BIO-COMPATIBLE POLYMERIC MATERIALS
This invention relates to bio-compatible polymeric materials and particularly, although not exclusively, provides a bio-compatible polymeric material, a method of producing such a material and the use of such a material in medical treatment, for example in a prosthesis.
Much research is being directed to the provision of materials to meet the growing need for prosthetic devices such as orthopaedic, dental or maxillofacial implants. For example, nearly half a million patients receive bone implants each year in the US with the majority being artifical hip and knee joints made from titanium or colbalt-chrome alloys. However, these materials are too stiff leading to bone resorption, loosening of the implant and, consequently, have lifetimes of less than 10 years. Additionally, medical devices or prostheses such as pacemakers, vascular grafts, stents, heart valves, catheters and dental implants that contact body tissues or fluids of living persons or animals have been developed and used clinically.
A major problem with medical devices such as those described is the susceptibility to foreign body reaction and possible rejection. Consequently, it is of great interest to the medical industry to develop materials from which medical devices can be made which are less prone to adverse biological reactions that typically accompany introduction of medical devices into humans or animals.
It is an object of the present invention to address the above described problems .
According to a first aspect of the invention, there is provided a bio-compatible polymeric material for use in medical applications, wherein said material comprises a polymer having bio-compatible moieties associated with chain ends thereof .
In the scientific literature there is inconsistency in the use of descriptions such as "bio-compatible", "bio- active" and "bio-materials" . In the context of the present specification, the term "bio-compatible" has generally been used to refer to a material which is compatible with use in medical applications, for example by not being toxic or otherwise harmful to living materials. It also encompasses materials which have a biological or physiological effect when associated with living materials.
"Bio-compatible moieties" referred to herein suitably refer to moieties which are compatible with use in medical applications, for example by not being toxic or otherwise harmful to living material. Such bio-compatible moieties may be arranged to bond (for example to form ionic or covaient bonds) or otherwise interact with materials present in human or animal bodies in order to improve their integration and acceptance by such bodies .
Except where otherwise stated, throughout this specification, any alkyl, akenyl or alkynyl moiety suitably has up to 8, preferably up to 6, more preferably up to 4, especially up to 2, carbon atoms and may be of straight chain or, where possible, of branched chain structure. Generally, methyl and ethyl are preferred
alkyl groups and C2 alkenyl and alkynyl groups are preferred.
Except where otherwise stated in this specification, optional substituents of an alkyl group may include halogen atoms, for example fluorine, chlorine, bromine and iodine atoms, and nitro, cyano, alkoxy, hydroxy, amino, alkylamino, sulphinyl, alkylsulphinyl, sulphonyl, alkylsulphonyl, amido, alkylamido, alkoxycarbonyl , haloalkoxycarbonyl and haloalkyl groups. Preferably,
-optionally—subst-i-tuted alkyl groups are unsubstituted.
Preferably, said bio-compatible polymeric material has improved or enhanced bio-compatibility compared to said polymer in the absence of bio-compatible moieties associated with chain ends thereof .
Bio-compatible moieties suitably include moieties arranged to reduce adverse biological reactions when the polymeric material is introduced into (or otherwise associated with) a human or animal body. For example, adverse biological reactions associated with introduction into a human or animal body of said polymer having said bio-compatible moieties may be less compared to use of the same polymer but which does not include associated bio- compatible moieties.
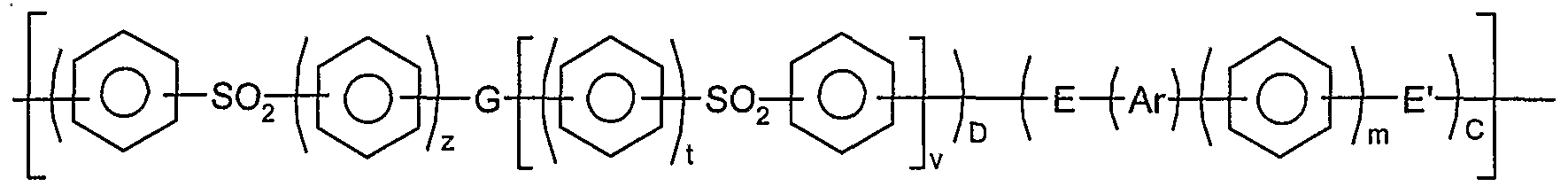
Preferably, said polymer has phenyl moieties; carbonyl or sulphone moieties; and ether or thioether moieties in the polymer backbone.
Preferably, said polymer has a moiety of formula
and/or a moiety of formula
and/or a moiety of formula
wherein the phenyl moieties in units I, II, and III are independently optionally substituted and optionally cross- linked; and wherein m,r,s,t,v,w and z independently represent zero or a positive integer, E and E' independently represent an oxygen or a sulphur atom or a direct link, G represents an oxygen or sulphur atom, a direct link or a -O-Ph-O- moiety where Ph represents a phenyl group and Ar is selected from one of the following moieties (i)*, (i)**, (i) to (x) which is bonded via one or more of its phenyl moieties to adjacent moieties
Unless otherwise stated in this specification, a phenyl moiety may have 1,4- or 1,3-, especially 1,4-, linkages to moieties to which it is bonded.
Said polymer may include more than one different type of repeat unit of formula I; more than one different type of repeat unit of formula II; and more than one different type of repeat unit of formula III. Preferably, however,
only one type of repeat unit of formula I, II and/or III is provided.
Said moieties I, II and III are suitably repeat units. In the polymer, units I, II and/or III are suitably bonded to one another - that is, with no other atoms or groups being bonded between units I, II, and III.
Where the phenyl moieties in units I, II or III are optionally substituted, they may be optionally substituted by one or more halogen, especially fluorine and chlorine, atoms or alkyl , cycloalkyl or phenyl groups . Preferred alkyl groups are Cι_ι0, especially Cι-4, alkyl groups. Preferred cycloalkyl groups include cyclohexyl and multicyclic groups, for example adamantyl .
Another group of optional substituents of the phenyl moieties in units I, II or III include alkyls, halogens, CyF2y+ι where y is an integer greater than zero, 0-Rq (where Rq is selected from the group consisting of alkyls, perfluoralkyls and aryls) , CF=CF2, CN, N02 and OH. Trifluormethylated phenyl moieties may be preferred in some circumstances .
Preferably, said phenyl moieties are not optionally- substituted as described.
Where said polymer is cross-linked, it is suitably cross-linked so as to improve its properties. Any suitable means may be used to effect cross-linking. For example, where E represents a sulphur atom, cross-linking between polymer chains may be effected via sulphur atoms on
respective chains. Preferably, said polymer is not optionally cross-linked as described.
Where w and/or z is/are greater than zero, the respective phenylene moieties may independently have 1,4- or 1,3-linkages to the other moieties in the repeat units of formulae II and/or III. Preferably, said phenylene moieties have 1,4- linkages.
Preferably, the polymeric chain of the polymer does not include a -S- moiety. Preferably, G represents a direct link.
Suitably, "a" represents the mole % of units of formula I in said polymer, suitably wherein each unit I is the same; "b" represents the mole % of units of formula II in said polymer, suitably wherein each unit II is the same; and "c" represents the mole % of units of formula III in said polymer, suitably wherein each unit III is the same. Preferably, a is in the range 45-100, more preferably in the range 45-55, especially in the range 48-52. Preferably, the sum of b and c is in the range 0-55, more preferably in the range 45-55, especially in the range 48- 52. Preferably, the ratio of a to the sum of b and c is in the range 0.9 to 1.1 and, more preferably, is about 1. Suitably, the sum of a, b and c is at least 90, preferably at least 95, more preferably at least 99, especially about 100. Preferably, said polymer consists essentially of moieties I, II and/or III.
Said polymer may be a homopolymer having a repeat unit of general formula
or a homopolymer having a repeat unit of general formula
or a random or block copolymer of at least two different units of IV and/or V
wherein A, B, C and D independently represent 0 or 1 and E,E ' ,G,Ar,m, r, s, t,v, w and z are as described in any statement herein.
As an alternative to a polymer comprising units IV and/or V discussed above, said polymer may be a homopolymer having a repeat unit of general formula
{(§M ©1 O-rf-∞-fO- - g
})-*
) or a homopolymer having a repeat unit of general formula
or a random or block copolymer of at least two different units of IV* and/or V*, wherein A, B, C, and D independently represent 0 or 1 and E, E
1, G, Ar, m, r, s, t, v, w and z are as described in any statement herein.
Preferably, m is in the range 0-3, more preferably 0-2, especially 0-1. Preferably, r is in the range 0-3, more preferably 0-2, especially 0-1. Preferably t is in the range 0-3, more preferably 0-2, especially 0-1. Preferably, s is 0 or 1. Preferably v is 0 or 1. Preferably, w is 0 or 1. Preferably z is 0 or 1.
Preferably, said polymer is a homopolymer having a repeat unit of general formula IV.
Preferably Ar is selected from the following moieties (xi)*, (xi)**,(xi) to (xxi) :
In (xi)*, the middle phenyl may be 1,4- or 1,3- substituted.
Preferably, (xv) is selected from a 1,2-, 1,3-, or a 1,5- moiety; (xvi) is selected from a 1,6-, 2,3-, 2,6- or a 2,7- moiety; and (xvii) is selected from a 1,2-, 1,4-, 1,5- , 1,8- or a 2,6- moiety.
One preferred class of polymers does not include any moieties of formula III, but suitably only includes moieties of formulae I and/or II . Where said polymer is a homopolymer or random or block copolymer as described, said homopolymer or copolymer suitably includes a repeat unit of general formula IV. Such a polymer may, in some embodiments, not include any repeat unit of general formula V.
Suitable moieties Ar are moieties (i)*, (i) , (ii) ,
(iii) and (iv) and, of these, moieties (i)*, (i) and (iv) are preferred. Other preferred moieties Ar are moieties
(xi)*, (xii) , (xi) , (xiii) and (xiv) and, of these, moieties (xi)*, (xi) and (xiv) are especially preferred.
An especially preferred class of polymers are polymers which consist essentially of phenyl moieties in conjunction with ketone and/or ether moieties. That is, in the preferred class, the polymer does not include repeat units which include -S-, -S02- or aromatic groups other than phenyl . Preferred polymers of the type described include :
(a) a polymer consisting essentially of units of formula IV wherein Ar represents moiety (iv) , E and E' represent oxygen atoms, m represents 0, w represents 1, G represents a direct link, s represents 0, and A and B represent 1 (i.e. polyetheretherketone) .
(b) a polymer consisting essentially of units of formula IV wherein E represents an oxygen atom, E' represents a direct link, Ar represents a moiety of structure (i) , m represents 0, A represents 1,
B represents 0 (i.e. polyetherketone) ;
(c) a polymer consisting essentially of units of formula IV wherein E represents an oxygen atom, Ar represents moiety (i)*, m represents 0, E' represents a direct link, A represents 1, B represents 0, (i.e. polyetherketoneketone) .
(d) a polymer consisting essentially of units of formula IV wherein Ar represents moiety (i) , E and
E' represent oxygen atoms, G represents a direct link, m represents 0, w represents 1, r represents 0, s represents 1 and A and B represent 1. (i.e. polyetherketoneetherketoneketone) .
(e) a polymer consisting essentially of units of formula IV, wherein Ar represents moiety (iv) , E and E' represents oxygen atoms, G represents a direct link, m represents 0, w represents 0, s, r, A and B represent 1
(i . e .polyetheretherketoneketone) .
Of the aforesaid, the polymers described in (a) and (b) are preferred, with the polymer described in (a) being especially preferred.
Said bio-compatible moieties are preferably associated with the surface of said bio-compatible polymeric material
and, suitably, do not substantially penetrate the bulk of the material. Preferably, in a bio-compatible polymeric material, moieties at the ends of chains of said polymer within the bulk thereof are different compared to bio- compatible moieties associated with ends of chains of said polymer at the surface of the bio-compatible polymeric material. Thus, the concentration of bio-compatible moieties at a surface of said bio-compatible polymeric material is preferably greater than the concentration in the bulk of said material. Consequently, said bio- compatible moieties are suitably associated with chain ends of said polymer which are at the surface of the polymer. The concentration of chain end groups at the surface may be greater than the concentration in the bulk. Chain ends of said polymer below the surface of the polymeric material preferably do not include associated bio-compatible moieties.
Thus, the invention extends to a bio-compatible polymeric material for use in medical applications, wherein said material comprises a polymer, wherein a surface of said material comprises said polymer having bio-compatible moieties associated with chain ends thereof and wherein the bulk of said material comprises said polymer without associated bio-compatible moieties. Thus, preferably, the concentration of bio-compatible moieties associated with chain ends at the surface of said polymer is greater than the concentration associated with ends in the bulk.
Said polymer having bio-compatible moieties associated with chain ends thereof preferably includes moieties other than fluorine atoms associated with its chain ends.
Since said polymer having bio-compatible moieties associated with ends thereof is suitably present only at a surface of said bio-compatible polymeric material and is present at a small fraction of the total weight of polymer (the majority of which will not include associated bio- compatible moieties) the existence of said bio-compatible moieties may have limited effect on the bulk properties of said bio-compatible polymeric material compared to said polymer in the absence of said associated bio-compatible moieties.— •■
The glass transition temperature (Tg) of said polymer, suitably the bulk thereof, (in the absence of associated bio-compatible moieties) may be at least 135°C, suitably at least 150°C, preferably at least 154°C, more preferably at least 160°C, especially at least 164°C. In some cases, the Tg may be at least 170°C, or at least 190°C or greater than 250°C or even 300°C.
Said polymer, suitably the bulk thereof, (in the absence of associated bio-compatible moieties) may have an inherent viscosity (IV) of at least 0.1, suitably at least 0.3, preferably at least 0.4, more preferably at least 0.6, especially at least 0.7 (which corresponds to a reduced viscosity (RV) of least 0.8) wherein RV is measured at 25°C on a solution of the polymer in concentrated sulphuric acid of density 1.84gcm"3, said solution containing lg of polymer per 100cm"3 of solution. IV is measured at 25°C on a solution of polymer in concentrated sulphuric acid of density 1.84gcm3, said solution containing 0. lg of polymer per 100cm3 of solution.
The measurements of both RV and IV both suitably employ a viscometer having a solvent flow time of approximately 2 minutes .
The main peak of the melting endotherm (Tm) for said polymer, suitably the bulk thereof, (if crystalline) may be at least 300°C.
Preferably, said polymer, suitably the bulk thereof, (in the absence of associated bio-compatible moieties) has —at—least some- crystailinity or -is—erysfeallisable. The existence and/or extent of crystallinity in a polymer is preferably measured by wide angle X-ray diffraction, for example as described by Blundell and Osborn (Polymer 24, 953, 1983). Alternatively, crystallinity may be assessed by Differential Scanning Calorimetry (DSC) .
Said polymer, suitably the bulk thereof, (in the absence of associated bio-compatible moieties) may have a number average molecular weight in the range 2000-80000.
Preferably said molecular weight is at least 14,000. The molecular weight may be less than 60,000.
Said bio-compatible polymeric material may consist essentially of a single polymer with associated bio- compatible moieties. Alternatively, said bio-compatible polymeric material may comprise a blend of polymers, suitably having different molecular weights. For example a blend may comprise a relatively low molecular weight polymer and a relatively high molecular weight polymer. A said low molecular weight polymer will have a greater proportion of ends per unit weight and, consequently, may be used to increase the concentration of ends in said bio-
compatible polymeric material and, accordingly, to increase the concentration of bio-compatible moieties of said bio-compatible polymer material.
Where a blend comprises a relatively low molecular weight polymer, the molecular weight of said low molecular weight polymer may be less than 14,000, but preferably greater than 2,000. The relatively high molecular weight polymer may have a molecular weight of at least 14,000 and suitably less than 80,000, especially less than 60,000.
Said bio-compatible polymeric material suitably has a tensile strength (according to ISO R527) of at least 80, preferably at least 90, especially at least 95 MPa. The tensile strength may be less than 360, suitably less than 250, preferably less than 140 MPa. It preferably has an elongate at break (according to ISO R527) of at least 40, preferably at least 50%. It preferably has a tensile modulus (according to ISO R527) of greater than 2.5, preferably greater than 3, especially greater than 3.5 GPa. The tensile modulus may be less than 40, suitably less than 30, preferably less than 20, more preferably less than 10 GPa. It preferably has a flexural strength (according to ASTM D695) of at least 100, more preferably at least 110, especially at least 115 MPa. The flexural strength may be less than 650, preferably less than 400, more preferably less than 260, especially less than 200 MPa. It preferably has a flexural modulus (according to ISO R178) of at least 3, preferably at least 3.5, especially at least 4 GPa. The flexural modulus may be less than 60 , suitably less than 25, preferably less than 20 especially less than 10 GPa. Advantageously, the aforementioned properties can be adjusted by appropriate
selection of polymers and/or any reinforcement means included in said support material to suit particular applications. For example, a continuous carbon fibre polyetheretherketone may typically have a tensile strength of about 350 MPa, a tensile modulus of 36 GPa, an elongation of 2%, a flexural modulus of 50 GPa and a flexural strength of 620 MPa. A polyaryletherketone with 30% of high performance fibres typically may have a tensile strength of 224 MPa, a tensile modulus of 13 GPa, a tensile elongation of 2%, a flexural modulus of 20 GPa and a—f-lexural- strength of 250 MPa. -- -
Said bio-compatible polymeric material may include one or more fillers for providing desired properties. Said material preferably incorporates an X-ray contrast medium. Fillers and/or said X-ray contrast medium is/are preferably distributed substantially uniformly throughout said material .
Where an X-ray contrast medium is provided it suitably comprises less than 25wt%, preferably less than 20wt%, more preferably less than 15wt%, especially less than 10wt% of said bio-compatible material. Where it is provided, at least 2wt% may be included. Preferred X-ray contrast mediums are particulate and preferably are inorganic. They preferably have low solubility in body fluids. They preferably also have a sufficient density compared to that of the polymer to create an image if a compounded mixture of the polymer and contrast medium are X-ray imaged. Barium sulphate and zirconium oxide are examples. Said particulate material is suitably physically held in position by entrapment within the polymer.
Preferably, said bio-compatible polymeric material includes a major amount of said polymer, especially one having moieties I, II and/or III, described according to said first aspect.
In the context of this specification, a "major" amount may mean greater than 50 wt%, suitably greater than 65 wt%, preferably greater than 80 wt%, more preferably greater than 95 wt%, especially greater than 98 wt% of the referenced material is present relative to the total
---weight of relevant- material present.
Where said bio-compatible polymeric material comprises a blend, said blend preferably includes at least two polymers of a type according to said first aspect. For example, said at least two polymers preferably include moieties I, II and/or III as described above. A said blend preferably includes a major amount of higher (or the highest) number average molecular weight polymer. Said bio-compatible polymeric material preferably includes a major amount of a higher molecular weight polymer.
A said bio-compatible moiety may be selected from an anticoagulant agent such as heparin and heparin sulfate, an antithrombotic agent, a clotting agent, a platelet agent, an anti-inflammatory agent, an antibody, an antigen, an immunoglobulin, a defence agent, an enzyme, a hormone, a growth factor, a neurotransmitter, a cytokine, a blood agent, a regulatory agent, a transport agent, a fibrous agent, a protein such as avidin, a glycoprotein, a globular protein, a structural protein, a membrane protein and a cell attachment protein, a peptide such as a glycopeptide, a structural peptide, a membrane peptide and
a cell attachment peptide, a proteoglycan, a toxin, an antibiotic agent, an antibacterial agent, an antimicrobial agent such as pencillin, ticarcillin, carbenicillin, ampicillin, oxacillian, cefazolin, bacitracin, cephalosporin, cephalothin, cefuroxime, cefoxitin, norfloxacin, perfloxacin and sulfadiazine, hyaluronic acid, a polysaccharide, a carbohydrate, a fatty acid, a catalyst, a drug, biotin, a vitamin, a DNA segment, a RNA segment, a nucleic acid, a nucleotide, a polynucleotide, a nucleoside, a lectin, a ligand and a dye (which acts as a biological ligand) , a radioisotope, a chelated radioisotope, a chelated metal, a metal salt, a sulphonic acid or salt thereof, a steroid, a non-steriod, a non- steroidal anti-inflammatory, an analgesic, an anti- histamine, a receptor binding agent, a chemotherapeutic agent, a hydrophilic polymer, (e.g. poly (ethylene glycol)
(PEG), poly (ethylene oxide) (PEO) , ethylene oxide- propylene oxide block co-polymers, poly (N-vinyl-2- pyrrolidone) (PNVP) , poly (2-hydroxyethyl methacrylate) (pHEMA) , HEMA co-polymers, poly(vinyl alcohol) (PVA), polyacrylamide, its derivatives, poly (methyl methacrylate)
(PMMA) , suitably having a PEG chain on each of the side groups, polysiloxanes (e.g. polydimethylsiloxanes (PDMS) ) , ionic water-soluble polymers like poly (acrylic acid) (PAAc) ) and a polyurethane . Examples of some of the aforesaid are provided in US5958430, US5925552, US5278063 and US5330911 and the contents of the aforementioned specifications are incorporated herein by reference.
In one embodiment, said bio-compatible moieties may comprise bone morphogenic protein (BMP) as described in US4563489 and patents cited therein and the contents of the aforesaid are incorporated herein. Said BMP may be
provided in combination, for example in admixture, with a physiologically acceptable biodegradable organic polymer and said biodegradable polymer may be associated with ends of said polymer of said bio-compatible polymeric material, for example by being covalently bonded to end groups . Thus, in this case, the combination of said biodegradable polymer and BMP defines said bio-compatible moieties. Said biodegradable polymer is preferably a biodegradable polylactic acid; or alternatively, other physiologically acceptable biodegradable organic polymers which are
-structurally equivalent to polylactic acid can be used as the delivery system for BMP. Examples include poly (hydroxy organic carboxylic acids) e.g. poly (hydroxy aliphatic carboxylic acids) , polyglycollic acid, polyglactin, polyglactic acid and poly adonic acids.
In another embodiment, said bio-compatible moieties may be selected from inorganic crystalline structures, inorganic amorphous structures, organic crystalline structures and organic amorphous structures. Preferred bio-compatible moieties are phosphorous based ceramics, for example calcium-phosphorous ceramics. Phosphates in general are suitable but calcium phosphates and calcium apatite are preferred. Especially preferred is hydroxyapatite, a synthetic Ca-P ceramic.
Linking moieties, for example linking atoms or groups may extend between repeat units of the polymer and said bio-compatible moieties. Said linking moieties may be covalently bonded to ends of respective repeat units of the polymer. Said linking moieties may be covalently bonded to said bio-compatible moieties or may otherwise be associated with said moieties.
A said linking moiety may be associated with a single bio-compatible moiety or, alternatively, a said linking moiety may be associated with more than one bio-compatible moiety. Thus, said linking moiety may be mono-func ional or multi-functional, for association with one or more bio- compatible moieties. Multi-functional linking moieties may be able advantageously to be associated with more bio- compatible moieties and may, therefore, provide a means to increase the concentration of bio-compatible moieties associated with said bio-compatible material.
Whilst said bio-compatible moieties may be associated with said chain ends of the polymer by any suitable means, for example covaient bond(s), hydrogen bond(s), encapsulation in a matrix which is bonded to or otherwise interacts with said end groups, or ionic interaction (s) , it is preferred that there are covaient bonds between the bio-compatible moieties and said chain ends or there are ionic interactions between said bio-compatible moieties and said ends .
Preferably, said bio-compatible moieties are associated with end groups of the polymer. The term "end group" suitably refers to moieties at the end of polymer chains. Thus, said bio-compatible polymeric material may be represented by the formula
PC-EG1.BM
where PC represents a polymer chain which suitably comprises repeat units and may, for example, include moieties I, II, III, IV, V, IV*, V* as described above;
EG' represents an end-group; and BM' represents a bio- compatible moiety.
EG' may represent said aforementioned linking moiety.
In some cases, EG' and- EG'.BM' may be the same, for example where an end group is itself bio-compatible. A -SO3H end group may fall into this category. Preferably, however, EG' and EG'.BM1 represent different moieties.
-The bond-between PC- and EG' is preferably a covaient bond. The interaction between EG' and BM' may be by any suitable means as described above. The interaction is preferably by means of a covaient bond or an ionic interaction.
According to a second aspect of the invention, there is provided a method of making a bio-compatible polymeric material for use in medical applications, the method including the step of causing bio-compatible moieties to become associated with ends, preferably with end groups, of a polymer.
Advantageously, the method may provide a relatively easy way of preparing a bio-compatible polymeric material which may avoid the need to post-functionalise repeat units of the polymer. Suitably, end groups may be produced in a polymerisation reaction in which the polymer is prepared and may then, optionally, be functionalised to enable association with bio-compatible moieties. In some embodiments, said end groups may themselves be bio- compatible. It is found that the concentration of end groups of the polymer at a surface of a solid material
made from the polymer is much greater than would be expected from a consideration of the molecular weight of the polymer and, accordingly, the concentration of bio- compatible moieties at the surface may be correspondingly greater.
The method preferably includes the step of treating said polymer with a material for providing bio-compatible moieties (hereinafter "BCM material") arranged to provide bio-compatible moieties for association with said chain ends, for example end groups-. Said BCM material may be arranged to provide any of the bio-compatible moieties described herein. Said polymer may be provided as a solid. Suitably, said bio-compatible moieties are caused to become associated with a surface of said solid, preferably with end groups present at a surface of said solid. Said solid is preferably shaped so as to represent at least a part of a device for use in medical applications. For example, said device may be a component of an implant for a human or animal body, for example an orthopaedic or dental implant or vascular graft . Said solid may be provided in a desired shape by any suitable means, for example by injection or compression moulding or by film formation techniques or extrusion. Thus, preferably, after association with said bio-compatible moieties, the bio-compatible polymeric material is not engineered or otherwise treated in a manner which may result in substantial depletion of the bio-compatible moieties associated with its surface.
Association of bio-compatible moieties with said chain ends may be effected in any suitable way which will depend on the nature of the BCM material and/or the identity of
chain ends of the polymer. In some embodiments, the method may include causing covaient bond formation between the polymer and said bio-compatible moieties. In other embodiments, association of chain ends of the polymer and bio-compatible moieties may be effected by other means, for example by ionic interactions .
Where EG' and EG'.BM' represent different moieties, the method may include the step of treating a polymer of general formula
PC-EG
wherein PC represents a polymer chain and EG represents an end group thereof with a material (BM) arranged to supply a bio-compatible material (BM') thereby to produce said bio-compatible polymeric material which may be represented by the formula
PC-EG' .BM'
wherein EG' .BM' represents an association between the end group and the bio-compatible material, wherein EG' represents a residue of end group EG or may represent EG, for example where there is no covaient bond formation between EG' and BM' ; and BM' represents a residue of bio- compatible material BM or may represent BM where there is no covaient bond formation between EG' and BM' .
EG may include any suitable functional groups arranged to become associated with suitable functional groups provided on BM. For example EG may include a functional group selected from the following: -OH, -CHO, -NR10 2,
preferably -NH2 or -NHR10, -SH, -CONH2, -CONHR10, -COOH, -COCl or -COOR10 group, a halogen atom, especially a fluorine, chlorine, bromine or iodine atom, -N02, -S03M, -S02R1:L, -S02NHR10, -SO2NR10 2 or -COOM groups, an anhydride, an epoxide, a cyanate, -CN, an isocyanate, a carbon-carbon double bond, for example a group -CR10=CR10 2 or a (Co-Cι0alk) acrylate (wherein "alk" refers to an alkyl group) such as -COOC(CH3) CH2 and -COOCHCH2, a carbon-carbon triple bond, for example a group -CR10 or an azide, wherein R10 represents a hydrogen atom or an optionally substituted alkyl group, wherein M represents a hydrogen atom or an alkali metal and R11 represents a halogen, especially a chlorine, atom.
BM may include any suitable functional group that is arranged to become associated with functional groups included in EG and may be selected from any of the functional groups referred to above for EG provided that a selected functional group provided by EG is capable of becoming associated with, suitably reacting, with a selected functional group provided by BM.
In some cases BM may be provided by reaction of EG with more than one functional group. For example a BM' may represent a polyurethane which may be prepared when EG provides a hydroxy group and BM provides a diisocyanate and a diol; or wherein EG provides an isocyanate group and BM provides a diisocyanate and a diol. In both cases, two different compounds BM may be used, (see Example 12 hereinafter) .
In some cases, BM may be provided by a monomer or monomers having a functional group arranged to react with
EG and being arranged to polymerise to provide a polymeric moiety BM' (see Example 11 hereinafter) .
In one embodiment, EG may be ionic in character, for example it may be -COOM or -S03M and such a group may be arranged to ionically associate with an ionic moiety provided by BM (see Example 9) .
In other embodiments, an amide bond may be formed between EG and BM (see Examples 8 and 13) .
In some cases, said group EG may be multi-functional, thereby enabling it to associate with a plurality of bio- compatible moieties. For example, multi-functionality may be provided by dendritic or hyperbranched end groups .
Preparation of said polymer of general formula PC-EG may include the step of making a polymer of general formula
PC-FG
• where FG represents a functional group at the end of the polymer chain PC. In some cases, FG may represent EG in which case the polymer having end groups which are caused to be associated with said bio-compatible moieties in the method may not need to be functionalised after polymer formation. However, where FG and EG are different, said polymer of formula PC-FG may be derivatised to provide the polymer which is caused to be associated with bio-compatible moieties in the method.
Where said end group EG is multi-functional and FG and EG are the same, an end-capper which includes EG may be contacted with monomers used in the preparation of said polymer. Example 5b hereinafter is of this type. Where, however, said end group EG is multi-functional but FG and EG are not the same, said polymer of formula PC-FG may be derivatised with a multi-functional material. For example, said multi-functional material may include a first functional group capable of reacting with said group FG to form a covaient bond and, additionally, includes at least two, preferably" at"- least three; other functional groups, suitably different to said first functional group, wherein said other functional groups, after optional derivatisation, may be caused to associate with bio- compatible moieties. Example 13 hereinafter is of this type.
Said moiety FG may include or consist of any of the functional groups described above which enable EG to become associated with functional groups provided by BM. Preferably, FG includes or consists of a group selected from -OH, -NR10 2, -N=CR10 2, -OR10, a halogen, especially a fluorine or chlorine atom, -N02, -CN and -SO3M1 wherein R10 represents a hydrogen atom or an optionally-substituted alkyl group and M represents an alkali metal and, of the aforesaid, fluorine atoms -NH2, -COOM, -OR10 (e.g. -OMe) , -N02 and -SO3M1 are especially preferred.
FG groups are suitably such that they can survive the polymerisation reaction wherein PC-FG is made and/or subsequent processing.
Where FG and EG represent the same atoms or groups, FG preferably represents a functional group that can both survive the polymerisation reaction and/or further processing and be caused to associate with BM. Preferred examples, of groups FG that may survive the polymerisation reaction and/or further processing include -OH, -NH2 and - S03M, especially where M is an alkali metal.
In a preferred embodiment, said polymer of formula PC- FG is made into a solid as described above. Then, optionally, the solid polymer may be treated so as to form
PC-EG and such treatment suitably involved a surface treatment of said solid. The treatment may be used to functionalise FG thereby to provide groups EG at the surface, wherein EG are of a type which would be less able to survive the polymerisation reaction and/or steps taken
(e.g. injection moulding) to form the solid. (Of course
FG and EG may be the same as described in which case no treatment of the type described needs to be undertaken) . Next, the EG groups at the surface of the solid are treated thereby to enable bio-compatible moieties to be associated therewith and, accordingly, with the surface of the bio-compatible polymeric material.
In a first type of method, said polymer of general formula PC-FG may be prepared by polycondensation of a difunctional monomer with itself, wherein said monomer includes a group of formula -FG.
In a second type of method, said polymer of formula PC-FG may be prepared by polycondensing first and second different difunctional monomers, wherein the monomers have at least two reactive groups each and wherein at least two
of the at least four reactive groups provided by the two monomers include a reactive group of formula -FG. Preferably, one of the monomers includes two FG groups. Preferably, in order to produce a polymer of formula PC-FG there is a slight molar excess of groups of formula -FG over the other groups involved in the polycondensation.
In a third type of method, said polymer of formula PC- FG may be prepared by polycondensing difunctional monomers in the presence of a monomer which includes only a single reactive grou — hich is able to participate in the polycondensation, but, additionally, incorporates a group of formula -FG (which in this case is suitably of a type which cannot polycondense with the other monomers) . For example, one embodiment of the third type of method may use a first difunctional monomer having two reactive groups of the same identity; a second difunctional monomer having two reactive groups of the same identity but which are different to those of said first difunctional monomers; and a third monomer having one reactive group which is able to polycondense with said first or second monomers, for example by virtue of it having one reactive group which is the same as a reactive group of the first or second monomers and a second group which is of formula -FG (which is suitably of a type which cannot polycondense with the other monomers) . In the third method, the third monomer acts as a chain terminator thereby leading to FG groups at the end of polymer chains .
More specifically, polymers of general formula PC-FG described herein may be prepared by:
(a) polycondensing a compound of general formula
with itself wherein Y1 represents a halogen atom or a group -EH and Y2 represents a halogen atom or, if Y1 represents a halogen atom, Y2 represents a group E'H; or
(b) polycondensing a compound of general formula
with a compound of formula
and/or with a compound of formula
wherein Y
1 represents a halogen atom or a group -EH (or -E'H if appropriate) and X
1 represents the other one of a halogen atom or group -EH (or -E'H if appropriate) and Y
2 represents a halogen atom or a group -E'H and X
2 represents the other one of a halogen atom or a group -E'H (or -EH if appropriate) .
(c) optionally copolymerizing a product of a process as described in paragraph (a) with a product of a process as described in paragraph (b) ;
wherein the phenyl moieties of units VI, VII and/or VIII are optionally substituted; and Ar, m, w, r, s, z, t, v, G, E and E' are as described above except that E and E' do not represent a direct link;
In some situations, repeat units of the polymer prepared, more particularly phenyl groups thereof, may be optionally substituted with the groups hereinabove described after polymer formation. Preferably, however, the repeats units are not functionalised.
Preferably, where Y1, Y2, X1 and/or X2 represent a halogen, especially a fluorine, atom, an activating group, especially a carbonyl or sulphone group, being arranged ortho- or para- to the halogen atom.
Preferred halogen atoms are fluorine and chlorine atoms, with fluorine atoms being especially preferred. Preferably, halogen atoms are arranged meta- or para- to activating groups, especially carbonyl groups.
Where the process described in paragraph (a) is carried out, preferably one of Y1 and Y2 represents a fluorine atom and the other represents an hydroxy group. More preferably in this case, Y1 represents a fluorine atom and Y2 represents an hydroxy group. Advantageously, the process described in paragraph (a) may be used when Ar represents a moiety of structure (i) and m represents 1.
When a process described in paragraph (b) is carried out, preferably, Y1 and Y2 each represent an hydroxy group. Preferably, X1 and X2 each represent a halogen atom, suitably the same halogen atom.
Compounds of general formula VI, VII and VIII are commercially available (eg from Sigma-Aldrich Co Ltd or
Aldrich Chemical Co) and/or may be prepared by standard techniques, generally involving Friedel-Crafts reactions, followed by appropriate derivatisation of functional groups . The preparations of some of the monomers described herein are described in P M Hergenrother, B J
Jensen and S J Havens, Polymer 29, 358 (1988) , H R
Kricheldorf and U Delius, Macromolecules 22, 517 (1989) and
P A Staniland, Bull, Soc, Chem, Belg., 98 (9-10), 667
(1989) . Other raw materials described herein are generally available from Aldrich as described. Hydroxytriphenylimine described in Example 6 may be prepared as described in J.
Poly. Sci. Part A Poly. Chem, 33, 779(1995).
Where the process described in paragraph (b) is carried out, suitably, "a*" represents the mole% of compound VI used in the process; »b*" represents the mole % of compound VII used in the process; and "c*" represents the mole % of compound VIII used in the process.
Preferably, a* is in the range 45-55, especially in the range 48-52. Preferably, the sum of b* and c* is in the range 45-55, especially in the range 48-52. Preferably, the sum of a*, b* and c* is 100.
Where the process described in paragraph (b) is carried out, preferably, one of either the total mole % of halogen atoms or groups -EH/-EΗ in compounds VI, VII and VIII is greater, for example by up to 10%, especially up to 5%, than the total mole % of the other one of either the total mole % of halogen atoms or groups -EH/-E'H in compounds VI, VII and VIII. Where the mole % of halogen atoms is greater, the polymer may have halogen end groups whereas when the mole % of groups -EH/-E'H is greater the polymer will have -EH/-EΗ end groups.
The molecular weight of the polymer can also be controlled by using an excess of halogen or hydroxy reactants. The excess may typically be in the range 0.1 to 5.0 mole %. The polymerisation reaction may be terminated by addition of one or more monofunctional reactants as end- cappers and such end-cappers may include said group FG.
The aforementioned processes for preparing the polymers are generally nucelophilic processes. Nonetheless, electrophilic processes can be used, by analogy to the processes described in US 5081215, US4808693, US4708448 and US5081215.
According to a third aspect of the present invention, there is provided a device for use in medical applications, wherein said device comprises a bio-
compatible polymeric material according to said first aspect or made in a method according to said second aspect .
Said device is preferably a prosthetic device, for example an implant such as an orthopaedic, dental or maxillofacila implant or a component thereof; or a device, for example a catheter, which is arranged to be temporarily associated with a human or animal body. Said device is preferably a prosthetic device as described. An orthopaedic device may be- an implant for a body joint, for example a hip or knee joint or spine fusion device.
A said device may include a part or parts made out of said bio-compatible polymeric material and a part or parts made out of other materials. Suitably, however, said device includes at least 50wt%, preferably at least 65wt%, more preferably at least 80wt%, especially at least 95wt% of said bio-compatible polymeric material. In some embodiments said device may consist essential of said bio- compatible polymeric material .
According to a fourth aspect, there is provided a method of making a device according to the third aspect, the method comprising: forming a material into a shape which represents or is a precursor of a device or a part of a device for use in medical applications wherein said material comprises a polymer; and treating material in said shape (preferably the surface thereof) thereby to cause bio-compatible moieties to associate with chain ends of said polymer (preferably chain ends present at or near the surface of said polymer) .
The invention extends to the use of a polymer having bio-compatible moieties associated with chain ends thereof in the manufacture of a device for use in a medical treatment, for example in surgery.
Any feature of any aspect of any invention or embodiment described herein may be combined with any feature of any aspect of any other invention or embodiment described herein.
-Specific embodiments-— of the invention will now be described, by way of example.
All chemicals referred to herein were used as received from Sigma-Aldrich Chemical Company, Dorset, U.K., unless otherwise stated.
Example 1 - Fluorine-terminated polyetheretherketone .
A 250ml, 3-necked, round-bottomed flask fitted with a stirrer/stirrer guide, nitrogen inlet and outlet and a thermocouple was charged with 4, 4 ' -difluorobenzophenone (22.26g, 0.102 mole), hydroquinone (ll.Olg, 0.10 mole) and diphenysulphone (60.0g) and purged with nitrogen for over 1 hour. The contents were then heated under a nitrogen blanket to between 140 and 150°C to form an almost colourless solution. While maintaining a nitrogen blanket, dried sodium carbonate (10.60g, 0.10 mole) and potassium carbonate (0.28g, 0.002 mole) was added. The temperature was raised to 175°C, held for 60 min; heated to 200 °C, held for 30mins; heated to 250°C, held for 30 mins; heated to 300°C and held for 120 mins .
The reaction mixture was allowed to cool, milled and washed with acetone and water. The resulting polymer was dried in an air oven at 120°C. The polymer had Inherent Viscosity (IV) of 0.71. IV is measured at 25°C on a solution of polymer in concentrated sulphuric acid of density 1.84gcm3, said solution containing 0. lg of polymer per 100cm3 of solution. A Melt Viscosity (MV) of 0.15kNsm"2 measured at 400°C on a ram extruder at a shear rate of 1000 s"1. A Number Average Molecular Weight 15000. Tg = 142°C and Tm = 342°C.
Example 2 - Disodium salt of 4-hydroxybenzoic acid
4-Hydroxybenzoic acid (30.36g, 0.22 mol) was added to a 500ml reaction flask containing a magnetic follower and sodium carbonate (46.64g, 0.440 mol) dissolved in deionised water (250ml) and heated at 80°C for 1 hour. The solution was cooled to room temperature, the insoluble material was removed by filtration, the water was evaporated and the residue was finely ground and dried under vacuum at 60°C overnight.
Example 3 - Carboxylic acid-surface modified polyetheretherketone .
An injection moulded specimen of the polymer of Example 1, 4cm x 1cm x 0.4cm, was placed in a 100ml round- bottomed flask fitted with a magnetic follower, reflux condenser and a nitrogen inlet and outlet and containing the disodium salt of 4-hydroxybenzoic acid of Example
2(1.82g, 0.01 mol) and N-methylpyrrolidone (50ml). The contents were stirred under an atmosphere of nitrogen at 150°C for 12hrs. The specimen was removed, washed with
deionised water, placed in a 100ml round-bottomed flask containing a magnetic follower and a 1M solution of hydrogen chloride in acetic acid (50ml) and stirred for 6 hours at room temperature .
Example 4 - Reaction of Carboxylic acid surface modified polyetheretherketone with the peptide GRGDS
The surface modified polyetheretherketone of Example 3 was stirred at 10°C for 1 hr under an atmosphere of nitrogen in an aqueous solution of the water soluble carbodiimide, 1-ethyl-3- (3-dimethylaminopropyl) - carbodiimide) (0.4g) dissolved in buffer at pH 4.5 (0.1M 2- (N-morpholino) ethanesulphonic acid) (40ml). The sample of polyetheretherketone was removed and washed with buffer solution.
The sample was stirred at 20°C for 24 hr under an atmosphere of nitrogen in a solution of the peptide GRGDS (80mg) in phosphate-buffered saline solution (40ml) (Na2HP04, 1.15g; KH2P04, 0.2g; NaCl. 8g; KCl, 0.2g; MgCl2, O.lg; CaCl2. 0. lg in 1 Litre of distilled water). The functionalised polyetherethereketone was washed successively with phosphate buffer and distilled water.
Example 5a - Hydroxy-terminated polyetheretherketone.
A 250ml, 3 -necked, round-bottomed flask fitted with a stirrer/stirrer guide, nitrogen inlet and outlet and a thermocouple was charged with 4, 4' -difluorobenzophenone (21.82g, 0.10 mole), hydroquinone (11.23g, 0.102 mole) and diphenysulphone (60.0g) and purged with nitrogen for over 1 hour. The contents were then heated under a nitrogen
blanket to between 140 and 150°C to form an almost colourless solution. While maintaining a nitrogen blanket, dried sodium carbonate (10.60g, 0.10 mole) and potassium carbonate (0.28g, 0.002 mole) was added. The temperature was raised to 175°C, held for 60 min; heated to 200 °C, held for 30mins; heated to 250°C, held for 30 mins; heated to 300°C and held for 120 mins.
The reaction mixture was allowed to cool, milled and washed with acetone and water. The resulting polymer was dried in an air oven at 120°C. The polymer had an IV of
0.79, and a Melt Viscosity (MV) of 0.17kNsm"2. Tg = 142°C and Tm = 342°C.
The hydroxy-terminated polymer was injection moulded into a specimen 4cm x 1cm x 0.4cm.
Example 5b - Dimethoxy-terminated polyetherethereketone .
A 250ml, 3 -necked, round-bottomed flask fitted with a stirrer/stirrer guide, nitrogen inlet and outlet and a thermocouple was charged with 4 , 4 ' -difluorobenzophenone (21.82g, 0.10 mole), hydroquinone (ll.Olg, 0.10 mole), 3, 5-dimethoxy-phenol (0.308g, 0.002mole) and diphenysulphone (60. Og) and purged with nitrogen for over 1 hour. The contents were then heated under a nitrogen blanket to between 140 and 150°C to form an almost colourless solution. While maintaining a nitrogen blanket, dried sodium carbonate (10.60g, 0.10 mole) and potassium carbonate (0.28g, 0.002 mole) was added. The temperature was raised to 175°C, held for 60 min; heated
to 200 °C, held for 30mins; heated to 250°C, held for 30 mins; heated to 300°C and held for 120 mins.
The reaction mixture was allowed to cool, milled and washed with acetone and water. The resulting polymer was dried in an air oven at 120°C. The polymer had an IV of 0.75.
The dimethoxy-terminated polymer was injection moulded into a specimen 4cm x 1cm x 0.4cm.
Example 5c - Dihydroxy-terminated polyetheretherketone .
The moulded sample from Example 5b was placed in a
700ml flanged flask fitted with a reflux condenser, a magnet follower and a nitrogen inlet and outlet and charged with glacial acetic acid (300ml) and 48% HBr
(180ml) . Under a nitrogen atmosphere and with continuous stirring the mixture was heated to reflux for 15 hrs. The reaction mixture was allowed to cool to room temperature and the PEEK sample was washed with ether (4 x 150ml) .
Example 6 - Amino-terminated polyetherketone .
A 250ml, 3 -necked, round-bottomed flask fitted with a stirrer/stirrer guide, nitrogen inlet and outlet and a thermocouple was charged with 4, 4' -difluorobenzophenone (22.04g, 0.101 mole), 4 , 4' -dihydroxbenzophenone (21.4g, 0.10 mole) hydroxytriphenylimine (0, 572g, 0.002 mole) and diphenysulphone (60. Og) and purged with nitrogen for over 1 hour. The contents were then heated under a nitrogen blanket to between 140 and 150°C to form an almost
colourless solution. While maintaining a nitrogen blanket, dried sodium carbonate (10.60g, 0.10 mole) and potassium carbonate (0.28g, 0.002 mole) was added. The temperature was raised to 175°C, held for 60 mins; heated to 200 °C, held for 30mins; heated to 250°C, held for 30 mins; heated to 300°C and held for 120 mins.
The reaction mixture was allowed to cool, milled and washed with acetone and water. The resulting polymer was dried in an air oven at 120°C. The polymer had a Tg = 142°C and Tm -= 339°C.
Example 7a - Sodium sulphonate-terminated polyetheretherketone .
A 250ml, 3-necked, round-bottomed flanged fitted with a stirrer/stirrer guide, nitrogen inlet and outlet and a thermocouple was charged with 4, 4' -difluorobenzophenone (22.04g, 0.101 mole), hydroquinone (ll.Olg, 0.10 mole) 4- hydroxybenzene sodium sulphonate dihydrate (0.23g, 0.001 mole) and diphenysulphone (60. Og) and purged with nitrogen for over 1 hour. The contents were then heated under a nitrogen blanket to between 140 and 150°C to form an almost colourless solution. While maintaining a nitrogen blanket, dried sodium carbonate (10.60g, 0.10 mole) and potassium carbonate (0.28g, 0.002 mole) was added. The temperature was raised to 175°C, held for 60 min; heated to 200 °C, held for 30mins; heated to 250°C, held for 30 mins; heated to 300°C and held for 120 mins.
The reaction mixture was allowed to cool, milled and washed with acetone and water. The resulting polymer was dried in an air oven at 120°C. The polymer had an IV of
0.81. Thereafter, the polymer was injection moulded into a specimen 4cm x 1cm x 0.4cm.
Example 7b - Sulphonic acid-terminated polyetheretherketone .
The specimen of Example 7a was placed in a 700ml flanged flask fitted with a reflux condenser, a magnetic follower and a nitrogen inlet and outlet and charged with 0.1M hydrochloric acid (500ml). Under a nitrogen atmosphere and with continuous stirring the contents were heated to 50°C for 6 hrs. The reaction mixture was allowed to cool to room temperature, the sample was removed, washed with deionised water until the pH was neutral and dried.
Example 7c - Sulphonyl chloride-terminated polyetheretherketone .
The plaque of Example 7b was placed in a 700ml flanged flask fitted with a reflux condenser, a magnetic follower and a nitrogen inlet and outlet and charged with thionyl chloride (250ml), dichloromethane (250ml) and dimethylformamide (30ml) . Under a nitrogen atmosphere and with continuous stirring the mixture was heated to under reflux for 15 hrs. The reaction mixture was allowed to cool to room temperature, the plaque was removed and washed with ether and dried in vacuo.
Example 8 - Reaction of Amino-surface modified polyetherketone with the peptide GRGDS
An injection moulded specimen of polymer of Example 6, 4cm x 1cm x 0.4cm, was placed in a 250ml round-bottomed flask fitted with a magnetic follower and a nitrogen inlet and outlet and containing N,N-dimethylacetamide (60ml) , and disuccinimidylsuberate (150mg) . The contents were stirred under an atmosphere of nitrogen at room temperature for 2hrs. The specimen was removed, washed with ether and dried in vacuo for lOhrs at 50°C. The dried sample was stirred at 20°C for 24 hr under an atmosphere of nitrogen in a solution of the peptide —GRGDS (-80mg) in--an- aqueous buffer solution (-40ml), pH 9. The functionalised PEK was washed successively with the buffer solution and ether.
Example 9 - Calcium Phosphate Deposition on a sulphonic acid and sodium sulphonate-surface modified polyetherethereketone .
A supersaturated calcium phosphate solution containing 5mM CaCl2, 1.5mM KH2P04 and 1.5mM Na2HP04 was prepared by mixing 0.1M Na2HP04 solution (1.5ml) and deionised water
(92ml), followed by the slow addition of 0.1M CaCl2 solution (5.0ml). The solution was stirred for 3 minutes and the injection moulded samples, 4cm x 1cm x 0.4cm, from Examples 7a and 7b were immersed in the solution for 1 hour. The specimens were washed with deionised water and blown dry with nitrogen. The process can be repeated several times to achieve a desired thickness.
Example 10 - Reaction of sulphonyl chloride-surface modified polyetheretherketone with 4-aminostyrene.
An injection moulded specimen of polymer from Example 7c, 4cm x 1cm x 0.4cm, was placed in a 250ml round- bottomed flask fitted with a magnetic follower and a nitrogen inlet and outlet and containing dichloromethane (50ml) and 4-aminostyrene (1.19g, 0. Olmole) . The contents were stirred under an atmosphere of nitrogen at 10°C for 16hrs. The specimen was removed, washed with ether and dried in vacuo for lOhrs at 40°C.
Example 11 - Reaction of Styrene-surface modified polyetheretherketone of Example 10 with hydroxyethylmethacrylate to produce polyetheretherketone with a surface coating of PHEMA.
The styrene-surface modified polyetheretherketone of Example 10 was placed in a 100ml round-bottomed flask, fitted with a magnetic follower, a nitrogen inlet and outlet and containing toluene (50ml) and hydroxyethylmethacrylate (3.25g) and benzoyl peroxide (0.005g). The mixture was stirred under nitrogen at 80°C for 2 hours. The sample of polyetheretherketone was removed washed with fresh toluene, followed by ethanol and water and dried in vacuum at 50°C for 24 hours.
Example 12 - Reaction of hydroxyl-surface modified polyetheretherketone with diphenylmethane-4, 4' - diisocyanate and diol to produce polyetheretherketone with a surface coating of polyurethane.
A 100ml round-bottomed flask, fitted with a magnetic follower, a nitrogen inlet and outlet was provided with THF (50ml), polyethylene glycol, PEG 1000 (lOg, 0.01 mol), diphenylmethane 4, 4' -diisocyanate (2.53g, 0.0101 mol) and
stannous octanoate (0. Olg) . The mixture was stirred under nitrogen at 50°C for 3 hours. The dihydroxy-modified polyetheretherketone of Example 5c was placed in flask and stirring was continued for a further 5 hours at 50°C. The sample of polyetheretherketone was removed washed with fresh THF and dried in a vacuum at 50°C for 24 hours.
Example 13 - An injection moulded specimen of polymer of Example 6, 4cm x 1cm x 0.4cm, was placed in a 100ml round-bottomed flask fitted with a magnetic follower
- n -a- nitrogen- i-n-l-et--and outlet and containing N,N- dimethylacetamide (50ml) , water soluble carbodiimide, 1- ethyl-3- (3-dimethylaminopropyl) -carbodiimide) (0.0625g) , ethylmorpholine (2ml) and MAP (Multiple Antigenic Peptide) , a commercially available tetravalent poly-lysine core (O.lmM) . The contents were stirred under an atmosphere of nitrogen at room temperature for 2hrs. The functionalised polyetherketone was washed successively with phosphate buffer and distilled water.
The reader's attention is directed to all papers and documents which are filed concurrently with or previous to this specification in connection with this application and which are open to public inspection with this specification, and the contents of all such papers and documents are incorporated herein by reference .
All of the features disclosed in this specification
(including any accompanying claims, abstract and drawings) , and/or all of the steps of any method or process so disclosed, may be combined in any combination, except combinations where at least some of such features and/or steps are mutually exclusive.
Each feature disclosed in this specification (including any accompanying claims, abstract and drawings) , may be replaced by alternative features serving the same, equivalent or similar purpose, unless expressly stated otherwise. Thus, unless expressly stated otherwise, each feature disclosed is one example only of a generic series of equivalent or similar features.
The invention is not restricted to the details of the
-foregoing embodiment (s) . The invention extend to any novel one, or any novel combination, of the features disclosed in this specification (including any accompanying claims, abstract and drawings) , or to any novel one, or any novel combination, of the steps of any method or process so disclosed.