affinity IgE receptors on mast cells. Therefore, allergens that are not recognized by pre-existing IgE are desirable.
- the methods of the invention provide methods by which one can obtain such allergen variants. Another improved property of interest is induction of broader immune responses, increased safety and efficacy.
- Synthesis of polyclonal and allergen-specific IgE requires multiple interactions between B cells, T cells and professional antigen-presenting cells (APC). Activation of naive, unprimed B cells is initiated when specific B cells recognize the allergen by cell surface immunoglobulin (slg). However, costimulatory molecules expressed by activated T cells in both soluble and membrane-bound forms are necessary for differentiation of B cells into IgE-secreting plasma cells. Activation of T helper cells requires recognition of an antigenic peptide in the context of MHC class II molecules on the plasma membrane of APC, such as monocytes, dendritic cells, Langerhans cells or primed B cells.
- APC professional antigen-presenting cells
- TCR T cell receptor
- activated B cells express CD80 (B7-1) and CD86 (B7-2, B70), which are the counter receptors for CD28 and which provide a costimulatory signal for T cell activation resulting in T cell proliferation and cytokine synthesis.
- allergen-specific T cells from atopic individuals generally belong to the T H 2 cell subset, activation of these cells also leads to production of IL-4 and IL-13, which, together with membrane-bound costimulatory molecules expressed by activated T helper cells, direct B cell differentiation into IgE- secreting plasma cells.
- Mast cells and eosinophils are key cells in inducing allergic symptoms in target organs. Recognition of specific antigen by IgE bound to high-affinity IgE receptors on mast cells, basophils or eosinophils results in crosslinking of the receptors leading to degranulation of the cells and rapid release of mediator molecules, such as histamine, prostaglandins and leukotrienes, causing allergic symptoms.
- Immunotherapy of allergic diseases currently includes hyposensibilization treatments using increasing doses of allergen injected to the patient. These treatments result skewing of immune responses towards T H I phenotype and increase the ratio of IgG/TgE antibodies specific for allergens.
- these treatments include significant risk of anaphylactic reactions.
- free circulating allergen is recognized by IgE molecules bound to high- affinity IgE receptors on mast cells and eosinophils.
- Recognition of the allergen results in crosslinking of the receptors leading to release of mediators, such as histamine, prostaglandins, and leukotrienes, which cause the allergic symptoms, and occasionally anaphylactic reactions.
- mediators such as histamine, prostaglandins, and leukotrienes
- the methods of the invention provide a means to obtain allergens that, when used in genetic vaccines, provide a means of circumventing the problems that have limited the usefulness of previously known hyposensibilization treatments. For example, by expressing antigens on the surface of cells, such as muscle cells, the risk of anaphylactic reactions is significantly reduced. This can be conveniently achieved by using genetic vaccine vectors that encode transmembrane forms of allergens.
- the allergens can also be modified in such a way that they are efficiently expressed in transmembrane forms, further reducing the risk of anaphylactic reactions.
- the genetic vaccines can include cytokines and accessory molecules which further direct the immune responses towards the T H I phenotype, thus reducing the amount of IgE antibodies produced and increasing the efficacy of the treatments.
- cytokines and accessory molecules which further direct the immune responses towards the T H I phenotype, thus reducing the amount of IgE antibodies produced and increasing the efficacy of the treatments.
- polynucleotides encoding known allergens, or homologs or fragments thereof are inserted into DNA vaccine vectors and used to immunize allergic and asthmatic individuals.
- the shuffled allergens are expressed in manufacturing cells, such as E. coli or yeast cells, and subsequently purified and used to treat the patients or prevent allergic disease.
- DNA shuffling or other recombination method can be used to obtain allergens that activate T cells but cannot induce anaphylactic reactions.
- a library of recombinant polynucleotides that encode allergen variants can be expressed in cells, such as antigen presenting cells, which are than contacted with PBMC or T cell clones from atopic patients.
- Those library members that efficiently activate T H cells from the atopic patients can be identified by assaying for T cell proliferation, or by cytokine synthesis (e.g., synthesis of IL-2, IL-4, EFN- ⁇ .
- cytokine synthesis e.g., synthesis of IL-2, IL-4, EFN- ⁇ .
- Those recombinant allergen variants that are positive in the in vitro tests can then be subjected to in vivo testing.
- allergies examples include, but are not limited to, allergies against house dust mite, grass pollen, birch pollen, ragweed pollen, hazel pollen, cockroach, rice, olive tree pollen, fungi, mustard, bee venom.
- Antigens of interest include those of animals, including the mite (e.g., Dermatophagoides pteronyssinus, Dermatophagoides farinae, Blomia tropicalis), such as the allergens der pi (Scobie et al. (1994) Biochem. Soc. Trans. 22: 448S; Yssel et al. (1992) J. Immunol. 148: 738-745), der p2 (Chua et al.
- bovine dermal/dander antigens BDA 1 1 (Rautiainen et al. (1995) J. Invest. Dermatol 105: 660-663) and BDA20 (Mantyjarvi et al. (1996) J. Allergy Clin. Immunol. 97: 1297-1303); the major horse allergen Equ cl (Gregoire et al. (1996) J. Biol. Chem. 271 : 32951-32959); Jumper ant pilosula allergen Myr p I and its homologous allergenic polypeptides Myr p2 (Donovan et al. ( 1996) Biochem. Mol. Biol. Int.
- Pollen and grass allergens are also useful in vaccines, particularly after optimization of the antigen by the methods of the invention.
- allergens include, for example, Hor v9 (Astwood and Hill (1996) Gene 182: 53-62, Lig vl (Batanero et al. (1996) Clin. Exp. Allergy 26: 1401-1410); Lol p 1 (Muller et al. (1996) Int. Arch. Allergy Immunol. 109: 352-355), Lol p II (Tamborini et al. (1995) Mol. Immunol. 32: 505-513), Lol pVA, Lol pVB (Ong et al. (1995) Mol Immunol.
- Vaccines against food allergens can also be developed using the methods of the invention.
- Suitable antigens for shuffling include, for example, profilin (Rihs et al.
- apple allergens such as the major allergen Mai d 1 (Vanek-Krebitz et al. (1995) Biochem. Biophys. Res. Commun. 214: 538-551); and peanut allergens, such as Ara h I (Burks et al. (1995) J. Clin. Invest. 96: 1715-1721).
- the methods of the invention can also be used to develop recombinant antigens that are effective against allergies to fungi.
- Fungal allergens useful in these vaccines include, but are not limited to, the allergen, Cla h III, of Cladosporium herbarum (Zhang et al. (1995) J. Immunol. 154: 710-717); the allergen Psi c 2, a fungal cyclophilin, from the basidiomycete Psilocybe cubensis (Horner et al. (1995) Int. Arch. Allergy Immunol.
- the invention also provides a solution to another shortcoming of vaccination as a treatment for allergy and asthma. While genetic vaccination primarily induces CD8 + T cell responses, induction of allergen-specific IgE responses is dependent on CD4 ⁇ T cells and their help to B cells. T H 2-type cells are particularly efficient in inducing IgE synthesis because they secrete high levels of IL-4, IL-5 and IL-13, which direct Ig isotype switching to IgE synthesis. IL-5 also induces eosinophilia.
- the methods of the invention can be used to develop recombinant antigens that efficiently induce CD4 + T cell responses, and direct differentiation of these cells towards the T H I phenotype.
- Autoimmune diseases are characterized by immune response that attacks tissues or cells of ones own body, or pathogen-specific immune responses that also are harmful for ones own tissues or cells, or non-specific immune activation which is harmful for ones own tissues or cells.
- autoimmune diseases include, but are not limited to, rheumatoid arthritis, SLE, diabetes mellitus, myasthenia gravis, reactive arthritis, ankylosing spondylitis, and multiple sclerosis.
- IBD psoriasis, pancreatitis, and various immunodeficiencies, can be treated using antigens that are optimized using the methods of the invention.
- autoimmune diseases including diabetes and rheumatoid arthritis, are linked to certain MHC haplotypes.
- Other autoimmune-type disorders such as reactive arthritis, have been shown to be triggered by bacteria such as Yersinia and Shigella, and evidence suggests that several other autoimmune diseases, such as diabetes, multiple sclerosis, rheumatoid arthritis, may also be initiated by viral or bacterial infections in genetically susceptible individuals.
- the invention provides methods of obtaining antigens having greater tolerogenicity and/or have improved antigenicity.
- the antigens prepared according to the invention exhibit improved induction of tolerance by oral delivery. Oral tolerance is characterized by induction of immunological tolerance after oral administration of large quantities of antigen.
- Expression library immunization (Barry et al. (1995) Nature 377: 632) is a particularly useful method of screening for optimal antigens for use in genetic vaccines. For example, to identify autoantigens present in Yersinia, Shigella, and the like, one can screen for induction of T cell responses in HLA-B27 positive individuals.
- Complexes that include epitopes of bacterial antigens and MHC molecules associated with autoimmune diseases, e.g., HLA-B27 in association with Yersinia antigens can be used in the prevention of reactive arthritis and ankylosing spondylitis in HLA-B27 positive individuals.
- Suitable models for various conditions include collagen induced arthritis, the NFS/sld mouse model of human Sjogren's syndrome; a 120 kD organ-specific autoantigen recently identified as an analog of human cytoskeletal protein ( ⁇ -fodrin (Haneji et al. (1997) Science 276: 604), the New Zealand Black/White FI hybrid mouse model of human SLE, NOD mice, a mouse model of human diabetes mellitus, fas/fas ligand mutant mice, which spontaneously develop autoimmune and lymphoproliferative disorders (Watanabe-Fukunaga et al. (1992) Nature 356: 314), and experimental autoimmune encephalomyelitis (EAE), in which myelin basic protein induces a disease that resembles human multiple sclerosis.
- EAE experimental autoimmune encephalomyelitis
- Autoantigens that can be shuffled according to the methods of the invention and used in vaccines for treating multiple sclerosis include, but are not limited to, myelin basic protein (Stinissen et al. (1996) J. Neurosci. Res. 45: 500-51 1) or a fusion protein of myelin basic protein and proteolipid protein (Elliott et al. (1996) J. Clin. Invest. 98: 1602- 1612), proteolipid protein (PLP) (Rosener et al. (1997) J. Neuroimmunol. 75: 28-34), 2',3'- cyclic nucleotide 3'-phosphodiesterase (CNPase) (Rosener et al.
- Target antigens that, after shuffling according to the methods of the invention, can be used to treat scleroderma, systemic sclerosis, and systemic lupus erythematosus include, for example, (-2-GPI, 50 kDa glycoprotein (Blank et al. (1994) J. Autoimmun. 7: 441-455), Ku (p70/p80) autoantigen, or its 80-kd subunit protein (Hong et al. (1994) Invest. Ophthalmol Vis. Sci. 35: 4023-4030; Wang et al. (1994) J. Cell Sci. 107: 3223-3233), the nuclear autoantigens La (SS-B) and Ro (SS-A) (Huang et al.
- Hepatic autoimmune disorders can also be treated using improved recombinant antigens that are prepared according to the methods described herein.
- antigens that are useful in such treatments are the cytochromes P450 and UDP- glucuronosyl-transferases (Obermayer-Straub and Manns (1996) Baillieres Clin. Gastroenterol. 10: 501-532), the cytochromes P450 2C9 and P450 1A2 (Bourdi et al. (1996) Chem. Res. Toxicol. 9: 1159-1166; Clemente et al. (1997) J. Clin. Endocrinol Metab. 82: 1353-1361), LC-1 antigen (Klein et al. (1996) J.
- useful antigens include, but are not limited to, the 450 kD human epidermal autoantigen (Fujiwara et al. (1996) J. Invest. Dermatol. 106: 1125-1130), the 230 kD and 180 kD bullous pemphigoid antigens (Hashimoto (1995) Keio J. Med. 44: 115-123; Murakami et al. (1996) J. Dermatol Sci.
- pemphigus foliaceus antigen (desmoglein 1), pemphigus vulgaris antigen (desmoglein 3), BPAg2, BPAgl, and type VII collagen
- pemphigus foliaceus antigen (desmoglein 1)
- pemphigus vulgaris antigen (desmoglein 3)
- BPAg2, BPAgl and type VII collagen
- a 168-kDa mucosal antigen in a subset of patients with cicatricial pemphigoid (Ghohestani et al. (1996) J. Invest. Dermatol. 107: 136-139)
- 218-kd nuclear protein (218-kd Mi-2)
- the methods of the invention are also useful for obtaining improved antigens for treating insulin dependent diabetes mellitus, using one or more of antigens which include, but are not limited to, insulin, proinsulin, GAD65 and GAD67, heat-shock protein 65 (hsp65), and islet-cell antigen 69 (ICA69) (French et al. (1997) Diabetes 46: 34-39; Roep (1996) Diabetes 45: 1 147-1 156; Schloot et al.
- antigens include, but are not limited to, insulin, proinsulin, GAD65 and GAD67, heat-shock protein 65 (hsp65), and islet-cell antigen 69 (ICA69)
- Diabetologia 40: 332-338 viral proteins homologous to GAD65 (Jones and Crosby (1996) Diabetologia 39: 1318-1324), islet cell antigen-related protein-tyrosine phosphatase (PTP) (Cui et al. (1996) J. Biol. Chem. 271: 24817-24823), GM2-1 ganglioside (Cavallo et al. (1996) J Endocrinol. 150: 113-120; Dotta et al. (1996) Diabetes 45: 1193-1196), glutamic acid decarboxylase (GAD) (Nepom (1995) Curr. Opin. Immunol.
- PTP protein-tyrosine phosphatase
- GAD glutamic acid decarboxylase
- an islet-cell protein tyrosine phosphatase and the 37-kDa autoantigen derived from it in type 1 diabetes including IA-2, IA-2) (La Gasse et al. (1997) Mol. Med. 3: 163-173), the 64 kDa protein from In-111 cells or human thyroid follicular cells that is immunoprecipitated with sera from patients with islet cell surface antibodies (ICSA) (Igawa et al. (1996) Endocr. J. 43: 299-306), phogrin, a homologue of the human transmembrane protein tyrosine phosphatase, an autoantigen of type 1 diabetes (Kawasaki et al. (1996) Biochem.
- ICSA islet cell surface antibodies
- carboxypeptidase H the human homologue of gp330, which is a renal epithelial glycoprotein involved in inducing Heymann nephritis in rats, and the 38-kD islet mitochondrial autoantigen (Arden et al. (1996) J. Clin. Invest. 97: 551-561.
- Rheumatoid arthritis is another condition that is treatable using optimized antigens prepared according to the present invention.
- Useful antigens for rheumatoid arthritis treatment include, but are not limited to, the 45 kDa DEK nuclear antigen, in particular onset juvenile rheumatoid arthritis and iridocyclitis (Murray et al. (1997) J. Rheumatol. 24: 560- 567), human cartilage glycoprotein-39, an autoantigen in rheumatoid arthritis (Verheijden et al. (1997) Arthritis Rheum. 40: 1115-1125), a 68k autoantigen in rheumatoid arthritis (Blass et al. (1997) Ann.
- autoimmune thyroid disorders include, for example, thyroid peroxidase and the thyroid stimulating hormone receptor (Tandon and Weetman (1994) J R. Coll Physicians Lond. 28: 10-18), thyroid peroxidase from human Graves' thyroid tissue (Gardas et al. (1997) Biochem. Biophys. Res. Commun. 234: 366-370; Zim er et al. (1997) Histochem. Cell. Biol.
- Other conditions and associated antigens include, but are not limited to, Sjogren's syndrome (-fodrin; Haneji et al. (1997) Science 276: 604-607), myastenia gravis (the human M2 acetylcholine receptor or fragments thereof, specifically the second extracellular loop of the human M2 acetylcholine receptor; Fu et al. (1996) Clin. Immunol. Immunopathol. 78: 203-207), vitiligo (tyrosinase; Fishman et al.
- Immunotherapy has great promise for the treatment of cancer and prevention of metastasis.
- the body's immune system can be enlisted to reduce or eliminate cancer.
- Improved antigens obtained using the methods of the invention provide cancer immunotherapies of increased effectiveness compared to those that are presently available.
- One approach to cancer immunotherapy is vaccination using vaccines that include or encode antigens that are specific for tumor cells or by injecting the patients with purified recombinant cancer antigens.
- the methods of the invention can be used for obtaining antigens that exhibit an enhancement of immune responses against known tumor- specific antigens, and also to search for novel protective antigenic sequences.
- Antigens having optimized expression, processing, and presentation can be obtained as described herein.
- the approach used for each particular cancer can vary.
- methods of the invention can be used to obtain optimized hormone antagonists.
- highly immunogenic tumors including melanoma, one can screen for recombinant antigens that optimally boost the immune response against the tumor.
- tumor-specific antigens that can be used in the antigen shuffling methods of the invention are: bullous pemphigoid antigen 2, prostate mucin antigen (PMA) (Beckett and Wright (1995) Int. J. Cancer 62: 703-710), tumor associated Thomsen- Friedenreich antigen (Dahlenborg et al. (1997) Int. J. Cancer 70: 63-71), prostate-specific antigen (PSA) (Dannull and Belldegrun (1997) Br. J. Urol.
- PMA prostate mucin antigen
- PSA prostate-specific antigen
- luminal epithelial antigen (LEA.135) of breast carcinoma and bladder transitional cell carcinoma (TCC) (Jones et al. (1997) Anticancer Res. 17: 685-687), cancer-associated serum antigen (CASA) and cancer antigen 125 (CA 125) (Kierkegaard et al. (1995) Gynecol Oncol. 59: 251-254), the epithelial glycoprotein 40 (EGP40) (Kievit et al. (1997) Int. J. Cancer 71 : 237-245), squamous cell carcinoma antigen (SCC) (Lozza et al. (1997) Anticancer Res.
- ECP40 epithelial glycoprotein 40
- SCC squamous cell carcinoma antigen
- Genetic vaccines that contain optimized antigens obtained by the methods of the invention are also useful for contraception.
- genetic vaccines can be obtained that encode sperm cell specific antigens, and thus induce anti-sperm immune responses.
- Vaccination can be achieved by, for example, administration of recombinant bacterial strains, e.g. Salmonella and the like, which express sperm antigen, as well as by induction of neutralizing anti-hCG antibodies by vaccination by DNA vaccines encoding human chorionic gonadotropin (hCG), or a fragment thereof.
- hCG human chorionic gonadotropin
- sperm antigens which can be used in the genetic vaccines include, for example, lactate dehydrogenase (LDH-C4), galactosyltransferase (GT), SP-10, rabbit sperm autoantigen (RSA), guinea pig (g)PH-20, cleavage signal protein (CS-1), HSA-63, human (h)PH-20, and AgX-1 (Zhu and Naz (1994) Arch. Androl 33: 141-144), the synthetic sperm peptide, P10G (O'Rand et al. (1993) J. Reprod. Immunol.
- LDH-C4 lactate dehydrogenase
- GT galactosyltransferase
- SP-10 rabbit sperm autoantigen
- RSA rabbit sperm autoantigen
- CS-1 cleavage signal protein
- HSA-63 human
- human (h)PH-20 human
- AgX-1 AgX-1
- the methods of the invention can also be used to obtain genetic vaccines that are expressed specifically in testis.
- polynucleotide sequences that direct expression of genes that are specific to testis can be used (e.g., fertilization antigen- 1 and the like).
- antigens expressed on oocytes or hormones regulating reproduction may be useful targets of contraceptive vaccines.
- genetic vaccines can be used to generate antibodies against gonadotropin releasing hormone (GnRH) or zona pellucida proteins (Miller et al. (1997) Vaccine 15:1858-1862). Vaccinations using these molecules have been shown to be efficacious in animal models (Miller et al. (1997) Vaccine 15:1858-1862).
- Another example of a useful component of a genetic contraceptive vaccine is the ovarian zona pellucida glycoprotein ZP3 (Tung et al. (1994) Reprod. Fertil Dev. 6:349-355).
- the library is subjected to selection and/or screening to identify those library members that encode antigenic peptides that have improved ability to induce an immune response to the pathogenic agent.
- Selection and screening of recombinant polynucleotides that encode polypeptides having an improved ability to induce an immune response can involve either in vivo and in vitro methods, but most often involves a combination of these methods.
- the members of a library of recombinant nucleic acids are picked, either individually or as pools.
- the clones can be subjected to analysis directly, or can be expressed to produce the corresponding polypeptides.
- an in vitro screen is performed to identify the best candidate sequences for the in vivo studies.
- the library can be subjected to in vivo challenge studies directly.
- the analyses can employ either the nucleic acids themselves (e.g., as genetic vaccines), or the polypeptides encoded by the nucleic acids.
- a schematic diagram of a typical strategy is shown in Figure 5. Both in vitro and in vivo methods are described in more detail below.
- recombinant segments are sometimes introduced into cells before the screening step.
- Recombinant segments can also be linked to an appropriate vector or other regulatory sequences before screening.
- products of recombination generated in vitro are sometimes packaged in viruses (e.g., bacteriophage) before screening.
- viruses e.g., bacteriophage
- recombination products can sometimes be screened in the cells in which recombination occurred.
- recombinant segments are extracted from the cells, and optionally packaged as viruses, before screening.
- Recursive sequence recombination can also be employed to achieve still further improvements in a desired property, or to bring about new (or "distinct") properties.
- Recursive sequence recombination entails successive cycles of recombination to generate molecular diversity. That is, one creates a family of nucleic acid molecules showing some sequence identity to each other but differing in the presence of mutations. In any given cycle, recombination can occur in vivo or in vitro, intracellularly or extracellularly.
- polynucleotides that encode optimized recombinant antigens are subjected to molecular backcrossing, which provides a means to breed the shuffled chimeras/mutants back to a parental or wild-type sequence, while retaining the mutations that are critical to the phenotype that provides the optimized immune responses.
- molecular backcrossing can also be used to characterize which of the many mutations in an improved variant contribute most to the improved phenotype.
- Screening/ selection can then be performed, for example, for recombinant segments that have increased ability to induce an immune response to a pathogenic agent without the need to attribute such improvement to any of the individual component sequences of the recombinant polynucleotide.
- initial round(s) of screening can sometimes be performed using bacterial cells due to high transfection efficiencies and ease of culture.
- test animals are used for library expression and screening.
- other types of screening which are not amenable to screening in bacterial or simple eukaryotic library cells, are performed in cells selected for use in an environment close to that of their intended use.
- Final rounds of screening can be performed in cells or organisms that are as close as possible to the precise cell type or organism of intended use. If further improvement in a property is desired, at least one, and usually a collection, of recombinant segments surviving a first round of screening/selection are subject to a further round of recombination.
- recombinant segments can be recombined with each other or with exogenous segments representing the original substrates or further variants thereof. Again, recombination can proceed in vitro or in vivo. If the previous screening step identifies desired recombinant segments as components of cells, the components can be subjected to further recombination in vivo, or can be subjected to further recombination in vitro, or can be isolated before performing a round of in vitro recombination. Conversely, if the previous screening step identifies desired recombinant segments in naked form or as components of viruses, these segments can be introduced into cells to perform a round of tn vivo recombination. The second round of recombination, irrespective how performed, generates further recombinant segments which encompass additional diversity than is present in recombinant segments resulting from previous rounds.
- the second round of recombination can be followed by a further round of screening/selection according to the principles discussed above for the first round.
- the stringency of screening/selection can be increased between rounds.
- the nature of the screen and the property being screened for can vary between rounds if improvement in more than one property is desired or if acquiring more than one new property is desired. Additional rounds of recombination and screening can then be performed until the recombinant segments have sufficiently evolved to acquire the desired new or improved property or function.
- the practice of this invention involves the construction of recombinant nucleic acids and the expression of genes in transfected host cells. Molecular cloning techniques to achieve these ends are known in the art.
- RNA polymerase mediated techniques e.g., NASBA
- PCR polymerase chain reaction
- LCR ligase chain reaction
- NASBA RNA polymerase mediated techniques
- Oligonucleotides for use as probes e.g., in in vitro amplification methods, for use as gene probes, or as shuffling targets (e.g., synthetic genes or gene segments) are typically synthesized chemically according to the solid phase phosphoramidite triester method described by Beaucage and Caruthers (1981) Tetrahedron Letts., 22(20): 1859-1862, e.g., using an automated synthesizer, as described in Needham-VanDevanter et al. (1984) Nucleic Acids Res., 12:6159-6168. Oligonucleotides can also be custom made and ordered from a variety of commercial sources known to persons of skill.
- nucleic acid with a known sequence can be custom ordered from any of a variety of commercial sources, such as The Midland Certified Reagent Company (mcrc@oligos.com), The Great American Gene Company (http://www.genco.com), ExpressGen Inc. (www.expressgen.com), Operon Technoloigies Inc. (Alameda, CA) and many others.
- peptides and antibodies can be custom ordered from any of a variety of sources, such as PeptidoGenic (pkim@ccnet.com), HTI Bio-products, Inc. (http://www.htibio.com), BMA Biomedicals Ltd (U.K.), Bio Synthesis, Inc., and many others.
- the resulting library of recombinant polynucleotides can be subjected to purification and preliminary analysis in vitro, in order to identify the most promising candidate recombinant nucleic acids.
- the assays can be practiced in a high-throughput format. For example, to purify individual shuffled recombinant antigens, clones can robotically picked into 96-well formats, grown, and, if desired, frozen for storage.
- the shuffled antigen-encoding polynucleotides are assayed as genetic vaccines.
- Genetic vaccine vectors containing the shuffled antigen sequences can be prepared using robotic colony picking and subsequent robotic plasmid purification. Robotic plasmid purification protocols are available that allow purification of 600-800 plasmids per day. The quantity and purity of the DNA can also be analyzed in 96- well plates, for example. In a presently preferred embodiment, the amount of DNA in each sample is robotically normalized, which can significantly reduce the variation between different batches of vectors.
- the proteins and/or nucleic acids can be subjected to any of a number of tn vitro analysis methods.
- screenings include, for example, phage display, flow cytometry, and ELISA assays to identify antigens that are efficiently expressed and have multiple epitopes and a proper folding pattern.
- the libraries may also be screened for reduced toxicity in mammalian cells.
- monoclonal antibodies for screening.
- a humoral immune response generally targets multiple regions of antigenic proteins. Accordingly, monoclonal antibodies can be raised against various regions of immunogenic proteins (Alving et al.
- Phage display provides a powerful method for selecting proteins of interest from large libraries (Bass et al. (1990) Proteins: Struct. Funct. Genet. 8: 309; Lowman and Wells (1991) Methods: A Companion to Methods Enz. 3(3);205-216. Lowman and Wells (1993) J. Mol. Biol. 234;564-578).
- Some recent reviews on the phage display technique include, for example, McGregor (1996) Mol Biotechnol. 6(2):155-62; Dunn (1996) Curr. Opin. Biotechnol. 7(5):547-53; Hill et al. (1996) Mol Microbiol 20(4):685-92; Phage Display of Peptides and Proteins: A Laboratory Manual. BK.
- Each phage particle displays a unique variant protein on its surface and packages the gene encoding that particular variant.
- the shuffled genes for the antigens are fused to a protein that is expressed on the phage surface, e.g., gene III of phage Ml 3, and cloned into phagemid vectors.
- a suppressible stop codon separates the genes so that in a suppressing strain of E. coli, the antigen-glllp fusion is produced and becomes incorporated into phage particles upon infection with Ml 3 helper phage.
- the same vector can direct production of the unfused antigen alone in a nonsuppressing E. coli for protein purification.
- the genetic packages most frequently used for display libraries are bacteriophage, particularly filamentous phage, and especially phage M13, Fd and FI. Most work has involved inserting libraries encoding polypeptides to be displayed into either gill or gVIII of these phage forming a fusion protein. See, e.g., Dower, WO 91/19818; Devlin, WO 91/18989; MacCafferty, WO 92/01047 (gene III); Huse, WO 92/06204; Kang, WO 92/18619 (gene VIII).
- Such a fusion protein comprises a signal sequence, usually but not necessarily, from the phage coat protein, a polypeptide to be displayed and either the gene III or gene VIII protein or a fragment thereof.
- Exogenous coding sequences are often inserted at or near the N-terminus of gene III or gene VIII although other insertion sites are possible.
- Eukaryotic viruses can be used to display polypeptides in an analogous manner. For example, display of human heregulin fused to gp70 of Moloney murine leukemia virus has been reported by Han et al., Proc. Natl. Acad. Sci. USA 92: 9747-9751 (1995). Spores can also be used as replicable genetic packages. In this case, polypeptides are displayed from the outer surface of the spore. For example, spores from B. subtilis have been reported to be suitable. Sequences of coat proteins of these spores are provided by
- Cells can also be used as replicable genetic packages.
- Polypeptides to be displayed are inserted into a gene encoding a cell protein that is expressed on the cells surface.
- Bacterial cells including Salmonella typhimurium, Bacillus subtilis, Pseudomonas aeruginosa, Vibrio cholerae, Klebsiella pneumonia, Neisseria gonorrhoeae, Neisseria meningitidis, Bacteroides nodosus, Moraxella bovis, and especially Escherichia coli are preferred. Details of outer surface proteins are discussed by Ladner et al., US 5,571,698 and references cited therein. For example, the lamB protein of E. coli is suitable.
- a basic concept of display methods that use phage or other replicable genetic package is the establishment of a physical association between DNA encoding a polypeptide to be screened and the polypeptide. This physical association is provided by the replicable genetic package, which displays a polypeptide as part of a capsid enclosing the genome of the phage or other package, wherein the polypeptide is encoded by the genome.
- the establishment of a physical association between polypeptides and their genetic material allows simultaneous mass screening of very large numbers of phage bearing different polypeptides. Phage displaying a polypeptide with affinity to a target, e.g., a receptor, bind to the target and these phage are enriched by affinity screening to the target.
- polypeptides displayed from these phage can be determined from their respective genomes. Using these methods a polypeptide identified as having a binding affinity for a desired target can then be synthesized in bulk by conventional means, or the polynucleotide that encodes the peptide or polypeptide can be used as part of a genetic vaccine.
- Variants with specific binding properties are easily enriched by panning with immobilized antibodies.
- Antibodies specific for a single family are used in each round of panning to rapidly select variants that have multiple epitopes from the antigen families.
- A-family specific antibodies can be used to select those shuffled clones that display A-specific epitopes in the first round of panning.
- a second round of panning with B-specific antibodies will select from the "A" clones those that display both A- and B-specific epitopes.
- a third round of panning with C- specific antibodies will select for variants with A, B, and C epitopes.
- a continual selection exists during this process for clones that express well in E. coli and that are stable throughout the selection. Improvements in factors such as transcription, translation, secretion, folding and stability are often observed and will enhance the utility of selected clones for use in vaccine production.
- Phage ⁇ LISA methods can be used to rapidly characterize individual variants. These assays provide a rapid method for quantitation of variants without requiring purification of each protein. Individual clones are arrayed into 96-well plates, grown, and frozen for storage. Cells in duplicate plates are infected with helper phage, grown overnight and pelleted by centrifugation. The supernatants containing phage displaying particular variants are incubated with immobilized antibodies and bound clones are detected by anti- M13 antibody conjugates. Titration series of phage particles, immobilized antigen, and/or soluble antigen competition binding studies are all highly effective means to quantitate protein binding. Variant antigens displaying multiple epitopes will be further studied in appropriate animal challenge models.
- the various display methods and ELISA assays can be used to screen for shuffled antigens with improved properties such as presentation of multiple epitopes, improved immunogenicity, increased expression levels, increased folding rates and efficiency, increased stability to factors such as temperature, buffers, solvents, improved purification properties, etc. Selection of shuffled antigens with improved expression, folding, stability and purification profile under a variety of chromatographic conditions can be very important improvements to incorporate for the vaccine manufacturing process.
- flow cytometry provides a method to efficiently analyze the functional properties of millions of individual cells.
- Very large numbers (>10 7 ) of cells can be evaluated in a single vial experiment, and the pool of the best individual sequences can be recovered from the sorted cells.
- These methods are particularly useful in the case of, for example, Hantaan virus glycoproteins, which are generally very poorly expressed in mammalian cells. This approach provides a general solution to improve expression levels of pathogen antigens in mammalian cells, a phenomenon that is critical for the function of genetic vaccines.
- polypeptides that are not expressed on the cell surface can be produced by using flow cytometry to analyze polypeptides that are not expressed on the cell surface.
- the polynucleotide is expressed as a fusion protein that has a region of amino acids which is targeted to the cell membrane.
- the region can encode a hydrophobic stretch of C-terminal amino acids which signals the attachment of a phosphoinositol-glycan (PIG) terminus on the expressed protein and directs the protein to be expressed on the surface of the transfected cell (Whitehorn et al. (1995) Biotechnology (N Y) 13:1215-9).
- PAG phosphoinositol-glycan
- an antigen that is naturally a soluble protein this method will likely not affect the three dimensional folding of the protein in this engineered fusion with a new C-terminus.
- an antigen that is naturally a transmembrane protein e.g., a surface membrane protein on pathogenic viruses, bacteria, protozoa or tumor cells
- the extracellular domain can be engineered to be in fusion with the C-terminal sequence for signaling PIG-linkage.
- the protein can be expressed in toto relying on the signalling of the host cell to direct it efficiently to the cell surface.
- the antigen for expression will have an endogenous PIG terminal linkage (e.g., some antigens of pathogenic protozoa).
- Those cells expressing the antigen can be identified with a fluorescent monoclonal antibody specific for the C-terminal sequence on PIG-linked forms of the surface antigen. FACS analysis allows quantitative assessment of the level of expression of the correct form of the antigen on the cell population. Cells expressing the maximal level of antigen are sorted and standard molecular biology methods are used to recover the plasmid DNA vaccine vector that conferred this reactivity.
- An alternative procedure that allows purification of all those cells expressing the antigen is to rosette or pan-purify the cells expressing surface antigen.
- Rosettes can be formed between antigen expressing cells and erythrocytes bearing covalently coupled antibody to the relevant antigen. These are readily purified by unit gravity sedimentation. Panning of the cell population over petri dishes bearing immobilized monoclonal antibody specific for the relevant antigen can also be used to remove unwanted cells.
- each well of a microtiter plate can be used to run a separate assay, or, if concentration or incubation time effects are to be observed, every 5-10 wells can test a single variant.
- a single standard microtiter plate can assay about 100 (e.g., 96) reactions. If 1536 well plates are used, then a single plate can easily assay from about 100 to about 1500 different reactions.
- library members e.g., cells, viral plaques, or the like
- solid media e.g., cells, viral plaques, or the like
- colonies or plaques are identified, picked, and up to 10,000 different mutants inoculated into 96 well microtiter dishes, optionally containing glass balls in the wells to prevent aggregation.
- the Q-bot does not pick an entire colony but rather inserts a pin through the center of the colony and exits with a small sampling of cells (or viruses in plaque applications).
- the uniform process of the Q-bot decreases human handling error and increases the rate of establishing cultures (roughly 10,000/4 hours). These cultures are then shaken in a temperature and humidity controlled incubator.
- the glass balls in the microtiter plates act to promote uniform aeration of cells dispersal of cells, or the like, similar to the blades of a fermentor.
- Clones from cultures of interest can be cloned by limiting dilution. Plaques or cells constituting libraries can also be screened directly for production of proteins, either by detecting hybridization, protein activity, protein binding to antibodies, or the like.
- the ability to detect a subtle increase in the performance of a shuffled library member over that of a parent strain relies on the sensitivity of the assay.
- the chance of finding the organisms having an improvement in ability to induce an immune response is increased by the number of individual mutants that can be screened by the assay.
- a prescreen that increases the number of mutants processed by 10-fold can be used. The goal of the prescreen will be to quickly identify mutants having equal or better product titers than the parent strain(s) and to move only these mutants forward to liquid cell culture for subsequent analysis.
- a number of well known robotic systems have also been developed for solution phase chemistries useful in assay systems. These systems include automated workstations like the automated synthesis apparatus developed by Takeda Chemical Industries, LTD. (Osaka, Japan) and many robotic systems utilizing robotic arms (Zymate II, Zymark Corporation, Hopkinton, Mass.; Orca, Hewlett-Packard, Palo Alto, Calif.) which mimic the manual synthetic operations performed by a scientist. Any of the above devices are suitable for use with the present invention, e.g., for high-throughput screening of molecules encoded by codon-altered nucleic acids. The nature and implementation of modifications to these devices (if any) so that they can operate as discussed herein with reference to the integrated system will be apparent to persons skilled in the relevant art.
- High throughput screening systems are commercially available (see, e.g., Zymark Corp., Hopkinton, MA; Air Technical Industries, Mentor, OH; Beckman Instruments, Inc. Fullerton, CA; Precision Systems, Inc., Natick, MA, etc.). These systems typically automate entire procedures including all sample and reagent pipetting, liquid dispensing, timed incubations, and final readings of the microplate in detector(s) appropriate for the assay. These configurable systems provide high throughput and rapid start up as well as a high degree of flexibility and customization.
- Zymark Corp. provides technical bulletins describing screening systems for detecting the modulation of gene transcription, ligand binding, and the like.
- Microfluidic approaches to reagent manipulation have also been developed, e.g., by Caliper Technologies (Palo Alto, CA).
- Optical images viewed (and, optionally, recorded) by a camera or other recording device are optionally further processed in any of the embodiments herein, e.g., by digitizing the image and/or storing and analyzing the image on a computer.
- the signals resulting from assays are florescent, making optical detection approaches appropriate in these instances.
- a variety of commercially available peripheral equipment and software is available for digitizing, storing and analyzing a digitized video or digitized optical image, e.g., using PC (Intel x86 or Pentium chip- compatible DOS , OS2 WINDOWS , WINDOWS NT or
- WINDOWS95 based machines WINDOWS95 based machines
- MACINTOSH MACINTOSH
- UNIX based e.g., SUN work station
- One conventional system carries light from the assay device to a cooled charge-coupled device (CCD) camera, in common use in the art.
- a CCD camera includes an array of picture elements (pixels). The light from the specimen is imaged on the CCD. Particular pixels corresponding to regions of the specimen (e.g., individual hybridization sites on an array of biological polymers) are sampled to obtain light intensity readings for each position. Multiple pixels are processed in parallel to increase speed.
- the apparatus and methods of the invention are easily used for viewing any sample, e.g., by fluorescent or dark field microscopic techniques.
- Integrated systems for analysis in the present invention typically include a digital computer with high-throughput liquid control software, image analysis software, data interpretation software, a robotic liquid control armature for transferring solutions from a source to a destination operably linked to the digital computer, an input device (e.g., a computer keyboard) for entering data to the digital computer to control high throughput liquid transfer by the robotic liquid control armature and, optionally, an image scanner for digitizing label signals from labeled assay component.
- the image scanner interfaces with the image analysis software to provide a measurement of optical intensity. Typically, the intensity measurement is interpreted by the data interpretation software to show whether the optimized recombinant antigenic polypeptide products are produced.
- antigen library immunization is used to identify optimized recombinant antigens that have improved immunogenicity.
- ALI involves introduction of the library of recombinant antigen-encoding nucleic acids, or the recombinant antigens encoded by the shuffled nucleic acids, into a test animal. The animals are then subjected to in vivo challenge using live pathogens. Neutralizing antibodies and cross-protective immune responses are studied after immunization with the entire libraries, pools and/or individual antigen variants.
- test animals are immunized twice or three times at two week intervals.
- the animals are challenged with live pathogens (or mixtures of pathogens), and the survival and symptoms of the animals is followed. Immunizations using test animal challenge are described in, for example, Roggenkamp et al. (1997) Infect. Immun. 65: 446; Woody et al. (1997) Vaccine 1: 133; Agren et al. (1997) J. Immunol. 158: 3936; Konishi et al. (1992) Virology 190: 454; Kinney et al (1988) J. Virol.
- the immunizations can be performed by injecting either the recombinant polynucleotides themselves, i.e., as a genetic vaccine, or by immunizing the animals with polypeptides encoded by the recombinant polynucleotides.
- Bacterial antigens are typically screened primarily as recombinant proteins, whereas viral antigens are preferably analyzed using genetic vaccinations.
- pooling and deconvolution As diagrammed in Figure 6. Pools of recombinant nucleic acids, or polypeptides encoded by the recombinant nucleic acids, are used to immunize test animals. Those pools that result in protection against pathogen challenge are then subdivided and subjected to additional analysis. The high throughput in vitro approaches described above can be used to identify the best candidate sequences for the in vivo studies.
- the challenge models that can be used to screen for protective antigens include pathogen and toxin models, such as Yersinia bacteria, bacterial toxins (such as
- Staphylococcal and Streptococcal enterotoxins E.coli/V. cholerae enterotoxins
- Venezuelan equine encephalitis virus VEE
- Flaviviruses Japanese encephalitis virus, Tick-borne encephalitis virus, Dengue virus
- Hantaan virus Herpes simplex, influenza virus (e.g., Influenza A virus), Vesicular Stomatitis Virus, Pseudomonas aeruginosa, Salmonella typhimurium, Escherichia coli, Klebsiella pneumoniae, Toxoplasma gondii, Plasmodium yoelii, Herpes simplex, influenza virus (e.g., Influenza A virus), and Vesicular Stomatitis Virus.
- test animals can also be challenged with tumor cells to enable screening of antigens that efficiently protect against malignancies.
- Individual shuffled antigens or pools of antigens are introduced into the animals intradermally, intramuscularly, intravenously, intratracheally, anally, vaginally, orally, or intraperitoneally and antigens that can prevent the disease are chosen, when desired, for further rounds of shuffling and selection.
- the most potent antigens based on in vivo data in test animals and comparative in vitro studies in animals and man, are chosen for human trials, and their capacity to prevent and treat human diseases is investigated.
- antigen library immunization and pooling of individual clones is used to immunize against a pathogen strain that was not included in the sequences that were used to generate the library.
- the level of crossprotection provided by different strains of a given pathogen can significantly.
- homologous titer is always higher than heterologous titer. Pooling and deconvolution is especially efficient in models where minimal protection is provided by the wild-type antigens used as starting material for shuffling (for example minimal protection by antigens A and B against strain C in Figure 3B). This approach can be taken, for example, when evolving the V-antigen of Yersinae or Hantaan virus glycoproteins.
- the desired screening involves analysis of the immune response based on immunological assays known to those skilled in the art.
- the test animals are first immunized and blood or tissue samples are collected for example one to two weeks after the last immunization.
- immune parameters that correlate to protective immunity, such as induction of specific antibodies (particularly IgG) and induction of specific T lymphocyte responses, in addition to determining whether an antigen or pools of antigens provides protective immunity.
- Spleen cells or peripheral blood mononuclear cells can be isolated from immunized test animals and measured for the presence of antigen-specific T cells and induction of cytokine synthesis.
- ELISA, ELISPOT and cytoplasmic cytokine staining, combined with flow cytometry, can provide such information on a single-cell level.
- Common immunological tests that can be used to identify the efficacy of immunization include antibody measurements, neutralization assays and analysis of activation levels or frequencies of antigen presenting cells or lymphocytes that are specific for the antigen or pathogen.
- the test animals that can be used in such studies include, but are not limited to, mice, rats, guinea pigs, hamsters, rabbits, cats, dogs, pigs and monkeys. Monkey is a particularly useful test animal because the MHC molecules of monkeys and humans are very similar.
- Virus neutralization assays are useful for detection of antibodies that not only specifically bind to the pathogen, but also neutralize the function of the virus.
- Virus neutralization assays provide means to screen for antigens that also provide protective immunity.
- shuffled antigens are screened for their capacity to induce T cell activation in vivo. More specifically, peripheral blood mononuclear cells or spleen cells from injected mice can be isolated and the capacity of cytotoxic T lymphocytes to lyse infected, autologous target cells is studied. The spleen cells can be reactivated with the specific antigen in vitro. In addition, T helper cell activation and differentiation is analyzed by measuring cell proliferation or production of T H I (IL-2 and IFN- ⁇ ) and T H 2 (IL- 4 and IL-5) cytokines by ELISA and directly in CD4 + T cells by cytoplasmic cytokine staining and flow cytometry.
- T H I IL-2 and IFN- ⁇
- T H 2 IL- 4 and IL-5
- T H 1/T H 2 differentiation Based on the cytokine production profile, one can also screen for alterations in the capacity of the antigens to direct T H 1/T H 2 differentiation (as evidenced, for example, by changes in ratios of IL-4/IFN- ⁇ , IL-4/IL-2, IL-5/IFN- ⁇ , IL-5/IL-2, IL- 13/IFN- ⁇ , IL-13/IL-2).
- the analysis of the T cell activation induced by the antigen variants is a very useful screening method, because potent activation of specific T cells in vivo correlates to induction of protective immunity.
- the frequency of antigen-specific CD8 + T cells in vivo can also be directly analyzed using tetramers of MHC class I molecules expressing specific peptides derived from the corresponding pathogen antigens (Ogg and McMichael, Curr. Opin. Immunol. 1998, 10:393-6; Altaian et al, Science 1996, 274:94-6).
- the binding of the tetramers can be detected using flow cytometry, and will provide information about the efficacy of the shuffled antigens to induce activation of specific T cells.
- flow cytometry and tetramer stainings provide an efficient method of identifying T cells that are specific to a given antigen or peptide.
- Another method involves panning using plates coated with tetramers with the specific peptides. This method allows large numbers of cells to be handled in a short time, but the method only selects for highest expression levels.
- APC antigen presenting cells
- Shuffled cancer antigens that induce cytotoxic T cells that have the capacity to kill cancer cells can be identified by measuring the capacity of T cells derived from immunized animals to kill cancer cells in vitro.
- the cancer cells are first labeled with radioactive isotopes and the release of radioactivity is an indication of tumor cell killing after incubation in the presence of T cells from immunized animals.
- cytotoxicity assays are known in the art.
- An indication of the efficacy of an antigen to activate T cells specific for, for example, cancer antigens, allergens or autoantigens is also the degree of skin inflammation when the antigen is injected into the skin of a patient or test animal. Strong inflammation is correlated with strong activation of antigen-specific T cells.
- T H 2 tumor-specific T cells
- T H I response a T H I response is desired.
- Skin biopsies can be taken, enabling detailed studies of the type of immune response that occurs at the sites of each injection (in mice and monkeys large numbers of injections/antigens can be analyzed). Such studies include detection of changes in expression of cytokines, chemokines, accessory molecules, and the like, by cells upon injection of the antigen into the skin.
- peripheral blood mononuclear cells from previously infected or immunized humans individuals can be used.
- MHC molecules that will present the antigenic peptides are human MHC molecules.
- Peripheral blood mononuclear cells or purified professional antigen-presenting cells can be isolated from previously vaccinated or infected individuals or from patients with acute infection with the pathogen of interest. Because these individuals have increased frequencies of pathogen- specific T cells in circulation, antigens expressed in PBMCs or purified APCs of these individuals will induce proliferation and cytokine production by antigen-specific CD4 + and CD8 + T cells.
- antigens that simultaneously harbor epitopes from several antigens can be recognized by their capacity to stimulate T cells from various patients infected or immunized with different pathogen antigens, cancer antigens, autoantigens or allergens.
- One buffy coat derived from a blood donor contains lymphocytes from 0.5 liters of blood, and up to 10 4 PBMC can be obtained, enabling very large screening experiments using T cells from one donor.
- EBV-transformed B cell lines When healthy vaccinated individuals (lab volunteers) are studied, one can make EBV-transformed B cell lines from these individuals. These cell lines can be used as antigen presenting cells in subsequent experiments using blood from the same donor; this reduces interassay and donor-to-donor variation. In addition, one can make antigen-specific T cell clones, after which antigen variants are introduced to EBV transformed B cells. The efficiency with which the transformed B cells induce proliferation of the specific T cell clones is then studied. When working with specific T cell clones, the proliferation and cytokine synthesis responses are significantly higher than when using total PBMCs, because the frequency of antigen-specific T cells among PBMC is very low.
- CTL epitopes can be presented by most cells types since the class I major histocompatibility complex (MHC) surface glycoproteins are widely expressed. Therefore, transfection of cells in culture by libraries of shuffled antigen sequences in appropriate expression vectors can lead to class I epitope presentation. If specific CTLs directed to a given epitope have been isolated from an individual, then the co-culture of the transfected presenting cells and the CTLs can lead to release by the CTLs of cytokines, such as IL-2,
- cytotoxic T cells that have the capacity to kill infected cells can also be identified by measuring the capacity of T cells derived from immunized animals to kill infected cells in vitro.
- the target cells are first labeled with radioactive isotopes and the release of radioactivity is an indication of target cell killing after incubation in the presence of T cells from immunized animals.
- cytotoxicity assays are known in the art.
- a second method for identifying optimized CTL epitopes does not require the isolation of CTLs reacting with the epitope.
- cells expressing class I MHC surface glycoproteins are transfected with the library of evolved sequences as above. After suitable incubation to allow for processing and presentation, a detergent soluble extract is prepared from each cell culture and after a partial purification of the MHC-epitope complex (perhaps optional) the products are submitted to mass spectrometry (Henderson et al. (1993) Proc. Nat'l. Acad. Sci. USA 90: 10275-10279). Since the sequence is known of the epitope whose presentation to be increased, one can calibrate the mass spectrogram to identify this peptide. In addition, a cellular protein can be used for internal calibration to obtain a quantitative result; the cellular protein used for internal calibration could be the MHC molecule itself. Thus one can measure the amount of peptide epitope bound as a proportion of the MHC molecules.
- the multivalent antigens of the invention are useful for treating and/or preventing the various diseases and conditions with which the respective antigens are associated.
- the multivalent antigens can be expressed in a suitable host cell and are administered in polypeptide form. Suitable formulations and dosage regimes for vaccine delivery are well known to those of skill in the art.
- the optimized recombinant polynucleotides that encode improved allergens are used in conjunction with a genetic vaccine vector.
- the choice of vector and components can also be optimized for the particular purpose of treating allergy.
- the polynucleotide that encodes the recombinant antigenic polypeptide can be placed under the control of a promoter, e.g., a high activity or tissue-specific promoter.
- the promoter used to express the antigenic polypeptide can itself be optimized using recombination and selection methods analogous to those described herein.
- the vector can contain immunostimulatory sequences such as are described in copending, commonly assigned US Patent Application Serial No. , entitled
- Genetic Vaccine Vector Engineering filed on February 10, 1999 as TTC Attorney Docket No. 18097-030100US. It is sometimes advantageous to employ a genetic vaccine that is targeted for a particular target cell type (e.g. , an antigen presenting cell or an antigen processing cell); suitable targeting methods are described in copending, commonly assigned
- Vaccine delivery vehicles can be delivered in vivo by administration to an individual patient, typically by systemic administration (e.g., intravenous, intraperitoneal, intramuscular, subdermal, intracranial, anal, vaginal, oral, buccal route or they can be inhaled) or they can be administered by topical application.
- systemic administration e.g., intravenous, intraperitoneal, intramuscular, subdermal, intracranial, anal, vaginal, oral, buccal route or they can be inhaled
- topical application e.g., intravenous, intraperitoneal, intramuscular, subdermal, intracranial, anal, vaginal, oral, buccal route or they can be inhaled
- vectors can be delivered to cells ex vivo, such as cells explanted from an individual patient (e.g., lymphocytes, bone marrow aspirates, tissue biopsy) or universal donor hematopoietic stem cells, followed by reimplantation of the cells into a patient, usually after selection for cells which have incorporated the vector.
- cells explanted from an individual patient e.g., lymphocytes, bone marrow aspirates, tissue biopsy
- tissue biopsy e.g., lymphocytes, bone marrow aspirates, tissue biopsy
- universal donor hematopoietic stem cells e.g., hematopoietic stem cells
- a large number of delivery methods are well known to those of skill in the art. Such methods include, for example liposome-based gene delivery (Debs and Zhu (1993) WO 93/24640; Mannino and Gould-Fogerite (1988) BioTechniques 6(7): 682-691; Rose U.S. Pat No. 5,279,833; Brigham (1991) WO 91/06309; and Feigner et al. (1987) Proc. Natl. Acad. Sci. USA 84: 7413-7414), as well as use of viral vectors (e.g., adenoviral (see, e.g., Be ⁇ is et al. (1995) Ann. NY Acad. Sci.
- adenoviral see, e.g., Be ⁇ is et al. (1995) Ann. NY Acad. Sci.
- DNA and/or RNA that comprises a genetic vaccine can be introduced directly into a tissue, such as muscle. See, e.g., USPN 5,580,859.
- Other methods such as “biolistic” or particle-mediated transformation (see, e.g., Sanford et al, USPN
- 4,945,050; USPN 5,036,006) are also suitable for introduction of genetic vaccines into cells of a mammal according to the invention. These methods are useful not only for in vivo introduction of DNA into a mammal, but also for ex vivo modification of cells for reintroduction into a mammal. As for other methods of delivering genetic vaccines, if necessary, vaccine administration is repeated in order to maintain the desired level of immunomodulation.
- Genetic vaccine vectors can be administered directly to the mammal for transduction of cells in vivo.
- the genetic vaccines obtained using the methods of the invention can be formulated as pharmaceutical compositions for administration in any suitable manner, including parenteral (e.g., subcutaneous, intramuscular, intradermal, or intravenous), topical, oral, rectal, intrathecal, buccal (e.g., sublingual), or local administration, such as by aerosol or transdermally, for prophylactic and/or therapeutic treatment.
- Pretreatment of skin for example, by use of hair-removing agents, may be useful in transdermal delivery.
- Suitable methods of administering such packaged nucleic acids are available and well known to those of skill in the art, and, although more than one route can be used to administer a particular composition, a particular route can often provide a more immediate and more effective reaction than another route.
- compositions of the present invention are determined in part by the particular composition being administered, as well as by the particular method used to administer the composition. Accordingly, there is a wide variety of suitable formulations of pharmaceutical compositions of the present invention.
- aqueous carriers can be used, e.g., buffered saline and the like. These solutions are sterile and generally free of undesirable matter.
- These compositions may be sterilized by conventional, well known sterilization techniques.
- the compositions may contain pharmaceutically acceptable auxiliary substances as required to approximate physiological conditions such as pH adjusting and buffering agents, toxicity adjusting agents and the like, for example, sodium acetate, sodium chloride, potassium chloride, calcium chloride, sodium lactate and the like.
- concentration of genetic vaccine vector in these formulations can vary widely, and will be selected primarily based on fluid volumes, viscosities, body weight and the like in accordance with the particular mode of administration selected and the patient's needs.
- Formulations suitable for oral administration can consist of (a) liquid solutions, such as an effective amount of the packaged nucleic acid suspended in diluents, such as water, saline or PEG 400; (b) capsules, sachets or tablets, each containing a predetermined amount of the active ingredient, as liquids, solids, granules or gelatin; (c) suspensions in an appropriate liquid; and (d) suitable emulsions.
- liquid solutions such as an effective amount of the packaged nucleic acid suspended in diluents, such as water, saline or PEG 400
- capsules, sachets or tablets each containing a predetermined amount of the active ingredient, as liquids, solids, granules or gelatin
- suspensions in an appropriate liquid such as water, saline or PEG 400
- Tablet forms can include one or more of lactose, sucrose, mannitol, sorbitol, calcium phosphates, com starch, potato starch, tragacanth, microcrystalline cellulose, acacia, gelatin, colloidal silicon dioxide, croscarmellose sodium, talc, magnesium stearate, stearic acid, and other excipients, colorants, fillers, binders, diluents, buffering agents, moistening agents, preservatives, flavoring agents, dyes, disintegrating agents, and pharmaceutically compatible carriers.
- Lozenge forms can comprise the active ingredient in a flavor, usually sucrose and acacia or tragacanth, as well as pastilles comprising the active ingredient in an inert base, such as gelatin and glycerin or sucrose and acacia emulsions, gels, and the like containing, in addition to the active ingredient, carriers known in the art.
- an inert base such as gelatin and glycerin or sucrose and acacia emulsions, gels, and the like containing, in addition to the active ingredient, carriers known in the art.
- the genetic vaccines when administered orally, must be protected from digestion. This is typically accomplished either by complexing the vaccine vector with a composition to render it resistant to acidic and enzymatic hydrolysis or by packaging the vector in an appropriately resistant carrier such as a liposome. Means of protecting vectors from digestion are well known in the art.
- the pharmaceutical compositions can be encapsulated, e.g., in liposomes, or in a formulation that provides for slow release of the active ingredient.
- the packaged nucleic acids alone or in combination with other suitable components, can be made into aerosol formulations (e.g., they can be "nebulized") to be administered via inhalation. Aerosol formulations can be placed into pressurized acceptable propellants, such as dichlorodifluoromethane, propane, nitrogen, and the like.
- Suitable formulations for rectal administration include, for example, suppositories, which consist of the packaged nucleic acid with a suppository base.
- Suitable suppository bases include natural or synthetic triglycerides or paraffin hydrocarbons.
- gelatin rectal capsules which consist of a combination of the packaged nucleic acid with a base, including, for example, liquid triglycerides, polyethylene glycols, and paraffin hydrocarbons.
- Formulations suitable for parenteral administration include aqueous and non-aqueous, isotonic sterile injection solutions, which can contain antioxidants, buffers, bacteriostats, and solutes that render the formulation isotonic with the blood of the intended recipient, and aqueous and non-aqueous sterile suspensions that can include suspending agents, solubilizers, thickening agents, stabilizers, and preservatives.
- compositions can be administered, for example, by intravenous infusion, orally, topically, intraperitoneally, intravesically or intrathecally.
- Parenteral administration and intravenous administration are the preferred methods of administration.
- the formulations of packaged nucleic acid can be presented in unit-dose or multi-dose sealed containers, such as ampoules and vials.
- Injection solutions and suspensions can be prepared from sterile powders, granules, and tablets of the kind previously described. Cells transduced by the packaged nucleic acid can also be administered intravenously or parenterally.
- the dose administered to a patient should be sufficient to effect a beneficial therapeutic response in the patient over time.
- the dose will be determined by the efficacy of the particular vector employed and the condition of the patient, as well as the body weight or vascular surface area of the patient to be treated.
- the size of the dose also will be determined by the existence, nature, and extent of any adverse side-effects that accompany the administration of a particular vector, or transduced cell type in a particular patient.
- the physician evaluates vector toxicities, progression of the disease, and the production of anti-vector antibodies, if any.
- the dose equivalent of a naked nucleic acid from a vector is from about 1 ⁇ g to 1 mg for a typical 70 kilogram patient, and doses of vectors used to deliver the nucleic acid are calculated to yield an equivalent amount of therapeutic nucleic acid. Administration can be accomplished via single or divided doses.
- compositions are administered to a patient suffering from a disease (e.g., an infectious disease or autoimmune disorder) in an amount sufficient to cure or at least partially arrest the disease and its complications.
- a disease e.g., an infectious disease or autoimmune disorder
- An amount adequate to accomplish this is defined as a "therapeutically effective dose.” Amounts effective for this use will depend upon the severity of the disease and the general state of the patient's health. Single or multiple administrations of the compositions may be administered depending on the dosage and frequency as required and tolerated by the patient. In any event, the composition should provide a sufficient quantity of the proteins of this invention to effectively treat the patient.
- compositions are administered to a human or other mammal to induce an immune response that can help protect against the establishment of an infectious disease or other condition.
- the toxicity and therapeutic efficacy of the genetic vaccine vectors provided by the invention are determined using standard pharmaceutical procedures in cell cultures or experimental animals.
- a typical pharmaceutical composition for intravenous administration would be about 0.1 to 10 mg per patient per day. Dosages from 0.1 up to about 100 mg per patient per day may be used, particularly when the drug is administered to a secluded site and not into the blood stream, such as into a body cavity or into a lumen of an organ. Substantially higher dosages are possible in topical administration. Actual methods for preparing parenterally administrable compositions will be known or apparent to those skilled in the art and are described in more detail in such publications as Remington 's Pharmaceutical Science, 15th ed., Mack Publishing Company, Easton, Pennsylvania (1980).
- the multivalent antigenic polypeptides of the invention, and genetic vaccines that express the polypeptides can be packaged in packs, dispenser devices, and kits for administering genetic vaccines to a mammal.
- packs or dispenser devices that contain one or more unit dosage forms are provided.
- instructions for administration of the compounds will be provided with the packaging, along with a suitable indication on the label that the compound is suitable for treatment of an indicated condition.
- the label may state that the active compound within the packaging is useful for treating a particular infectious disease, autoimmune disorder, tumor, or for preventing or treating other diseases or conditions that are mediated by, or potentially susceptible to, a mammalian immune response.
- This Example describes the use of DNA shuffling to develop immunogens that produce strong cross-protective immune responses against a variety of Yersinia strains. Passive immunization with anti-V-antigen antibodies or active immunization with purified V-antigen can provide protection from challenge with a virulent autologous Yersinia species. However, protection against heterologous species is limited (Motin et al. (1994) Infect. Immun. 62: 4192).
- V-antigen genes from a variety of Yersinia strains including serotypes of Y. pestis, Y. enterocolitica, and Y. pseudotuberculosis are subjected to DNA shuffling as described herein.
- the Yersinia pestis V antigen coding sequence for example, is used as a query in a database search to identify homologous genes that can be used in a family shuffling format to obtain improved antigens.
- Results for a BLAST search of GenBank and EMBL databases are shown in Table 1 , in which each line represents a unique sequence entry listing the database, accession number, locus name, bit score and E value. See, Altschul et al. (1997) Nucleic Acids Res.
- Homologous antigens have been cloned and sequenced from a number of related yet distinct Yersinia strains and additional natural diversity is obtained by cloning antigen genes from other strains. These genes and others or fragments thereof are cloned by methods such as PCR, shuffled and screened for improved antigens.
- Shuffled clones are selected by phage display and/or screened by ELISA to identify those recombinant nucleic acids that encode polypeptides that have multiple epitopes corresponding to the different serotypes.
- the shuffled antigen genes are cloned into a filamentous phage genome for polyvalent phage display or a suitable phagemid vector for monovalent phage display.
- a typical protocol for panning antigens by phage display is as follows.
- Phage ELISA assays are a useful method to rapidly evaluate single clones after panning of libraries. Single colonies are picked in individual wells of a multiwell plate containing 2YT media and grown as a master plate. A replicate plate is infected with helper phage and grown so that phage from a single well will display a single antigen variant.
- a suitable protocol for phage ELISA assays is as follows.
- ELISA assays can also be used to screen for individual antigens with multiple epitopes or increased expression levels. Single colonies are picked in individual wells of a multiwell plate containing appropriate media and grown as a master plate so that antigens produced from a single well are a single antigen variant. A replicate plate is grown and induced for protein production, e.g., by addition of 0.5 mM IPTG for Lac repressor-based systems and grown for an appropriate time for the antigen to be produced. At this point a crude antigen preparation is made which depends on the antigen and where it is produced. Secreted proteins can be evaluated by assaying the cell supernatants after centrifugation. Periplasmic proteins are often readily released from cells by simple extraction into hyper- or hypo-tonic buffers. Intracellularly produced proteins will require some form of cell lysis such as detergent treatment to release them. A suitable protocol for ELISA assays is as follows.
- Antibodies specific for many of the various antigens are commercially available (e.g., Toxin Technology, Inc, Sarasota, FL) or can be generated by immunizing suitable animals with purified antigens. Protein A or Protein G Sepharose (Pharmacia) can be used to purify immunoglobulins from the serum. Various affinity purification schemes can be used to further purify family-specific antibodies if needed such as immobilization of specific antigens to NHS-, CNBr-, or epoxy-activated sepharose beads. Other related antigens may be included soluble form to prevent binding and immobilization of cross- reactive antibodies.
- the multivalent polypeptides that are identified by the initial screening protocol are purified and subjected to in vivo screening.
- the shuffled antigens selected by a combination of any or none of these methods are purified and used to immunize animals, initially mice, which are then evaluated for improved immune responses.
- 10 micrograms of protein is injected to a suitable location with or without appropriate adjuvant, e.g., Alhydrogel (EM Seargent Pulp and Chemical, Inc.) and the animals are boosted with an additional dose after 2-4 weeks.
- adjuvant e.g., Alhydrogel (EM Seargent Pulp and Chemical, Inc.)
- serum samples is drawn and evaluated by ELISA assay for the presence of antibodies that cross-react against multiple parental antigens.
- the antigens are coated onto multiwell plates, then serial dilutions of each sera is allowed to bind. After washing unbound antibodies, a secondary HRP- or AP- conjugated antibody directed against the appropriate test antibody constant region, e.g., goat anti-mouse IgG Fc (Sigma) is bound. After another washing, the appropriate substrate is added, e.g., O-phenylenediamine (Sigma). The absorbance of each well is read by a plate reader at the appropriate wavelength (e.g., 490 nm for OPD) and those producing high antibody titers to multiple antigens are selected for further evaluation.
- a secondary HRP- or AP- conjugated antibody directed against the appropriate test antibody constant region e.g., goat anti-mouse IgG Fc (Sigma) is bound.
- the appropriate substrate e.g., O-phenylenediamine (Sigma).
- the absorbance of each well is read by a plate reader at the appropriate wavelength (e.g.,
- antigens to generate neutralizing antibodies can be evaluated in an appropriate system.
- Antigen variants that elicit a broad cross-reactive response are evaluated further in a virulent challenge model with the appropriate pathogenic organism.
- the multivalent polypeptides are used to immunize mice, which are then challenged with live Yersinia bacteria. Those multivalent polypeptides that protect against the challenge are identified and purified.
- This Example describes the use of DNA shuffling to obtain multivalent polypeptides that are effective in inducing an immune response against a broad spectrum of bacterial toxins.
- the Group A Streptococci which can cause diseases such as food poisoning, toxic shock syndrome, and autoimmune disorders, are highly toxic by inhalation.
- the family of Group A Streptococcus toxins numbers about 30 related members, making this group a suitable target for family shuffling. Accordingly, this Example describes the use of family DNA shuffling to create chimeric proteins that are capable of eliciting broad spectrum protection.
- Nucleic acids that encode many diverse attenuated toxins are subjected to DNA shuffling as described herein.
- Table 2 shows the output of a BLAST search of GenBank, PDL, EMBL, and Swissprot using the S. aureus enterotoxin B protein to identify homologous genes that may be used in a family shuffling format to obtain improved antigens.
- Shuffled recombinant clones are initially selected by phage display and/or screened by ELISA for the presence of multiple epitopes from the different families.
- Variant proteins with multiple epitopes are purified and used to for in vivo screening as described above.
- the mouse sera are analyzed for antibodies specific for different toxin subtypes and variants that elicit broadly cross-reactive responses will be evaluated further in challenge models.
- This Example describes the use of DNA shuffling to obtain cross-reactive multivalent polypeptides that induce an immune response against the E. coli heat-labile toxin (LT), cholera toxin (CT), and verotoxin (VT).
- Nucleic acids that encode cholera and LT toxin B-chains are subjected to DNA shuffling.
- Table 3 shows the results of a BLAST search using the V. cholerae toxin B-chain to identify homologous genes that can be used in a family shuffling format to obtain improved antigens.
- Homologous antigens have been cloned and sequenced from a number of related yet distinct Vibrio and E. coli strains, and additional natural diversity can be obtained by cloning antigen genes from other strains. These genes and others or fragments thereof can be cloned by methods such as PCR, shuffled and screened for improved antigens.
- chimeric toxins that elicit high levels of neutralizing antibodies against both toxins and have improved adjuvant properties are identified. For example, shuffled clones are selected by phage display and/or screened by ELISA assays for the presence of epitopes from the different parental B-chains. Variants with multiple epitopes are purified and further studied for their capacity to act as adjuvants and to elicit cross-protective immune responses in challenge models.
- Lyme disease is currently one of the fastest-growing infectious diseases in the United States. It is caused by infection of the spirochete bacterium Borrelia burgdorferi, which is carried and spread by the bite of infected ticks. Early signs of infection include skin rash and flu-like symptoms. If left untreated Lyme disease can cause arthritis, heart abnormalities, and facial paralysis. Treatment of early Lyme disease with antibiotics can stop the infection, but a lasting immunity may not develop making reinfection possible. A current vaccine requires three immunizations over a 1 -year period to acquire immunity.
- OspA outer surface protein A
- OspC outer surface protein C
- the ospA genes from these and other strains provide a source of diversity for family shuffling to obtain improved antigens for the prevention of Lyme disease. These genes are cloned by methods such as PCR, shuffled and screened for improved antigens.
- a BLAST search with the B. burgdorferi OspC protein gene revealed over 200 related entries. Entries for one hundred sequences sharing at least 82% DNA sequence identity are shown in Table 5 below that provide a source of diversity for family shuffling to obtain improved therapeutics in the treatment of Lyme disease. These genes are cloned by methods such as PCR, shuffled and screened for improved antigens.
- Tuberculosis is an ancient bacterial disease caused by Mycobacterium tuberculosis that continues to be an important public health problem worldwide and calls are being made for an improved effort in eradication (Morb. Mortal Wkly Rep (1998 Aug 21; 47(RR-13): 1-6). It infects over 50 million people and over 3 million people will die from tuberculosis this year.
- the currently available vaccine, Bacille Calmette-Guerin (BCG) is found to be less effective in developing countries and an increasing number of multidrug- resistant (MDR) strains are being isolated.
- the major immunodominant antigen of M. tuberculosis is the 30-35 kDa (a.k.a. antigen 85, alpha-antigen) which is normally a lipoglycoprotein on the cell surface.
- Other protective antigens include a 65-kDa heat shock protein, and a 36-kDa proline-rich antigen (Tascon et al. (1996) Nat. Med. 1: 888-92).
- Table 6 shows the output of a BLAST search using the 30-35 kDa major M. Tuberculosis antigen (a.k.a. antigen 85, alpha-antigen) coding sequence to identify homologous genes that may be used in a family shuffling format to obtain improved antigens. Many homologous antigens have been cloned and sequenced from a large number of related yet distinct mycobacterial strains. These genes are cloned by methods such as PCR, shuffled and screened for improved antigens.
- Tuberculosis antigen a.k.a. antigen 85, alpha-antigen
- Chronic infection of the gastroduodenal mucosae by Helicobacter pylori bacteria is responsible for chronic active gastritis, peptic ulcers, and gastric cancers such as adenocarcinoma and low-grade B-cell lymphoma.
- An increasing occurrence of antibiotic- resistant strains is limiting this therapy.
- the use of vaccines to both prevent and treat ongoing infections is being actively pursued (Crabtree JE (1998) Gut 43: 7-8; Axon AT (1998) Gut 43 Suppl 1 : S70-3; Dubois et al. (1998) Infect. Immun. 66: 4340-6; Tytgat GN ( ⁇ 99S) Aliment. Pharmacol. Ther.
- Identification of appropriate Helicobacter antigens for use in preventive and therapeutic vaccines can include two-dimensional gel electrophoresis, sequence analysis, and serum profiling (McAtee et al. (1998) Clin. Diagn. Lab. Immunol. 5:537-42; McAtee et al. (1998) Helicobacter 3: 163-9).
- Antigenic differences between related Helicobacter species and strains can limit the use of vaccines for prevention and treatment of infections (Keenan et al. ( 1998) FEMS Microbiol Lett. 161 : 21-7).
- DNA family shuffling of related yet immunologically distinct antigens allows for the isolation of complex chimeric antigens that can provide a broad cross-reactive protection against many related strains and species of Helicobacter.
- Mouse models of persistent infection by mouse-adapted H. pylori strains that have been used to evaluate therapeutic use of vaccines against infection are used to evaluate shuffled antigens (Crabtree JE (1998) Gut 43: 7-8; Axon AT (1998) Gut 43 Suppl 1.S70-3).
- Table 7 shows the results of a BLAST search using the H. pylori VacA gene to identify homologous genes that can be used in a family shuffling format to obtain improved antigens.
- Homologous antigens have been cloned and sequenced from a number of related yet distinct H. pylori strains and additional natural diversity can be obtained by cloning antigen genes from other strains. These genes and others or fragments thereof are cloned by methods such as PCR, shuffled and screened for improved antigens. Table 7
- Table 8 shows the results of a BLAST search using the H. pylori CagA gene to identify homologous genes that can be used in a family shuffling format to obtain improved antigens.
- Homologous antigens have been cloned and sequenced from a number of related yet distinct H pylori strains and additional natural diversity can be obtained by cloning antigen genes from other strains. These genes and others or fragments thereof are be cloned by methods such as PCR, shuffled and screened for improved antigens.
- This Example describes the use of DNA shuffling to generate improved vaccines against malaria infection.
- An excellent target for evolution by DNA shuffling is the Plasmodium falciparum merozoite surface protein, MSP 1 (Hui et al. (1996) Infect. Immun.
- MSP 1 is expressed on the surface of merozoites as an integral membrane protein. It is cleaved by parasite proteases just before and concomitant with rupture and release from infected cells. The cleavage appears to be obligatory for full function in MSP 1 binding to RBC receptors. The cleaved fragments remain attached to the membrane of the merozoite. Other membrane proteins on merozoites also participate in the attachment and specific invasion events. MSP1 is a proven candidate for inclusion in a vaccine against the asexual blood stage of malaria.
- MSP 1 genes encoding MSP 1 can be isolated from various isolates of
- Plasmodium falciparum merozoites by PCR technology Plasmodium falciparum merozoites by PCR technology.
- Related naturally existing genes can be additionally used to increase the diversity of the starting genes.
- MSP1 genes is generated by DNA shuffling, and this library is screened for induction of efficient immune responses.
- the screening can be done by injecting individual variants into test animals, such as mice or monkeys. Either purified recombinant proteins, or DNA vaccines or viral vectors encoding the relevant genes are injected. Typically, a booster injection is given 2-3 weeks after the first injection. Thereafter, the sera of the test animals are collected and these sera are analyzed for the presence of antibodies that reduce invasion of merozoites into uninfected erythrocytes (RBC).
- RBC uninfected erythrocytes
- RBC are infected by the merozoite, immediately inside the RBC, the merozoite differentiates into a ring and this matures to a schizont that contains several nascent daughter merozoites, which then burst out of the infected cell, destroying it, and go on to attach and invade another RBC.
- the merozoite is likely only extracellular for seconds. In vitro, any blockade of this event can dramatically reduce the level of reinfection.
- Antibodies against MSP 1 bind to the surface of merozoites that are released from schizont infected RBC when they rupture and thereby reduce the ability of these merozoites to attach and engage cognate RBC receptors on the uninfected RBC surface.
- This Example describes the use of DNA shuffling to obtain vaccines that can induce an immune response against multiple isolates of viral pathogens.
- VEE Venezuelan equine encephalitis virus
- VEE belongs to the alphavirus genus, which are generally transmitted by mosquitoes.
- VEE is an unusual alphavirus in that it is also highly infectious by aerosol inhalation for both humans and rodents. The disease manifestations in humans range from subclinical or mild febrile disease to serious infection and inflammation of the central nervous system. Virus clearance coincides that of production of specific anti-VEE antibodies, which are believed to be the primary mediators of protective immune responses (Schmaljohn et al. (1982) Nature 297: 70).
- VEE is an unusual virus also because its primary target outside the central nervous system is the lymphoid tissue, and therefore, replication defective variants may provide means to target vaccines or pharmaceutically useful proteins to the immune system.
- VEE At least seven subtypes of VEE are known that can be identified genetically and serologically. Based on epidemiological data, the virus isolates fall into two main categories: I-AB and I-C strains, which are associated with VEE epizootics/epidemics, and the remaining serotypes, which are associated primarily with enzootic vertebrate-mosquito cycles and circulate in specific ecological zones (Johnston and Peters, In Fields Virology, Third Edition, eds. B.N. Fields et al, Lippincott-Raven Publishers, Philadelphia, 1996).
- the envelope protein (E) appears to be the major antigen in inducing neutralizing Abs.
- DNA shuffling is used to obtain a library of recombinant E proteins by shuffling the corresponding genes derived from various strains of VEE. These libraries and individuals chimeras/mutants thereof are subsequently screened for their capacity to induce widely cross-reacting and protective Ab responses.
- Japanese encephalitis virus (JE), Tick-borne encephalitis virus (TBE) and Dengue virus are arthropod-borne viruses belonging to the Flavivirus family, which comprises 69 related viruses.
- the heterogeneity of the viruses within the family is a major challenge for vaccine development.
- non-neutralizing antibodies induced by infection or vaccination by one Dengue virus may cause enhancement of the disease during a subsequent infection by another serotype. Therefore, cross-protective, broad spectrum vaccines for TBE and JE would provide significant improvements to the existing vaccines.
- the ability of DNA shuffling to efficiently generate chimeric and mutated genes is used to generate cross-protective vaccines.
- Japanese encephalitis virus is a prototype of the JE antigenic complex, which comprises St. Louis encephalitis virus, Murray Valley encephalitis virus, Kunjin virus and West Nile virus (Monath and Heinz, In Fields Virology, Third Edition, eds. B.N. Fields et al, Lippincott-Raven Publishers, Philadelphia, pp 961-1034, 1996). Infections caused by JE are relatively rare, but the case-fatality is 5-40% because no specific treatment is available. JE is widely distributed in China, Japan, Philippines, far-eastern Russia and India providing a significant threat to those traveling in these areas. Currently available JE vaccine is produced from brain tissues of mice infected with single virus isolate. Side effects are observed in 10% to 30% of the vaccinees.
- DNA shuffling is performed on viral envelope genes.
- the amino acid identity within the JE complex varies between 72% and 93%.
- significant antigenic variation has been observed among JE strains by neutralization assays, agar gel diffusion, antibody absorption and monoclonal antibody analysis (Oda (1976) Kobe J. Med. Sci. 22: 123; Kobyashi et al. (1984) Infect. Immun. 44: 117).
- the amino acid divergence of the envelope protein gene among 13 strains from different Asian countries is as much as 4.2% (Ni and Barrett (1995) J. Gen. Virol. 76: 401).
- the resulting library of recombinant polypeptides encoded by the shuffled genes is screened to identify those that provide a cross-protective immune response.
- tick-bome encephalitis virus complex comprises 14 antigenically related viruses, eight of which cause human disease, including Powassan, Louping ill and Tick- bome encephalitis virus (TBE) (Monath and Heinz, In Fields Virology, Third Edition, eds. B.N. Fields et al, Lippincott-Raven Publishers, Philadelphia, pp. 961-1034, 1996).
- TBE has been recognized in all Central and Eastern European countries, Scandinavia and Russia, whereas Powassan occurs in Russia, Canada and the United States.
- the symptoms vary from flu-like illness to severe meningitis, meningoencephalitis and meningoencephalitis with a fatality rate of 1% to 2% (Gresikova and Calisher, In Monath ed., The arboviruses: ecology and epidemiology, vol. IV, Boca Raton, FL, CRC Press, pp. 177-203, 1988).
- Family DNA shuffling is used to generate chimeric envelope proteins derived from the TBE complex to generate crossprotective antigens.
- the envelope proteins within the family are 77-96% homologous, and viruses can be distinguished by specific mAbs (Holzmann et al, Vaccine, 10, 345, 1992).
- the envelope protein of Powassan is 78% identical at the amino acid level with that of TBE, and cross-protection is unlikely, although epidemiological data is limited.
- Langat virus is used as a model system to analyze protective immune responses in vivo (lacono-Connors et al. (1996) Virus Res. 43: 125). Langat virus belongs to the TBE complex, and can be used in challenge studies in BSL3 facilities. Serological studies based on recombinant envelope proteins are performed to identify immunogen variants that induce high levels of antibodies against envelope proteins derived from most or all viruses of the TBE complex.
- Dengue viruses Dengue viruses are transmitted though mosquito bites, posing a significant threat to troops and civilian populations particularly in tropical areas. There are four major serotypes of Dengue virus, namely Dengue 1, 2, 3 and 4. A tetravalent vaccine that induces neutralizing antibodies against all four strains of Dengue is required to avoid antibody- mediated enhancement of the disease when the individual encounters the virus of the other strain.
- the envelope protein of Dengue virus has been shown to provide an immune response that protects from a future challenge with the same strain of virus.
- the levels of neutralizing antibodies produced are relatively low and protection from live virus challenge is not always observed.
- mice injected with genetic vaccines encoding envelope protein of Dengue- 2 virus developed neutralizing antibodies when analyzed by in vitro neutralization assays, but the mice did not survive the challenge with live Dengue-2 virus (Kochel et al. (1997) Vaccine 15: 547-552).
- protective immune responses were observed in mice immunized with recombinant vaccinia virus expressing Dengue 4 virus structural proteins (Bray et al. (1989) J. Virol. 63: 2853).
- DNA shuffling is performed on the genes encoding the envelope (E) protein from all four Dengue viruses and their antigenic variants.
- Family DNA shuffling is used to generate chimeric E protein variants that induce high titer neutralizing antibodies against all serotypes of Dengue.
- the E proteins of the different dengue viruses share 62% to 77% of their amino acids.
- Dengue 1 and Dengue 3 are most closely related (77% homologous), followed by Dengue 2 (69%) and Dengue 4 (62%). These homologies are well in the range that allows efficient family shuffling (Crameri et al. (1998) Nature 391: 288-291).
- the shuffled antigen sequences are incorporated into genetic vaccine vectors, the plasmids purified, and subsequently injected into mice.
- the sera are collected from the mice and analyzed for the presence of high levels of cross-reactive antibodies.
- the best antigens are selected for further studies using in vivo challenge models to screen for chimeras/mutants that induce cross-protection against all strains of Dengue.
- Hantaan virus belongs to the Bunyavirus family.
- a characteristic feature of this family is that their glycoproteins typically accumulate at the membranes of the Golgi apparatus when expressed by cloned cDNAs, thereby reducing the efficacy of corresponding genetic vaccines (Matsuoka et al. (1991) Curr. Top. Microb. Immunol. 169: 161-179). Poor expression of Hantaan virus glycoproteins on the cell surface is also one explanation for poor immune responses following injections of Hantaan virus genetic vaccines.
- family DNA shuffling is used to generate recombinant Hantaan virus derived glycoproteins that are efficiently expressed in human cells and that can induce protective immune responses against the wild-type pathogen.
- Nucleic acids that encode the Hantaan virus glycoprotein are shuffled with genes that encode other homologous Bunyavirus glycoproteins.
- the resulting library is screened to identify proteins that are readily expressed in human cells. The screening is performed using a dual marker expression vector that enables simultaneous analysis of transfection efficiency and expression of fusion proteins that are PIG-linked to the cell surface (Whitehorn et al. (1995) Biotechnology (N Y) 13: 1215-9).
- Flow cytometry based cell sorting is used to select Hantaan virus glycoprotein variants that are efficiently expressed in mammalian cells. The corresponding sequences are then obtained by PCR or plasmid recovery. These chimeras/mutants are further analyzed for their capacity to protect wild mice against Hantaan virus infections.
- HSV-1 glycoprotein D
- gD glycoprotein D polypeptides that exhibit improved ability to induce protective immune responses upon administration to a mammal.
- Epidemiological studies have shown that prior infections with HSV-1 give partial protection against infections with HSV-2, indicating existence of cross-reactive immune responses.
- the main immunogenic glycoproteins in HSV appear to be gB and gD, which are encoded by 2.7 kb and 1.2 kb genes, respectively.
- the gB and gD genes of HSV- 1 are about 85% identical to the corresponding gene of HSV-2, and the gB genes of each share little sequence identity with the gD genes.
- Baboon HSV-2 gB is appr. 75% identical to human HSV-1 or -2 gB, with rather long stretches of almost 90% identity. In addition, 60- 75%) identity is found in portions of the genes of equine and bovine herpesviruses.
- Family shuffling is employed using as substrates nucleic acids that encode gB and/or gD from HSV-1 and HSV-2.
- homologous genes are obtained from HSVs of various strains.
- An alignment of gD nucleotide sequences from HSV-1 and two strains of HSV-2 is shown in Figure 7.
- Antigens encoded by the shuffled nucleic acids are expressed and analyzed in vivo. For example, one can screen for improved induction of neutralizing antibodies and/or CTL responses against HSV-1 /HSV-2.
- One can also detect protective immunity by challenging mice or guinea pigs with the viruses. Screening can be done using pools or individuals clones.
- This Example describes the use of DNA shuffling to generate immunogens that crossreact among different strains of viruses, unlike the wild-type immunogens.
- Shuffling two kinds of envelope sequences can generate immunogens that induce neutralizing antibodies against a third strain.
- Antibody-mediated neutralization of HIV- 1 is strictly type-specific. Although neutralizing activity broadens in infected individuals over time, induction of such antibodies by vaccination has been shown to be extremely difficult. Antibody-mediated protection from HIV-1 infection in vivo correlates with antibody-mediated neutralization of virus in vitro.
- Figure 8 illustrates the generation of libraries of shuffled gpl20 genes.
- gpl20 genes derived from HIV-1DH12 and HIV-1IIIB(NL43) are shuffled.
- the chimeric/mutant gpl20 genes are then analyzed for their capacity to induce antibodies that have broad spectrum capacity to neutralize different strains of HIV.
- Individual shuffled gpl20 genes are incorporated into genetic vaccine vectors, which are then introduced to mice by injection or topical application onto the skin. These antigens can also be delivered as purified recombinant proteins.
- the immune responses are measured by analyzing the capacity of the mouse sera to neutralize HIV growth in vitro. Neutralization assays are performed against HIV-1DH12, HIV-1IIIB and HIV-189.6.
- the chimeras/mutants that demonstrate broad spectrum neutralization are chosen for further rounds of shuffling and selection. Additional studies are performed in monkeys to illustrate the capacity of the shuffled gpl20 genes to provide protection for subsequent infection with immuno
- the Hepatitis B virus is one of a member of a family of viruses called hepadnaviruses. This Example describes the use of genomes and individual genes from this family are used for DNA shuffling, which results in antigens having improved properties.
- the envelope protein of the HBV assembles to form particles that carry the antigenic structures collectively known as the Hepatitis B surface antigen (HBsAg; this term is also used to designate the protein itself).
- Antibodies to the major antigenic site designated the "a" epitope (which is found in the envelope domain called S), are capable of neutralizing the virus. Immunization with the HBsAg-bearing protein thus serves as a vaccine against viral infection.
- the HBV envelope also contains other antigenic sites that can protect against viral infection and are potentially vital components of an improved vaccine.
- the epitopes are part of the envelope protein domains known as preSl and preS2 ( Figure 9). DNA shuffling of the envelope gene from several members of the hepadnavirus family is used to obtain more immunogenic proteins. Specifically, the genes from the following hepatitis viruses are shuffled:
- genes from other genotypes of the human virus are available for inclusion in the DNA shuffling reactions.
- other animal hepadnaviruses are available.
- some artificial genes are made:
- a synthetic gene is made that contains the HBV envelope sequences, except for those codons which specify amino acids found in the chimpanzee and gibbon genes. For those codons, the chimpanzee or gibbon sequence is used.
- a synthetic gene is synthesized in which the preS2 gene sequence from the human HBV adw2 strain is fused with the woodchuck S region.
- all the oligonucleotides required to chemically synthesize each of the hepadnavirus envelope genes are mixed in approximately equal quantities and allowed to anneal to form a library of sequences. After DNA shuffling of the hepadnavirus envelope genes, either or both of two strategies are used to obtain improved HBsAg antigens.
- Antigens are screened by immunizing mice using two possible methods.
- the genes are injected in the form of DNA vaccines, i.e., shuffled envelope genes carried by a plasmid that comprises the genetic regulatory elements required for expression of the envelope proteins.
- the protein is prepared from the shuffled genes and used as the immunogen.
- the individually optimized genes are then used as a combination vaccine for the induction of optimal responses to preSl-, preS2- and S-bom epitopes.
- Strategy B After isolation of the individually optimized genes as in Strategy
- the preSl, preS2 and S candidates are shuffled together, or in a pairwise fashion, in further rounds to obtain genes which encode proteins that demonstrate improved immunogenicity for at least two regions containing HBsAg epitopes (Figure 11).
- HBsAg Use of HBsAg to carry epitopes from unrelated antigens from unrelated antigens Several of the characteristics of the HBsAg make it a useful protein to carry epitopes drawn from other, unrelated antigens.
- the epitopes can be either B epitopes (which induce antibodies) or T epitopes drawn from the class I type (which stimulate CD8 + T lymphocytes and induced cytotoxic cells) or class II type (which induce helper T lymphocytes and are important in providing immunological memory responses. 1.
- B cell epitopes which induce antibodies
- T epitopes drawn from the class I type which stimulate CD8 + T lymphocytes and induced cytotoxic cells
- class II type which induce helper T lymphocytes and are important in providing immunological memory responses.
- Amino acid sequences of potential B epitopes are chosen from any pathogen. Such sequences are often known to induce antibodies, but the immunogenicity is weak or otherwise unsatisfactory for preparation of a vaccine. These sequences can also be mimotopes, which have been selected based on their ability to have a certain antigenicity or immunogenicity.
- the amino acid coding sequences are added to a hepadnavirus envelope gene.
- the heterologous sequences can either replace certain envelope sequences, or be added in addition to all the envelope sequences.
- the heterologous epitope sequences can be placed at any position in the envelope gene. A preferred position is the region of the envelope gene that encodes the major "a" epitope of the HBsAg ( Figure 12). This region is likely to be exposed on the external side of the particles formed by the envelope protein, and thus will expose the heterologous epitopes.
- DNA shuffling is carried out on the envelope gene sequences, keeping the sequence of the heterologous epitopes constant. Screening is carried out to choose candidates that are secreted into the culture medium after transfection of plasmids from the shuffled library into cells in tissue culture.
- Clones that encode a secreted protein are then tested for immunogenicity in mice either as a DNA vaccine or as a protein antigen, as described above. Clones that give an improved induction of antibodies to the heterologous epitopes are chosen for further rounds of DNA shuffling. The process is continued until the immunogenicity of the heterologous epitope is sufficient for use as a vaccine against the pathogen from which the heterologous epitopes were derived.
- Class I epitopes MHC Class I epitopes are relatively short, linear peptide sequences that are generally between 6 and 12 amino acids amino acids in length, most often 9 amino acids in length. These epitopes are processed by antigen-presenting cells either after synthesis of the epitope within the cell (usually as part of a larger protein) or after uptake of soluble protein by the cells. Polynucleotide sequences that encode one or more class I epitopes are inserted into the sequence of a hepadnavirus envelope gene either by replacing certain envelope sequences, or by inserting the epitope sequences into the envelope gene.
- heterologous class I epitopes are placed into different positions in the several hepadnavirus genes used for the DNA shuffling reaction. This will optimize the chances for finding chimeric gene carrying the epitopes in an optimal position for efficient presentation.
- Class II epitopes MHC Class II epitopes are generally required to be part of a protein which is taken up by antigen presenting cells, rather than synthesized within the cell.
- a carrier protein such as the HBV envelope that can be produced in a soluble form or which can be secreted if the gene is delivered in the form of a DNA vaccine.
- Polynucleotides that encode heterologous class II epitopes are inserted into regions of the hepadnavirus envelope genes that are not involved in the transmembrane structure of the protein. DNA shuffling is performed to obtain a secreted protein that also carries the class II epitopes.
- the protein can be taken up by antigen presenting cells for processing of the class II epitopes.
- HCV Hepatitis C Virus
- HCV envelope genes which encode envelope proteins El and E2 have been shown to induce both antibody and lymphoproliferative responses against these antigens (Lee et al. (1998) J. Virol. 72: 8430-6), and these responses can be optimized by
- HVR1 hypervariable region 1 of the envelope protein E2 of HCV is the most variable antigenic fragment in the whole viral genome and is primarily responsible for the large inter- and intra-individual heterogeneity of the infecting virus (Puntoriero et al. (1998) EMBO J. 17: 3521-33). Therefore, the gene encoding E2 is a particularly useful target for evolution by DNA shuffling.
- El, E2 provides a means to generate multivalent HCV vaccines that simultaneously protect against several strains of HCV. These antigens are shuffled using the family DNA shuffling approach.
- the starting genes will be obtained from various natural isolates of HCV.
- related genes from other viruses can be used to increase the number of different recombinants that are generated.
- a library of related, chimeric variants of HCV antigens are then generated and this library will be screened for induction of widely crossreactive immune responses.
- the screening can be done directly in vivo by injecting individual variants into test animals, such as mice or monkeys. Either purified recombinant proteins or DNA vaccines encoding the relevant genes are injected. Typically, a booster injection is given 2-3 weeks after the first injection. Thereafter, the sera of the test animals are collected and these sera are tested for the presence of antibodies that react against multiple HCV virus isolates.
- the antigens can be pre-enriched in vitro for antigens that are recognized by polyclonal antisera derived from previously infected patients or test animals.
- monoclonal antibodies that are specific for various strains of HCV are used.
- the screening is performed using phage display or ELISA assays.
- the antigen variants are expressed on bacteriophage Ml 3 and the phage are then incubated on plates coated with antisera derived from patients or test animals infected with various HCV isolates. The phage that bind to the antibodies are then eluted and further analyzed in test animals for induction of crossreactive antibodies.
- Example 11 Evolution of chimeric allergens that induce broad immune responses and have reduced risk of inducing anaphylactic reactions
- Specific immunotherapy of allergy is performed by injecting increasing amounts of the given allergens into the patients.
- the therapy typically alters the types of allergen-specific immune responses from a dominating T helper 2 (T H 2) type response to a dominating T helper 1 (T H I) type response.
- T H 2 dominating T helper 2
- T H I dominating T helper 1
- allergic patients have increased levels of IgE antibodies specific for the allergens, the immunotherapy of allergy involves a risk of IgE receptor mediated anaphylactic reactions.
- T helper (T H ) cells are capable of producing a large number of different cytokines, and based on their cytokine synthesis pattern T H cells are divided into two subsets (Paul and Seder (1994) Cell 76: 241-251).
- T H 1 cells produce high levels of IL-2 and IFN- gamma and no or minimal levels of IL-4, IL-5 and IL-13.
- T H 2 cells produce high levels of IL-4, IL-5 and IL-13, whereas IL-2 and IFN-gamma production is minimal or absent.
- T H I cells activate macrophages, dendritic cells and augment the cytolytic activity of CD8+ cytotoxic T lymphocytes and NK cells (Id.), whereas T H 2 cells provide efficient help for B cells and they also mediate allergic responses due to the capacity of T H 2 cells to induce IgE isotype switching and differentiation of B cells into IgE secreting cell (De Vries and Punnonen (1996) In Cytokine regulation of humoral immunity: basic and clinical aspects. Eds. Snapper, CM., John Wiley & Sons, Ltd., West Wales, UK, pp. 195-215
- This Example describes methods to generate chimeric allergens that can broadly modulate allergic immune responses. This can be achieved by DNA shuffling of related allergen genes to generate chimeric genes. In addition, chimeric/mutated allergens are less likely to be recognized by preexisting IgE antibodies of the patients. Importantly, allergen variants that are not recognized by IgE antibodies can be selected using patient sera and negative selection ( Figure 13). As one example, chimeric allergen variants of Der p2, Der f2, Tyr p2 Lep d2 and Gly d2 allergens are generated. These house dust mite allergens are very common in exacerbating allergic and asthmatic symptoms, and improved means to downregulate such allergic immune responses are desired.
- House dust mites can be used as sources of the genes.
- the corresponding genes are shuffled using family DNA shuffling and a shuffled library is generated.
- Phage display is used to exclude allergens that are recognized by antibodies from allergic individuals. It is particularly important is to exclude variants that are recognized by IgE antibodies. Phage expressing the allergen variants are incubated with pools of sera derived from allergic individuals. The phage that are recognized by IgE antibodies are removed, and the remaining allergens are further tested in vitro and in vivo for their capacity to activate allergen-specific human T cells (Figure 14).
- the optimal allergen variants are then further tested in vivo by studying skin responses after injections to the skin.
- a strong inflammatory response around the injection site is an indication of efficient T cell activation, and the allergen variants that induce the most efficient delayed type T cell response (typically observed 24 hours after the injection) are the best candidates for further studies in vivo to identify allergens that effectively downregulate allergic immune responses. Accordingly, these allergen variants re analyzed for their capacity to inhibit allergic responses in allergic, atopic and asthmatic individuals.
- the screening of allergen variants is further illustrated in Figure 13 and Figure 14.
- cancer cells express antigens that are present at significantly higher levels on the malignant cells than on other cells in the body. Such antigens provide excellent targets for preventive cancer vaccines and immunotherapy of cancer.
- the immunogenicity of such antigens can be improved by DNA shuffling.
- DNA shuffling provides means to improve expression levels of cancer antigens.
- This Example describes methods to generate cancer antigens that can efficiently induce anti-tumor immune responses by DNA shuffling of related cancer antigen genes.
- Libraries of shuffled melanoma-associated glycoprotein (gpl00/pmell7) genes (Huang et al. (1998) J. Invest. Dermatol. I l l : 662-7) are generated.
- the genes can be isolated from melanoma cells obtained from various patients, who may have mutations of the gene, increasing the diversity in the starting genes.
- a gplOO gene can be isolated from other mammalian species to further increase the diversity of starting genes.
- a typical method for the isolation of the genes is RT-PCR.
- the corresponding genes are shuffled using single gene DNA shuffling or family DNA shuffling and a shuffled library is generated.
- the shuffled gplOO variants are subsequently injected into test animals, and the immune responses are studied (Figure 15).
- the shuffled antigens are either expressed in E. coli and recombinant, purified proteins are injected, or the antigen genes are used as components of DNA vaccines.
- the immune response can be analyzed for example by measuring anti-gplOO antibodies, as previous studies indicate that the antigen can induce specific antibody responses (Huang et al., supra.).
- the test animals that can be challenged by malignant cells expressing gplOO.
- cytotoxic T cells specific for gp 100 and will survive the challenge, whereas in non-immunized or poorly immunized animals the malignant cells will efficiently grow eventually resulting in lethal expansion of the cells.
- antigens that induce cytotoxic T cells that have the capacity to kill cancer cells can be identified by measuring the capacity of T cells derived from immunized animals to kill cancer cells in vitro. Typically the cancer cells are first labeled with radioactive isotopes and the release of radioactivity is an indication of tumor cell killing after incubation in the presence of T cells from immunized animals. Such cytotoxicity assays are known in the art.
- the antigens that induce highest levels of specific antibodies and/or can protect against the highest number of malignant cells can be chosen for additional rounds of shuffling and screening.
- Mice are useful test animals because large numbers of antigens can be studied.
- monkeys are a preferred test animal, because the MHC molecules of monkeys are very similar to those of humans.
- peripheral blood mononuclear cells from previously infected or immunized humans individuals can be used. This is a particularly useful method, because the MHC molecules that will present the antigenic peptides are human MHC molecules.
- Shuffled cancer antigens that induce cytotoxic T cells that have the capacity to kill cancer cells can be identified by measuring the capacity of T cells derived from immunized animals to kill cancer cells in vitro. Typically the cancer cells are first labeled with radioactive isotopes and the release of radioactivity is an indication of tumor cell killing after incubation in the presence of T cells from immunized animals. Such cytotoxicity assays are known in the art.
- Example 13 Evolution of autoantigens that induce efficient immune responses
- Autoimmune diseases are characterized by an immune response directed against self antigens expressed by the host.
- Autoimmune responses are generally mediated by T H 1 cells that produce high levels of IL-2 and IFN-gamma.
- Vaccines that can direct autoantigen specific T cells towards T H 2 phenotype producing increased levels of IL-4 and IL-5 would be beneficial.
- the vaccine antigens have to be able to efficiently activate specific T cells.
- DNA shuffling can be used to generate antigens that have such properties.
- cytokines that have been shown to enhance TH2 cell activation and differentiation, such as IL-4 (Racke et al. (1994) J. Exp. Med. 180: 1961-66).
- This Example describes methods for generating autoantigens that can efficiently induce immune responses.
- DNA shuffling is performed on related autoantigen genes. For example, libraries of shuffled myelin basic proteins, or fragments thereof (Zamvil and Steinman (1990) Ann. Rev. Immunol 8: 579-621); Brocke et al. (1996) Nature 379: 343- 46) are generated.
- MBP is considered to be an important autoantigen in patients with multiple sclerosis (MS).
- the genes encoding MBP from at least bovine, mouse, rat, guinea pig and human have been isolated providing an excellent starting point for family shuffling.
- a typical method for the isolation of the genes is RT-PCR.
- the shuffled MBP variants are subsequently injected into test animals, and the immune responses are studied.
- the shuffled antigens are either expressed in E. coli and recombinant, purified proteins are injected, or the antigen genes are used as components of DNA vaccines or viral vectors.
- the immune response can be analyzed for example by measuring anti-MBP antibodies by ELISA.
- the lymphocytes derived from immunized test animals are activated with MBP, and the T cell proliferation or cytokine synthesis is studies.
- a sensitive assays for cytokine synthesis is ELISPOT (McCutcheon et al. (1997) J. Immunol. Methods 210: 149-66). Mice are useful test animals because large numbers of antigens can be studied. However, monkeys are a preferred test animal, because the MHC molecules of monkeys are very similar to those of humans.
- peripheral blood mononuclear cells from patients with MS can also be used. This is a particularly useful method, because the MHC molecules that will present the antigenic peptides are human MHC molecules.
- Shuffled antigens that activate MBP specific T cells can be identified by measuring the capacity of T cells derived from MS patients to proliferate or produce cytokines upon culture in the presence of the antigen variants. Such assays are known in the art. One such assay is ELISPOT (McCutcheon et al, supra.).
- An indication of the efficacy of an MBP variant to activate specific T cells is also the degree of skin inflammation when the antigen is injected into the skin of a patient with MS. Strong inflammation is correlated with strong activation of antigen-specific T cells. Improved activation of MBP specific T cells, particularly in the presence of IL-4, is likely to result in enhanced T H 2 cell responses, which are beneficial in the treatment of MS patients.
- This Example describes methods by which the envelope protein sequence of the hepatitis B virus can be evolved to provide a more immunogenic surface antigen.
- Such a protein is important for vaccination of low responders and for immunotherapy of chronic hepatitis B.
- HBV vaccines are based on the immunogenicity of the viral envelope protein and contain the Major (or Small) form of the envelope protein produced as particles in yeast. These particles induce antibodies to the major surface antigen (HBsAg) which can protect against infection when antibody levels are at least 10 milli- International Units per milliliter (mU/ml). These recombinant protein preparations are not capable of inducing humoral immunity in chronic carriers (some 300 million cases worldwide) the induction of which would be important to control virus spread. Moreover, certain individuals respond poorly to the vaccine (up to 30-50% of vaccinees in some groups) and do not develop protective levels of antibody.
- HBsAg major surface antigen
- the inclusion of the natural epitope sequences contained in the Middle or Large forms of the viral envelope protein has been used as a method to increase the immunogenicity of vaccine preparations.
- An alternative method is to introduce new (i.e., not present in the natural virus sequence) helper T-cell epitopes into the HBsAg sequence using DNA shuffling technology.
- DNA sequences of HBsAg from different subtypes of HBV are prepared for shuffling. Comparison of the genes encoding these proteins suggests that recombination would occur at least ten times within 850 base pairs when shuffling the ayw and woodchuck hepatitis virus (WHV) DNA sequences. Nucleotide and amino acid sequences of portions of different subtypes of HBV are shown in Figure 17. The sequence of the main HBsAg B-cell antigenic site (the "a" epitope) can be retained in the protein sequence by including the coding sequences of the external "a" loop in the final protein preparation.
- HBV woodchuck hepatitis virus
- B or T helper or CTL
- the availability of the "a" loop on the HBsAg may provide a region of the envelope protein into which other artificial antigens or mimotopes could be included.
- HBV envelope sequence is prepared to include a specific epitope (from HBV, another pathogen or a tumor cell)
- shuffling of the surrounding sequences in the HBV envelope will serve to optimize expression of the protein and help to ensure that the immune response is directed to the desired epitope.
- HBsAg sequences are evaluated for their ability to induce an immune response to the clinically relevant HBsAg epitopes. This can be done using mice of the H-2s and H-2f haplotypes, which respond poorly or not at all to HBsAg protein immunization. In these experiments, one can verify that antibodies are generated to the main "a" epitope in the S protein, and a second protective epitope in the PreS2 region (a linear sequence).
- the PreS2 and S coding sequences for the envelope protein (HBsAg) from the HBV ayw subtype (plasmid pCAG-M-Kan; Whalen) and the WHV (plasmid pWHV8 from ATCC) are amplified from the two plasmids by PCR and shuffled. Examples of suitable primers for PCR amplification are shown in Figure 18.
- the shuffled library of sequences is cloned into an HBsAg-expression vector and individual colonies are chosen for preparation of plasmid DNA. The DNA is administered to the test animals and vectors which induce the desired immune response are identified and recovered.
- This example describes methods of using the evolved HBsAg protein to present natural HBsAg CTL epitopes. Shuffling is used to increase overall immunogenicity of the HBsAg protein, as discussed above. However, some of the evolved HBsAg sequences are replaced with class I or class II epitope sequences from the natural HBsAg protein in order to stimulate immunoreactivity specifically to these natural viral epitopes.
- the natural viral epitopes can be added to the evolved protein without loss of immunogenicity of the evolved HBsAg.
- This example describes methods of using the evolved HBsAg protein is used to express tumor-derived CTL epitopes.
- the overall immunogenicity of the HBsAg protein is increased by shuffling.
- some of the evolved HBsAg sequences are replaced with class I or class II epitope sequences from tumor cells in order to stimulate immunoreactivity specifically to these natural viral epitopes.
- the tumor cells epitopes can be added to the evolved protein without loss of immunogenicity of the evolved HBsAg.
- This example describes the use of an evolved HBsAg protein for expression of mimotope sequences.
- the evolved HBsAg protein is used to increase overall immunogenicity of the protein.
- some of the evolved HBsAg sequences are replaced with mimotope sequences to stimulate immunoreactivity specifically to the natural sequence which cross reacts with the mimotope.
- the mimotope sequences can be added to the evolved protein without loss of immunogenicity of the evolved HBsAg.
- Fusion Proteins Of The HBsAg Polypeptide and HIV gpl20 Protein This Example describes the preparation of fusion proteins ("chimeras") formed from the HBsAg polypeptide and the extracellular fragment gpl20 of the HIV envelope protein, and their use as vaccines.
- a fusion is prepared between gpl20 sequences (on the N-terminus of the fusion) and HBsAg sequences (on the C-terminal of the fusion).
- the N-terminal peptide sequence of the S region of the HBsAg polypeptide is a transmembrane structure which is locked into the membrane of the endoplasmic reticulum.
- the actual N-terminus of the S region as well as the preS2 sequences are located in the lumenal part of the ER. They are found on the outside of the final HBsAg particles.
- the gpl20 sequences are also located on the outside of the particles.
- the gpl20 molecules can thus be brought together in three-dimensional space to interact as in the virus.
- DNA shuffling is employed.
- the sequences of the HBsAg polypeptide, which functions as a scaffold, and of gpl20 are both shuffled. Screening of the shuffled products can be performed by ELISA assay using antibodies (polyclonal or monoclonal) which have previously been determined to have virus neutralizing activity.
- sequences encoding the gpl20 fragment of the HIV envelope protein are preferably prepared as a synthetic gene to include codons which are optimal for gene expression in mammals.
- the gpl20 sequence will typically include a signal sequence on its N-terminal end.
- the gpl20 sequences are inserted into the preS2 region of an HBsAg- expressing plasmid.
- an EcoRI site and anXhol site are available for cloning.
- the gpl20 sequences can be inserted between these two sites, which brings the gpl20 closer to the start of the S coding sequences, or into the EcoRI site alone, which leaves a spacer sequence of about 50 amino acids between the gpl20 sequence and the start of the S region of the HBsAg.
- DNA shuffling of the entire chimeric sequence is carried out.
- Family shuffling is prefened; this involves the preparation of several gpl20-HBsAg fusion proteins in which different gpl20 and HBsAg (or WHV) sequences are used.
- An alignment of HBsAg nucleotide sequences is shown in Figure 19.
- the products are cloned into an expression vector such as pMKan. Pools of clones from the library of shuffled products are transfected into cultured cells and the secretion of chimeric proteins is assayed with broadly reactive antibodies to gpl20.
- Positive clones can be further evaluated with particular antibodies that have demonstrated HIV neutralizing activity, for example the anti-CD4 binding domain recombinant human monoclonal antibody, IgGlbl2 (Kessler et al. (1997) AIDS Res. Hum. Retroviruses . 1 : 13: 575-582; Roben et al. (1994) J. Virol. 68: 4821-4828).
- IgGlbl2 the anti-CD4 binding domain recombinant human monoclonal antibody
- Candidate clones can then be used to immunize mice and the antiserum obtained is evaluated for HIV virus-neutralizing activity in in vitro assays.
- the gpl20 molecule (approx. 1100 amino acids) is larger in size than the monomeric HBsAg preS2+S protein (282 amino acids), it is likely that not every HBsAg monomer in an aggregated particle will contain a gpl20 sequence. Internal initiation of protein synthesis can take place on the HBsAg coding sequences at the initiator methionine that marks the beginning of the S region. Thus, the chimeric molecule (which contains the gpl20 sequences) will be mixed in the cell with the S region and the multimeric particles should assemble with an appropriate number of chimeric polypeptides and native HBsAg S monomers.
- an S-expressing plasmid can be mixed with the plasmid expressing the chimera, or a single plasmid which expresses the chimera and the S form can be constructed.
- a diagram of the resulting particles is shown in Figure 20.
- This Example describes the use of DNA shuffling to obtain HSV glycoprotein B (gB) and glycoprotein D (gD) polypeptides that exhibit improved ability to induce protective immune responses upon administration to a mammal. Epidemiological studies have shown that prior infections with HSV- 1 give partial protection against infections with
- HSV-2 indicating existence of cross-reactive immune responses.
- the main immunogenic glycoproteins in HSV appear to be gB and gD, which are encoded by 2.7 kb and 1.2 kb genes, respectively.
- HSV-2 gB are about 85% identical to the conesponding gene of HSV-2, and the gB genes of each share little sequence identity with the gD genes.
- Baboon HSV-2 gB is appr. 75% identical to human HSV-1 or -2 gB, with rather long stretches of almost 90% identity.
- This Example describes the use of DNA shuffling to generate immunogens that crossreact among different strains of viruses, unlike the wild-type immunogens. Shuffling two kinds of envelope sequences can generate immunogens that induce neutralizing antibodies against a third strain.
- Antibody-mediated neutralization of HIV- 1 is strictly type-specific. Although neutralizing activity broadens in infected individuals over time, induction of such antibodies by vaccination has been shown to be extremely difficult. Antibody-mediated protection from HIV-1 infection in vivo correlates with antibody-mediated neutralization of virus in vitro.
- Figure 8 illustrates the generation of libraries of shuffled gpl20 genes.
- gpl20 genes derived from HIV-1DH12 and HIV-1IIIB(NL43) are shuffled.
- the chimeric/mutant gpl20 genes are then analyzed for their capacity to induce antibodies that have broad spectrum capacity to neutralize different strains of HIV.
- Individual shuffled gpl20 genes are incorporated into genetic vaccine vectors, which are then introduced to mice by injection or topical application onto the skin. These antigens can also be delivered as purified recombinant proteins.
- the immune responses are measured by analyzing the capacity of the mouse sera to neutralize HIV growth in vitro. Neutralization assays are performed against HIV-1DH12, HIV-1IIIB and HIV-189.6.
Abstract
Description













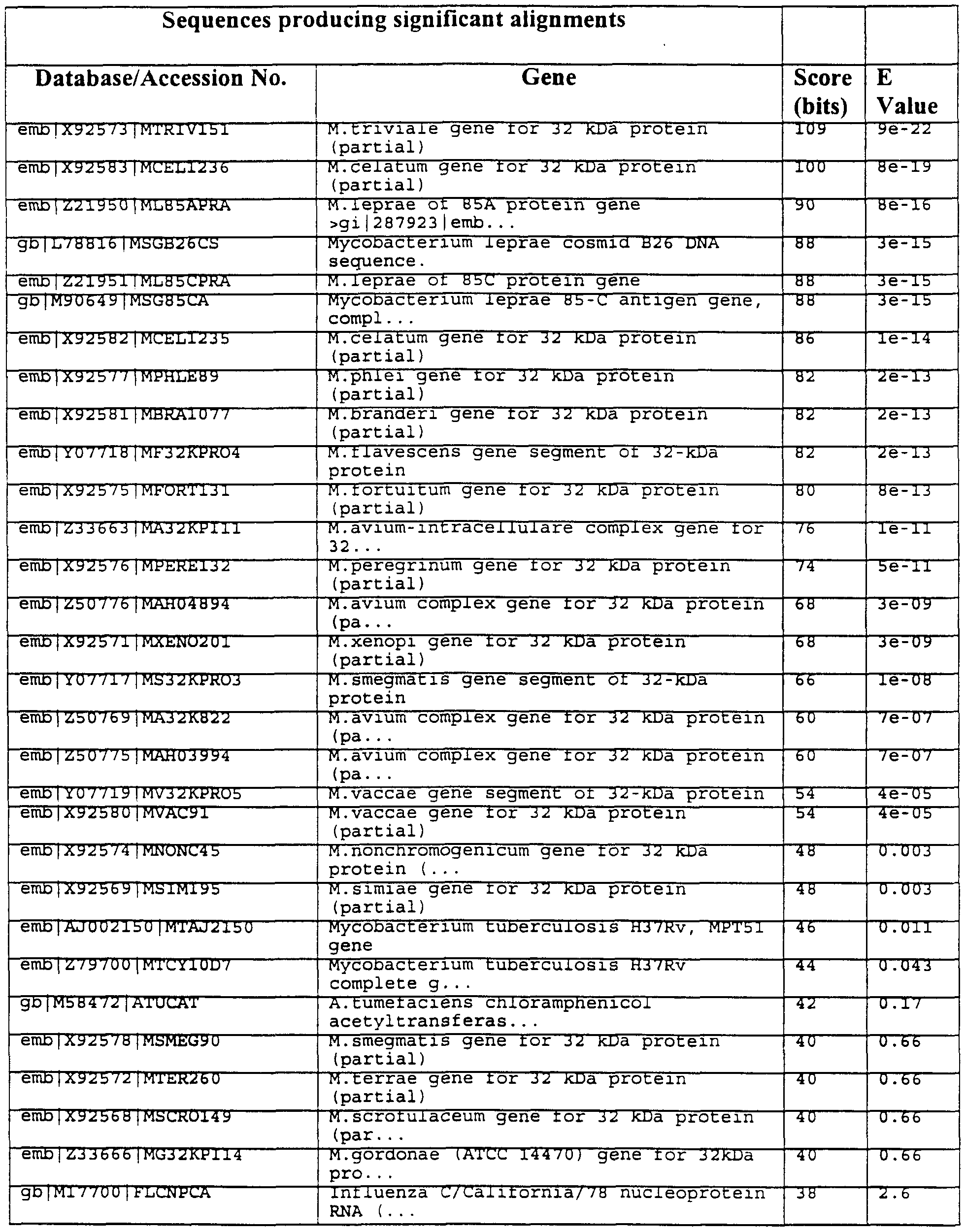






Claims
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA 2320958 CA2320958A1 (en) | 1998-02-11 | 1999-02-10 | Antigen library immunization |
| AU32891/99A AU3289199A (en) | 1998-02-11 | 1999-02-10 | Antigen library immunization |
| JP2000531564A JP2002507393A (en) | 1998-02-11 | 1999-02-10 | Antigen library immunization |
| EP19990932510 EP1054973A1 (en) | 1998-02-11 | 1999-02-10 | Antigen library immunization |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US2176998A | 1998-02-11 | 1998-02-11 | |
| US7429498P | 1998-02-11 | 1998-02-11 | |
| US09/021,769 | 1998-02-11 | ||
| US60/074,294 | 1998-02-11 | ||
| US10550998P | 1998-10-23 | 1998-10-23 | |
| US60/105,509 | 1998-10-23 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO1999041383A1 true WO1999041383A1 (en) | 1999-08-19 |
| WO1999041383A8 WO1999041383A8 (en) | 1999-09-30 |
Family
ID=27361726
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1999/002944 WO1999041383A1 (en) | 1998-02-11 | 1999-02-10 | Antigen library immunization |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP1054973A1 (en) |
| JP (1) | JP2002507393A (en) |
| AU (1) | AU3289199A (en) |
| CA (1) | CA2320958A1 (en) |
| WO (1) | WO1999041383A1 (en) |
Cited By (116)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000028009A1 (en) * | 1998-11-11 | 2000-05-18 | Melbourne Health | Biological compositions, components thereof and uses therefor |
| WO2001022083A2 (en) * | 1999-09-21 | 2001-03-29 | GSF-Forschungszentrum für Umwelt und Gesundheit GmbH | Method for identifying mhc-restricted antigens |
| WO2001040803A1 (en) * | 1999-12-03 | 2001-06-07 | Diversys Limited | Direct screening method |
| WO2001044828A1 (en) * | 1999-12-17 | 2001-06-21 | Maxygen, Inc. | Methods for parallel detection of compositions having desired characteristics by means of mri spectroscopy |
| US6287862B1 (en) | 1997-01-17 | 2001-09-11 | Maxygen, Inc. | Evolution of whole cells and organisms by recursive sequence recombination |
| US6319714B1 (en) | 1999-01-19 | 2001-11-20 | Maxygen, Inc. | Oligonucleotide mediated nucleic acid recombination |
| US6337186B1 (en) | 1998-06-17 | 2002-01-08 | Maxygen, Inc. | Method for producing polynucleotides with desired properties |
| US6399383B1 (en) | 1997-10-28 | 2002-06-04 | Maxygen, Inc. | Human papilloma virus vectors |
| US6436675B1 (en) | 1999-09-28 | 2002-08-20 | Maxygen, Inc. | Use of codon-varied oligonucleotide synthesis for synthetic shuffling |
| US6483011B1 (en) | 1998-11-10 | 2002-11-19 | Maxygen, Inc. | Modified ADP-glucose pyrophosphorylase for improvement and optimization of plant phenotypes |
| US6500639B2 (en) | 1998-10-07 | 2002-12-31 | Maxygen, Inc. | DNA shuffling to produce nucleic acids for mycotoxin detoxification |
| US6531316B1 (en) | 1999-03-05 | 2003-03-11 | Maxyag, Inc. | Encryption of traits using split gene sequences and engineered genetic elements |
| US6596539B1 (en) | 1997-10-31 | 2003-07-22 | Maxygen, Inc. | Modification of virus tropism and host range by viral genome shuffling |
| US6605430B1 (en) | 1998-08-12 | 2003-08-12 | Maxygen, Inc. | DNA shuffling of monooxygenase genes for production of industrial chemicals |
| WO2003075129A2 (en) | 2002-03-01 | 2003-09-12 | Maxygen, Inc. | Methods, systems, and software for identifying functional bio-molecules |
| WO2003078583A2 (en) | 2002-03-09 | 2003-09-25 | Maxygen, Inc. | Optimization of crossover points for directed evolution |
| WO2003087128A2 (en) * | 2002-04-09 | 2003-10-23 | Rmf Dictagene S.A. | Secretory signal sequences and uses thereof |
| US6686515B1 (en) | 1999-11-23 | 2004-02-03 | Maxygen, Inc. | Homologous recombination in plants |
| WO2004041200A2 (en) * | 2002-11-04 | 2004-05-21 | Picoscript Ltd, Llp | Dna based plasmid formulations and vaccines and prophylactics containing the same |
| WO2004090099A2 (en) | 2003-04-04 | 2004-10-21 | Diversa Corporation | Pectate lyases, nucleic acids encoding them and methods for making and using them |
| US6858422B2 (en) | 2000-07-13 | 2005-02-22 | Codexis, Inc. | Lipase genes |
| WO2005021714A2 (en) | 2003-08-11 | 2005-03-10 | Diversa Corporation | Laccases, nucleic acids encoding them and methods for making and using them |
| JP2005510533A (en) * | 2001-11-21 | 2005-04-21 | ザ ボード オブ トラスティーズ オブ レランド スタンフォード ジュニア ユニバーシティ | Polynucleotide therapy |
| WO2005123929A2 (en) | 2004-06-09 | 2005-12-29 | Pioneer Hi-Bred International, Inc. | Plastid transit peptides |
| WO2006009676A2 (en) | 2004-06-16 | 2006-01-26 | Diversa Corporation | Compositions and methods for enzymatic decolorization of chlorophyll |
| US6998263B2 (en) | 2000-02-09 | 2006-02-14 | Genvec, Inc. | Methods of preparing and using a viral vector library |
| EP1678203A2 (en) * | 2003-09-25 | 2006-07-12 | Hadasit Medical Research Services And Development Ltd. | Multiepitope polypeptides for cancer immunotherapy |
| US7094875B2 (en) | 2000-06-23 | 2006-08-22 | Maxygen, Inc. | Co-stimulatory polypeptides |
| WO2006101584A2 (en) | 2005-03-15 | 2006-09-28 | Diversa Corporation | Cellulases, nucleic acids encoding them and methods for making and using them |
| WO2006125983A1 (en) * | 2005-05-23 | 2006-11-30 | Oxxon Therapeutics Ltd | Compositions for inducing an immune response against hepatitis b |
| WO2007095398A2 (en) | 2006-02-14 | 2007-08-23 | Verenium Corporation | Xylanases, nucleic acids encoding them and methods for making and using them |
| WO2008036863A2 (en) | 2006-09-21 | 2008-03-27 | Verenium Corporation | Phospholipases, nucleic acids encoding them and methods for making and using them |
| US7367155B2 (en) | 2000-12-20 | 2008-05-06 | Monsanto Technology Llc | Apparatus and methods for analyzing and improving agricultural products |
| US7384387B1 (en) | 1999-02-11 | 2008-06-10 | Maxygen, Inc. | High throughput mass spectrometry |
| WO2008080093A2 (en) | 2006-12-21 | 2008-07-03 | Verenium Corporation | Amylases and glucoamylases, nucleic acids encoding them and methods for making and using them |
| US7407661B2 (en) | 1997-06-09 | 2008-08-05 | Oxxon Therapeutics Limited | Methods and reagents that generate a CD8 T cell immune response |
| WO2008095033A2 (en) | 2007-01-30 | 2008-08-07 | Verenium Corporation | Enzymes for the treatment of lignocellulosics, nucleic acids encoding them and methods for making and using them |
| WO2008098198A2 (en) | 2007-02-08 | 2008-08-14 | The California Institute Of Technology | Alkane oxidation by modified hydroxylases |
| US7476390B2 (en) | 2002-02-26 | 2009-01-13 | Maxygen, Inc. | Flavivirus antigens |
| WO2009045627A2 (en) | 2007-10-03 | 2009-04-09 | Verenium Corporation | Xylanases, nucleic acids encoding them and methods for making and using them |
| EP1996220A4 (en) * | 2006-03-06 | 2009-04-29 | Amunix Inc | Unstructured recombinant polymers and uses thereof |
| WO2009088949A1 (en) | 2008-01-03 | 2009-07-16 | Verenium Corporation | Transferases and oxidoreductases, nucleic acids encoding them and methods for making and using them |
| US7600642B2 (en) | 2003-09-23 | 2009-10-13 | Monsanto Technology, Llc | High throughput automated seed analysis system |
| EP2112227A1 (en) | 2006-03-07 | 2009-10-28 | Cargill, Incorporated | Aldolases, nucleic acids encoding them and methods for making and using them |
| JP2009543541A (en) * | 2006-03-06 | 2009-12-10 | アムニクス, インコーポレイテッド | Non-structural recombinant polymers and uses thereof |
| WO2010025395A2 (en) | 2008-08-29 | 2010-03-04 | Verenium Corporation | Hydrolases, nucleic acids encoding them and methods for making and using them |
| US7685768B2 (en) | 2004-08-26 | 2010-03-30 | Monsanto Technology Llc | Automated testing of seeds |
| EP2194133A2 (en) | 2003-03-06 | 2010-06-09 | Verenium Corporation | Amylases, nucleic acids encoding them and methods for making and using them |
| EP2216403A2 (en) | 2006-02-02 | 2010-08-11 | Verenium Corporation | Esterases and related nucleic acids and methods |
| EP2253704A1 (en) | 1999-01-19 | 2010-11-24 | Maxygen, Inc. | Oligonucleotide mediated nucleic acid recombination |
| WO2010135588A2 (en) | 2009-05-21 | 2010-11-25 | Verenium Corporation | Phytases, nucleic acids encoding them and methods for making and using them |
| US7846445B2 (en) | 2005-09-27 | 2010-12-07 | Amunix Operating, Inc. | Methods for production of unstructured recombinant polymers and uses thereof |
| US7855279B2 (en) | 2005-09-27 | 2010-12-21 | Amunix Operating, Inc. | Unstructured recombinant polymers and uses thereof |
| EP2275536A1 (en) | 2002-08-06 | 2011-01-19 | Verdia, Inc. | AP1 amine oxidase variants |
| WO2011022597A1 (en) | 2009-08-20 | 2011-02-24 | Pioneer Hi-Bred International, Inc. | Functional expression of shuffled yeast nitrate transporter (ynti) in maize to improve nitrate uptake under low nitrate environment |
| EP2298871A1 (en) | 2002-04-19 | 2011-03-23 | Verenium Corporation | Phospholipases, nucleic acids encoding them and methods for making and using them |
| WO2011046815A1 (en) | 2009-10-16 | 2011-04-21 | Bunge Oils, Inc. | Oil degumming methods |
| WO2011046812A1 (en) | 2009-10-16 | 2011-04-21 | Verenium Corporation | Phospholipases, nucleic acids encoding them and methods for making and using them |
| US7934600B2 (en) | 2002-04-04 | 2011-05-03 | Monsanto Technology Llc | Automated picking, weighing and sorting system for particulate matter |
| EP2322629A2 (en) | 2003-04-29 | 2011-05-18 | Pioneer Hi-Bred International Inc. | Novel glyphosate-n-acetyltransferase (GAT) genes |
| WO2011082304A1 (en) | 2009-12-31 | 2011-07-07 | Pioneer Hi-Bred International, Inc. | Engineering plant resistance to diseases caused by pathogens |
| EP2385108A1 (en) | 2006-03-07 | 2011-11-09 | Verenium Corporation | Aldolases, nucleic acids encoding them and methods for making and using them |
| EP2397486A1 (en) | 2006-09-21 | 2011-12-21 | Verenium Corporation | Phytases, nucleic acids encoding them and methods for making and using them |
| EP2404929A1 (en) | 2003-07-02 | 2012-01-11 | Verenium Corporation | Glucanases, nucleic acids encoding them and methods for making and using them |
| EP2415864A1 (en) | 2006-02-10 | 2012-02-08 | Verenium Corporation | Oligomerase-2 (or beta-xylosidase) enzymes, nucleic acids encoding them and methods for making and using them |
| WO2012021785A1 (en) | 2010-08-13 | 2012-02-16 | Pioneer Hi-Bred International, Inc. | Compositions and methods comprising sequences having hydroxyphenylpyruvate dioxygenase (hppd) activity |
| EP2444413A1 (en) | 2006-08-04 | 2012-04-25 | Verenium Corporation | Methods for oil or gas well drilling, washing and/or fracturing |
| US8282935B2 (en) | 2001-07-30 | 2012-10-09 | Isis Innovation Limited | Materials and methods relating to improved vaccination strategies |
| EP2559703A1 (en) | 2007-02-08 | 2013-02-20 | Domantis Limited | Antibody single variable domains against serum albumin |
| WO2013166113A1 (en) | 2012-05-04 | 2013-11-07 | E. I. Du Pont De Nemours And Company | Compositions and methods comprising sequences having meganuclease activity |
| EP2706122A2 (en) | 2008-01-03 | 2014-03-12 | Verenium Corporation | Isomerases, nucleic acids encoding them and methods for making and using them |
| US8673860B2 (en) | 2009-02-03 | 2014-03-18 | Amunix Operating Inc. | Extended recombinant polypeptides and compositions comprising same |
| WO2014153242A1 (en) | 2013-03-14 | 2014-09-25 | Pioneer Hi-Bred International, Inc. | Compositions having dicamba decarboxylase activity and methods of use |
| WO2014150914A2 (en) | 2013-03-15 | 2014-09-25 | Pioneer Hi-Bred International, Inc. | Phi-4 polypeptides and methods for their use |
| WO2014153234A1 (en) | 2013-03-14 | 2014-09-25 | Pioneer Hi-Bred International, Inc. | Compositions having dicamba decarboxylase activity and methods of use |
| WO2014159714A1 (en) * | 2013-03-14 | 2014-10-02 | Altravax, Inc. | Hepatitis b virus vaccines |
| US8933197B2 (en) | 2007-08-15 | 2015-01-13 | Amunix Operating Inc. | Compositions comprising modified biologically active polypeptides |
| WO2015023846A2 (en) | 2013-08-16 | 2015-02-19 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| WO2015038734A2 (en) | 2013-09-13 | 2015-03-19 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| EP2853593A1 (en) | 2003-03-07 | 2015-04-01 | DSM IP Assets B.V. | Hydrolases, nucleic acids encoding them and mehods for making and using them |
| EP2865754A1 (en) | 1999-06-14 | 2015-04-29 | BP Corporation North America Inc. | Synthetic ligation reassembly in directed evolution |
| US9027278B2 (en) | 2006-03-02 | 2015-05-12 | Monsanto Technology Llc | Automated contamination-free seed sampler and methods of sampling, testing and bulking seeds |
| EP2886658A1 (en) | 2005-03-10 | 2015-06-24 | BASF Enzymes LLC | Lyase enzymes, nucleic acids encoding them and methods for making and using them |
| WO2015120270A1 (en) | 2014-02-07 | 2015-08-13 | Pioneer Hi Bred International, Inc. | Insecticidal proteins and methods for their use |
| WO2015120276A1 (en) | 2014-02-07 | 2015-08-13 | Pioneer Hi Bred International Inc | Insecticidal proteins and methods for their use |
| WO2016038895A1 (en) * | 2014-09-11 | 2016-03-17 | Vlp Therapeutics, Llc | Flavivirus virus like particle |
| WO2016061206A1 (en) | 2014-10-16 | 2016-04-21 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| US9376672B2 (en) | 2009-08-24 | 2016-06-28 | Amunix Operating Inc. | Coagulation factor IX compositions and methods of making and using same |
| US9387518B2 (en) | 2006-06-28 | 2016-07-12 | Monsanto Technology Llc | Small object sorting system and method |
| WO2016144688A1 (en) | 2015-03-11 | 2016-09-15 | Pioneer Hi Bred International Inc | Insecticidal combinations of pip-72 and methods of use |
| WO2016186986A1 (en) | 2015-05-19 | 2016-11-24 | Pioneer Hi Bred International Inc | Insecticidal proteins and methods for their use |
| EP3115459A2 (en) | 2008-05-23 | 2017-01-11 | E. I. du Pont de Nemours and Company | Novel dgat genes for increased seed storage lipid production and altered fatty acid profiles in oilseed plants |
| WO2017023486A1 (en) | 2015-08-06 | 2017-02-09 | Pioneer Hi-Bred International, Inc. | Plant derived insecticidal proteins and methods for their use |
| WO2017105987A1 (en) | 2015-12-18 | 2017-06-22 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| EP3190182A1 (en) | 2004-03-08 | 2017-07-12 | DSM IP Assets B.V. | Phospholipases, nucleic acids encoding them and methods for making and using them |
| WO2017192560A1 (en) | 2016-05-04 | 2017-11-09 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| WO2018005411A1 (en) | 2016-07-01 | 2018-01-04 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins from plants and methods for their use |
| WO2018084936A1 (en) | 2016-11-01 | 2018-05-11 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| US9969986B2 (en) | 2014-08-08 | 2018-05-15 | Vlp Therapeutics, Llc | Virus like particle comprising modified envelope protein E3 |
| US9986699B2 (en) | 2004-08-26 | 2018-06-05 | Monsanto Technology Llc | Methods of seed breeding using high throughput nondestructive seed sampling |
| WO2018111551A1 (en) | 2016-12-14 | 2018-06-21 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| WO2018118811A1 (en) | 2016-12-22 | 2018-06-28 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| WO2018148001A1 (en) | 2017-02-08 | 2018-08-16 | Pioneer Hi-Bred International Inc | Insecticidal combinations of plant derived insecticidal proteins and methods for their use |
| WO2018208882A1 (en) | 2017-05-11 | 2018-11-15 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| EP3460061A1 (en) | 2008-09-26 | 2019-03-27 | Tocagen Inc. | Recombinant replication competent murine leukemia gamma-retrovirus (mlv) for the expression of heat-stabilized cytosine deaminase |
| US10370430B2 (en) | 2012-02-15 | 2019-08-06 | Bioverativ Therapeutics Inc. | Recombinant factor VIII proteins |
| US10385101B2 (en) | 2014-08-08 | 2019-08-20 | Vlp Therapeutics, Llc | Virus like particle comprising modified envelope protein E3 |
| US10421798B2 (en) | 2012-02-15 | 2019-09-24 | Bioverativ Therapeutics Inc. | Factor VIII compositions and methods of making and using same |
| US10464986B2 (en) | 2013-07-12 | 2019-11-05 | Vlp Therapeutics, Llc | Virus like particle comprising PD-1 antigen or PD-1 ligand antigen |
| US10542661B2 (en) | 2006-03-02 | 2020-01-28 | Monsanto Technology Llc | Automated high-throughput seed sampler and methods of sampling, testing and bulking seeds |
| US10548953B2 (en) | 2013-08-14 | 2020-02-04 | Bioverativ Therapeutics Inc. | Factor VIII-XTEN fusions and uses thereof |
| US10745680B2 (en) | 2015-08-03 | 2020-08-18 | Bioverativ Therapeutics Inc. | Factor IX fusion proteins and methods of making and using same |
| US10925950B2 (en) | 2015-08-03 | 2021-02-23 | University Of Washington | Immunogenic compositions, antigen screening methods, and methods of generating immune responses |
| CN113624953A (en) * | 2015-05-11 | 2021-11-09 | 亿明达股份有限公司 | Platform for discovery and analysis of therapeutic agents |
| WO2022015619A2 (en) | 2020-07-14 | 2022-01-20 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| US11345726B2 (en) | 2012-02-16 | 2022-05-31 | VLP Theranentics. Inc. | Chikungunya virus (CHIKV) or Venezuelan equine encephalitis virus (VEEV) virus-like particles comprising heterologous antigens inserted into the envelope protein |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2166830B1 (en) | 2007-05-31 | 2013-08-07 | Monsanto Technology, LLC | Seed sorter |
| EP2000531A1 (en) * | 2007-06-06 | 2008-12-10 | Biomay AG | Antigen presenting cells |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997020078A1 (en) * | 1995-11-30 | 1997-06-05 | Maxygen, Inc. | Methods for generating polynucleotides having desired characteristics by iterative selection and recombination |
| US5691170A (en) * | 1989-04-18 | 1997-11-25 | Therion Biologics | Generation of hybrid genes and proteins by virus-mediated recombination |
-
1999
- 1999-02-10 JP JP2000531564A patent/JP2002507393A/en not_active Withdrawn
- 1999-02-10 EP EP19990932510 patent/EP1054973A1/en not_active Withdrawn
- 1999-02-10 AU AU32891/99A patent/AU3289199A/en not_active Abandoned
- 1999-02-10 CA CA 2320958 patent/CA2320958A1/en not_active Abandoned
- 1999-02-10 WO PCT/US1999/002944 patent/WO1999041383A1/en not_active Application Discontinuation
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5691170A (en) * | 1989-04-18 | 1997-11-25 | Therion Biologics | Generation of hybrid genes and proteins by virus-mediated recombination |
| WO1997020078A1 (en) * | 1995-11-30 | 1997-06-05 | Maxygen, Inc. | Methods for generating polynucleotides having desired characteristics by iterative selection and recombination |
Non-Patent Citations (1)
| Title |
|---|
| ANDREAS CRAMERI ET AL.: "DNA shuffling of a family of genes from diverse species accelerates directed evolution", NATURE, vol. 391, no. 6664, 15 January 1998 (1998-01-15), LONDON GB, pages 288 - 291, XP002106084 * |
Cited By (202)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6287862B1 (en) | 1997-01-17 | 2001-09-11 | Maxygen, Inc. | Evolution of whole cells and organisms by recursive sequence recombination |
| US7514087B2 (en) | 1997-06-09 | 2009-04-07 | Oxxon Therapeutics Limited | Methods and reagents for immunization which generate a CD8 T cell immune response |
| US7407661B2 (en) | 1997-06-09 | 2008-08-05 | Oxxon Therapeutics Limited | Methods and reagents that generate a CD8 T cell immune response |
| US6399383B1 (en) | 1997-10-28 | 2002-06-04 | Maxygen, Inc. | Human papilloma virus vectors |
| US6596539B1 (en) | 1997-10-31 | 2003-07-22 | Maxygen, Inc. | Modification of virus tropism and host range by viral genome shuffling |
| US8377681B2 (en) | 1998-01-16 | 2013-02-19 | Codexis Mayflower Holdings, Llc | Evolution of whole cells and organisms by recursive sequence recombination |
| US6337186B1 (en) | 1998-06-17 | 2002-01-08 | Maxygen, Inc. | Method for producing polynucleotides with desired properties |
| US6605430B1 (en) | 1998-08-12 | 2003-08-12 | Maxygen, Inc. | DNA shuffling of monooxygenase genes for production of industrial chemicals |
| US6500639B2 (en) | 1998-10-07 | 2002-12-31 | Maxygen, Inc. | DNA shuffling to produce nucleic acids for mycotoxin detoxification |
| US6483011B1 (en) | 1998-11-10 | 2002-11-19 | Maxygen, Inc. | Modified ADP-glucose pyrophosphorylase for improvement and optimization of plant phenotypes |
| WO2000028009A1 (en) * | 1998-11-11 | 2000-05-18 | Melbourne Health | Biological compositions, components thereof and uses therefor |
| US6319714B1 (en) | 1999-01-19 | 2001-11-20 | Maxygen, Inc. | Oligonucleotide mediated nucleic acid recombination |
| US6426224B1 (en) | 1999-01-19 | 2002-07-30 | Maxygen, Inc. | Oligonucleotide mediated nucleic acid recombination |
| US6423542B1 (en) | 1999-01-19 | 2002-07-23 | Maxygen, Inc. | Oligonucleotide mediated nucleic acid recombination |
| EP2253704A1 (en) | 1999-01-19 | 2010-11-24 | Maxygen, Inc. | Oligonucleotide mediated nucleic acid recombination |
| US7384387B1 (en) | 1999-02-11 | 2008-06-10 | Maxygen, Inc. | High throughput mass spectrometry |
| US6531316B1 (en) | 1999-03-05 | 2003-03-11 | Maxyag, Inc. | Encryption of traits using split gene sequences and engineered genetic elements |
| EP2865754A1 (en) | 1999-06-14 | 2015-04-29 | BP Corporation North America Inc. | Synthetic ligation reassembly in directed evolution |
| WO2001022083A2 (en) * | 1999-09-21 | 2001-03-29 | GSF-Forschungszentrum für Umwelt und Gesundheit GmbH | Method for identifying mhc-restricted antigens |
| WO2001022083A3 (en) * | 1999-09-21 | 2001-10-11 | Gsf Forschungszentrum Umwelt | Method for identifying mhc-restricted antigens |
| US6436675B1 (en) | 1999-09-28 | 2002-08-20 | Maxygen, Inc. | Use of codon-varied oligonucleotide synthesis for synthetic shuffling |
| US6686515B1 (en) | 1999-11-23 | 2004-02-03 | Maxygen, Inc. | Homologous recombination in plants |
| EP2000804A3 (en) * | 1999-12-03 | 2009-06-03 | Domantis Limited | Direct screening method |
| WO2001040803A1 (en) * | 1999-12-03 | 2001-06-07 | Diversys Limited | Direct screening method |
| WO2001044828A1 (en) * | 1999-12-17 | 2001-06-21 | Maxygen, Inc. | Methods for parallel detection of compositions having desired characteristics by means of mri spectroscopy |
| US6998263B2 (en) | 2000-02-09 | 2006-02-14 | Genvec, Inc. | Methods of preparing and using a viral vector library |
| US7094875B2 (en) | 2000-06-23 | 2006-08-22 | Maxygen, Inc. | Co-stimulatory polypeptides |
| US7612170B2 (en) | 2000-06-23 | 2009-11-03 | Maxygen, Inc. | Co-stimulatory polypeptides |
| US6858422B2 (en) | 2000-07-13 | 2005-02-22 | Codexis, Inc. | Lipase genes |
| US7407786B2 (en) | 2000-07-13 | 2008-08-05 | Codexis, Inc. | Lipase genes |
| US7367155B2 (en) | 2000-12-20 | 2008-05-06 | Monsanto Technology Llc | Apparatus and methods for analyzing and improving agricultural products |
| US8282935B2 (en) | 2001-07-30 | 2012-10-09 | Isis Innovation Limited | Materials and methods relating to improved vaccination strategies |
| JP2005510533A (en) * | 2001-11-21 | 2005-04-21 | ザ ボード オブ トラスティーズ オブ レランド スタンフォード ジュニア ユニバーシティ | Polynucleotide therapy |
| US8168772B2 (en) | 2002-02-26 | 2012-05-01 | Altravax, Inc. | Nucleic acids encoding novel chimeric C15/prM/E dengue virus immunogens prepared by DNA shuffling |
| US9186397B2 (en) | 2002-02-26 | 2015-11-17 | Altravax, Inc. | Recombinant dengue virus antigen comprising the capsid protein leader sequence, full-length prM protein, and full-length E protein |
| US8158131B2 (en) | 2002-02-26 | 2012-04-17 | Altravax, Inc. | Recombinant dengue virus antigens comprising the C15 signal peptide, full-length PRM protein, and full-length E protein |
| US8715694B2 (en) | 2002-02-26 | 2014-05-06 | Altravax, Inc. | Recombinant dengue virus antigen comprising the capsid protein leader sequence, full-length prM protein, and full-length E protein |
| US7476390B2 (en) | 2002-02-26 | 2009-01-13 | Maxygen, Inc. | Flavivirus antigens |
| EP2315145A1 (en) | 2002-03-01 | 2011-04-27 | Codexis Mayflower Holdings, LLC | Methods, systems, and software for identifying functional biomolecules |
| EP2278509A1 (en) | 2002-03-01 | 2011-01-26 | Maxygen Inc. | Methods, systems, and software for identifying functional biomolecules |
| EP2390803A1 (en) | 2002-03-01 | 2011-11-30 | Codexis Mayflower Holdings, LLC | Methods, systems, and software for identifying functional biomolecules |
| WO2003075129A2 (en) | 2002-03-01 | 2003-09-12 | Maxygen, Inc. | Methods, systems, and software for identifying functional bio-molecules |
| WO2003078583A2 (en) | 2002-03-09 | 2003-09-25 | Maxygen, Inc. | Optimization of crossover points for directed evolution |
| US8752712B2 (en) | 2002-04-04 | 2014-06-17 | Monsanto Technology Llc | Automated picking, weighing and sorting system for particulate matter |
| US7934600B2 (en) | 2002-04-04 | 2011-05-03 | Monsanto Technology Llc | Automated picking, weighing and sorting system for particulate matter |
| US8281935B2 (en) | 2002-04-04 | 2012-10-09 | Monsanto Technology Llc | Automated picking, weighing and sorting system for particulate matter |
| WO2003087128A2 (en) * | 2002-04-09 | 2003-10-23 | Rmf Dictagene S.A. | Secretory signal sequences and uses thereof |
| WO2003087128A3 (en) * | 2002-04-09 | 2004-12-29 | Rmf Dictagene Sa | Secretory signal sequences and uses thereof |
| EP2298871A1 (en) | 2002-04-19 | 2011-03-23 | Verenium Corporation | Phospholipases, nucleic acids encoding them and methods for making and using them |
| EP2275536A1 (en) | 2002-08-06 | 2011-01-19 | Verdia, Inc. | AP1 amine oxidase variants |
| WO2004041200A2 (en) * | 2002-11-04 | 2004-05-21 | Picoscript Ltd, Llp | Dna based plasmid formulations and vaccines and prophylactics containing the same |
| WO2004041200A3 (en) * | 2002-11-04 | 2004-08-05 | Picoscript Ltd Llp | Dna based plasmid formulations and vaccines and prophylactics containing the same |
| EP3023498A1 (en) | 2003-03-06 | 2016-05-25 | BASF Enzymes LLC | Amylases, nucleic acids encoding them and methods for making and using them |
| EP3508578A1 (en) | 2003-03-06 | 2019-07-10 | BASF Enzymes, LLC | Amylases, nucleic acids encoding them and methods for making and using them |
| EP2194133A2 (en) | 2003-03-06 | 2010-06-09 | Verenium Corporation | Amylases, nucleic acids encoding them and methods for making and using them |
| EP2853593A1 (en) | 2003-03-07 | 2015-04-01 | DSM IP Assets B.V. | Hydrolases, nucleic acids encoding them and mehods for making and using them |
| WO2004090099A2 (en) | 2003-04-04 | 2004-10-21 | Diversa Corporation | Pectate lyases, nucleic acids encoding them and methods for making and using them |
| EP2341136A1 (en) | 2003-04-04 | 2011-07-06 | Verenium Corporation | Pectate lyases, Nucleic Acids encoding them and methods for making and using them |
| EP2535414A1 (en) | 2003-04-29 | 2012-12-19 | Pioneer Hi-Bred International Inc. | Novel glyphosate-n-acetyltransferase (gat) genes |
| EP2322629A2 (en) | 2003-04-29 | 2011-05-18 | Pioneer Hi-Bred International Inc. | Novel glyphosate-n-acetyltransferase (GAT) genes |
| EP2409981A1 (en) | 2003-07-02 | 2012-01-25 | Verenium Corporation | Glucanases, nucleic acids encoding them and methods for making and using them |
| EP2404929A1 (en) | 2003-07-02 | 2012-01-11 | Verenium Corporation | Glucanases, nucleic acids encoding them and methods for making and using them |
| EP2404928A1 (en) | 2003-07-02 | 2012-01-11 | Verenium Corporation | Glucanases, nucleic acids encoding them and methods for making and using them |
| EP2404931A1 (en) | 2003-07-02 | 2012-01-11 | Verenium Corporation | Glucanases, nucleic acids encoding them and methods for making and using them |
| EP2404930A1 (en) | 2003-07-02 | 2012-01-11 | Verenium Corporation | Glucanases, nucleic acids encoding them and methods for making and using them |
| WO2005021714A2 (en) | 2003-08-11 | 2005-03-10 | Diversa Corporation | Laccases, nucleic acids encoding them and methods for making and using them |
| US7600642B2 (en) | 2003-09-23 | 2009-10-13 | Monsanto Technology, Llc | High throughput automated seed analysis system |
| EP1678203A2 (en) * | 2003-09-25 | 2006-07-12 | Hadasit Medical Research Services And Development Ltd. | Multiepitope polypeptides for cancer immunotherapy |
| EP3190182A1 (en) | 2004-03-08 | 2017-07-12 | DSM IP Assets B.V. | Phospholipases, nucleic acids encoding them and methods for making and using them |
| WO2005123929A2 (en) | 2004-06-09 | 2005-12-29 | Pioneer Hi-Bred International, Inc. | Plastid transit peptides |
| WO2006009676A2 (en) | 2004-06-16 | 2006-01-26 | Diversa Corporation | Compositions and methods for enzymatic decolorization of chlorophyll |
| EP2468853A1 (en) | 2004-06-16 | 2012-06-27 | Verenium Corporation | Composition and methods for enzymatic decolorization of chlorophyll |
| US11006593B2 (en) | 2004-08-26 | 2021-05-18 | Monsanto Technology Llc | Methods of seed breeding using high throughput nondestructive seed sampling |
| US7685768B2 (en) | 2004-08-26 | 2010-03-30 | Monsanto Technology Llc | Automated testing of seeds |
| US9986699B2 (en) | 2004-08-26 | 2018-06-05 | Monsanto Technology Llc | Methods of seed breeding using high throughput nondestructive seed sampling |
| EP2886658A1 (en) | 2005-03-10 | 2015-06-24 | BASF Enzymes LLC | Lyase enzymes, nucleic acids encoding them and methods for making and using them |
| WO2006101584A2 (en) | 2005-03-15 | 2006-09-28 | Diversa Corporation | Cellulases, nucleic acids encoding them and methods for making and using them |
| EP2949756A2 (en) | 2005-03-15 | 2015-12-02 | BP Corporation North America Inc. | Cellulases, nucleic acids encoding them and methods for making and using them |
| WO2006125983A1 (en) * | 2005-05-23 | 2006-11-30 | Oxxon Therapeutics Ltd | Compositions for inducing an immune response against hepatitis b |
| US7855279B2 (en) | 2005-09-27 | 2010-12-21 | Amunix Operating, Inc. | Unstructured recombinant polymers and uses thereof |
| US9938331B2 (en) | 2005-09-27 | 2018-04-10 | Amunix Operating Inc. | Biologically active proteins having increased in vivo and/or in vitro stability |
| US7846445B2 (en) | 2005-09-27 | 2010-12-07 | Amunix Operating, Inc. | Methods for production of unstructured recombinant polymers and uses thereof |
| EP2216403A2 (en) | 2006-02-02 | 2010-08-11 | Verenium Corporation | Esterases and related nucleic acids and methods |
| EP2444490A1 (en) | 2006-02-10 | 2012-04-25 | Verenium Corporation | Cellulolytic enzymes, nucleic acids encoding them and methods for making and using them |
| EP2450439A1 (en) | 2006-02-10 | 2012-05-09 | Verenium Corporation | Cellulolytic enzymes, nucleic acids encoding them and methods for making and using them |
| EP2420570A1 (en) | 2006-02-10 | 2012-02-22 | Verenium Corporation | Arabinofuranosidase enzymes, nucleic acids encoding them and methods for making and using them |
| EP2444487A1 (en) | 2006-02-10 | 2012-04-25 | Verenium Corporation | Cellulolytic enzymes, nucleic acids encoding them and methods for making and using them |
| EP2444489A1 (en) | 2006-02-10 | 2012-04-25 | Verenium Corporation | Cellucloytic enzymes, nucleic acids encoding them and methods for making and using them |
| EP2444488A1 (en) | 2006-02-10 | 2012-04-25 | Verenium Corporation | Cellucloytic enzymes, nucleic acids encoding them and methods for making and using them |
| EP2415864A1 (en) | 2006-02-10 | 2012-02-08 | Verenium Corporation | Oligomerase-2 (or beta-xylosidase) enzymes, nucleic acids encoding them and methods for making and using them |
| EP2447363A1 (en) | 2006-02-10 | 2012-05-02 | Verenium Corporation | Cellucloytic enzymes, nucleic acids encoding them and methods for making and using them |
| EP2548955A1 (en) | 2006-02-14 | 2013-01-23 | Verenium Corporation | Xylanases, nucleic acids encoding them and methods for making and using them |
| EP2548954A1 (en) | 2006-02-14 | 2013-01-23 | Verenium Corporation | Xylanases, nucleic acids encoding them and methods for making and using them |
| WO2007095398A2 (en) | 2006-02-14 | 2007-08-23 | Verenium Corporation | Xylanases, nucleic acids encoding them and methods for making and using them |
| EP2548956A1 (en) | 2006-02-14 | 2013-01-23 | Verenium Corporation | Xylanases, nucleic acids encoding them and methods for making and using them |
| US10254200B2 (en) | 2006-03-02 | 2019-04-09 | Monsanto Technology Llc | Automated contamination-free seed sampler and methods of sampling, testing and bulking seeds |
| US11293840B2 (en) | 2006-03-02 | 2022-04-05 | Monsanto Technology Llc | Automated contamination-free seed sampler and methods of sampling, testing and bulking seeds |
| US9027278B2 (en) | 2006-03-02 | 2015-05-12 | Monsanto Technology Llc | Automated contamination-free seed sampler and methods of sampling, testing and bulking seeds |
| US11357159B2 (en) | 2006-03-02 | 2022-06-14 | Monsanto Technology Llc | Automated high-throughput seed sampler and methods of sampling, testing and bulking seeds |
| US10542661B2 (en) | 2006-03-02 | 2020-01-28 | Monsanto Technology Llc | Automated high-throughput seed sampler and methods of sampling, testing and bulking seeds |
| JP2009543541A (en) * | 2006-03-06 | 2009-12-10 | アムニクス, インコーポレイテッド | Non-structural recombinant polymers and uses thereof |
| EP1996220A4 (en) * | 2006-03-06 | 2009-04-29 | Amunix Inc | Unstructured recombinant polymers and uses thereof |
| EP2112227A1 (en) | 2006-03-07 | 2009-10-28 | Cargill, Incorporated | Aldolases, nucleic acids encoding them and methods for making and using them |
| EP3153580A2 (en) | 2006-03-07 | 2017-04-12 | BASF Enzymes LLC | Aldolases, nucleic acids encoding them and methods for making and using them |
| EP2388316A2 (en) | 2006-03-07 | 2011-11-23 | Verenium Corporation | Aldolases, nucleic acids encoding them and methods for making and using them |
| EP2322643A1 (en) | 2006-03-07 | 2011-05-18 | Cargill, Incorporated | Aldolases, nucleic acids encoding them and methods for making and using them |
| EP2316962A1 (en) | 2006-03-07 | 2011-05-04 | Cargill, Incorporated | Aldolases, nucleic acids encoding them and methods for making and using them |
| EP2385108A1 (en) | 2006-03-07 | 2011-11-09 | Verenium Corporation | Aldolases, nucleic acids encoding them and methods for making and using them |
| US11084064B2 (en) | 2006-06-28 | 2021-08-10 | Monsanto Technology Llc | Small object sorting system and method |
| US11897003B2 (en) | 2006-06-28 | 2024-02-13 | Monsanto Technology Llc | Small object sorting system and method |
| US9387518B2 (en) | 2006-06-28 | 2016-07-12 | Monsanto Technology Llc | Small object sorting system and method |
| EP2444413A1 (en) | 2006-08-04 | 2012-04-25 | Verenium Corporation | Methods for oil or gas well drilling, washing and/or fracturing |
| EP2617817A2 (en) | 2006-09-21 | 2013-07-24 | Verenium Corporation | Phytases, nucleic acids encoding them and methods for making and using them |
| EP2617728A2 (en) | 2006-09-21 | 2013-07-24 | Verenium Corporation | Phytases, nucleic acids encoding them and methods for making and using them |
| EP2617729A2 (en) | 2006-09-21 | 2013-07-24 | Verenium Corporation | Phytases, nucleic acids encoding them and methods for making and using them |
| EP2617821A2 (en) | 2006-09-21 | 2013-07-24 | Verenium Corporation | Phytases, nucleic acids encoding them and methods for making and using them |
| EP2617815A2 (en) | 2006-09-21 | 2013-07-24 | Verenium Corporation | Phytases, nucleic acids encoding them and methods for making and using them |
| EP2617820A2 (en) | 2006-09-21 | 2013-07-24 | Verenium Corporation | Phytases, nucleic acids encoding them and methods for making and using them |
| EP2620495A2 (en) | 2006-09-21 | 2013-07-31 | Verenium Corporation | Phytases, nucleic acids encoding them and methods for making and using them |
| EP2617816A2 (en) | 2006-09-21 | 2013-07-24 | Verenium Corporation | Phytases, nucleic acids encoding them and methods for making and using them |
| WO2008036863A2 (en) | 2006-09-21 | 2008-03-27 | Verenium Corporation | Phospholipases, nucleic acids encoding them and methods for making and using them |
| EP2617819A2 (en) | 2006-09-21 | 2013-07-24 | Verenium Corporation | Phytases, nucleic acids encoding them and methods for making and using them |
| EP2617823A2 (en) | 2006-09-21 | 2013-07-24 | Verenium Corporation | Phytases, nucleic acids encoding them and methods for making and using them |
| EP2397486A1 (en) | 2006-09-21 | 2011-12-21 | Verenium Corporation | Phytases, nucleic acids encoding them and methods for making and using them |
| EP2479267A1 (en) | 2006-12-21 | 2012-07-25 | Verenium Corporation | Amylases and glucoamylases, nucleic acids encoding them and methods for making and using them |
| EP3540053A1 (en) | 2006-12-21 | 2019-09-18 | BASF Enzymes, LLC | Amylases and glucoamylases, nucleic acids encoding them and methods for making and using them |
| EP3101128A1 (en) | 2006-12-21 | 2016-12-07 | BASF Enzymes LLC | Amylases and glucoamylases, nucleic acids encoding them and methods for making and using them |
| WO2008080093A2 (en) | 2006-12-21 | 2008-07-03 | Verenium Corporation | Amylases and glucoamylases, nucleic acids encoding them and methods for making and using them |
| EP2479266A1 (en) | 2006-12-21 | 2012-07-25 | Verenium Corporation | Amylases and glucoamylases, nucleic acids encoding them and methods for making and using them |
| WO2008095033A2 (en) | 2007-01-30 | 2008-08-07 | Verenium Corporation | Enzymes for the treatment of lignocellulosics, nucleic acids encoding them and methods for making and using them |
| WO2008098198A2 (en) | 2007-02-08 | 2008-08-14 | The California Institute Of Technology | Alkane oxidation by modified hydroxylases |
| EP2559702A1 (en) | 2007-02-08 | 2013-02-20 | Domantis Limited | Antibody single variable domains against serum albumin |
| EP2559704A1 (en) | 2007-02-08 | 2013-02-20 | Domantis Limited | Antibody single variable domains against serum albumin |
| EP2559703A1 (en) | 2007-02-08 | 2013-02-20 | Domantis Limited | Antibody single variable domains against serum albumin |
| US8933197B2 (en) | 2007-08-15 | 2015-01-13 | Amunix Operating Inc. | Compositions comprising modified biologically active polypeptides |
| WO2009045627A2 (en) | 2007-10-03 | 2009-04-09 | Verenium Corporation | Xylanases, nucleic acids encoding them and methods for making and using them |
| EP2708602A2 (en) | 2007-10-03 | 2014-03-19 | Verenium Corporation | Xylanases, nucleic acids encoding them and methods for making and using them |
| WO2009088949A1 (en) | 2008-01-03 | 2009-07-16 | Verenium Corporation | Transferases and oxidoreductases, nucleic acids encoding them and methods for making and using them |
| EP2865750A2 (en) | 2008-01-03 | 2015-04-29 | BASF Enzymes LLC | Transferases and oxidoreductases, nucleic acids encoding them and methods for making and using them |
| EP2706122A2 (en) | 2008-01-03 | 2014-03-12 | Verenium Corporation | Isomerases, nucleic acids encoding them and methods for making and using them |
| EP3115459A2 (en) | 2008-05-23 | 2017-01-11 | E. I. du Pont de Nemours and Company | Novel dgat genes for increased seed storage lipid production and altered fatty acid profiles in oilseed plants |
| WO2010025395A2 (en) | 2008-08-29 | 2010-03-04 | Verenium Corporation | Hydrolases, nucleic acids encoding them and methods for making and using them |
| EP3460061A1 (en) | 2008-09-26 | 2019-03-27 | Tocagen Inc. | Recombinant replication competent murine leukemia gamma-retrovirus (mlv) for the expression of heat-stabilized cytosine deaminase |
| US9371369B2 (en) | 2009-02-03 | 2016-06-21 | Amunix Operating Inc. | Extended recombinant polypeptides and compositions comprising same |
| US9926351B2 (en) | 2009-02-03 | 2018-03-27 | Amunix Operating Inc. | Extended recombinant polypeptides and compositions comprising same |
| US10961287B2 (en) | 2009-02-03 | 2021-03-30 | Amunix Pharmaceuticals, Inc | Extended recombinant polypeptides and compositions comprising same |
| US8673860B2 (en) | 2009-02-03 | 2014-03-18 | Amunix Operating Inc. | Extended recombinant polypeptides and compositions comprising same |
| EP2698374A1 (en) | 2009-05-21 | 2014-02-19 | Verenium Corporation | Phytases, nucleic acids encoding them and methods for making and using them |
| WO2010135588A2 (en) | 2009-05-21 | 2010-11-25 | Verenium Corporation | Phytases, nucleic acids encoding them and methods for making and using them |
| WO2011022597A1 (en) | 2009-08-20 | 2011-02-24 | Pioneer Hi-Bred International, Inc. | Functional expression of shuffled yeast nitrate transporter (ynti) in maize to improve nitrate uptake under low nitrate environment |
| US9376672B2 (en) | 2009-08-24 | 2016-06-28 | Amunix Operating Inc. | Coagulation factor IX compositions and methods of making and using same |
| US9758776B2 (en) | 2009-08-24 | 2017-09-12 | Amunix Operating Inc. | Coagulation factor IX compositions and methods of making and using same |
| US9045712B2 (en) | 2009-10-16 | 2015-06-02 | Bunge Global Innovation, Llc | Oil degumming methods |
| WO2011046815A1 (en) | 2009-10-16 | 2011-04-21 | Bunge Oils, Inc. | Oil degumming methods |
| WO2011046812A1 (en) | 2009-10-16 | 2011-04-21 | Verenium Corporation | Phospholipases, nucleic acids encoding them and methods for making and using them |
| WO2011082304A1 (en) | 2009-12-31 | 2011-07-07 | Pioneer Hi-Bred International, Inc. | Engineering plant resistance to diseases caused by pathogens |
| WO2012021785A1 (en) | 2010-08-13 | 2012-02-16 | Pioneer Hi-Bred International, Inc. | Compositions and methods comprising sequences having hydroxyphenylpyruvate dioxygenase (hppd) activity |
| US10370430B2 (en) | 2012-02-15 | 2019-08-06 | Bioverativ Therapeutics Inc. | Recombinant factor VIII proteins |
| US11685771B2 (en) | 2012-02-15 | 2023-06-27 | Bioverativ Therapeutics Inc. | Recombinant factor VIII proteins |
| US10421798B2 (en) | 2012-02-15 | 2019-09-24 | Bioverativ Therapeutics Inc. | Factor VIII compositions and methods of making and using same |
| US11345726B2 (en) | 2012-02-16 | 2022-05-31 | VLP Theranentics. Inc. | Chikungunya virus (CHIKV) or Venezuelan equine encephalitis virus (VEEV) virus-like particles comprising heterologous antigens inserted into the envelope protein |
| WO2013166113A1 (en) | 2012-05-04 | 2013-11-07 | E. I. Du Pont De Nemours And Company | Compositions and methods comprising sequences having meganuclease activity |
| WO2014153234A1 (en) | 2013-03-14 | 2014-09-25 | Pioneer Hi-Bred International, Inc. | Compositions having dicamba decarboxylase activity and methods of use |
| WO2014159714A1 (en) * | 2013-03-14 | 2014-10-02 | Altravax, Inc. | Hepatitis b virus vaccines |
| WO2014153242A1 (en) | 2013-03-14 | 2014-09-25 | Pioneer Hi-Bred International, Inc. | Compositions having dicamba decarboxylase activity and methods of use |
| US9353158B2 (en) | 2013-03-14 | 2016-05-31 | Altravax, Inc. | Hepatitis B virus vaccines |
| EP2970394A4 (en) * | 2013-03-14 | 2016-08-10 | Altravax Inc | Hepatitis b virus vaccines |
| WO2014150914A2 (en) | 2013-03-15 | 2014-09-25 | Pioneer Hi-Bred International, Inc. | Phi-4 polypeptides and methods for their use |
| US10464986B2 (en) | 2013-07-12 | 2019-11-05 | Vlp Therapeutics, Llc | Virus like particle comprising PD-1 antigen or PD-1 ligand antigen |
| US10548953B2 (en) | 2013-08-14 | 2020-02-04 | Bioverativ Therapeutics Inc. | Factor VIII-XTEN fusions and uses thereof |
| WO2015023846A2 (en) | 2013-08-16 | 2015-02-19 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| EP3692786A1 (en) | 2013-09-13 | 2020-08-12 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| WO2015038734A2 (en) | 2013-09-13 | 2015-03-19 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| EP4159028A1 (en) | 2013-09-13 | 2023-04-05 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| WO2015120270A1 (en) | 2014-02-07 | 2015-08-13 | Pioneer Hi Bred International, Inc. | Insecticidal proteins and methods for their use |
| WO2015120276A1 (en) | 2014-02-07 | 2015-08-13 | Pioneer Hi Bred International Inc | Insecticidal proteins and methods for their use |
| EP3705489A1 (en) | 2014-02-07 | 2020-09-09 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| US10385101B2 (en) | 2014-08-08 | 2019-08-20 | Vlp Therapeutics, Llc | Virus like particle comprising modified envelope protein E3 |
| US9969986B2 (en) | 2014-08-08 | 2018-05-15 | Vlp Therapeutics, Llc | Virus like particle comprising modified envelope protein E3 |
| US10098943B2 (en) | 2014-09-11 | 2018-10-16 | Vlp Therapeutics, Llc | Flavivirus virus like particle |
| WO2016038895A1 (en) * | 2014-09-11 | 2016-03-17 | Vlp Therapeutics, Llc | Flavivirus virus like particle |
| WO2016061206A1 (en) | 2014-10-16 | 2016-04-21 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| WO2016144688A1 (en) | 2015-03-11 | 2016-09-15 | Pioneer Hi Bred International Inc | Insecticidal combinations of pip-72 and methods of use |
| CN113624953A (en) * | 2015-05-11 | 2021-11-09 | 亿明达股份有限公司 | Platform for discovery and analysis of therapeutic agents |
| WO2016186986A1 (en) | 2015-05-19 | 2016-11-24 | Pioneer Hi Bred International Inc | Insecticidal proteins and methods for their use |
| US10745680B2 (en) | 2015-08-03 | 2020-08-18 | Bioverativ Therapeutics Inc. | Factor IX fusion proteins and methods of making and using same |
| US10925950B2 (en) | 2015-08-03 | 2021-02-23 | University Of Washington | Immunogenic compositions, antigen screening methods, and methods of generating immune responses |
| US11738073B2 (en) | 2015-08-03 | 2023-08-29 | University Of Washington | Immunogenic compositions, antigen screening methods, and methods of generating immune responses |
| EP3943602A1 (en) | 2015-08-06 | 2022-01-26 | Pioneer Hi-Bred International, Inc. | Plant derived insecticidal proteins and methods for their use |
| WO2017023486A1 (en) | 2015-08-06 | 2017-02-09 | Pioneer Hi-Bred International, Inc. | Plant derived insecticidal proteins and methods for their use |
| WO2017105987A1 (en) | 2015-12-18 | 2017-06-22 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| EP3960863A1 (en) | 2016-05-04 | 2022-03-02 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| WO2017192560A1 (en) | 2016-05-04 | 2017-11-09 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| EP3954202A1 (en) | 2016-07-01 | 2022-02-16 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins from plants and methods for their use |
| WO2018005411A1 (en) | 2016-07-01 | 2018-01-04 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins from plants and methods for their use |
| EP4050021A1 (en) | 2016-11-01 | 2022-08-31 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| WO2018084936A1 (en) | 2016-11-01 | 2018-05-11 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| WO2018111551A1 (en) | 2016-12-14 | 2018-06-21 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| WO2018118811A1 (en) | 2016-12-22 | 2018-06-28 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| WO2018148001A1 (en) | 2017-02-08 | 2018-08-16 | Pioneer Hi-Bred International Inc | Insecticidal combinations of plant derived insecticidal proteins and methods for their use |
| WO2018208882A1 (en) | 2017-05-11 | 2018-11-15 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| WO2022015619A2 (en) | 2020-07-14 | 2022-01-20 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2002507393A (en) | 2002-03-12 |
| EP1054973A1 (en) | 2000-11-29 |
| WO1999041383A8 (en) | 1999-09-30 |
| AU3289199A (en) | 1999-08-30 |
| CA2320958A1 (en) | 1999-08-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6569435B1 (en) | Flavivirus and alphavirus recombinant antigen libraries | |
| WO1999041383A1 (en) | Antigen library immunization | |
| JP2023533228A (en) | Stabilized coronavirus spike (S) protein immunogens and related vaccines | |
| US6479258B1 (en) | Non-stochastic generation of genetic vaccines | |
| CA3176481A1 (en) | Coronavirus vaccine | |
| JP2003524392A (en) | Non-stochastic generation of genetic vaccines and enzymes | |
| US20220233678A1 (en) | Zika virus chimeric polyepitope comprising non-structural proteins and its use in an immunogenic composition | |
| US9284356B2 (en) | Identification of a west nile virus CD4 T cell epitope and use thereof | |
| WO2004007664A2 (en) | Nucleic acid vectors | |
| Dupuy et al. | Directed molecular evolution improves the immunogenicity and protective efficacy of a Venezuelan equine encephalitis virus DNA vaccine | |
| EP1555316A2 (en) | Antigen library immunization | |
| WO2023160654A1 (en) | Preparation and use of recombinant multicomponent sars-cov-2 trimeric protein vaccine capable of inducing broad-spectrum neutralizing activity | |
| US20230034467A1 (en) | Engineered HCV E2 Immunogens and Related Vaccine Compositions | |
| AU2761502A (en) | Antigen library immunization | |
| MXPA00007892A (en) | Antigen library immunization | |
| US11738079B2 (en) | Zika virus vaccine | |
| Zhang et al. | Severe acute respiratory syndrome: vaccine on the way | |
| Nadeem et al. | Individual expression and processing of hepatitis C virus E1/E2 epitopes-based DNA vaccine candidate in healthy humans’ peripheral blood mononuclear cells | |
| Tang et al. | Development of a novel virus-like particle-based vaccine for preventing tick-borne encephalitis virus infection | |
| CN115867312A (en) | Method for generating vaccine compositions eliciting human leukocyte antigen class I-restricted CD8T cell responses against non-virion constituent derived epitopes of viruses | |
| KR20220068617A (en) | VACCINE COMPOSITION FOR PREVENTING OR TREATING SARS-CoV-2 DIESEASE | |
| GB2621127A (en) | Vaccine constructs comprising tuberculosis antigens | |
| RU2275937C1 (en) | Method for presentation of viral antigen in immune system of host based on proteasome-mediated technology designated for development of methods of biological protection | |
| WO2024023790A1 (en) | Vaccine constructs comprising tuberculosis antigens | |
| Shimony | Identification and Characterization of new candidate proteins for vaccination against leishmaniasis using a novel approach |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| AK | Designated states |
Kind code of ref document: C1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: C1 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| CFP | Corrected version of a pamphlet front page | ||
| CR1 | Correction of entry in section i |
Free format text: PAT. BUL. 33/99 UNDER (51) REPLACE THE EXISTING SYMBOLS BY "C12N 15/31, 15/36, 15/48, 15/51, C07K 19/00, 14/02, 14/16, 14/24, 14/035, 14/18, 14/31, 14/315, 14/28, 14/245, 14/35, 14/445, 14/435, C12N 15/10, 15/86" |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2320958 Country of ref document: CA Ref country code: JP Ref document number: 2000 531564 Kind code of ref document: A Format of ref document f/p: F Ref country code: CA Ref document number: 2320958 Kind code of ref document: A Format of ref document f/p: F |
|
| NENP | Non-entry into the national phase |
Ref country code: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2000/007892 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 32891/99 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1999932510 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1999932510 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1999932510 Country of ref document: EP |