WO2004084939A2 - Hiv-peptide-carrier-conjugates - Google Patents
Hiv-peptide-carrier-conjugates Download PDFInfo
- Publication number
- WO2004084939A2 WO2004084939A2 PCT/EP2004/003163 EP2004003163W WO2004084939A2 WO 2004084939 A2 WO2004084939 A2 WO 2004084939A2 EP 2004003163 W EP2004003163 W EP 2004003163W WO 2004084939 A2 WO2004084939 A2 WO 2004084939A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- seq
- amino acid
- acid sequence
- hiv
- sequence
- Prior art date
Links
- 108090000765 processed proteins & peptides Proteins 0.000 claims abstract description 293
- 102000004196 processed proteins & peptides Human genes 0.000 claims abstract description 247
- 239000000427 antigen Substances 0.000 claims abstract description 236
- 108091007433 antigens Proteins 0.000 claims abstract description 228
- 102000036639 antigens Human genes 0.000 claims abstract description 228
- 239000002245 particle Substances 0.000 claims abstract description 208
- 108091034117 Oligonucleotide Proteins 0.000 claims abstract description 146
- 230000003308 immunostimulating effect Effects 0.000 claims abstract description 138
- 239000000126 substance Substances 0.000 claims abstract description 116
- 229960005486 vaccine Drugs 0.000 claims abstract description 77
- 230000028993 immune response Effects 0.000 claims abstract description 61
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 claims abstract description 51
- 230000005867 T cell response Effects 0.000 claims abstract description 30
- 230000003612 virological effect Effects 0.000 claims abstract description 27
- 210000003719 b-lymphocyte Anatomy 0.000 claims abstract description 13
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 9
- 229920001184 polypeptide Polymers 0.000 claims description 235
- 101710132601 Capsid protein Proteins 0.000 claims description 210
- 239000000203 mixture Substances 0.000 claims description 195
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 134
- 238000000034 method Methods 0.000 claims description 130
- 101710125418 Major capsid protein Proteins 0.000 claims description 125
- 108090000623 proteins and genes Proteins 0.000 claims description 105
- 230000000890 antigenic effect Effects 0.000 claims description 89
- 102000004169 proteins and genes Human genes 0.000 claims description 85
- 150000007523 nucleic acids Chemical class 0.000 claims description 84
- 235000018102 proteins Nutrition 0.000 claims description 84
- 102000039446 nucleic acids Human genes 0.000 claims description 70
- 108020004707 nucleic acids Proteins 0.000 claims description 70
- NYHBQMYGNKIUIF-UUOKFMHZSA-N Guanosine Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O NYHBQMYGNKIUIF-UUOKFMHZSA-N 0.000 claims description 64
- 239000012634 fragment Substances 0.000 claims description 59
- 108020004414 DNA Proteins 0.000 claims description 56
- 241000829111 Human polyomavirus 1 Species 0.000 claims description 54
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 claims description 53
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 claims description 53
- 102000002067 Protein Subunits Human genes 0.000 claims description 52
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 claims description 52
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 claims description 52
- 241000700605 Viruses Species 0.000 claims description 50
- 108091081548 Palindromic sequence Proteins 0.000 claims description 49
- 108010001267 Protein Subunits Proteins 0.000 claims description 49
- 241001465754 Metazoa Species 0.000 claims description 48
- 230000027455 binding Effects 0.000 claims description 44
- 210000004027 cell Anatomy 0.000 claims description 40
- 108091035707 Consensus sequence Proteins 0.000 claims description 37
- 239000002773 nucleotide Substances 0.000 claims description 34
- 125000003729 nucleotide group Chemical group 0.000 claims description 34
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 claims description 33
- MIKUYHXYGGJMLM-GIMIYPNGSA-N Crotonoside Natural products C1=NC2=C(N)NC(=O)N=C2N1[C@H]1O[C@@H](CO)[C@H](O)[C@@H]1O MIKUYHXYGGJMLM-GIMIYPNGSA-N 0.000 claims description 32
- NYHBQMYGNKIUIF-UHFFFAOYSA-N D-guanosine Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(CO)C(O)C1O NYHBQMYGNKIUIF-UHFFFAOYSA-N 0.000 claims description 32
- 229940029575 guanosine Drugs 0.000 claims description 32
- 230000004048 modification Effects 0.000 claims description 32
- 238000012986 modification Methods 0.000 claims description 32
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 claims description 29
- 241000282414 Homo sapiens Species 0.000 claims description 27
- 239000002671 adjuvant Substances 0.000 claims description 26
- 241001515965 unidentified phage Species 0.000 claims description 26
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 claims description 23
- 230000002708 enhancing effect Effects 0.000 claims description 22
- 239000013612 plasmid Substances 0.000 claims description 22
- 230000003252 repetitive effect Effects 0.000 claims description 22
- 208000002672 hepatitis B Diseases 0.000 claims description 19
- 230000003053 immunization Effects 0.000 claims description 19
- 108091028043 Nucleic acid sequence Proteins 0.000 claims description 18
- 241000700721 Hepatitis B virus Species 0.000 claims description 16
- 101710192141 Protein Nef Proteins 0.000 claims description 14
- 241001534160 Escherichia virus Qbeta Species 0.000 claims description 13
- 102000004895 Lipoproteins Human genes 0.000 claims description 13
- 108090001030 Lipoproteins Proteins 0.000 claims description 13
- 229940115272 polyinosinic:polycytidylic acid Drugs 0.000 claims description 13
- 229920002477 rna polymer Polymers 0.000 claims description 12
- 125000003396 thiol group Chemical group [H]S* 0.000 claims description 12
- 102000053602 DNA Human genes 0.000 claims description 10
- 102000004127 Cytokines Human genes 0.000 claims description 9
- 108090000695 Cytokines Proteins 0.000 claims description 9
- 241000709738 Enterobacteria phage fr Species 0.000 claims description 9
- 101710177291 Gag polyprotein Proteins 0.000 claims description 9
- 241000124008 Mammalia Species 0.000 claims description 9
- 102000002689 Toll-like receptor Human genes 0.000 claims description 9
- 108020000411 Toll-like receptor Proteins 0.000 claims description 9
- 125000003277 amino group Chemical group 0.000 claims description 9
- 230000001939 inductive effect Effects 0.000 claims description 9
- 241001672158 Acinetobacter phage AP205 Species 0.000 claims description 8
- 229940031689 heterologous vaccine Drugs 0.000 claims description 8
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 claims description 7
- 241000702670 Rotavirus Species 0.000 claims description 7
- 239000002253 acid Substances 0.000 claims description 7
- 230000036755 cellular response Effects 0.000 claims description 7
- 230000004044 response Effects 0.000 claims description 7
- 241000709737 Enterobacteria phage GA Species 0.000 claims description 6
- 241000709743 Enterobacteria phage SP Species 0.000 claims description 6
- 241000709744 Enterobacterio phage MS2 Species 0.000 claims description 6
- 230000003213 activating effect Effects 0.000 claims description 6
- 241001430294 unidentified retrovirus Species 0.000 claims description 6
- 229940021995 DNA vaccine Drugs 0.000 claims description 5
- 241001278075 Enterobacteria phage MX1 Species 0.000 claims description 5
- 241001278054 Enterobacteria phage NL95 Species 0.000 claims description 5
- 241000710198 Foot-and-mouth disease virus Species 0.000 claims description 5
- 241000712079 Measles morbillivirus Species 0.000 claims description 5
- 241000714209 Norwalk virus Species 0.000 claims description 5
- 241000709749 Pseudomonas phage PP7 Species 0.000 claims description 5
- 108010015780 Viral Core Proteins Proteins 0.000 claims description 5
- 239000003085 diluting agent Substances 0.000 claims description 5
- 239000002158 endotoxin Substances 0.000 claims description 5
- 229920006008 lipopolysaccharide Polymers 0.000 claims description 5
- RHKWIGHJGOEUSM-UHFFFAOYSA-N 3h-imidazo[4,5-h]quinoline Chemical class C1=CN=C2C(N=CN3)=C3C=CC2=C1 RHKWIGHJGOEUSM-UHFFFAOYSA-N 0.000 claims description 4
- 108010041986 DNA Vaccines Proteins 0.000 claims description 4
- 241000709739 Enterobacteria phage f2 Species 0.000 claims description 4
- 241000701806 Human papillomavirus Species 0.000 claims description 4
- 108091005804 Peptidases Proteins 0.000 claims description 4
- 239000004365 Protease Substances 0.000 claims description 4
- 150000007513 acids Chemical class 0.000 claims description 4
- 210000001165 lymph node Anatomy 0.000 claims description 4
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 4
- 230000037452 priming Effects 0.000 claims description 4
- 241000702188 Enterobacteria phage M1 Species 0.000 claims description 3
- 108010061833 Integrases Proteins 0.000 claims description 3
- 230000028327 secretion Effects 0.000 claims description 3
- MJNIWUJSIGSWKK-UHFFFAOYSA-N Riboflavine 2',3',4',5'-tetrabutanoate Chemical compound CCCC(=O)OCC(OC(=O)CCC)C(OC(=O)CCC)C(OC(=O)CCC)CN1C2=CC(C)=C(C)C=C2N=C2C1=NC(=O)NC2=O MJNIWUJSIGSWKK-UHFFFAOYSA-N 0.000 claims 10
- 102100034343 Integrase Human genes 0.000 claims 6
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 claims 2
- 201000005505 Measles Diseases 0.000 claims 1
- 208000003154 papilloma Diseases 0.000 claims 1
- 230000002163 immunogen Effects 0.000 abstract description 15
- 238000002255 vaccination Methods 0.000 abstract description 11
- 201000010099 disease Diseases 0.000 abstract description 9
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 9
- 230000001225 therapeutic effect Effects 0.000 abstract description 5
- 239000003814 drug Substances 0.000 abstract description 4
- 230000000069 prophylactic effect Effects 0.000 abstract description 4
- 206010020751 Hypersensitivity Diseases 0.000 abstract description 2
- 230000007815 allergy Effects 0.000 abstract description 2
- 230000001684 chronic effect Effects 0.000 abstract description 2
- 241000725303 Human immunodeficiency virus Species 0.000 description 186
- 101710094648 Coat protein Proteins 0.000 description 113
- 101710141454 Nucleoprotein Proteins 0.000 description 113
- 101710083689 Probable capsid protein Proteins 0.000 description 113
- 102100021181 Golgi phosphoprotein 3 Human genes 0.000 description 109
- CTMZLDSMFCVUNX-VMIOUTBZSA-N cytidylyl-(3'->5')-guanosine Chemical group O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@H](OP(O)(=O)OC[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C3=C(C(N=C(N)N3)=O)N=C2)O)[C@@H](CO)O1 CTMZLDSMFCVUNX-VMIOUTBZSA-N 0.000 description 109
- 235000001014 amino acid Nutrition 0.000 description 90
- 229940024606 amino acid Drugs 0.000 description 90
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 73
- 150000001413 amino acids Chemical class 0.000 description 60
- 210000000234 capsid Anatomy 0.000 description 58
- 238000005859 coupling reaction Methods 0.000 description 45
- 230000008878 coupling Effects 0.000 description 35
- 238000010168 coupling process Methods 0.000 description 35
- 230000004927 fusion Effects 0.000 description 32
- 239000013256 coordination polymer Substances 0.000 description 27
- 102000037865 fusion proteins Human genes 0.000 description 27
- 108020001507 fusion proteins Proteins 0.000 description 27
- 239000004971 Cross linker Substances 0.000 description 24
- 229940046168 CpG oligodeoxynucleotide Drugs 0.000 description 23
- 108020002230 Pancreatic Ribonuclease Proteins 0.000 description 23
- 102000005891 Pancreatic ribonuclease Human genes 0.000 description 23
- 238000003780 insertion Methods 0.000 description 23
- 230000037431 insertion Effects 0.000 description 23
- 125000000539 amino acid group Chemical group 0.000 description 21
- 210000004899 c-terminal region Anatomy 0.000 description 20
- 239000013598 vector Substances 0.000 description 19
- 208000015181 infectious disease Diseases 0.000 description 18
- 241000588724 Escherichia coli Species 0.000 description 17
- 210000001744 T-lymphocyte Anatomy 0.000 description 16
- 238000012217 deletion Methods 0.000 description 16
- 230000037430 deletion Effects 0.000 description 16
- 238000007792 addition Methods 0.000 description 14
- 210000004443 dendritic cell Anatomy 0.000 description 14
- 238000000502 dialysis Methods 0.000 description 14
- 238000002649 immunization Methods 0.000 description 14
- 238000006467 substitution reaction Methods 0.000 description 14
- 238000006243 chemical reaction Methods 0.000 description 13
- 239000003795 chemical substances by application Substances 0.000 description 13
- 239000003446 ligand Substances 0.000 description 13
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 12
- 239000000178 monomer Substances 0.000 description 12
- 238000011282 treatment Methods 0.000 description 12
- 235000018417 cysteine Nutrition 0.000 description 11
- 230000029087 digestion Effects 0.000 description 11
- 230000003993 interaction Effects 0.000 description 11
- 239000000047 product Substances 0.000 description 11
- 108090000565 Capsid Proteins Proteins 0.000 description 10
- 102100023321 Ceruloplasmin Human genes 0.000 description 10
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 10
- 239000007771 core particle Substances 0.000 description 10
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 10
- 239000000499 gel Substances 0.000 description 10
- 238000011534 incubation Methods 0.000 description 10
- 239000002243 precursor Substances 0.000 description 10
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 9
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 9
- 241000699670 Mus sp. Species 0.000 description 9
- 108010076039 Polyproteins Proteins 0.000 description 9
- 238000013459 approach Methods 0.000 description 9
- 238000003491 array Methods 0.000 description 9
- 230000002458 infectious effect Effects 0.000 description 9
- 238000000746 purification Methods 0.000 description 9
- 229960005542 ethidium bromide Drugs 0.000 description 8
- 238000002523 gelfiltration Methods 0.000 description 8
- 230000005012 migration Effects 0.000 description 8
- 238000013508 migration Methods 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 7
- QFVHZQCOUORWEI-UHFFFAOYSA-N 4-[(4-anilino-5-sulfonaphthalen-1-yl)diazenyl]-5-hydroxynaphthalene-2,7-disulfonic acid Chemical compound C=12C(O)=CC(S(O)(=O)=O)=CC2=CC(S(O)(=O)=O)=CC=1N=NC(C1=CC=CC(=C11)S(O)(=O)=O)=CC=C1NC1=CC=CC=C1 QFVHZQCOUORWEI-UHFFFAOYSA-N 0.000 description 7
- 239000004471 Glycine Substances 0.000 description 7
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical group SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 7
- 241001505332 Polyomavirus sp. Species 0.000 description 7
- 230000004913 activation Effects 0.000 description 7
- 230000001580 bacterial effect Effects 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 150000001875 compounds Chemical class 0.000 description 7
- 230000000694 effects Effects 0.000 description 7
- 238000001493 electron microscopy Methods 0.000 description 7
- ZMMJGEGLRURXTF-UHFFFAOYSA-N ethidium bromide Chemical compound [Br-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CC)=C1C1=CC=CC=C1 ZMMJGEGLRURXTF-UHFFFAOYSA-N 0.000 description 7
- 210000000987 immune system Anatomy 0.000 description 7
- 238000004519 manufacturing process Methods 0.000 description 7
- -1 phosphoester Chemical class 0.000 description 7
- 230000003362 replicative effect Effects 0.000 description 7
- 241000701447 unidentified baculovirus Species 0.000 description 7
- 241000894006 Bacteria Species 0.000 description 6
- 241000699666 Mus <mouse, genus> Species 0.000 description 6
- 108010083644 Ribonucleases Proteins 0.000 description 6
- 102000006382 Ribonucleases Human genes 0.000 description 6
- 108091008874 T cell receptors Proteins 0.000 description 6
- 210000000612 antigen-presenting cell Anatomy 0.000 description 6
- 235000009697 arginine Nutrition 0.000 description 6
- 125000000637 arginyl group Chemical class N[C@@H](CCCNC(N)=N)C(=O)* 0.000 description 6
- 239000002299 complementary DNA Substances 0.000 description 6
- 238000005516 engineering process Methods 0.000 description 6
- 230000006870 function Effects 0.000 description 6
- 229930182817 methionine Natural products 0.000 description 6
- 244000005700 microbiome Species 0.000 description 6
- 238000004806 packaging method and process Methods 0.000 description 6
- 238000012545 processing Methods 0.000 description 6
- 230000009467 reduction Effects 0.000 description 6
- 238000001338 self-assembly Methods 0.000 description 6
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 6
- 241000894007 species Species 0.000 description 6
- 231100000331 toxic Toxicity 0.000 description 6
- 230000002588 toxic effect Effects 0.000 description 6
- 241000388186 Deltapapillomavirus 4 Species 0.000 description 5
- 241000282412 Homo Species 0.000 description 5
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 5
- 108060003951 Immunoglobulin Proteins 0.000 description 5
- 102100034349 Integrase Human genes 0.000 description 5
- 241000712899 Lymphocytic choriomeningitis mammarenavirus Species 0.000 description 5
- 229920002684 Sepharose Polymers 0.000 description 5
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 238000010367 cloning Methods 0.000 description 5
- 239000013078 crystal Substances 0.000 description 5
- 230000003247 decreasing effect Effects 0.000 description 5
- 238000001212 derivatisation Methods 0.000 description 5
- 125000000524 functional group Chemical group 0.000 description 5
- 102000018358 immunoglobulin Human genes 0.000 description 5
- 238000001727 in vivo Methods 0.000 description 5
- 230000006698 induction Effects 0.000 description 5
- 125000001360 methionine group Chemical group N[C@@H](CCSC)C(=O)* 0.000 description 5
- 102000040430 polynucleotide Human genes 0.000 description 5
- 108091033319 polynucleotide Proteins 0.000 description 5
- 230000003389 potentiating effect Effects 0.000 description 5
- 102000005962 receptors Human genes 0.000 description 5
- 108020003175 receptors Proteins 0.000 description 5
- 238000010186 staining Methods 0.000 description 5
- 239000006228 supernatant Substances 0.000 description 5
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 4
- 108020000946 Bacterial DNA Proteins 0.000 description 4
- 208000035473 Communicable disease Diseases 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- 241000709747 Enterobacteria phage R17 Species 0.000 description 4
- 241000238631 Hexapoda Species 0.000 description 4
- 102100024065 Inhibitor of growth protein 1 Human genes 0.000 description 4
- 239000004472 Lysine Substances 0.000 description 4
- 101710107921 Secreted protein BARF1 Proteins 0.000 description 4
- 108010034546 Serratia marcescens nuclease Proteins 0.000 description 4
- 230000024932 T cell mediated immunity Effects 0.000 description 4
- 239000007983 Tris buffer Substances 0.000 description 4
- 239000011543 agarose gel Substances 0.000 description 4
- 238000000246 agarose gel electrophoresis Methods 0.000 description 4
- FPIPGXGPPPQFEQ-OVSJKPMPSA-N all-trans-retinol Chemical compound OC\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-OVSJKPMPSA-N 0.000 description 4
- 230000004071 biological effect Effects 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 238000010276 construction Methods 0.000 description 4
- 230000028996 humoral immune response Effects 0.000 description 4
- 230000002209 hydrophobic effect Effects 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- 230000001965 increasing effect Effects 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 210000002540 macrophage Anatomy 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 244000052769 pathogen Species 0.000 description 4
- 230000001717 pathogenic effect Effects 0.000 description 4
- YBYRMVIVWMBXKQ-UHFFFAOYSA-N phenylmethanesulfonyl fluoride Chemical compound FS(=O)(=O)CC1=CC=CC=C1 YBYRMVIVWMBXKQ-UHFFFAOYSA-N 0.000 description 4
- 239000002157 polynucleotide Substances 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 229930000044 secondary metabolite Natural products 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 4
- MPVDXIMFBOLMNW-ISLYRVAYSA-N 7-hydroxy-8-[(E)-phenyldiazenyl]naphthalene-1,3-disulfonic acid Chemical compound OC1=CC=C2C=C(S(O)(=O)=O)C=C(S(O)(=O)=O)C2=C1\N=N\C1=CC=CC=C1 MPVDXIMFBOLMNW-ISLYRVAYSA-N 0.000 description 3
- 241000271566 Aves Species 0.000 description 3
- 101150083464 CP gene Proteins 0.000 description 3
- 108091026890 Coding region Proteins 0.000 description 3
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 208000031886 HIV Infections Diseases 0.000 description 3
- 241000713340 Human immunodeficiency virus 2 Species 0.000 description 3
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 3
- 108091054437 MHC class I family Proteins 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 108020004511 Recombinant DNA Proteins 0.000 description 3
- 241000710960 Sindbis virus Species 0.000 description 3
- PZBFGYYEXUXCOF-UHFFFAOYSA-N TCEP Chemical compound OC(=O)CCP(CCC(O)=O)CCC(O)=O PZBFGYYEXUXCOF-UHFFFAOYSA-N 0.000 description 3
- 108020005038 Terminator Codon Proteins 0.000 description 3
- 241000700618 Vaccinia virus Species 0.000 description 3
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 3
- 108010067390 Viral Proteins Proteins 0.000 description 3
- 230000004075 alteration Effects 0.000 description 3
- 230000005875 antibody response Effects 0.000 description 3
- 210000001185 bone marrow Anatomy 0.000 description 3
- 230000001413 cellular effect Effects 0.000 description 3
- 238000003776 cleavage reaction Methods 0.000 description 3
- 238000004132 cross linking Methods 0.000 description 3
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical class NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 238000013461 design Methods 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- 229940088598 enzyme Drugs 0.000 description 3
- 239000013604 expression vector Substances 0.000 description 3
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 3
- 125000003630 glycyl group Chemical group [H]N([H])C([H])([H])C(*)=O 0.000 description 3
- 210000002865 immune cell Anatomy 0.000 description 3
- 229940072221 immunoglobulins Drugs 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 210000005007 innate immune system Anatomy 0.000 description 3
- 239000012160 loading buffer Substances 0.000 description 3
- 230000035800 maturation Effects 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 238000010369 molecular cloning Methods 0.000 description 3
- 229940046166 oligodeoxynucleotide Drugs 0.000 description 3
- 230000008520 organization Effects 0.000 description 3
- 239000008188 pellet Substances 0.000 description 3
- 239000008194 pharmaceutical composition Substances 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 230000010076 replication Effects 0.000 description 3
- 210000003705 ribosome Anatomy 0.000 description 3
- 230000007017 scission Effects 0.000 description 3
- 239000000243 solution Substances 0.000 description 3
- 238000001228 spectrum Methods 0.000 description 3
- 230000004936 stimulating effect Effects 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 229920001059 synthetic polymer Polymers 0.000 description 3
- 230000014616 translation Effects 0.000 description 3
- PIEPQKCYPFFYMG-UHFFFAOYSA-N tris acetate Chemical compound CC(O)=O.OCC(N)(CO)CO PIEPQKCYPFFYMG-UHFFFAOYSA-N 0.000 description 3
- 238000005199 ultracentrifugation Methods 0.000 description 3
- 241000701161 unidentified adenovirus Species 0.000 description 3
- 239000013603 viral vector Substances 0.000 description 3
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 3
- WCMOHMXWOOBVMZ-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 6-[3-(2,5-dioxopyrrol-1-yl)propanoylamino]hexanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCCCNC(=O)CCN1C(=O)C=CC1=O WCMOHMXWOOBVMZ-UHFFFAOYSA-N 0.000 description 2
- FPIPGXGPPPQFEQ-UHFFFAOYSA-N 13-cis retinol Natural products OCC=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-UHFFFAOYSA-N 0.000 description 2
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- 241000282693 Cercopithecidae Species 0.000 description 2
- 108020004705 Codon Proteins 0.000 description 2
- 241000701022 Cytomegalovirus Species 0.000 description 2
- SHIBSTMRCDJXLN-UHFFFAOYSA-N Digoxigenin Natural products C1CC(C2C(C3(C)CCC(O)CC3CC2)CC2O)(O)C2(C)C1C1=CC(=O)OC1 SHIBSTMRCDJXLN-UHFFFAOYSA-N 0.000 description 2
- 238000011510 Elispot assay Methods 0.000 description 2
- 241000196324 Embryophyta Species 0.000 description 2
- 241000709661 Enterovirus Species 0.000 description 2
- 108010040721 Flagellin Proteins 0.000 description 2
- 101710121996 Hexon protein p72 Proteins 0.000 description 2
- 102000014150 Interferons Human genes 0.000 description 2
- 108010050904 Interferons Proteins 0.000 description 2
- 108010065805 Interleukin-12 Proteins 0.000 description 2
- UQSXHKLRYXJYBZ-UHFFFAOYSA-N Iron oxide Chemical compound [Fe]=O UQSXHKLRYXJYBZ-UHFFFAOYSA-N 0.000 description 2
- 102000043129 MHC class I family Human genes 0.000 description 2
- 241001529936 Murinae Species 0.000 description 2
- 108700026244 Open Reading Frames Proteins 0.000 description 2
- 101710190786 PI protein Proteins 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 229930012538 Paclitaxel Natural products 0.000 description 2
- 241001631646 Papillomaviridae Species 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 108091036414 Polyinosinic:polycytidylic acid Proteins 0.000 description 2
- 241000125945 Protoparvovirus Species 0.000 description 2
- 241000235070 Saccharomyces Species 0.000 description 2
- 239000012506 Sephacryl® Substances 0.000 description 2
- 241000700584 Simplexvirus Species 0.000 description 2
- 108091081024 Start codon Proteins 0.000 description 2
- 108010090804 Streptavidin Proteins 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 108010008038 Synthetic Vaccines Proteins 0.000 description 2
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical class OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 2
- 241000723873 Tobacco mosaic virus Species 0.000 description 2
- 108010003533 Viral Envelope Proteins Proteins 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 2
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 2
- 235000011130 ammonium sulphate Nutrition 0.000 description 2
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 2
- 229960000723 ampicillin Drugs 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 230000002238 attenuated effect Effects 0.000 description 2
- 239000011324 bead Substances 0.000 description 2
- 229960002685 biotin Drugs 0.000 description 2
- 235000020958 biotin Nutrition 0.000 description 2
- 239000011616 biotin Substances 0.000 description 2
- AIYUHDOJVYHVIT-UHFFFAOYSA-M caesium chloride Chemical compound [Cl-].[Cs+] AIYUHDOJVYHVIT-UHFFFAOYSA-M 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 238000001516 cell proliferation assay Methods 0.000 description 2
- 230000007969 cellular immunity Effects 0.000 description 2
- ZPUCINDJVBIVPJ-LJISPDSOSA-N cocaine Chemical compound O([C@H]1C[C@@H]2CC[C@@H](N2C)[C@H]1C(=O)OC)C(=O)C1=CC=CC=C1 ZPUCINDJVBIVPJ-LJISPDSOSA-N 0.000 description 2
- 230000000139 costimulatory effect Effects 0.000 description 2
- 210000000805 cytoplasm Anatomy 0.000 description 2
- 231100000433 cytotoxic Toxicity 0.000 description 2
- 230000001472 cytotoxic effect Effects 0.000 description 2
- 238000002784 cytotoxicity assay Methods 0.000 description 2
- 231100000263 cytotoxicity test Toxicity 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- 239000007857 degradation product Substances 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- 229940029030 dendritic cell vaccine Drugs 0.000 description 2
- QONQRTHLHBTMGP-UHFFFAOYSA-N digitoxigenin Natural products CC12CCC(C3(CCC(O)CC3CC3)C)C3C11OC1CC2C1=CC(=O)OC1 QONQRTHLHBTMGP-UHFFFAOYSA-N 0.000 description 2
- SHIBSTMRCDJXLN-KCZCNTNESA-N digoxigenin Chemical compound C1([C@@H]2[C@@]3([C@@](CC2)(O)[C@H]2[C@@H]([C@@]4(C)CC[C@H](O)C[C@H]4CC2)C[C@H]3O)C)=CC(=O)OC1 SHIBSTMRCDJXLN-KCZCNTNESA-N 0.000 description 2
- 239000000539 dimer Substances 0.000 description 2
- 230000008034 disappearance Effects 0.000 description 2
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 2
- 229910000397 disodium phosphate Inorganic materials 0.000 description 2
- 235000019800 disodium phosphate Nutrition 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 239000012636 effector Substances 0.000 description 2
- 230000008030 elimination Effects 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 210000003527 eukaryotic cell Anatomy 0.000 description 2
- 230000001747 exhibiting effect Effects 0.000 description 2
- 229960002143 fluorescein Drugs 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 2
- 210000002443 helper t lymphocyte Anatomy 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 229910052588 hydroxylapatite Inorganic materials 0.000 description 2
- 150000003949 imides Chemical class 0.000 description 2
- 230000000951 immunodiffusion Effects 0.000 description 2
- 230000005847 immunogenicity Effects 0.000 description 2
- 229940079322 interferon Drugs 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 238000011005 laboratory method Methods 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 239000006166 lysate Substances 0.000 description 2
- 125000005439 maleimidyl group Chemical group C1(C=CC(N1*)=O)=O 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 229910021645 metal ion Inorganic materials 0.000 description 2
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 2
- 230000000813 microbial effect Effects 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 238000001823 molecular biology technique Methods 0.000 description 2
- 238000013365 molecular weight analysis method Methods 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- 229920005615 natural polymer Polymers 0.000 description 2
- 238000006384 oligomerization reaction Methods 0.000 description 2
- 210000001672 ovary Anatomy 0.000 description 2
- 229960001592 paclitaxel Drugs 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- XYJRXVWERLGGKC-UHFFFAOYSA-D pentacalcium;hydroxide;triphosphate Chemical compound [OH-].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O XYJRXVWERLGGKC-UHFFFAOYSA-D 0.000 description 2
- 229940023041 peptide vaccine Drugs 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 150000004713 phosphodiesters Chemical group 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 230000000770 proinflammatory effect Effects 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 238000011321 prophylaxis Methods 0.000 description 2
- 239000012474 protein marker Substances 0.000 description 2
- 238000001742 protein purification Methods 0.000 description 2
- 230000012743 protein tagging Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 108091008146 restriction endonucleases Proteins 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- 229960003471 retinol Drugs 0.000 description 2
- 235000020944 retinol Nutrition 0.000 description 2
- 239000011607 retinol Substances 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 description 2
- 238000007086 side reaction Methods 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 2
- 238000013519 translation Methods 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- SNICXCGAKADSCV-JTQLQIEISA-N (-)-Nicotine Chemical compound CN1CCC[C@H]1C1=CC=CN=C1 SNICXCGAKADSCV-JTQLQIEISA-N 0.000 description 1
- JWDFQMWEFLOOED-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-(pyridin-2-yldisulfanyl)propanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCSSC1=CC=CC=N1 JWDFQMWEFLOOED-UHFFFAOYSA-N 0.000 description 1
- AUHDWARTFSKSAC-HEIFUQTGSA-N (2S,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)-2-(6-oxo-1H-purin-9-yl)oxolane-2-carboxylic acid Chemical compound [C@]1([C@H](O)[C@H](O)[C@@H](CO)O1)(N1C=NC=2C(O)=NC=NC12)C(=O)O AUHDWARTFSKSAC-HEIFUQTGSA-N 0.000 description 1
- ASWBNKHCZGQVJV-UHFFFAOYSA-N (3-hexadecanoyloxy-2-hydroxypropyl) 2-(trimethylazaniumyl)ethyl phosphate Chemical compound CCCCCCCCCCCCCCCC(=O)OCC(O)COP([O-])(=O)OCC[N+](C)(C)C ASWBNKHCZGQVJV-UHFFFAOYSA-N 0.000 description 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- DIYPCWKHSODVAP-UHFFFAOYSA-N 1-[3-(2,5-dioxopyrrol-1-yl)benzoyl]oxy-2,5-dioxopyrrolidine-3-sulfonic acid Chemical compound O=C1C(S(=O)(=O)O)CC(=O)N1OC(=O)C1=CC=CC(N2C(C=CC2=O)=O)=C1 DIYPCWKHSODVAP-UHFFFAOYSA-N 0.000 description 1
- CULQNACJHGHAER-UHFFFAOYSA-N 1-[4-[(2-iodoacetyl)amino]benzoyl]oxy-2,5-dioxopyrrolidine-3-sulfonic acid Chemical compound O=C1C(S(=O)(=O)O)CC(=O)N1OC(=O)C1=CC=C(NC(=O)CI)C=C1 CULQNACJHGHAER-UHFFFAOYSA-N 0.000 description 1
- UFBJCMHMOXMLKC-UHFFFAOYSA-N 2,4-dinitrophenol Chemical compound OC1=CC=C([N+]([O-])=O)C=C1[N+]([O-])=O UFBJCMHMOXMLKC-UHFFFAOYSA-N 0.000 description 1
- ASNTZYQMIUCEBV-UHFFFAOYSA-N 2,5-dioxo-1-[6-[3-(pyridin-2-yldisulfanyl)propanoylamino]hexanoyloxy]pyrrolidine-3-sulfonic acid Chemical compound O=C1C(S(=O)(=O)O)CC(=O)N1OC(=O)CCCCCNC(=O)CCSSC1=CC=CC=N1 ASNTZYQMIUCEBV-UHFFFAOYSA-N 0.000 description 1
- VDCRFBBZFHHYGT-IOSLPCCCSA-N 2-amino-9-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-enyl-3h-purine-6,8-dione Chemical compound O=C1N(CC=C)C=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O VDCRFBBZFHHYGT-IOSLPCCCSA-N 0.000 description 1
- ASJSAQIRZKANQN-CRCLSJGQSA-N 2-deoxy-D-ribose Chemical compound OC[C@@H](O)[C@@H](O)CC=O ASJSAQIRZKANQN-CRCLSJGQSA-N 0.000 description 1
- GNTFNWAZTKLCRE-UHFFFAOYSA-N 4-amino-4-bromo-1,3-dihydropyrimidin-2-one Chemical compound NC1(Br)NC(=O)NC=C1 GNTFNWAZTKLCRE-UHFFFAOYSA-N 0.000 description 1
- OZFPSOBLQZPIAV-UHFFFAOYSA-N 5-nitro-1h-indole Chemical compound [O-][N+](=O)C1=CC=C2NC=CC2=C1 OZFPSOBLQZPIAV-UHFFFAOYSA-N 0.000 description 1
- 102100032123 AMP deaminase 1 Human genes 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- 241000701242 Adenoviridae Species 0.000 description 1
- 241000701386 African swine fever virus Species 0.000 description 1
- 229920000936 Agarose Polymers 0.000 description 1
- 102100027211 Albumin Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 241000710929 Alphavirus Species 0.000 description 1
- 102000004400 Aminopeptidases Human genes 0.000 description 1
- 108090000915 Aminopeptidases Proteins 0.000 description 1
- 241000239223 Arachnida Species 0.000 description 1
- 241000712892 Arenaviridae Species 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 108090001008 Avidin Proteins 0.000 description 1
- 238000011725 BALB/c mouse Methods 0.000 description 1
- 241000702628 Birnaviridae Species 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- CIUUIPMOFZIWIZ-UHFFFAOYSA-N Bropirimine Chemical compound NC1=NC(O)=C(Br)C(C=2C=CC=CC=2)=N1 CIUUIPMOFZIWIZ-UHFFFAOYSA-N 0.000 description 1
- 101100228196 Caenorhabditis elegans gly-4 gene Proteins 0.000 description 1
- 241000282836 Camelus dromedarius Species 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 101710169873 Capsid protein G8P Proteins 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 241001275954 Cortinarius caperatus Species 0.000 description 1
- 241000186216 Corynebacterium Species 0.000 description 1
- 241000723655 Cowpea mosaic virus Species 0.000 description 1
- 241000709687 Coxsackievirus Species 0.000 description 1
- 241000938605 Crocodylia Species 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- 238000011238 DNA vaccination Methods 0.000 description 1
- 230000004568 DNA-binding Effects 0.000 description 1
- 241000710829 Dengue virus group Species 0.000 description 1
- 101000797456 Dictyostelium discoideum AMP deaminase Proteins 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 241001115402 Ebolavirus Species 0.000 description 1
- 241001466953 Echovirus Species 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 102100038132 Endogenous retrovirus group K member 6 Pro protein Human genes 0.000 description 1
- 102000004533 Endonucleases Human genes 0.000 description 1
- 108010042407 Endonucleases Proteins 0.000 description 1
- 241000991587 Enterovirus C Species 0.000 description 1
- 101710091045 Envelope protein Proteins 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 208000000832 Equine Encephalomyelitis Diseases 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- 108060002716 Exonuclease Proteins 0.000 description 1
- 206010063827 Extramedullary haemopoiesis Diseases 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 241000724791 Filamentous phage Species 0.000 description 1
- 241000711950 Filoviridae Species 0.000 description 1
- 241000723754 Flock house virus Species 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 241000287828 Gallus gallus Species 0.000 description 1
- 208000005577 Gastroenteritis Diseases 0.000 description 1
- YLJHCWNDBKKOEB-IHRRRGAJSA-N Glu-Glu-Phe Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC1=CC=CC=C1)C(O)=O YLJHCWNDBKKOEB-IHRRRGAJSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 241000856850 Goose coronavirus Species 0.000 description 1
- 241001506229 Goose reovirus Species 0.000 description 1
- 102000004457 Granulocyte-Macrophage Colony-Stimulating Factor Human genes 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 208000037357 HIV infectious disease Diseases 0.000 description 1
- 229940033330 HIV vaccine Drugs 0.000 description 1
- 206010061192 Haemorrhagic fever Diseases 0.000 description 1
- 241000150562 Hantaan orthohantavirus Species 0.000 description 1
- 102100021410 Heat shock 70 kDa protein 14 Human genes 0.000 description 1
- 102000002812 Heat-Shock Proteins Human genes 0.000 description 1
- 108010004889 Heat-Shock Proteins Proteins 0.000 description 1
- 241000700739 Hepadnaviridae Species 0.000 description 1
- 208000005176 Hepatitis C Diseases 0.000 description 1
- 208000005331 Hepatitis D Diseases 0.000 description 1
- 241000709721 Hepatovirus A Species 0.000 description 1
- GVGLGOZIDCSQPN-PVHGPHFFSA-N Heroin Chemical compound O([C@H]1[C@H](C=C[C@H]23)OC(C)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4OC(C)=O GVGLGOZIDCSQPN-PVHGPHFFSA-N 0.000 description 1
- 241000700586 Herpesviridae Species 0.000 description 1
- 102000008949 Histocompatibility Antigens Class I Human genes 0.000 description 1
- 101000775844 Homo sapiens AMP deaminase 1 Proteins 0.000 description 1
- 101001041756 Homo sapiens Heat shock 70 kDa protein 14 Proteins 0.000 description 1
- 101000763579 Homo sapiens Toll-like receptor 1 Proteins 0.000 description 1
- 101000763537 Homo sapiens Toll-like receptor 10 Proteins 0.000 description 1
- 101000831567 Homo sapiens Toll-like receptor 2 Proteins 0.000 description 1
- 101000831496 Homo sapiens Toll-like receptor 3 Proteins 0.000 description 1
- 101000669447 Homo sapiens Toll-like receptor 4 Proteins 0.000 description 1
- 101000669460 Homo sapiens Toll-like receptor 5 Proteins 0.000 description 1
- 101000669406 Homo sapiens Toll-like receptor 6 Proteins 0.000 description 1
- 101000669402 Homo sapiens Toll-like receptor 7 Proteins 0.000 description 1
- 101000800483 Homo sapiens Toll-like receptor 8 Proteins 0.000 description 1
- 241000701085 Human alphaherpesvirus 3 Species 0.000 description 1
- 241000701828 Human papillomavirus type 11 Species 0.000 description 1
- 241000341655 Human papillomavirus type 16 Species 0.000 description 1
- 241000709701 Human poliovirus 1 Species 0.000 description 1
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 1
- 206010061598 Immunodeficiency Diseases 0.000 description 1
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 1
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 1
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 1
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 1
- UGQMRVRMYYASKQ-KQYNXXCUSA-N Inosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(O)=C2N=C1 UGQMRVRMYYASKQ-KQYNXXCUSA-N 0.000 description 1
- GRSZFWQUAKGDAV-UHFFFAOYSA-N Inosinic acid Natural products OC1C(O)C(COP(O)(O)=O)OC1N1C(NC=NC2=O)=C2N=C1 GRSZFWQUAKGDAV-UHFFFAOYSA-N 0.000 description 1
- 102000002227 Interferon Type I Human genes 0.000 description 1
- 108010014726 Interferon Type I Proteins 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- 241000701377 Iridoviridae Species 0.000 description 1
- 239000007836 KH2PO4 Substances 0.000 description 1
- 241000701646 Kappapapillomavirus 2 Species 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- 241000713666 Lentivirus Species 0.000 description 1
- 239000000232 Lipid Bilayer Substances 0.000 description 1
- 208000008771 Lymphadenopathy Diseases 0.000 description 1
- 241000282553 Macaca Species 0.000 description 1
- 101710156564 Major tail protein Gp23 Proteins 0.000 description 1
- 241000283923 Marmota monax Species 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 241000711386 Mumps virus Species 0.000 description 1
- 102000016943 Muramidase Human genes 0.000 description 1
- 108010014251 Muramidase Proteins 0.000 description 1
- 108010062010 N-Acetylmuramoyl-L-alanine Amidase Proteins 0.000 description 1
- 108091060545 Nonsense suppressor Proteins 0.000 description 1
- 101710163270 Nuclease Proteins 0.000 description 1
- 108010038807 Oligopeptides Proteins 0.000 description 1
- 102000015636 Oligopeptides Human genes 0.000 description 1
- 241000712464 Orthomyxoviridae Species 0.000 description 1
- 241000150218 Orthonairovirus Species 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- 241000711504 Paramyxoviridae Species 0.000 description 1
- 208000002606 Paramyxoviridae Infections Diseases 0.000 description 1
- 241000237988 Patellidae Species 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091093037 Peptide nucleic acid Proteins 0.000 description 1
- 241000713137 Phlebovirus Species 0.000 description 1
- 241000709664 Picornaviridae Species 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 241000700625 Poxviridae Species 0.000 description 1
- 208000024777 Prion disease Diseases 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- 108090000708 Proteasome Endopeptidase Complex Proteins 0.000 description 1
- 102000004245 Proteasome Endopeptidase Complex Human genes 0.000 description 1
- 108010076504 Protein Sorting Signals Proteins 0.000 description 1
- 101710188315 Protein X Proteins 0.000 description 1
- 230000004570 RNA-binding Effects 0.000 description 1
- 241000711798 Rabies lyssavirus Species 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 241000702247 Reoviridae Species 0.000 description 1
- 241000725643 Respiratory syncytial virus Species 0.000 description 1
- 241000712907 Retroviridae Species 0.000 description 1
- 241000710799 Rubella virus Species 0.000 description 1
- 241000235346 Schizosaccharomyces Species 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 108020004682 Single-Stranded DNA Proteins 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 206010041660 Splenomegaly Diseases 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- 229910052776 Thorium Inorganic materials 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- RKEITGVZZHXKON-SKAWGCAZSA-N Thymidine glycol Chemical compound O=C1NC(=O)C(C)(O)C(O)N1[C@@H]1O[C@H](CO)[C@@H](O)C1 RKEITGVZZHXKON-SKAWGCAZSA-N 0.000 description 1
- 241000710924 Togaviridae Species 0.000 description 1
- 102000008235 Toll-Like Receptor 9 Human genes 0.000 description 1
- 108010060818 Toll-Like Receptor 9 Proteins 0.000 description 1
- 102100027010 Toll-like receptor 1 Human genes 0.000 description 1
- 102100027009 Toll-like receptor 10 Human genes 0.000 description 1
- 102100024333 Toll-like receptor 2 Human genes 0.000 description 1
- 102100024324 Toll-like receptor 3 Human genes 0.000 description 1
- 102100039360 Toll-like receptor 4 Human genes 0.000 description 1
- 102100039357 Toll-like receptor 5 Human genes 0.000 description 1
- 102100039387 Toll-like receptor 6 Human genes 0.000 description 1
- 102100039390 Toll-like receptor 7 Human genes 0.000 description 1
- 102100033110 Toll-like receptor 8 Human genes 0.000 description 1
- 239000007875 V-40 Substances 0.000 description 1
- 206010046865 Vaccinia virus infection Diseases 0.000 description 1
- 241000700647 Variola virus Species 0.000 description 1
- 241000711975 Vesicular stomatitis virus Species 0.000 description 1
- 101000936049 Vibrio cholerae serotype O1 (strain ATCC 39315 / El Tor Inaba N16961) Outer membrane lipoprotein Blc Proteins 0.000 description 1
- 108700005077 Viral Genes Proteins 0.000 description 1
- 241000120645 Yellow fever virus group Species 0.000 description 1
- 229920000392 Zymosan Polymers 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 230000021736 acetylation Effects 0.000 description 1
- 238000006640 acetylation reaction Methods 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 230000004721 adaptive immunity Effects 0.000 description 1
- 108700010877 adenoviridae proteins Proteins 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 101150115889 al gene Proteins 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 229930013930 alkaloid Natural products 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 239000001166 ammonium sulphate Substances 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 230000001775 anti-pathogenic effect Effects 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 210000004436 artificial bacterial chromosome Anatomy 0.000 description 1
- 210000001106 artificial yeast chromosome Anatomy 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 244000309743 astrovirus Species 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 229950009494 bropirimine Drugs 0.000 description 1
- 238000010805 cDNA synthesis kit Methods 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 238000002619 cancer immunotherapy Methods 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 230000006037 cell lysis Effects 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 238000002659 cell therapy Methods 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 238000010382 chemical cross-linking Methods 0.000 description 1
- 150000005829 chemical entities Chemical class 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229960003920 cocaine Drugs 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 238000004590 computer program Methods 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 230000009260 cross reactivity Effects 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- UFULAYFCSOUIOV-UHFFFAOYSA-N cysteamine Chemical compound NCCS UFULAYFCSOUIOV-UHFFFAOYSA-N 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 230000000120 cytopathologic effect Effects 0.000 description 1
- 229940104302 cytosine Drugs 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 230000000368 destabilizing effect Effects 0.000 description 1
- 230000001627 detrimental effect Effects 0.000 description 1
- 229960002069 diamorphine Drugs 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 206010014599 encephalitis Diseases 0.000 description 1
- 210000001163 endosome Anatomy 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 239000006167 equilibration buffer Substances 0.000 description 1
- 230000008029 eradication Effects 0.000 description 1
- 238000012869 ethanol precipitation Methods 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 230000000763 evoking effect Effects 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 102000013165 exonuclease Human genes 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000013265 extended release Methods 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 210000003495 flagella Anatomy 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 108060003552 hemocyanin Proteins 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 208000029570 hepatitis D virus infection Diseases 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 description 1
- 230000005965 immune activity Effects 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 230000002998 immunogenetic effect Effects 0.000 description 1
- 230000016784 immunoglobulin production Effects 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 230000002584 immunomodulator Effects 0.000 description 1
- 238000009169 immunotherapy Methods 0.000 description 1
- 230000001976 improved effect Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000015788 innate immune response Effects 0.000 description 1
- 239000002054 inoculum Substances 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 235000013902 inosinic acid Nutrition 0.000 description 1
- 229940028843 inosinic acid Drugs 0.000 description 1
- 239000004245 inosinic acid Substances 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 210000001911 interdigitating cell Anatomy 0.000 description 1
- 125000000741 isoleucyl group Chemical group [H]N([H])C(C(C([H])([H])[H])C([H])([H])C([H])([H])[H])C(=O)O* 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 210000002664 langerhans' cell Anatomy 0.000 description 1
- 239000004816 latex Substances 0.000 description 1
- 229920000126 latex Polymers 0.000 description 1
- 125000001909 leucine group Chemical group [H]N(*)C(C(*)=O)C([H])([H])C(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 229950005634 loxoribine Drugs 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 208000018555 lymphatic system disease Diseases 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 239000004325 lysozyme Substances 0.000 description 1
- 229960000274 lysozyme Drugs 0.000 description 1
- 235000010335 lysozyme Nutrition 0.000 description 1
- 108010026228 mRNA guanylyltransferase Proteins 0.000 description 1
- 108010051618 macrophage stimulatory lipopeptide 2 Proteins 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 201000006512 mast cell neoplasm Diseases 0.000 description 1
- 208000006971 mastocytoma Diseases 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 229960003151 mercaptamine Drugs 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- WSFSSNUMVMOOMR-NJFSPNSNSA-N methanone Chemical compound O=[14CH2] WSFSSNUMVMOOMR-NJFSPNSNSA-N 0.000 description 1
- 244000000010 microbial pathogen Species 0.000 description 1
- 239000013586 microbial product Substances 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 230000002297 mitogenic effect Effects 0.000 description 1
- 238000009126 molecular therapy Methods 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- 230000000877 morphologic effect Effects 0.000 description 1
- DAZSWUUAFHBCGE-KRWDZBQOSA-N n-[(2s)-3-methyl-1-oxo-1-pyrrolidin-1-ylbutan-2-yl]-3-phenylpropanamide Chemical compound N([C@@H](C(C)C)C(=O)N1CCCC1)C(=O)CCC1=CC=CC=C1 DAZSWUUAFHBCGE-KRWDZBQOSA-N 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 229940097496 nasal spray Drugs 0.000 description 1
- 210000000822 natural killer cell Anatomy 0.000 description 1
- 229960002715 nicotine Drugs 0.000 description 1
- SNICXCGAKADSCV-UHFFFAOYSA-N nicotine Natural products CN1CCCC1C1=CC=CN=C1 SNICXCGAKADSCV-UHFFFAOYSA-N 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 125000006501 nitrophenyl group Chemical group 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 230000009871 nonspecific binding Effects 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 230000030648 nucleus localization Effects 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 239000000668 oral spray Substances 0.000 description 1
- 229940041678 oral spray Drugs 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000006174 pH buffer Substances 0.000 description 1
- 244000045947 parasite Species 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N phenylalanine group Chemical group N[C@@H](CC1=CC=CC=C1)C(=O)O COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical group [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 229920001983 poloxamer Polymers 0.000 description 1
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 1
- 229920000447 polyanionic polymer Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 108010094020 polyglycine Proteins 0.000 description 1
- 229920000232 polyglycine polymer Polymers 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 108010000222 polyserine Proteins 0.000 description 1
- GNSKLFRGEWLPPA-UHFFFAOYSA-M potassium dihydrogen phosphate Chemical compound [K+].OP(O)([O-])=O GNSKLFRGEWLPPA-UHFFFAOYSA-M 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 210000004986 primary T-cell Anatomy 0.000 description 1
- 238000011809 primate model Methods 0.000 description 1
- 210000001236 prokaryotic cell Anatomy 0.000 description 1
- 235000019419 proteases Nutrition 0.000 description 1
- 238000001243 protein synthesis Methods 0.000 description 1
- 239000002510 pyrogen Substances 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- BXNMTOQRYBFHNZ-UHFFFAOYSA-N resiquimod Chemical compound C1=CC=CC2=C(N(C(COCC)=N3)CC(C)(C)O)C3=C(N)N=C21 BXNMTOQRYBFHNZ-UHFFFAOYSA-N 0.000 description 1
- 230000001177 retroviral effect Effects 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- 125000000548 ribosyl group Chemical group C1([C@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 238000002864 sequence alignment Methods 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 231100000245 skin permeability Toxicity 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- ULARYIUTHAWJMU-UHFFFAOYSA-M sodium;1-[4-(2,5-dioxopyrrol-1-yl)butanoyloxy]-2,5-dioxopyrrolidine-3-sulfonate Chemical compound [Na+].O=C1C(S(=O)(=O)[O-])CC(=O)N1OC(=O)CCCN1C(=O)C=CC1=O ULARYIUTHAWJMU-UHFFFAOYSA-M 0.000 description 1
- VUFNRPJNRFOTGK-UHFFFAOYSA-M sodium;1-[4-[(2,5-dioxopyrrol-1-yl)methyl]cyclohexanecarbonyl]oxy-2,5-dioxopyrrolidine-3-sulfonate Chemical compound [Na+].O=C1C(S(=O)(=O)[O-])CC(=O)N1OC(=O)C1CCC(CN2C(C=CC2=O)=O)CC1 VUFNRPJNRFOTGK-UHFFFAOYSA-M 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 229940043517 specific immunoglobulins Drugs 0.000 description 1
- 210000004989 spleen cell Anatomy 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 150000003505 terpenes Chemical class 0.000 description 1
- 235000007586 terpenes Nutrition 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 150000003568 thioethers Chemical class 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 230000009258 tissue cross reactivity Effects 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 241000724775 unclassified viruses Species 0.000 description 1
- 241001529453 unidentified herpesvirus Species 0.000 description 1
- 241000712461 unidentified influenza virus Species 0.000 description 1
- 208000007089 vaccinia Diseases 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 230000007501 viral attachment Effects 0.000 description 1
- 230000029812 viral genome replication Effects 0.000 description 1
- 210000000605 viral structure Anatomy 0.000 description 1
- 229940023147 viral vector vaccine Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/117—Nucleic acids having immunomodulatory properties, e.g. containing CpG-motifs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
- A61K39/21—Retroviridae, e.g. equine infectious anemia virus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/39—Medicinal preparations containing antigens or antibodies characterised by the immunostimulating additives, e.g. chemical adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/005—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/005—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from viruses
- C07K14/08—RNA viruses
- C07K14/15—Retroviridae, e.g. bovine leukaemia virus, feline leukaemia virus human T-cell leukaemia-lymphoma virus
- C07K14/155—Lentiviridae, e.g. visna-maedi virus, equine infectious virus, FIV, SIV
- C07K14/16—HIV-1 ; HIV-2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
- C07K14/4701—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals not used
- C07K14/4748—Tumour specific antigens; Tumour rejection antigen precursors [TRAP], e.g. MAGE
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/525—Virus
- A61K2039/5258—Virus-like particles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55561—CpG containing adjuvants; Oligonucleotide containing adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/60—Medicinal preparations containing antigens or antibodies characteristics by the carrier linked to the antigen
- A61K2039/6031—Proteins
- A61K2039/6075—Viral proteins
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/18—Type of nucleic acid acting by a non-sequence specific mechanism
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/20011—Papillomaviridae
- C12N2710/20022—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2730/00—Reverse transcribing DNA viruses
- C12N2730/00011—Details
- C12N2730/10011—Hepadnaviridae
- C12N2730/10111—Orthohepadnavirus, e.g. hepatitis B virus
- C12N2730/10122—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2730/00—Reverse transcribing DNA viruses
- C12N2730/00011—Details
- C12N2730/10011—Hepadnaviridae
- C12N2730/10111—Orthohepadnavirus, e.g. hepatitis B virus
- C12N2730/10123—Virus like particles [VLP]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2730/00—Reverse transcribing DNA viruses
- C12N2730/00011—Details
- C12N2730/10011—Hepadnaviridae
- C12N2730/10111—Orthohepadnavirus, e.g. hepatitis B virus
- C12N2730/10134—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/16011—Human Immunodeficiency Virus, HIV
- C12N2740/16211—Human Immunodeficiency Virus, HIV concerning HIV gagpol
- C12N2740/16222—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/16011—Human Immunodeficiency Virus, HIV
- C12N2740/16211—Human Immunodeficiency Virus, HIV concerning HIV gagpol
- C12N2740/16234—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2760/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses negative-sense
- C12N2760/00011—Details
- C12N2760/10011—Arenaviridae
- C12N2760/10022—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2760/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses negative-sense
- C12N2760/00011—Details
- C12N2760/10011—Arenaviridae
- C12N2760/10034—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2795/00—Bacteriophages
- C12N2795/00011—Details
- C12N2795/18011—Details ssRNA Bacteriophages positive-sense
- C12N2795/18111—Leviviridae
- C12N2795/18122—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2795/00—Bacteriophages
- C12N2795/00011—Details
- C12N2795/18011—Details ssRNA Bacteriophages positive-sense
- C12N2795/18111—Leviviridae
- C12N2795/18123—Virus like particles [VLP]
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- the present invention is related to the fields of vaccinology, immunology and medicine.
- the invention provides compositions and methods for enhancing immunological responses against HIV-peptides which are coupled, fused or attached otherwise to virus-like particles (VLPs) by binding, preferably by packaging immunostimulatory substances, in particular immunostimulatory nucleic acids, and even more particular oligonucleotides containing at least one non-methylated CpG sequence, into the VLPs.
- VLPs virus-like particles
- the invention can be used to induce strong and sustained T cell responses particularly useful for the treatment of HIV viral diseases.
- the essence of the immune system is built on two separate foundation pillars: one is specific or adaptive immunity which is characterized by relatively slow response- kinetics and the ability to remember; the other is non-specific or innate immunity exhibiting rapid response-kinetics but lacking memory.
- viruses Unlike isolated proteins, viruses induce prompt and efficient immune responses in the absence of any adjuvants both with and without T-cell help (Bachmann & Zinkernagel, Ann. Rev. Immunol. 15:235-270 (1997)). Many viruses exhibit a quasi- crystalline surface that displays a regular array of epitopes which efficiently crosslinks epitope-specific immunoglobulins on B cells (Bachmann & Zinkernagel, Immunol. Today 17:553-558 (1996)). Viral structure is even linked to the generation of anti-antibodies in autoimmune disease and as a part of the natural response to pathogens (see Fehr, T., et al., J. Exp. Med. 185:1785-1792 (1997)). Thus, antigens on viral particles that are organized in an ordered and repetitive array are highly immunogenic since they can directly activate B cells and induce the generation of a cytotoxic T cell response, another crucial arm of the immune system.
- cytotoxic T cells are particularly important for the elimination of non- cytopathic viruses such as HIV or Hepatitis B virus and for the eradication of tumors. Cytotoxic T cells do not recognize native antigens but rather recognize their degradation products in association with MHC class I molecules (Townsend & Bodmer, Ann. Rev. Immunol. 7:601-624 (1989)). Macrophages and dendritic cells are able to take up and process exogenous viral particles (but not their soluble, isolated components) and present the generated degradation product to cytotoxic T cells, leading to their activation and proliferation (Kovacsovics-Bankowski et al, Proc. Natl. Acad. Sci. USA 90:4942-4946 (1993); Bachmann et al, Eur. J. Immunol. 26:2595-2600 (1996)).
- Viral particles as antigens exhibit two advantages over their isolated components: (1) due to their highly repetitive surface structure, they are able to directly activate B cells, leading to high antibody titers and long-lasting B cell memory; and (2) viral particles, but not soluble proteins, have the potential to induce a cytotoxic T cell response, even if the viruses are non-infectious and adjuvants are absent.
- VLPs virus-like particles
- WO 94/15585 discloses synthetic polymer particles carrying on the surface one or more proteins covalently bonded thereto; and a core particle with a non-covalently bound coating, which at least partially covers the surface of said core particle, and at least one biologically active agent in contact with said coated core particle.
- virus-like particles are being exploited in the area of vaccine production because of both their structural properties and their non- infectious nature.
- VLPs are supermolecular structures built in a symmetric manner from many protein molecules of one or more types. They lack the viral genome and, therefore, are noninfectious. VLPs can often be produced in large quantities by heterologous expression and can be easily be purified.
- CpG non-methylated CG motifs
- DNA oligomers rich in CpG motifs can exhibit immunostimulatory capacity, their efficiency is often limited, since they are unstable in vitro and in vivo. Thus, they exhibit unfavorable pharmacokinetics. In order to render CpG-oligonucleotides more potent, it is therefore usually necessary to stabilize them by introducing phosphorothioate modifications of the phosphate backbone.
- immunostimulatory CpG-oligodeoxynucleotides induce strong side effects by causing extramedullary hemopoiesis accomponied by splenomegaly and lymphadenopathy in mice (Sparigan et al, J. Immunol. (1999), 162:2368-74 and Example 18).
- HIV is a retrovirus and belongs to the family of the lentiviruses. Two types of HIV viruses have been discovered, HIV-1 and HIV-2. HIV-2 is mainly found in the countries of Western Africa, while HIV-1 is the most common form of HIV elsewhere.
- DNA immunisation may lead to integration of DNA into the genome, plasmid DNA may contain resistance genes, viral promoters are used, and antibodies to DNA may be elicited in the host. Furthermore, large amounts of DNA are required.
- live attenuated or replication deficient viruses always bears the risk of recombination, which might lead to more virulent species, which is a concern particularly in immunocompromised individuals.
- the use of viral vectors is expected to lead to the infection of a large number of different cell types in the body, and indeed infection is required for the efficacy of the vaccine.
- the use of adenoviral vectors may be inefficient or lead to side effects in patients sero-positive for adenovirus. There is therefore a need for a safe and immunogenic vaccine technology to induce strong and potent CTL responses against HIV.
- This invention is based on the surprising finding that particular HIV polypeptides, when bound to a core particle having a structure with an inherent repetitive organization, and hereby in particular to virus-like-particles (VLPs) and subunits of VLPs, respectively, which VLPs are packaged with immunostimulatory substances (ISSs) such as DNA oligonucleotides, represent potent immunogens for the induction of specific antibodies.
- VLPs virus-like-particles
- ISSs immunostimulatory substances
- DNA oligonucleotides represent potent immunogens for the induction of specific antibodies.
- the invention is further based on the finding that immunostimulatory substances such as DNA oligonucleotides can be packaged into VLPs which renders them more immunogenic.
- nucleic acids and oligonucleotides, respectively, present in VLPs can be replaced specifically by the immunostimulatory substances and DNA- oligonucleotides containing CpG motifs, respectively.
- these packaged immunostimulatory substances in particular immunostimulatory nucleic acids such as unmethylated CpG-containing oligonucleotides retained their immunostimulatory capacity without widespread activation of the innate immune system.
- the compositions comprising VLP's and the immunostimulatory substances in accordance with the present invention, and in particular the CpG- VLPs are dramatically more immunogenic than their CpG-free counterparts and induce enhanced B and T cell responses.
- the immune response against HIV polypeptides optionally coupled, fused or attached otherwise to the VLPs is similarly enhanced as the immune response against the VLP itself.
- the T cell responses against both the VLPs and HIV polypeptides are especially directed to the Thl type. HIV polypeptides attached to CpG-loaded VLPs may therefore be ideal vaccines for prophylactic or therapeutic vaccination against HIV.
- the invention provides a composition, typically and preferabyl for enhancing an immune response in an animal, comprising a virus-like particle, an immunostimulatory substance, preferably an immunostimulatory nucleic acid, and even more preferably an unmethylated CpG-containing oligonucleotide, and at least one antigen or antigenic determinant, where the immunostimulatory substance, nucleic acid or oligonucleotide is coupled, fused, or otherwise attached to or enclosed by, i.e., bound, to the virus-like particle and wherein said antigen or antigenic determinant is bound to said virus-like particle and wherein said antigen comprises, alternatively consists essentially of, or alternatively consists of a HIV polypeptide.
- the immunostimulatory nucleic acids in particular the unmethylated CpG-containing oligonucleotides are stabilized by phosphorothioate modifications of the phosphate backbone.
- the immunostimulatory nucleic acids, in particular the unmethylated CpG- containing oligonucleotides are packaged into the VLPs by digestion of RNA within the VLPs and simultaneous addition of the DNA oligonucleotides containing CpGs of choice.
- the VLPs can be disassembled before they are reassembled in the presence of CpGs.
- the immunostimulatory nucleic acids do not contain CpG motifs but nevertheless exhibit immunostimulatory activities.
- Such nucleic acids are described in WO 01/22972. All sequences described therein are hereby incorporated by way of reference.
- the virus-like particle is a recombinant viruslike particle.
- the virus-like particle is free of a lipoprotein envelope.
- the recombinant virus-like particle comprises, or alternatively consists of, recombinant proteins of Hepatitis B virus, BK virus or other human Polyoma virus, measles virus, Sindbis virus, Rotavirus, Foot-and-Mouth-Disease virus, Retrovirus, Norwalk virus or human Papilloma virus, RNA-phages, Q ⁇ -phage, GA-phage, fr-phage and Ty.
- the virus-like particle comprises, or alternatively consists of, one or more different Hepatitis B virus core (capsid) proteins (HBcAgs).
- the virus-like particle comprises recombinant proteins, or fragments thereof, of a RNA-phage.
- Preferred RNA-phages are Q ⁇ -phage, AP 205-phage, GA-phage, fr-phage
- the antigen comprises, or alternatively consists of, a cytotoxic T cell epitope.
- the virus-like particle comprises the Hepatitis B virus core protein and the cytotoxic T cell epitope is fused to the C-terminus of said Hepatitis B virus core protein. In one embodiment, they are fused by a leucine linking sequence.
- a method of enhancing an immune response in a human or other animal species comprising introducing into the animal a composition comprising a virus-like particle, an immunostimulatory substance, preferably an immunostimulatory nucleic acid, and even more preferably an unmethylated CpG-containing oligonucleotide, and at least one antigen or antigenic determinant, where the immunostimulatory substance, preferably the nucleic acid, and even more preferally the oligonucleotide is bound (i.e. coupled, attached or enclosed) to the virus-like particle.
- an immunostimulatory substance preferably an immunostimulatory nucleic acid, and even more preferably an unmethylated CpG-containing oligonucleotide, and at least one antigen or antigenic determinant
- the composition further comprises an antigen bound to the viruslike particle, and wherein said antigen comprises, alternatively consists essentially of, or alternatively consists of a HIV polypeptide, and wherein said antigen or antigenic determinant is bound to said virus-like particle.
- the composition is introduced into an animal subcutaneously, intramuscularly, intranasally, intradermally, intravenously or directly into a lymph node.
- the immune enhancing composition is applied locally, near a tumor or local viral reservoir against which one would like to vaccinate.
- the immune response is a T cell response, and the T cell response against the antigen is enhanced.
- the T cell response is a cytotoxic T cell response, and the cytotoxic T cell response against the HIV polypeptide is enhanced.
- the present invention also relates to a vaccine comprising an immunologically effective amount of the immune enhancing composition of the present invention together with a pharmaceutically acceptable diluent, carrier or excipient.
- the vaccine further comprises at least one adjuvant.
- the invention also provides a method of immunizing and/or treating an animal comprising administering to the animal an immunologically effective amount of the disclosed vaccine.
- the immunostimulatory substance- containing VLPs preferably the immunostimulatory nucleic acid-containing VLP's, an even more preferably the unmethylated CpG-containing oligonucleotide VLPs are used for vaccination of animals, typically and preferably humans, against HIV polypeptides coupled, fused or attached otherwise to the VLP.
- the modified VLPs can typically and preferably be used to vaccinate against HIV viral disease. The vaccination can be for prophylactic or therapeutic purposes, or both.
- the desired immune response will be directed against HIV polypeptides coupled, fused or attached otherwise to the immunostimulatory substance- containing VLPs, preferably the immunostimulatory nucleic acid-containing VLP's, an even more preferably the unmethylated CpG-containing oligonucleotide VLPs.
- the route of injection is preferably subcutaneous or intramuscular, but it would also be possible to apply the CpG-containing VLPs intradermally, intranasally, intravenously or directly into the lymph node.
- the CpG-containing HIV polypeptide-coupled or free VLPs are applied locally, near a local viral reservoir against which one would like to vaccinate.
- FIG. 1 shows the virus titers after immunizing mice with Qbx33 packaged with poly (I:C), G3-6, or G6.
- C57B16 mice were immunized by injecting either 100 ⁇ g Qbx33, 100 ⁇ g Qb VLPs packaged with poly (I:C) and coupled to ⁇ 33 (Qb-pIC-33, also termed QbxZnxpolyICxp33GGC) , 90 ⁇ g Qbx33 packaged with G3-6 (Qbx33/G3-6), or 90 ⁇ g Qbx33 packaged with G6 (Qbx33/G6).
- Qb-pIC-33 also termed QbxZnxpolyICxp33GGC
- mice were challenged with 1.5 xl06 plaque forming units Vaccinia virus, carrying the LCMV-p33 epitope.
- mice were sacrificed and the ovaries were collected.
- a single cell suspension from the ovaries was prepared and added to BCS40 cells in serial dilutions.
- the cell layer was stained with a solution containing 50% Ethanol, 2% formaldehyde, 0.8% NaCI and 0.5% Crystal violet) and the viral plaques were counted.
- Figure 2 shows the SDS-PAGE analysis of the coupling reaction of Q ⁇ VLP to gag-G50 peptide. The samples were run under reducing conditions on a 12% NuPage gel (Invitrogen).
- Lane 1 is the protein marker, with corresponding molecular weights indicated on the left border of the gel; lane 2, derivatized Q ⁇ VLP; lane 3, the supernatant of the coupling reaction of Q ⁇ capsid protein to the gag-G50 peptide; lane 4, the pellet of the coupling reaction of Q ⁇ capsid protein to the gag-G50 peptide.
- Coupling products corresponding to the coupling of a peptide on a Q ⁇ monomer or Q ⁇ dimer are indicated by arrows in the Figure.
- Figure 3 shows the SDS-PAGE analysis of the coupling reaction of Q ⁇ VLP to nef-N56 peptide.
- the samples were run under reducing conditions on a 12% NuPage gel (Invitrogen).
- Lane 1 is the protein marker, with corresponding molecular weights indicated on the left border of the gel;
- lane 4 the pellet of the coupling reaction of Q ⁇ capsid protein to the nef-N56 peptide.
- Coupling products corresponding to the coupling of a peptide on a Q ⁇ monomer or Q ⁇ dimer are indicated by arrows in the Figure.
- Amino acid linker An "amino acid linker”, or also just termed “linker” within this specification, as used herein, either associates the antigen or antigenic determinant with the second attachment site, or more preferably, already comprises or contains the second attachment site, typically - but not necessarily - as one amino acid residue, preferably as a cysteine residue.
- amino acid linker does not intend to imply that such an amino acid linker consists exclusively of amino acid residues, even if an amino acid linker consisting of amino acid residues is a preferred embodiment of the present invention.
- amino acid residues of the amino acid linker are, preferably, composed of naturally occuring amino acids or unnatural amino acids known in the art, all-L or all-D or mixtures thereof.
- an amino acid linker comprising a molecule with a sulfhydryl group or cysteine residue is also encompassed within the invention.
- Such a molecule comprise preferably a Cl -C6 alkyl-, cycloalkyl (C5,C6), aryl or heteroaryl moiety.
- a linker comprising preferably a C1-C6 alkyl-, cycloalkyl- (C5,C6), aryl- or heteroaryl- moiety and devoid of any amino acid(s) shall also be encompassed within the scope of the invention.
- Association between the antigen or antigenic determinant or optionally the second attachment site and the amino acid linker is preferably by way of at least one covalent bond, more preferably by way of at least one peptide bond.
- animal As used herein, the term “animal” is meant to include, for example, humans, sheep, horses, cattle, pigs, dogs, cats, rats, mice, mammals, birds, reptiles, fish, insects and arachnids.
- Antibody refers to molecules which are capable of binding an epitope or antigenic determinant.
- the term is meant to include whole antibodies and antigen-binding fragments thereof, including single-chain antibodies.
- the antibodies are human antigen binding antibody fragments and include, but are not limited to, Fab, Fab' and F(ab')2, Fd, single-chain Fvs (scFv), single-chain antibodies, disulfide-linked Fvs (sdFv) and fragments comprising either a VL or VH domain.
- the antibodies can be from any animal origin including birds and mammals.
- the antibodies are human, murine, rabbit, goat, guinea pig, camel, horse or chicken.
- human antibodies include antibodies having the amino acid sequence of a human immunoglobulin and include antibodies isolated from human immunoglobulin libraries or from animals transgenic for one or more human immunoglobulins and that do not express endogenous immunoglobulins, as described, for example, in U.S. Patent No. 5,939,598 by Kucherlapati et al.
- Antigen refers to a molecule capable of being bound by an antibody or a T cell receptor (TCR) if presented by MHC molecules.
- TCR T cell receptor
- An antigen is additionally capable of being recognized by the immune system and/or being capable of inducing a humoral immune response and/or cellular immune response leading to the activation of B- and/or T-lymphocytes. This may, however, require that, at least in certain cases, the antigen contains or is linked to a T helper cell epitope (Th cell epitope) and is given in adjuvant.
- An antigen can have one or more epitopes (B- and T- epitopes). The specific reaction referred to above is meant to indicate that the antigen will preferably react, typically in a highly selective manner, with its corresponding antibody or TCR and not with the multitude of other antibodies or TCRs which may be evoked by othier antigens.
- Antigens as used herein may also be mixtures of several individual antigens.
- a "microbial antigen” as used herein is an antigen of a microorganism arid includes, but is not limited to, infectious virus, infectious bacteria, parasites and infectious fungi.
- Such antigens include the intact microorganism as well as natural isolate s and fragments or derivatives thereof and also synthetic or recombinant compounds which are identical to or similar to natural microorganism antigens and induce an immune response specific for that microorganism.
- a compound is similar to a natural microorganism antigen if it induces an immune response (humoral and/or cellular) to a natural microorganism antigen.
- Such antigens are used routinely in the art and are well known to the skilled artisan.
- Retroviridae e.g. human immunodeficiency viruses, such as HIV-1 £also referred to as HTLV-III, LAV or HTLV-III/LAV, or HIV-III
- other isolates such as HIV-LP
- Picornaviridae e.g. polio viruses, hepatitis A virus; enteroviruses, human Coxsackie viruses, rhinoviruses, echoviruses
- Calciviridae e.g. strains that cause gastroenteritis
- Togaviridae e.g. equine encephalitis viruses, rubella viruses
- Flaviridae e.g.
- Coronoviridae e.g. coronaviruses
- Rhabdoviradae e.g. vesicular stomatitis viruses, rabies viruses
- Filoviridae e.g. ebola viruses
- Paramyxoviridae e.g. parainfluenza viruses, mumps virus, measles virus, respiratory syncytial virus
- Orthomyxoviridae e.g. influenza viruses
- Bungaviridae e.g. Hantaan viruses, bunga viruses, phleboviruses and Nairo viruses
- Arena viridae hemorrhagic fever viruses
- Reoviridae e.g.
- reoviruses reoviruses, orbiviurses and rotaviruses
- Birnaviridae Hepadnaviridae (Hepatitis B virus); Parvovirida (parvoviruses); Papovaviridae (papilloma viruses, polyoma viruses); Adenoviridae (most adenoviruses); Herpesviridae (herpes simplex virus (HSV) 1 and 2, varicella zoster virus, cytomegalovirus (CMV), herpes virus); Poxviridae (variola viruses, vaccinia viruses, pox viruses); and Iridoviridae (e.g. African swine fever virus); and unclassified viruses (e.g.
- Antigenic determinant As used herein, the term “antigenic determinant” is meant to refer to that portion of an antigen that is specifically recognized by either B- or T- lymphocytes. B-lymphocytes respond to foreign antigenic determinants via antibody production, whereas T-lymphocytes are the mediator of cellular immunity. Thus, antigenic determinants or epitopes are those parts of an antigen that are recognized by antibodies, or in the context of an MHC, by T-cell receptors.
- Antigen presenting cell As used herein, the term “antigen presenting cell” is meant to refer to a heterogenous population of leucocytes or bone marrow derived cells which possess an immunostimulatory capacity.
- these cells are capable of generating peptides bound to MHC molecules that can be recognized by T cells.
- the term is synonymous with the term "accessory cell” and includes, for example, Langerhans' cells, interdigitating cells, B cells, macrophages and dendritic cells. Under some conditions, epithelial cells, endothelial cells and other, non-bone marrow derived cells may also serve as antigen presenting cells.
- Association As used herein, the term "association" as it applies to the first and second attachment sites, refers to the binding of the first and second attachment sites that is preferably by way of at least one non-peptide bond.
- association may be covalent, ionic, hydrophobic, polar or any combination thereof, preferably the nature of the association is covalent, and again more preferably the association is through at least one, preferably one, non-peptide bond.
- association as it applies to the first and second attachment sites, not only encompass the direct binding or association of the first and second attachment site forming the compositions of the invention but also, alternatively and preferably, the indirect association or binding of the first and second attachment site leading to the compositions of the invention, and hereby typically and preferably by using a heterobifunctional cross-linker.
- first attachment site refers to an element of non-natural or natural origin, typically and preferably being comprised by the virus-like particle, to which the second attachment site typically and preferably being comprised by the HIV polypeptide may associate.
- the first attachment site may be a protein, a polypeptide, an amino acid, a peptide, a sugar, a polynucleotide, a natural or synthetic polymer, a secondary metabolite or compound (biotin, fluorescein, retinol, digoxigenin, metal ions, phenylmethylsulfonylfluoride), or a combination thereof, or a chemically reactive group thereof.
- the first attachment site is located, typically and preferably on the surface, of the virus-like particle. Multiple first attachment sites are present on the surface of virus-like particle typically in a repetitive configuration.
- the first attachment site is an amino acid or a chemically reactive group thereof.
- Second attachment site refers to an element associated with, typically and preferably being comprised by, the
- the second attachment site of HIV polypeptide may be a protein, a polypeptide, a peptide, a sugar, a polynucleotide, a natural or synthetic polymer, a secondary metabolite or compound (biotin, fluorescein, retinol, digoxigenin, metal ions, phenylmethylsulfonylfluoride), or a combination thereof, or a chemically reactive group thereof.
- At least one second attachment site is present on the HIV polypeptide.
- HIV polypeptide with at least one second attachment site refers, therefore, to an antigen or antigenic construct comprising at least the HIV polypeptide and the second attachment site.
- these antigen or antigenic constructs comprise an "amino acid linker".
- bound refers to binding that may be covalent, e.g., by chemically coupling, or non-covalent, e.g., ionic interactions, hydrophobic interactions, hydrogen bonds, etc. Covalent bonds can be, for example, ester, ether, phosphoester, amide, peptide, imide, carbon-sulfur bonds, carbon-phosphorus bonds, and the like.
- bound is broader than and includes terms such as “coupled”, “fused,” “associated” and “attached”.
- bound also includes the enclosement, or partial enclosement, of the immunostimulatory substance.
- the term "bound” is broader than and includes terms such as “coupled,” “fused,” “enclosed”, “packaged” and “attached.”
- the immunostimulatory substance such as the unmethylated CpG-containing oligonucleotide can be enclosed by the VLP without the existence of an actual binding, neither covalently nor non-covalently.
- Coat protein(s) refers to the protein(s) of a bacteriophage or a RNA-phage capable of being incorporated within the capsid assembly of the bacteriophage or the RNA-phage.
- the term "CP” is used.
- the specific gene product of the coat protein gene of RNA-phage Q ⁇ is referred to as "Q ⁇ CP”
- the "coat proteins” of bacteriophage Q ⁇ comprise the "Q ⁇ CP” as well as the Al protein.
- the capsid of Bacteriophage Q ⁇ is composed mainly of the Q ⁇ CP, with a minor content of the Al protein.
- the VLP Q ⁇ coat protein contains mainly Q ⁇ CP, with a minor content of Al protein.
- Coupled refers to attachment by covalent bonds or by strong non-covalent interactions. With respect to the coupling of the antigen to the virus-like particle the term “coupled” preferably refers to attachment by covalent bonds. Moreover, with respect to the coupling of the antigen to the virus-like particle the term “coupled” preferably refers to association and attachment, respectively, by at least one non-peptide bond. Any method normally used by those skilled in the art for the coupling of biologically active materials can be used in the present invention. Fusion: As used herein, the term “fusion” refers to the combination of amino acid sequences of different origin in one polypeptide chain by in-frame combination of their coding nucleotide sequences. The term “fusion” explicitly encompasses internal fusions, i.e., insertion of sequences of different origin within a polypeptide chain, in addition to fusion to one of its termini.
- CpG refers to an oligonucleotide which contains at least one unmethylated cytosine, guanine dinucleotide sequence (e.g. "CpG DNA” or DNA containing a cytosine followed by guanosine and linked by a phosphate bond) and stimulates/activates, e.g. has a mitogenic effect on, or induces or increases cytokine expression by, a vertebrate cell.
- CpGs can be useful in activating B cells, NK cells and antigen-presenting cells, such as monocytes, dendritic cells and macrophages, and T cells.
- the CpGs can include nucleotide analogs such as analogs containing phosphorothioester bonds and can be double-stranded or single-stranded. Generally, double-stranded molecules are more stable in vivo, while single-stranded molecules have increased immune activity.
- Epitope refers to portions of a polypeptide having antigenic or immunogenic activity in an animal, preferably a mammal, and most preferably in a human.
- An "immunogenic epitope,” as used herein, is defined as a portion of a polypeptide that elicits an antibody response or induces a T-cell response in an animal, as determined by any method known in the art. (See, for example, Geysen et al., Proc. Natl. Acad. Sci. USA 81:3998 4002 (1983)).
- antigenic epitope is defined as a portion of a protein to which an antibody can immunospecifically bind its antigen as determined by any method well known in the art. Immunospecific binding excludes non specific binding but does not necessarily exclude cross reactivity with other antigens. Antigenic epitopes need not necessarily be immunogenic. Antigenic epitopes can also be T-cell epitopes, in which case they can be bound immunospecifically by a T-cell receptor within the context of an MHC molecule.
- An epitope can comprise 3 amino acids in a spatial conformation which is unique to the epitope. Generally, an epitope consists of at least about 5 such amino acids, and more usually, consists of at least about 8-10 such amino acids. If the epitope is an organic molecule, it may be as small as Nitrophenyl. Preferred epitopes are the HIV polypeptides of the invention.
- a "HIV polypeptide” as used herein shall include a polypeptide, a polyprotein, a peptide, a polyepitope, an epitope of HIV. In a preferred embodiment of the present invention, the term "HIV polypeptide” as used herein shall refer to a sequence corresponding to a HIV consensus sequence.
- HIV polypeptide as used herein shall refer to a polypeptide of HIV comprising, or alternatively consisting essentially of, or alternatively consisting of an epitope of HIV.
- Preferred epitopes of the present invention are epitopes with a sequence derived from a consensus HIV sequence.
- the HIV polypeptide comprises, or alternatively consists essentially of, or alternatively consists of a polyepitope of HIV.
- polyepitope of HIV shall refer to a combination of at least two HIV polypeptides, wherein said at least two HIV polypeptides are bound directly or by way of a linking sequence.
- Immune response refers to a humoral immune response and/or cellular immune response leading to the activation or proliferation of B- and/or T-lymphocytes. In some instances, however, the immune responses may be of low intensity and become detectable only when using at least one substance in accordance with the invention.
- Immunogenic refers to an agent used to stimulate the immune system of a living organism, so that one or more functions of the immune system are increased and directed towards the immunogenic agent.
- An "immunogenic polypeptide” is a polypeptide that elicits a cellular and/or humoral immune response, whether alone or linked to a carrier in the presence or absence of an adjuvant.
- Immunostimulatory nucleic acid refers to a nucleic acid capable of inducing and/or enhancing an immune response.
- Immunostimulatory nucleic acids comprise ribonucleic acids and in particular deoxyribonucleic acids.
- immunostimulatory nucleic acids contain at least one CpG motif e.g. a CG dinucleotide in which the C is unmethylated.
- the CG dinucleotide can be part of a palindromic sequence or can be encompassed within a non-palindromic sequence.
- Immunostimulatory nucleic acids not containing CpG motifs as described above encompass, by way of example, nucleic acids lacking CpG dinucleotides, as well as nucleic acids containing CG motifs with a methylated CG dinucleotide.
- the term "immunostimulatory nucleic acid” as used herein should also refer to nucleic acids that contain modified bases such as 4-bromo-cytosine.
- Immunostimulatory substance refers to a substance capable of inducing and/or enhancing an immune response.
- Immunostimulatory substances include, but are not limited to, toll-like receptor activing substances and substances inducing cytokine secretion.
- Toll-like receptor activating substances include, but are not limited to, immunostimulatory' nucleic acids, peptideoglycans, lipopolysaccharides, lipoteichonic acids, imidazoquinoline compounds, flagellins, lipoproteins, and immunostimulatory organic substances such as taxol.
- Natural origin As used herein, the term “natural origin” means that the whole or parts thereof are not synthetic and exist or are produced in nature.
- Non-natural As used herein, the term generally means not from nature, more specifically, the term means from the hand of man.
- Non-natural origin As used herein, the term “non-natural origin” generally means synthetic or not from nature; more specifically, the term means from the hand of man.
- Ordered and repetitive antigen or antigenic determinant array generally refers to a repeating pattern of antigen or antigenic determinant, characterized by a typically and preferably uniform spacial arrangement of the antigens or antigenic determinants with respect to the core particle and virus-like particle, respectively.
- the repeating pattern may be a geometric pattern.
- suitable ordered and repetitive antigen or antigenic determinant arrays are those which possess strictly repetitive paracrystalline orders of antigens or antigenic determinants, preferably with spacings of 0.5 to 30 nanometers, more preferably 3 to 15 nanometers, even more preferably 3 to 8 nanometers.
- Oligonucleotide refers to a nucleic acid sequence comprising 2 or more nucleotides, generally at least about 6 nucleotides to about 100,000 nucleotides, preferably about 6 to about 2000 nucleotides, and more preferably about 6 to about 300 nucleotides, even more preferably about 20 to about 300 nucleotides, and even more preferably about 20 to about 100 nucleotides.
- oligonucleotide or “oligomer” also refer to a nucleic acid sequence comprising more than 100 to about 2000 nucleotides, preferably more than 100 to about 1000 nucleotides, and more preferably more than 100 to about 500 nucleotides.
- Oligonucleotide also generally refers to any polyribonucleotide or polydeoxribonucleotide, which may be unmodified RNA or DNA or modified RNA or DNA.
- Oligonucleotide includes, without limitation, single- and double-stranded DNA, DNA that is a mixture of single- and double-stranded regions, single- and double-stranded RNA, and RNA that is mixture of single- and double-stranded regions, hybrid molecules comprising DNA and RNA that may be single-stranded or, more typically, double- stranded or a mixture of single- and double-stranded regions.
- oligonucleotide refers to triple-stranded regions comprising RNA or DNA or both RNA and DNA.
- an oligonucleotide can be synthetic, genomic or recombinant, e.g., ⁇ - DNA, cosmid DNA, artificial bacterial chromosome, yeast artificial chromosome and filamentous phage such as Ml 3.
- the oligonucleotide is a synthetic oligonucleotide.
- oligonucleotide also includes DNAs or RNAs containing one or more modified bases and DNAs or RNAs with backbones modified for stability or for other reasons.
- suitable nucleotide modifications/analogs include peptide nucleic acid, inosin, tritylated bases, phosphorothioates, alkylphosphorothioates, 5-nitroindole deoxyribofuranosyl, 5-methyldeoxycytosine and 5,6-dihydro-5,6- dihydroxydeoxythymidine.
- oligonucleotide embraces chemically, enzymatically or metabolically modified forms of polynucleotides as typically found in nature, as well as the chemical forms of DNA and RNA characteristic of viruses and cells. Other nucleotide analogs/modifications will be evident to those skilled in the art.
- the term "packaged” as used herein refers to the state of an immunostimulatory substance, preferably of an immunostimulatory nucleic acid in relation to the VLP.
- the term “packaged” as used herein includes binding that may be covalent, e.g., by chemically coupling, or non-covalent, e.g., ionic interactions, hydrophobic interactions, hydrogen bonds, etc. Covalent bonds can be, for example, ester, ether, phosphoester, amide, peptide, imide, carbon-sulfur bonds such as thioether bonds, carbon-phosphorus bonds, and the like.
- the term also includes the enclosement, or partial enclosement, of a substance.
- the term “packaged” includes tenns such as “coupled, "enclosed” and “attached.”
- the immunostimulatory substance such as the unmethylated CpG-containing oligonucleotide can be enclosed by the VLP without the existence of an actual binding, neither covalently nor non-covalently.
- the term “packaged” indicates that the immunostimulatory nucleic acid in a packaged state is not accessible to DNAse or RNAse hydrolysis.
- the immunostimulatory nucleic acid is packaged inside the VLP capsids, most preferably in a non-covalent manner.
- compositions of the invention can be combined, optionally, with a pharmaceutically-acceptable carrier.
- pharmaceutically-acceptable carrier means one or more compatible solid or liquid fillers, diluents or encapsulating substances which are suitable for administration into a human or other animal.
- carrier denotes an organic or inorganic ingredient, natural or synthetic, with which the active ingredient is combined to facilitate the application.
- Peptide The term "peptide” as used herein, and in particular with respect to the HIV peptide shall refer to a molecule composed of monomers (amino acids), typically and preferably linearly, linked by amide bonds (also known as peptide bonds). It indicates a molecular chain of amino acids and does not refer to a specific length of the product.
- Organic molecule As used herein, the term “organic molecule” refers to any chemical entity of natural or synthetic origin.
- organic molecule encompasses, for example, any molecule being a member of the group of nucleotides, lipids, carbohydrates, polysaccharides, lipopolysaccharides, steroids, alkaloids, terpenes and fatty acids, being either of natural or synthetic origin.
- organic molecule encompasses molecules such as nicotine, cocaine, heroin or other pharmacologically active molecules contained in drugs of abuse.
- an organic molecule contains or is modified to contain a chemical functionality allowing its coupling, binding or other method of attachment to the virus-like particle in accordance with the invention.
- Polypeptide refers to a molecule composed of monomers (amino acids) linearly linked by amide bonds (also known as peptide bonds). It indicates a molecular chain of amino acids and does not refer to a specific length of the product. Thus, peptides, oligopeptides and proteins are included within the definition of polypeptide. This term is also intended to refer to post-expression modifications of the polypeptide, for example, glycosolations, acetylations, phosphorylations, and the like. A recombinant or derived polypeptide is not necessarily translated from a designated nucleic acid sequence. It may also be generated in any manner, including chemical synthesis.
- a substance which "enhances" an immune response refers to a substance in which an immune response is observed that is greater or intensified or deviated in any way with the addition of the substance when compared to the same immune response measured without the addition of the substance.
- the T-cell response induced upon vaccination with HIV polypeptides of the invention can be assessed e.g. in proliferation assays (for Th cell response, Belshe R.B. et al., J. Inf. Dis. 183: 1343-1352 (2001)), in ELISPOT assays (Oxenius, A. et al., Proc. Natl. Acad. Sci. USA 99: 13747-13752 (2002)), or in Cytotoxicity assays (Belshe R.B. et al, J. Inf. Dis. 183: 1343-1352 (2001).
- Effective Amount refers to an amount necessary or sufficient to realize a desired biologic effect.
- An effective amount of the composition would be the amount that achieves this selected result, and such an amount could be determined as a matter of routine by a person skilled in the art.
- an effective amount for treating an immune system deficiency could be that amount necessary to cause activation of the immune system, resulting in the development of an antigen specific immune response upon exposure to antigen.
- the term is also synonymous with "sufficient amount.”
- the effective amount for any particular application can vary depending on such factors as the disease or condition being treated, the particular composition being administered, the size of the subject, and/or the severity of the disease or condition.
- One of ordinary skill in the art can empirically determine the effective amount of a particular composition of the present invention without necessitating undue experimentation.
- Self antigen refers to proteins encoded by the host's genome or DNA and products generated by proteins or RNA encoded by the host's genome or DNA are defined as self.
- the tern "self antigen”, as used herein refers to proteins encoded by the human genome or DNA and products generated by proteins or RNA encoded by the human genome or DNA are defined as self.
- inventive compositions, pharmaceutical compositions and vaccines comprising self antigens are in particular capable of breaking tolerance against a self antigen when applied to the host.
- breaking tolerance against a self antigen shall refer to enhancing an immune response, as defined herein, and preferably enhancing a B or a T cell response, specific for the self antigen when applying the inventive compositions, pharmaceutical compositions and vaccines comprising the self antigen to the host.
- proteins that result from a combination of two or several self-molecules or that represent a fraction of a self-molecule and proteins that have a high, homology two self- molecules as defined above may also be considered self.
- treatment refers to prophylaxis and/or therapy.
- the term refers to a prophylactic treatment which increas s the resistance of a subject to infection with a pathogen or, in other words, decreases tine likelihood that the subject will become infected with the pathogen or will show signs of illness attributable to the infection, as well as a treatment after the subject has become infected in order to fight the infection, e.g., reduce or eliminate the infection or prevent it from becoming worse.
- the term "vaccine” refers to a formulation which contains the composition of the present invention and which is in a form that is capable of being administered to an animal.
- the vaccine comprises a conventional saline or buffered aqueous solution medium in which the composition of the present invention is suspended or dissolved.
- the composition of the present invention can be used conveniently to prevent, ameliorate, or otherwise treat a condition.
- the vaccine Upon introduction into a host, the vaccine is able to provoke an immune response including, but not limited to, the production of antibodies, cytokines and/or the activation of cytotoxic T cells, antigen presenting cells, helper T cells, dendritic cells and/or other cellular responses.
- the vaccine of the present invention additionally includes an adjuvant which can be present in either a minor or major proportion relative to the compound of the present invention.
- adjuvant refers to non-specific stimulators of the immune response or substances that allow generation of a depot in the host which when combined with the vaccine of the present invention provide for an even more enhanced immune response.
- adjuvants can be used. Examples include incomplete Freund's adjuvant, aluminum hydroxide and modified n uramyldipeptide.
- adjuvant as used herein also refers to typically specific stimulators of the immune response which when combined with the vaccine of the present invention provide for an even more enhanced and typically specific immune response. Examples include, but limited to, GM-CSF, IL-2, IL-12, IFN ⁇ . Further examples are within the knowledge of the person skilled in the art.
- virus-like particle refers to a structure resembling a virus particle but which has not been demonstrated to be pathogenic.
- a virus-like particle in accordance with the invention does not carry genetic information encoding for the proteins of the virus-like particle.
- viruslike particles lack the viral genome and, therefore, are noninfectious.
- virus-like particles can often be produced in large quantities by heterologous expression and can be easily purified.
- Some virus-like particles may contain nucleic acid distinct from their genome.
- a virus-like particle in accordance with the invention is non replicative and noninfectious since it lacks all or part of the viral genome, in particular the replicative and infectious components of the viral genome. A.
- virus-like particle in accordance with the invention may contain nucleic acid distinct from their genome.
- a typical and preferred embodiment of a virus-like particle in accordance with the present invention is a viral capsid such as the viral capsid of the corresponding virus, bacteriophage, or RNA-phage.
- viral capsid or “capsid”, as interchangeably used herein, refer to a macromolecular assembly composed of viral protein subunits. Typically and preferably, the viral protein subunits assemble into a viral capsid and capsid, respectively, having a structure with an inherent repetitive organization, wherein said structure is, typically, spherical or tubular.
- capsids of RNA-phages or HBcAg's have a spherical form of icosahedral symmetry.
- capsid-like structure refers to a macromolecular assembly composed of viral protein subunits reproducing the capsid morphology in the above defined sense but deviating from the typical symmetrical assembly while maintaining a sufficient degree of order and repetitiveness.
- virus-like particle of a bacteriophage refers to a virus-like particle resembling the structure of a bacteriophage, being non replicative and noninfectious, and lacking at least the gene or genes encoding for the replication machinery of the bacteriopliage, and typically also lacking the gene or genes encoding the protein or proteins responsible for viral attachment to or entry into the host.
- This definition should, however, also encompass virus-like particles of bacteriophages, in which the aforementioned gene or genes are still present but inactive, and, therefore, also leading to non-replicative and noninfectious virus-like particles of a bacteriophage.
- VLP of RNA phage coat protein The capsid structure formed from the self- assembly of 180 subunits of RNA phage coat protein and optionally containing host RNA is referred to as a "VLP of RNA phage coat protein".
- VLP of RNA phage coat protein A specific example is the VLP of Q ⁇ coat protein.
- the VLP of Q ⁇ coat protein may either be assembled exclusively from Q ⁇ CP subunits (SEQ ID: No 10) generated by expression of a Q ⁇ CP gene containing, for example, a TAA stop codon precluding any expression of the longer Al protein through suppression, see Kozlovska, T.M., et al., Intervirology 39: 9-15 (1996)), or additionally contain Al protein subunits (SEQ ID: No 11) in the capsid assembly.
- the readthrough process has a low efficiency and is leading to an only very low amount of Al protein in the VLPs. An extensive number of examples have been performed with different combinations of ISS packaged and antigen coupled.
- VLPs of Q ⁇ coat protein assembled exclusively from Q ⁇ CP subunits or VLPs of Q ⁇ coat protein containing additionally Al protein subunits in the capsids were used. Furthermore, no difference of the immune response between these Q ⁇ VLP preparations was observed. Therefore, for the sake of clarity the term "Q ⁇ VLP” is used throughout the description of the examples either for VLPs of Q ⁇ coat protein assembled exclusively from Q ⁇ CP subunits or VLPs of Q ⁇ coat protein containing additionally Al protein subunits in the capsids.
- virus particle refers to the morphological form of a virus. In some virus types it comprises a genome surrounded by a protein capsid; others have additional structures (e.g., envelopes, tails, etc.).
- Non-enveloped viral particles are made up of a proteinaceous capsid that surrounds and protects the viral genome. Enveloped viruses also have a capsid structure surrounding the genetic material of the virus but, in addition, have a lipid bilayer envelope that surrounds the capsid.
- the VLP's are free of a lipoprotein envelope or a lipoprotein-containing envelope. In a further preferred embodiment, the VLP's are free of an envelope altogether.
- certain embodiments of the invention involve the use of recombinant nucleic acid technologies such as cloning, polymerase chain reaction, the purification of DNA and RNA, the expression of recombinant proteins in prokaryotic and eukaryotic cells, etc.
- recombinant nucleic acid technologies such as cloning, polymerase chain reaction, the purification of DNA and RNA, the expression of recombinant proteins in prokaryotic and eukaryotic cells, etc.
- Such methodologies are well known to those skilled in the art and can be conveniently found in published laboratory methods manuals (e.g., Sambrook, J. et al, eds., MOLECULAR CLONING, A LABORATORY MANUAL, 2nd. edition, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. (1989); Ausubel, F.
- compositions and Methods for Enhancing an Immune Response The disclosed invention provides compositions and methods for enhancing an immune response against one or more antigens in an animal.
- Compositions of the invention comprise, or alternatively consist essentially of, or alternatively consist of, a virus-like particle, at least one immunostimulatory substance, preferably an immunostimulatory nucleic acid, and even more preferably an unmethylated CpG- containing oligonucleotide, and at least one antigen or antigenic determinant, wherein the immunostimulatory substance, the immunostimulatory nucleic acid or the oligonucleotide is bound to the virus-like particle, and wherein said antigen or antigenic determinant is bound to said virus-like particle and wherein said antigen comprises, alternatively consists essentially of, or alternatively consists of a HIV polypeptide.
- virus-like particles in the context of the present application refer to structures resembling a virus particle but which are not pathogenic. In general, virus-like particles lack the viral genome and, therefore, are noninfectious. Also, virus-like particles can be produced in large quantities by heterologous expression and can be easily purified. In a preferred embodiment, the virus-like particle is a recombinant virus-like particle. The skilled artisan can produce VLPs using recombinant DNA technology and virus coding sequences which are readily available to the public.
- the coding sequence of a virus envelope or core protein can be engineered for expression in a baculovirus expression vector using a commercially available baculovirus vector, under the regulatory control of a virus promoter, with appropriate modifications of the sequence to allow functional linkage of the coding sequence to the regulatory sequence.
- the coding sequence of a virus envelope or core protein can also be engineered for expression in a bacterial expression vector, for example.
- VLPs include, but are not limited to, the capsid proteins of Hepatitis B virus, measles virus, Sindbis virus, rotavirus, foot-and-mouth-disease virus, Norwalk virus, the retroviral GAG protein, the retrotransposon Ty protein pi, the surface protein of Hepatitis B virus, human papilloma virus, human polyoma virus, , BK virus (BKV), RNA phages, Ty, fr-phage, GA-phage, AP 205-phage and, in particular, Q ⁇ -phage.
- capsid proteins of Hepatitis B virus measles virus, Sindbis virus, rotavirus, foot-and-mouth-disease virus, Norwalk virus
- the retroviral GAG protein the retrotransposon Ty protein pi
- the surface protein of Hepatitis B virus human papilloma virus, human polyoma virus, , BK virus (BKV)
- RNA phages Ty
- the VLP of the invention is not limited to any specific form.
- the particle can be synthesized chemically or through a biological process, which can be natural or non-natural.
- this type of embodiment includes a virus-like particle or a recombinant form thereof.
- the VLP can comprise, or alternatively consist of, recombinant polypeptides of Rotavirus; recombinant polypeptides of Norwalk virus; recombinant polypeptides of Alphavirus; recombinant proteins which form bacterial pili or pilus like structures; recombinant polypeptides of Foot and Mouth Disease virus; recombinant polypeptides of measles virus, recombinant polypeptides of Sindbis virus, recombinant polypeptides of Retrovirus; recombinant polypeptides of Hepatitis B virus (e.g., a HBcAg); recombinant polypeptides of Tobacco mosaic virus; recombinant polypeptides of Flock House Virus; recombinant polypeptides of human Papillomavirus; recombinant polypeptides of Polyoma virus and, in particular, recombinant polypeptides of human Polyoma
- the virus-like particle can further comprise, or alternatively consist of, one or more fragments of such polypeptides, as well as variants of such polypeptides.
- Variants of polypeptides can share, for example, at least 80%, 85%, 90%, 95%, 97%, or 99% identity at the amino acid level with their wild type counterparts.
- the virus-like particle comprises, consists essentially of, or alternatively consists of recombinant proteins, or fragments thereof, of a RNA- phage.
- the RNA-phage is selected from the group consisting of a) bacteriophage Q ⁇ ; b) bacteriophage R17; c) bacteriophage fr; d) bacteriophage GA; e) bacteriophage SP; f) bacteriophage MS2; g) bacteriophage Mi l; h) bacteriophage MX1; i) bacteriophage NL95; k) bacteriophage G; 1) bacteriophage PP7; and m) bacteriophage AP205.
- the virus-like particle comprises, or alternatively consists essentially of, or alternatively consists of recombinant proteins, or fragments thereof, of the RNA-bacteriophage Q ⁇ or of the RNA- bacteriophage fr or of the RNA-bacteriophage AP205.
- the recombinant proteins comprise, or alternatively consist essentially of, or alternatively consist of coat proteins of RNA phages.
- RNA-phage coat proteins forming capsids or VLPs, or fragments of the bacteriophage coat proteins compatible with self-assembly into a capsid or a VLP are, therefore, further preferred embodiments of the present invention.
- Bacteriophage Q ⁇ coat proteins for example, can be expressed recombinantly in E. coli. Further, upon such expression these proteins spontaneously form capsids. Additionally, these capsids form a structure with an inherent repetitive organization.
- bacteriophage coat proteins which can be used to prepare compositions of the invention include the coat proteins of RNA bacteriophages such as bacteriophage Q ⁇ (SEQ ID NO: 10; PIR Database, Accession No. VCBPQ ⁇ referring to Q ⁇ CP and SEQ ID NO: 11 ; Accession No. AAA16663 referring to Q ⁇ Al protein), bacteriophage R17 (PIR Accession No. VCBPR7), bacteriophage fr (SEQ ID NO:13; PIR Accession No. VCBPFR), bacteriophage GA (SEQ ID NO:14; GenBank Accession No. NP-040754), bacteriophage SP (GenBank Accession No.
- bacteriophage Q ⁇ SEQ ID NO: 10; PIR Database, Accession No. VCBPQ ⁇ referring to Q ⁇ CP and SEQ ID NO: 11 ; Accession No. AAA16663 referring to Q ⁇ Al protein
- bacteriophage R17 PIR Accession No. VCBPR7
- bacteriophage MS2 PIR Accession No. VCBPM2
- bacteriophage Ml 1 GenBank Accession No. AAC06250
- bacteriophage MX1 GenBank Accession No. AAC14699
- bacteriophage NL95 GenBank Accession No. AAC14704
- bacteriophage f2 GenBank Accession No. P03611
- bacteriophage PP7 SEQ ID NO: 22
- bacteriophage AP205 SEQ ID NO: 31.
- the Al protein of bacteriophage Q ⁇ or C-terminal truncated forms missing as much as 100, 150 or 180 amino acids from its C-terminus may be incorporated in a capsid assembly of Q ⁇ coat proteins.
- the percentage of Q ⁇ Al protein relative to Q ⁇ CP in the capsid assembly will be limited, in order to ensure capsid formation.
- Further specific examples of bacteriophage coat proteins are described in WO 02/056905 on page 45 and 46 incorporated herein by way of reference.
- Further preferred virus-like particles of RNA-phages, in particular of Q ⁇ in accordance of this invention are disclosed in WO 02/056905, the disclosure of which is herewith incorporated by reference in its entirety.
- the virus-like particle comprises, or alternatively consists essentially of, or alternatively consists of recombinant proteins, or fragments thereof, of a RNA-phage, wherein the recombinant proteins comprise, consist essentially of or alternatively consist of mutant coat proteins of a RNA phage, preferably of mutant coat proteins of the RNA phages mentioned above.
- the mutant coat proteins of the RNA phage have been modified by removal of at least one lysine residue by way of substitution, or by addition of at least one lysine residue by way of substitution; alternatively, the mutant coat proteins of the RNA phage have been modified by deletion of at least one lysine residue, or by addition of at least one lysine residue by way of insertion.
- the deletion, substitution or addition of at least one lysine residue allows varying the degree of coupling, i.e. the amount of HIV polypeptides per subunits of the VLP of the RNA-phages, in particular, to match and tailor the requirements of the vaccine.
- At least 1.0 HIV peptide per subunit are linked to the VLP of the RNA-phage. This value is calculated as an average over all the subunits or monomers of the VLP of the RNA-phage.
- at least 0.1, preferrably 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9 or at least 2.0 HIV polypeptides are linked to the VLP of the RNA-phages as being calculated as a coupling average over all the subunits or monomers of the VLP of the RNA-phage.
- the virus-like particle comprises, or alternatively consists essentially of, or alternatively consists of recombinant proteins, or fragments thereof, of the RNA-bacteriophage Q ⁇ , wherein the recombinant proteins comprise, or alternatively consist essentially of, or alternatively consist of coat proteins having an amino acid sequence of SEQ ID NO: 10, or a mixture of coat proteins having amino acid sequences of SEQ ID NO: 10 and of SEQ ID NO: 11 or mutants of SEQ ID NO: 11 and wherein the N-terminal methionine is preferably cleaved.
- the virus-like particle comprises, consists essentially of or alternatively consists of recombinant proteins of Q ⁇ , or fragments thereof, wherein the recombinant proteins comprise, or alternatively consist essentially of, or alternatively consist of mutant Q ⁇ coat proteins.
- these mutant coat proteins have been modified by removal of at least one lysine residue by way of substitution, or by addition of at least one lysine residue by way of substitution.
- these mutant coat proteins have been modified by deletion of at least one lysine residue, or by addition of at least one lysine residue by way of insertion.
- Q ⁇ mutants for which exposed lysine residues are replaced by arginines can also be used for the present invention.
- the following Q ⁇ coat protein mutants and mutant Q ⁇ VLPs can, thus, be used in the practice of the invention: "Q ⁇ -240" (Lysl3-Arg; SEQ ID NO:20), "Q ⁇ -243" (Asn 10-Lys; SEQ ID NO:21), "Q ⁇ -250” (Lys 2-Arg, Lysl3-Arg; SEQ ID NO:22), “Q ⁇ -251” (SEQ ID NO:23) and "Q ⁇ -259” (Lys 2-Arg, Lysl6-Arg; SEQ ID NO:24).
- the virus-like particle comprises, consists essentially of or alternatively consists of recombinant proteins of mutant Q ⁇ coat proteins, which comprise proteins having an amino acid sequence selected from the group of a) the amino acid sequence of SEQ ID NO: 20; b) the amino acid sequence of SEQ ID NO: 21; c) the amino acid sequence of SEQ ID NO: 22; d) the amino acid sequence of SEQ ID NO:23; and e) the amino acid sequence of SEQ ID NO: 24.
- mutant Q ⁇ coat protein VLPs and capsids are disclosed in WO02/056905. In particular is hereby referred to Example 18 of above mentioned application.
- the virus-like particle comprises, or alternatively consists essentially of, or alternatively consists of recombinant proteins of Q ⁇ , or fragments thereof, wherein the recombinant proteins comprise, consist essentially of or alternatively consist of a mixture of either one of the foregoing Q ⁇ mutants and the corresponding Al protein.
- the virus-like particle comprises, or alternatively essentially consists of, or alternatively consists of recombinant proteins, or fragments thereof, of RNA-phage AP205.
- the AP205 genome consists of a maturation protein, a coat protein, a replicase and two open reading frames not present in related phages; a lysis gene and an open reading frame playing a role in the translation of the maturation gene (Klovins ., et al., J. Gen. Virol. 83: 1523-33 (2002)).
- AP205 coat protein can be expressed from plasmid pAP283- 58 (SEQ ID NO: 30), which is a derivative of pQblO (Kozlovska, T. M. et al., Gene
- Vectors pQbl 0 and pQbl 85 are vectors derived from pGEM vector, and expression of the cloned genes in these vectors is controlled by the tip promoter (Kozlovska, T. M. et al., Gene 137:133-37 (1993)).
- Plasmid pAP283-58 (SEQ ID NO:30) comprises a putative AP205 ribosomal binding site in the following sequence, which is downstream of the Xbal site, and immediately upstream of the ATG start codon of the AP205 coat protein: tctagaATTTTCTGCGCACCCAT
- the vector pQbl85 comprises a Shine Delagarno sequence downstream from the Xbal site and upstream of the start codon (tctagaTTAACCCAACGCGTAGGAGTCAGGCCatg (SEQ ID NO: 50), Shine Delagarno sequence underlined).
- the virus-like particle comprises, or alternatively essentially consists of, or alternatively consists of recombinant coat proteins, or fragments thereof, of the RNA-phage AP205.
- This preferred embodiment of the present invention thus, comprises AP205 coat proteins that form capsids. Such proteins are recombinantly expressed, or prepared from natural sources. AP205 coat proteins produced in bacteria spontaneously form capsids, as evidenced by Electron Microscopy (EM) and immunodiffusion. The structural properties of the capsid formed by the AP205 coat protein (SEQ ID NO: 31) and those formed by the coat protein of the AP205 RNA phage are nearly indistinguishable when seen in EM.
- AP205 VLPs are highly immunogenic, and can be linked with antigens and/or antigenic determinants to generate vaccine constructs displaying the antigens and/or antigenic determinants oriented in a repetitive manner. High titers are elicited against the so displayed antigens showing that bound antigens and/or antigenic determinants are accessible for interacting with antibody molecules and are immunogenic.
- the virus-like particle comprises, or alternatively essentially consists of, or alternatively consists of recombinant mutant coat proteins, or fragments thereof, of the RNA-phage AP205.
- Assembly-competent mutant forms of AP205 VLPs including AP205 coat protein with the subsitution of proline at amino acid 5 to threonine (SEQ ID NO: 32), may also be used in the practice of the invention and leads to a further preferred embodiment of the invention.
- These VLPs, AP205 VLPs derived from natural sources, or AP205 viral particles may be bound to antigens to produce ordered repetitive arrays of the antigens in accordance with the present invention.
- AP205 P5-T mutant coat protein can be expressed from plasmid pAP281-32 (SEQ ID No. 33), which is derived directly from pQbl85, and which contains the mutant AP205 coat protein gene instead of the Q ⁇ coat protein gene.
- Vectors for expression of the AP205 coat protein are transfected into E. coli for expression of the AP205 coat protein. Methods for expression of the coat protein and the mutant coat protein, respectively, leading to self-assembly into VLPs are described in WO 04/007538 which is incorporated by reference in its entirety.
- Suitable E. coli strains include, but are not limited to, E. coli K802, JM 109, RR1.
- Suitable vectors and strains and combinations thereof can be identified by testing expression of the coat protein and mutant coat protein, respectively, by SDS-PAGE and capsid formation and assembly by optionally first purifying the capsids by gel filtration and subsequently testing them in an immunodiffusion assay (Ouchterlony test) or Electron Microscopy (Kozlovska, T. M. et al., Gene 137:133-37 (1993)).
- AP205 coat proteins expressed from the vectors pAP283-58 and pAP281-32 may be devoid of the initial Methionine amino-acid, due to processing in the cytoplasm of E. coli.
- the virus-like particle comprises, or alternatively essentially consists of, or alternatively consists of a mixture of recombinant coat proteins, or fragments thereof, of the RNA-phage AP205 and of recombinant mutant coat proteins, or fragments thereof, of the RNA-phage AP205.
- the virus-like particle comprises, or alternatively essentially consists of, or alternatively consists of fragments of recombinant coat proteins or recombinant mutant coat proteins of the RNA-phage AP205.
- Recombinant AP205 coat protein fragments capable of assembling into a VLP and a capsid, respectively are also useful in the practice of the invention. These fragments may be generated by deletion, either internally or at the termini of the coat protein and mutant coat protein, respectively. Insertions in the coat protein and mutant coat protein sequence or fusions of antigen sequences to the coat protein and mutant coat protein sequence, and compatible with assembly into a VLP, are further embodiments of the invention and lead to chimeric AP205 coat proteins, and particles, respectively. The outcome of insertions, deletions and fusions to the coat protein sequence and whether it is compatible with assembly into a VLP can be determined by electron microscopy.
- the particles fonned by the AP205 coat protein, coat protein fragments and chimeric coat proteins described above, can be isolated in pure form by a combination of fractionation steps by precipitation and of purification steps by gel filtration using e.g. Sepharose CL-4B, Sepharose CL-2B, Sepharose CL-6B columns and combinations thereof as described in WO 04/007538 which is incorporated by reference in its entirety.
- Other methods of isolating virus-like particles are known in the art, and may be used to isolate the virus-like particles (VLPs) of bacteriophage AP205.
- VLPs virus-like particles
- the use of ultracentrifugation to isolate VLPs of the yeast retrotransposon Ty is described in U.S. Patent No. 4,918,166, which is incorporated by reference herein in its entirety.
- the crystal structure of several RNA bacteriophages has been determined
- modified RNA bacteriophages as well as variants of proteins and N- and C terminal truncation mutants which fo ⁇ n capsids or capsid like structures, as well as methods for preparing such compositions and vaccine compositions, respectively are described in WO 02/056905 on page 50-52.
- the invention thus includes compositions and vaccine compositions prepared from proteins which form capsids or VLPs, methods for preparing these compositions from individual protein subunits and VLPs or capsids, methods for preparing these individual protein subunits, nucleic acid molecules which encode these subunits, and methods for vaccinating and/or eliciting immunological responses in individuals using these compositions of the present invention.
- Fragments of VLPs which retain the ability to induce an immune response can comprise, or alternatively consist of, polypeptides which are about 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 250, 300, 350, 400, 450 or 500 amino acids in length, but will obviously depend on the length of the sequence of the subunit composing the VLP. Examples of such fragments include fragments of proteins discussed herein which are suitable for the preparation of the immune response enhancing composition.
- the VLP's are free of a lipoprotein envelope or a lipoprotein-containing envelope. In a further preferred embodiment, the VLP's are free of an envelope altogether.
- the lack of a lipoprotein envelope or lipoprotein-containing envelope and, in particular, the complete lack of an envelope leads to a more defined virus-like particle in its structure and composition. Such more defined virus-like particles, therefore, may minimize side-effects.
- the lack of a lipoprotein-containing envelope or, in particular, the complete lack of an envelope avoids or minimizes incorporation of potentially toxic molecules and pyrogens within the virus-like particle.
- the invention provides a vaccine composition of the invention comprising a virus-like particle, wherein preferably said virus-like particle is a recombinant virus-like particle.
- the virus-like particle comprises, or alternatively consist essentially of, or alternatively consists of, recombinant proteins, or fragments thereof, of a RNA-phage, preferably of coat proteins of RNA phages.
- the recombinant proteins of the virus-like particle of the vaccine composition of the invention comprise, or alternatively consist essentially of, or alternatively consist of mutant coat proteins of RNA phages, wherein the RNA-phage is selected from the group consisting of: (a) bacteriophage Q ⁇ ; (b) bacteriophage R17; (c) bacteriophage fr; (d) bacteriophage GA; (e) bacteriophage SP; (f) bacteriophage MS2; (g) bacteriophage Ml 1; (h) bacteriophage MX1; (i) bacteriophage NL95; (k) bacteriophage f2; (1) bacteriophage PP7; and (m) bacteriophage AP205.
- the mutant coat proteins of said RNA phage have been modified by removal, or by addition of at least one lysine residue by way of substitution. In another preferred embodiment, the mutant coat proteins of said RNA phage have been modified by deletion of at least one lysine residue or by addition of at least one lysine residue by way of insertion.
- the virus-like particle comprises recombinant proteins or fragments thereof, of RNA-phage Q ⁇ , RNA-phage fr, or of RNA- phage AP205.
- the invention includes virus-like particles or recombinant forms thereof. Skilled artisans have the knowledge to produce such particles and attach antigens thereto. Further preferred embodiments of the present invention hereto are disclosed in the Example Section.
- the virus-like particle comprises, or alternatively consists essentially of, or alternatively consists of recombinant proteins, or fragments thereof, of the BK virus (BKV), wherein the recombinant proteins comprise, or alternatively consist essentially of, or alternatively consist of proteins having an amino acid sequence of SEQ ID NO: 12.
- BK virus (BKV) is a non-enveloped double stranded DNA virus belonging to the polyoma virus subfamily of the papovaviridae.
- VP1 is the major capsid protein of BKV.
- VP1 has 362 amino acids (SEQ ID NO: 12, Gene Bank entry: AAA46882) and is 42 kDa in size.
- capsid structures (Salunke D.M., et al., Cell 46(6):895-904 (1986); Sasnauskas, K., et al, Biol. Chem. 380(3):381-6 (1999); Sasnauskas, K., et al, 3rd International Workshop "Virus-like particles as vaccines” Berlin, September 26-29 (2001); Touze, A., et al., J Gen Virol. 82(Pt 12):3005-9 (2001).
- the capsid is organized in 72 VPl pentamers forming an icosahedral structure.
- the capsids have a diameter of approximately 45 nm.
- the particles used in compositions of the invention are composed of a Hepatitis B capsid (core) protein (HBcAg) or a fragment of a HBcAg which has been modified to either eliminate or reduce the number of free cysteine residues.
- core particles suitable for use in compositions of the invention include those comprising modified HBcAgs, or fragments thereof, in which one or more of the naturally resident cysteine residues have been either deleted or substituted with another amino acid residue (e.g., a serine residue).
- the HBcAg is a protein generated by the processing of a Hepatitis B core antigen precursor protein.
- a number of isotypes of the HBcAg have been identified and their amino acids sequences are readily available to those skilled in the art.
- the HBcAg protein having the amino acid sequence shown in SEQ ID NO: 16 is 185 amino acids in length and is generated by the processing of a 212 amino acid Hepatitis B core antigen precursor protein. This processing results in the removal of 29 amino acids from the N terminus of the Hepatitis B core antigen precursor protein.
- the HBcAg protein that is 185 amino acids in length is generated by the processing of a 214 amino acid Hepatitis B core antigen precursor protein.
- vaccine compositions of the invention will be prepared using the processed form of a HBcAg (i.e., a HBcAg from which the N terminal leader sequence of the Hepatitis B core antigen precursor protein have been removed).
- the HBcAgs when HBcAgs are produced under conditions where processing will not occur, the HBcAgs will generally be expressed in "processed" form.
- bacterial systems such as E. coli, generally do not remove the leader sequences, also referred to as "signal peptides," of proteins which are normally expressed in eukaryotic cells.
- leader sequences also referred to as “signal peptides”
- these proteins will generally be expressed such that the N terminal leader sequence of the Hepatitis B core antigen precursor protein is not present.
- Hepatitis B virus-like particles which can be used for the present invention, is disclosed, for example, in WO 00/32227, and hereby in particular in Examples 17 to 19 and 21 to 24, as well as in WO 01/85208, and hereby in particular in Examples 17 to 19, 21 to 24, 31 and 41, and in WO 02/056905. For the latter application, it is in particular referred to Example 23, 24, 31 and 51. All three documents are explicitly incorporated herein by reference.
- the present invention also includes HBcAg variants which have been modified to delete or substitute one or more additional cysteine residues.
- the vaccine compositions of the invention include compositions comprising HBcAgs in which cysteine residues not present in the amino acid sequence shown in SEQ ID NO: 16 have been deleted.
- side reactions include disulfide exchanges, reaction with chemical substances or metabolites that are, for example, injected or formed in a combination therapy with other substances, or direct oxidation and reaction with nucleotides upon exposure to UV light.
- Toxic adducts could thus be generated, especially considering the fact that HBcAgs have a strong tendency to bind nucleic acids.
- the toxic adducts would thus be distributed between a multiplicity of species, which individually may each be present at low concentration, but reach toxic levels when together.
- HBcAgs in vaccine compositions which have been modified to remove naturally resident cysteine residues is that sites to which toxic species can bind when antigens or antigenic determinants are attached would be reduced in number or eliminated altogether.
- HBcAg variants suitable for use in the practice of the present invention have been identified. Yuan et al., (J. Virol. 73:10122 10128 (1999)), for example, describe variants in which the isoleucine residue at position corresponding to position 97 in SEQ ID NO:25 is replaced with either a leucine residue or a phenylalanine residue.
- HBcAg variants differ in amino acid sequence at a number of positions, including amino acid residues which corresponds to the amino acid residues located at positions 12, 13, 21, 22, 24, 29, 32, 33, 35, 38, 40, 42, 44, 45, 49, 51, 57, 58, 59, 64, 66, 67, 69, 74, 77, 80, 81, 87, 92, 93, 97, 98, 100, 103, 105, 106, 109, 113, 116, 121, 126, 130, 133, 135, 141, 147, 149, 157, 176, 178, 182 and 183 in SEQ ID NO:28.
- HBcAgs suitable for use in the present invention can be derived from any organism so long as they are able to enclose or to be coupled or otherwise attached to, in particular as long as they are capable of packaging, an unmethylated CpG-containing oligonucleotide and induce an immune response.
- HBcAgs As noted above, generally processed HBcAgs (i.e., those which lack leader sequences) will be used in the vaccine compositions of the invention.
- the present invention includes vaccine compositions, as well as methods for using these compositions, which employ the above described variant HBcAgs. Further included within the scope of the invention are additional HBcAg variants which are capable of associating to form dimeric or multimeric structures.
- the invention further includes vaccine compositions comprising HBcAg polypeptides comprising, or alternatively consisting of, amino acid sequences which are at least 80%, 85%, 90%, 95%», 97% or 99% identical to any of the wild-type amino acid sequences, and forms of these proteins which have been processed, where appropriate, to remove the N terminal leader sequence.
- the amino acid sequence of a polypeptide has an amino acid sequence that is at least 80%, 85%, 90%, 95%, 97% or 99% identical to one of the wild-type amino acid sequences, or a subportion thereof, can be determined conventionally using known computer programs such the Bestfit program.
- Bestfit or any other sequence alignment program to determine whether a particular sequence is, for instance, 95% identical to a reference amino acid sequence, the parameters are set such that the percentage of identity is calculated over the full length of the reference amino acid sequence and that gaps in homology of up to 5% of the total number of amino acid residues in the reference sequence are allowed.
- amino acid sequences of the hereinabove mentioned HBcAg variants and precursors are relatively similar to each other.
- reference to an amino acid residue of a HBcAg variant located at a position which corresponds to a particular position in SEQ ID NO:28 refers to the amino acid residue which is present at that position in the amino acid sequence shown in SEQ ID NO:28.
- the homology between these HBcAg variants is for the most part high enough among Hepatitis B viruses that infect mammals so that one skilled in the art would have little difficulty reviewing both the amino acid sequence shown in SEQ ID NO:28 and in SEQ ID NO: 16, respectively, and that of a particular HBcAg variant and identifying "corresponding" amino acid residues.
- the HBcAg amino acid sequence shown in SEQ ID NO:27 which shows the amino acid sequence of a HBcAg derived from a virus which infect woodchucks, has enough homology to the HBcAg having the amino acid sequence shown in SEQ ID NO:28 that it is readily apparent that a three amino acid residue insert is present in SEQ ID NO:27 between amino acid residues 155 and 156 of SEQ ID NO:28.
- the invention also includes vaccine compositions which comprise HBcAg variants of Hepatitis B viruses which infect birds, as wells as vaccine compositions which comprise fragments of these HBcAg variants.
- vaccine compositions which comprise HBcAg variants of Hepatitis B viruses which infect birds, as wells as vaccine compositions which comprise fragments of these HBcAg variants.
- cysteine residues naturally present in these polypeptides could be either substituted with another amino acid residue or deleted prior to their inclusion in vaccine compositions of the invention.
- cysteine residues of the Hepatitis B virus capsid protein have been either deleted or substituted with another amino acid residue.
- Expression and purification of an HBcAg- Lys variant has been described in Example 24 of WO 02/056905 and the construction of a HBcAg devoid of free cysteine residues and containing an inserted lysine residue has been described in Example 31 of WO 02/056905.
- compositions and vaccine compositions, respectively, of the invention will contain HBcAgs from which the C terminal region (e.g., amino acid residues 145 185 or 150 185 of SEQ ID NO: 28) has been removed.
- additional modified HBcAgs suitable for use in the practice of the present invention include C terminal truncation mutants. Suitable truncation mutants include HBcAgs where 1, 5, 10, 15, 20, 25, 30, 34, 35, amino acids have been removed from the C terminus.
- HBcAgs suitable for use in the practice of the present invention also include N terminal truncation mutants. Suitable truncation mutants include modified HBcAgs where 1, 2, 5, 7, 9, 10, 12, 14, 15, or 17 amino acids have been removed from the N terminus.
- HBcAgs suitable for use in the practice of the present invention include N and C terminal truncation mutants.
- Suitable truncation mutants include HBcAgs where 1, 2, 5, 7, 9, 10, 12, 14, 15, and 17 amino acids have been removed from the N terminus and 1, 5, 10, 15, 20, 25, 30, 34, 35 amino acids have been removed from the C terminus.
- the invention further includes compositions and vaccine compositions, respectively, comprising HBcAg polypeptides comprising, or alternatively essentially consisting of, or alternatively consisting of, amino acid sequences which are at least 80%, 85%, 90%, 95%, 97%, or 99% identical to the above described truncation mutants.
- a lysine residue is introduced into a HBcAg polypeptide, to mediate the binding of the HIV polypeptide of the invention to the VLP of HBcAg.
- compositions of the invention are prepared using a HBcAg comprising, or alternatively consisting of, amino acids 1-144, or 1-149, 1- 185 of SEQ ID NO: 28, which is modified so that the amino acids corresponding to positions 79 and 80 are replaced with a peptide having the amino acid sequence of Gly- Gly-Lys-Gly-Gly (SEQ ID NO: 18) resulting in the HBcAg polypeptide having the sequence shown in SEQ ID NO:29).
- SEQ ID NO: 18 amino acid sequence of Gly- Gly-Lys-Gly-Gly
- compositions comprising the corresponding polypeptides having amino acid sequences shown in any of the hereinabove mentioned -Hepatitis B core antigen precursor variants which also have above noted amino acid alterations.
- additional HBcAg variants which are capable of associating to form a capsid or VLP and have the above noted amino acid alterations.
- compositions and vaccine compositions comprising HBcAg polypeptides which comprise, or alternatively consist of, amino acid sequences which are at least 80%, 85%, 90%, 95%, 97%o or 99% identical to any of the wild-type amino acid sequences, and forms of these proteins which have been processed, where appropriate, to remove the N terminal leader sequence and modified with above noted alterations.
- compositions or vaccine compositions of the invention may comprise mixtures of different HBcAgs.
- these vaccine compositions may be composed of HBcAgs which differ in amino acid sequence.
- vaccine compositions could be prepared comprising a "wild type" HBcAg and a modified HBcAg in which one or more amino acid residues have been altered (e.g., deleted, inserted or substituted).
- preferred vaccine compositions of the invention are those which present highly ordered and repetitive antigen arrays, wherein the antigen is a HIV polypeptide.
- the invention is partly based on the surprising finding that immunostimulatory substances, preferably immunostimulatory nucleic acids and even more preferably DNA oligonucleotides or alternatively poly (I:C) can be packaged into VLPs.
- immunostimulatory substances preferably immunostimulatory nucleic acids and even more preferably DNA oligonucleotides or alternatively poly (I:C)
- the nucleic acids present in VLPs can be replaced specifically by the immunostimulatory substances, preferably by the immunostimulatory nucleic acids and even more preferably by the DNA-oligonucleotides containing CpG motifs or poly (I:C).
- the CpG- VLPs are more immunogenic and elicit more specific effects than their CpG-free counterparts and induce enhanced B and T cell responses.
- the immune response against antigens coupled, fused or attached otherwise to the VLPs is similarly enhanced as the immune response against the VLP itself.
- the T cell responses against both the VLPs and antigens are especially directed to the Thl type.
- the packaged nucleic acids and CpGs, respectively are protected from degradation, i.e., they are more stable.
- non-specific activation of cells from the innate immune system is dramatically reduced.
- the innate immune system has the capacity to recognize invariant molecular pattern shared by microbial pathogens. Recent studies have revealed that this recognition is a crucial step in inducing effective immune responses.
- the main mechanism by which microbial products augment immune responses is to stimulate APC, expecially dendritic cells to produce proinflammatory cytokines and to express high levels costimulatory molecules for T cells. These activated dendritic cells subsequently initiate primary T cell responses and dictate the type of T cell-mediated effector function.
- RNA synthesized by various types of viruses represent important members of the microbial components that enhance immune responses.
- Synthetic double stranded (ds) RNA such as polyinosinic-polycytidylic acid (poly I:C) are capable of inducing dendritic cells to produce proinflammatory cytokines and to express high levels of costimulatory molecules.
- Preferred ribonucleic acid encompass polyinosinic-polycytidylic acid double- stranded RNA (poly I:C). Ribonucleic acids and modifications thereof as well as methods for their production have been described by Levy, H.B (Methods Enzymol. 1981, 78:242- 251), DeClercq, E (Methods Enzymol. 1981,78:227-236) and Torrence, P.F. (Methods Enzymol 1981;78:326-331) and references therein.
- ribonucleic acids comprise polynucleotides of inosinic acid and cytidiylic acid such poly (IC) of which two strands forms double stranded RNA.
- Ribonucleic acids can be isolated from organisms. Ribonucleic acids also encompass further synthetic ribonucleic acids, in particular synthetic poly (I:C) oligonucleotides that have been rendered nuclease resistant by modification of the phosphodiester backbone, in particular by phosphorothioate modifications. In a further embodiment the ribose backbone of poly (I:C) is replaced by a deoxyribose. Those skilled in the art know procedures how to synthesize synthetic oligonucleotides.
- TLR active toll-like receptors
- TLR2 is activated by peptidoglycans, lipoproteins, lipopolysacchrides, lipoteichonic acid and Zymosan, and macrophage-activating lipopeptide MALP-2
- TLR3 is activated by double-stranded RNA such as poly (I:C)
- TLR4 is activated by lipopolysaccharide, lipoteichoic acids and taxol and heat-shock proteins such as heat shock protein HSP-60 and Gp96
- TLR5 is activated by bacterial flagella, especially the flagellin protein
- TLR6 is activated by peptidoglycans
- TLR7 is activated by imiquimoid and imidazoquinoline compounds, such as R-848, loxoribine and bropirimine
- TLR9 is activated by a ligands.
- TLR2 is activated by peptidoglycans, lipoproteins, lipopoly
- Ligands for TLR1, TLR8 and TLR10 are not known so far. However, recent reports indicate that same receptors can react with different ligands and that further receptors are present. The above list of ligands is not exhaustive and further ligands are within the knowledge of the person skilled in the art.
- the unmethylated CpG-containing oligonucleotide comprises the sequence:
- the oligonucleotide can comprise about 6 to about 100,000 nucleotides, preferably about 6 to about 2000 nucleotides, more preferably about 20 to about 2000 nucleotides, and even more preferably comprises about 20 to about 300 nucleotides.
- the oligonucleotide can comprise more than 100 to about 2000 nucleotides, preferably more than 100 to about 1000 nucleotides, and more preferably more than 100 to about 500 nucleotides.
- the CpG-containing oligonucleotide contains one or more phosphorothioate modifications of the phosphate backbone.
- a CpG- containing oligonucleotide having one or more phosphate backbone modifications or having all of the phosphate backbone modified and a CpG-containing oligonucleotide wherein one, some or all of the nucleotide phosphate backbone modifications are phosphorothioate modifications are included within the scope of the present invention.
- at least one of the nucleotide XI, X2, X3, and X4 has a phosphate backbone modification.
- the immunostimulatory substance is an unmethylated CpG-containing oligonucleotide, wherein said unmethylated CpG-containing oligonucleotide has a nucleic acid sequence selected without limitation from the group consisting of (a)
- GGGGACGATCGTCGGGGGG ((SEQ ID NO: 2); and typically abbreviated herein as G3-6)
- GGGGGACGATCGTCGGGGGG ((SEQ ID NO: 3); and typically abbreviated herein as G4-6)
- GGGGGGACGATCGTCGGGGGG (SEQ ID NO: 4); and typically abbreviated herein as G5-6)
- GGGGGGGGACGATCGTCGGGGGGGG ((SEQ ID NO: 5); and typically abbreviated herein as G6-6)
- GGGGGGGGACGATCGTCGGGGGGGGG ((SEQ ID NO: 6); and typically abbreviated herein as G7-7)
- GGGGGGGGGACGATCGTCGGGGGGGGGG ((SEQ ID NO: 7); and typically abbreviated herein as G8-8)
- G9-9
- GGGGGGCGACGACGATCGTCGTCGGGGGGG ((SEQ ID NO: 9); and typically abbreviated herein as G6), (i) tccatgacgttcctgaataat ((SEQ ID NO: 34); and typically abbreviated herein as CyCpGpt), 0) TCCATGACGTTCCTGAATAAT ((SEQ ID NO: 35); and typically abbreviated herein CyCpG), (k) tccatgacgttcctgacgtt ((SEQ ID NO: 36); and typically abbreviated herein as B-CpGpt), (1) TCCATGACGTTCCTGACGTT ((SEQ ID NO: 37); and typically abbreviated herein as B-CpG), (m) ggggtcaacgttgaggggg ((SEQ ID NO: 38); and typically abbreviated herein as NKCpGpt), (n) GGGGTCAA
- the immunostimulatory substance is an unmethylated CpG-containing oligonucleotide, wherein said unmethylated CpG-containing oligonucleotide has a nucleic acid sequence of GGGGGGGGGGGACGATCGTCGGGGGGGGGG ((SEQ ID NO: 41); and typically abbreviated herein as gl Ogacga-PO or Gl 0-PO).
- the CpG-containing oligonucleotide can also be recombinant, genomic, synthetic, cDNA, plasmid-derived and single or double stranded.
- the nucleic acids can be synthesized de novo using any of a number of procedures well known in the art. For example, the b-cyanoethyl phosphoramidite method (Beaucage, S. L., and Caruthers, M. H., Tet. Let. 22: 1859 (1981); nucleoside H-phosphonate method (Garegg et al., Tet. Let. 27:4051-4054 (1986); Froehler et al, Nucl. Acid. Res.
- CpGs can be produced on a large scale in plasmids, (see Sambrook, T., et al., "Molecular Cloning: A Laboratory Manual,” Cold Spring Harbor laboratory Press, New York, 1989) which after being administered to a subject are degraded into oligonucleotides.
- Oligonucleotides can be prepared from existing nucleic acid sequences (e.g., genomic or cDNA) using known techniques, such as those employing restriction enzymes, exonucleases or endonucleases.
- the immunostimulatory substances, the immunostimulatory nucleic acids as well as the unmethylated CpG-containing oligonucleotide can be bound to the VLP by any way known is the art provided the composition enhances an immune response in an animal.
- the oligonucleotide can be bound either covalently or non- covalently.
- the VLP can enclose, fully or partially, the immunostimulatory substances, the immunostimulatory nucleic acids as well as the unmethylated CpG- containing oligonucleotide.
- the immunostimulatory nucleic acid as well as the unmethylated CpG-containing oligonucleotide can be bound to a VLP site such as an oligonucleotide binding site (either naturally or non-naturally occurring), a DNA binding site or a RNA binding site.
- the VLP site comprises an arginine- rich repeat or a lysine-rich repeat.
- compositions of the invention are to activate dendritic cells for the purpose of enhancing a specific immune response against antigens.
- the immune response can be enhanced using ex vivo or in vivo techniques.
- the ex vivo procedure can be used on autologous or heterologous cells, but is preferably used on autologous cells.
- the dendritic cells are isolated from peripheral blood or bone marrow, but can be isolated from any source of dendritic cells. Ex vivo manipulation of dendritic cells for the purposes of cancer immunotherapy have been described in several references in the art, including Engleman, E.
- the dendritic cells can also be contacted with the inventive compositions using in vivo methods.
- the CpGs are administered in combination with the VLP optionally coupled, fused or otherwise attached to an antigen directly to a subject in need of immunotherapy.
- the VLPs/CpGs be administered in the local region of the tumor, which can be accomplished in any way known in the art, e.g., direct injection into the tumor.
- a preferred embodiment of the present invention is to provide a composition for enhancing an immune response in an animal comprising (a) a virus-like particle; (b) at least one immunostimulatory substance; and (c) at least one antigen or antigenic determinant; wherein said antigen or said antigenic determinant is bound to said virus-like particle and wherein said antigen comprises, alternatively consists essentially of, or alternatively consists of a HIV polypeptide, and wherein said immunostimulatory substance is bound to said virus-like particle, and wherein said immunostimulatory substance is an unmethylated CpG-containing oligonucleotide, wherein the CpG motif of said unmethylated CpG-containing oligonucleotide is part of a palindromic sequence, wherein said palindromic sequence is GACGATCGTC (SEQ ID NO: 1), and wherein said palindromic sequence is flanked at its 3 '-terminus and at its 5 '-terminus by more than two and less than 11
- the inventive immunostimulatory substances i.e. the unmethylated CpG-containing oligonucleotides, wherein the CpG motif of said unmethylated CpG- containing oligonucleotides are part of a palindromic sequence, wherein the palindromic sequence is GACGATCGTC (SEQ ID NO: 1), and wherein the palindromic sequence is flanked at its 3 '-terminus and at its 5 '-terminus by more than two and less than 11 guanosine entities or, more preferably by 8-10 guanosine entities, or, most preferably by 10 guanosine entities, are, in particular, effective at stimulating immune cells in vitro.
- the palindromic sequence comprises, or alternatively consist essentially of, or alternatively consists of or is GACGATCGTC (SEQ ID NO: 1), wherein said palindromic sequence is flanked at its 5'- terminus by at least 3 and at most 10 guanosine entities and wherein said palindromic sequence is flanked at its 3 '-terminus by at least 6 and at most 10 guanosine entities.
- the palindromic sequence is flanked at its 5 '-terminus by at least 3 and at most 10 guanosine entities and wherein said palindromic sequence is flanked at its 3 '-terminus by at least 6 and at most 10 guanosine entities.
- the immunostimulatory substance is an unmethylated CpG-containing oligonucleotide, wherein the CpG motif of said unmethylated CpG-containing oligonucleotide is part of a palindromic sequence, wherein said unmethylated CpG-containing oligonucleotide has a nucleic acid sequence selected from (a) GGGGACGATCGTCGGGGGG ((SEQ ID NO: 2); and typically abbreviated herein as G3-6), (b) GGGGGACGATCGTCGGGGGG ((SEQ ID NO: 3); and typically abbreviated herein as G4-6), (c) GGGGGGACGATCGTCGGGGGG ((SEQ ID NO: 4); and typically abbreviated herein as G5-6), (d) GGGGGGGACGATCGTCGGGGGG ((SEQ ID NO: 5); and typically abbreviated herein as G6-6), (e) GGGGGGGGACGATC
- GGGGGGGGGACGATCGTCGGGGGGGG ((SEQ ID NO: 7); and typically abbreviated herein as G8-8), (g) GGGGGGGGGGACGATCGTCGGGGGGGGG ((SEQ ID NO: 8); and typically abbreviated herein as G9-9), and (h) GGGGGGCGACGACGATCGTCGTCGGGGGGG ((SEQ ID NO: 9); and typically abbreviated herein as G6), and (i) GGGGGGGGGGGACGATCGTCGGGGGGGGGG ((SEQ ID NO: 41); and typically abbreviated herein as G10-PO).
- the immunostimulatory substance is an unmethylated CpG-containing oligonucleotide, wherein the CpG motif of said unmethylated CpG-containing oligonucleotide is part of a palindromic sequence, wherein said palindromic sequence is GACGATCGTC (SEQ ID NO: 1), and wherein said palindromic sequence is flanked at its 5 '-terminus by at least 4 and at most 9 guanosine entities and wherein said palindromic sequence is flanked at its 3 '-terminus by at least 6 and at most 9 guanosine entities.
- the immunostimulatory substance is an unmethylated CpG-containing oligonucleotide, wherein the CpG motif of said unmethylated CpG-containing oligonucleotide is part of a palindromic sequence, wherein said unmethylated CpG-containing oligonucleotide has a nucleic acid sequence selected from (a) GGGGGACGATCGTCGGGGGG ((SEQ ID NO: 3); and typically abbreviated herein as G4-6), (b) GGGGGGACGATCGTCGGGGGG ((SEQ ID NO: 4); and typically abbreviated herein as G5-6), (c) GGGGGGGACGATCGTCGGGGGG ((SEQ ID NO: 5); and typically abbreviated herein as G6-6), (d)
- GGGGGGGGACGATCGTCGGGGGGGGG ((SEQ ID NO: 6); and typically abbreviated herein as G7-7), (e) GGGGGGGGGACGATCGTCGGGGGGGG ((SEQ ID NO: 7); and typically abbreviated herein as G8-8), (f) GGGGGGGGGGACGATCGTCGGGGGGGGG ((SEQ ID NO: 8); and typically abbreviated herein as G9-9); and (g) GGGGGGGGGGGACGATCGTCGGGGGGGGGG ((SEQ ID NO: 41); and typically abbreviated herein as G10-PO).
- the immunostimulatory substance is an unmethylated CpG-containing oligonucleotide, wherein the CpG motif of said unmethylated CpG-containing oligonucleotide is part of a palindromic sequence, wherein said palindromic sequence is GACGATCGTC (SEQ ID NO: 1), and wherein said palindromic sequence is flanked at its 5 '-terminus by at least 5 and at most 8 guanosine entities and wherein said palindromic sequence is flanked at its 3 '-terminus by at least 6 and at most 10 guanosine entities.
- the experimental data show that the ease of packaging of the preferred inventive immunostimulatory substances, i.e. the guanosine flanked, palindromic and unmethylated CpG-containing oligonucleotides, wherein the palindromic sequence is GACGATCGTC (SEQ ID NO: 1), and wherein the palindromic sequence is flanked at its 3 '-terminus and at its 5'-terminus by less thanl 1 or less than 10 guanosine entities, into VLP's increases if the palindromic sequences are flanked by fewer guanosine entities.
- decreasing the number of guanosine entities flanking the palindromic sequences leads to a decrease of stimulating blood cells in vitro.
- packagability is paid by decreased biological activity of the indicated inventive immunostimulatory substances.
- the present preferred embodiments represent, thus, a compromise between packagability and biological activity.
- the immunostimulatory substance is an unmethylated CpG-containing oligonucleotide, wherein the CpG motif of said unmethylated CpG-containing oligonucleotide is part of a palindromic sequence, wherein said unmethylated CpG-containing oligonucleotide has a nucleic acid sequence selected from (a) GGGGGGACGATCGTCGGGGGG ((SEQ ID NO: 4); and typically abbreviated herein as G5-6), (b) GGGGGGGACGATCGTCGGGGGG ((SEQ ID NO: 5); and typically abbreviated herein as G6-6), (c) GGGGGGGGGGACGATCGTCGGGGGGG ((SEQ ID NO: 6); and typically abbreviated herein as G7-7), (d) GGGGGGGGGACGATCGTCGGGGGG ((SEQ ID NO: 7); and typically abbreviated herein as G8-8); and (e) GGGGGGGGGACGATCGTC
- the immunostimulatory substance is an unmethylated CpG-containing oligonucleotide, wherein the CpG motif of said unmethylated CpG-containing oligonucleotide is part of a palindromic sequence, wherein said unmethylated has the nucleic acid sequence of SEQ ID NO: 7, i.e.the immunostimulatory substance is G8-8, or of SEQ ID NO: 41, i.e. G10-PO.
- the immunostimulatory substance is an unmethylated CpG-containing oligonucleotide, wherein the CpG motif of said unmethylated CpG-containing oligonucleotide is part of a palindromic sequence, wherein said unmethylated has the nucleic acid sequence of SEQ ID NO: 41, i.e.the immunostimulatory substance is G10-PO.
- the present invention provides a composition for enhancing an immune response in an animal comprising (a) a virus-like particle; (b) at least one immunostimulatory substance; and (c) at least one antigen or antigenic determinant; wherein said antigen is bound to said viruslike particle and wherein said antigen comprises, alternatively consists essentially of, or alternatively consists of a HIV polypeptide, and wherein said immunostimulatory substance is bound to said virus-like particle, and wherein said immunostimulatory substance is an unmethylated CpG-containing oligonucleotide, wherein the CpG motif of said unmethylated CpG-containing oligonucleotide is part of a palindromic sequence, wherein said palindromic sequence is GACGATCGTC (SEQ ID NO: 1), and wherein said palindromic sequence is flanked at its 3 '-terminus and at its 5 '-terminus by 10 guanosine entities.
- the optimal sequence used to package into VLPs is a compromise between packagability and biological activity.
- the G8-8 immunostimulatoy substance is a preferred, and the G10-PO immunostimulatory substance a very preferred embodiment of the present invention since they are biologically highly active while still reasonably well packaged.
- the inventive composition further comprise an HIV peptide analogue of the invention bound to the virus-like particle.
- the at least one HIV polypeptide is fused to the virus-like particle.
- a VLP is typically composed of at least one subunit assembling into a VLP.
- the HIV polypeptide is fused to at least one subunit of the virus-like particle or of a protein capable of being incorporated into a VLP generating a chimeric VLP-subunit-antigen fusion.
- Fusion of the HIV polypeptide can be effected by insertion into the VLP subunit sequence, or by fusion to either the N- or C-terminus of the VLP-subunit or protein capable of being incorporated into a VLP.
- fusion proteins of a peptide to a VLP subunit the fusion to either ends of the subunit sequence or internal insertion of the peptide within the subunit sequence are encompassed.
- Fusion may also be effected by inserting HIV polypeptide sequences into a variant of a VLP subunit where part of the subunit sequence has been deleted, that are further referred to as truncation mutants.
- Truncation mutants may have N- or C-terminal, or internal deletions of part of the sequence of the VLP subunit.
- the specific VLP HBcAg with, for example, deletion of amino acid residues 79 to 81 is a truncation mutant with an internal deletion. Fusion of antigens or antigenic determinants to either the N- or C-terminus of the truncation mutants VLP-subunits also lead to embodiments of the invention.
- fusion of an epitope into the sequence of the VLP subunit may also be effected by substitution, where for example for the specific VLP HBcAg, amino acids 79-81 are replaced with a foreign epitope.
- fusion as referred to hereinafter, may be effected by insertion of the HIV polypeptide sequence in the sequence of a VLP subunit, by substitution of part of the sequence of the VLP subunit with the HIV polypeptide, or by a combination of deletion, substitution or insertions.
- the chimeric HIV polypeptide -VLP subunit will be in general capable of self- assembly into a VLP.
- VLP displaying epitopes fused to their subunits are also herein referred to as chimeric VLPs.
- the virus-like particle comprises or alternatively is composed of at least one VLP subunit.
- the virus-like particle comprises or alternatively is composed of a mixture of chimeric VLP subunits and non-chimeric VLP subunits, i.e. VLP subunits not having an antigen fused thereto, leading to so called mosaic particles. This may be advantageous to ensure formation of, and assembly to a VLP.
- the proportion of chimeric VLP-subunits may be 1, 2, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 95% or higher. Flanking amino acid residues may be added to either end of the sequence of the peptide or epitope to be fused to either end of the sequence of the subunit of a VLP, or for internal insertion of such peptidic sequence into the sequence of the subunit of a VLP.
- Glycine and serine residues are particularly favored amino acids to be used in the flanking sequences added to the peptide to be fused. Glycine residues confer additional flexibility, which may diminish the potentially destabilizing effect of fusing a foreign sequence into the sequence of a VLP subunit.
- the VLP is a Hepatitis B core antigen
- VLP VLP. Fusion proteins to either the N-terminus of a HBcAg (Neyrinck, S. et al., Nature Med. 5:1157-1163 (1999)) or insertions in the so called major immunodominant region (MIR) have been described (Pumpens, P. and Grens, E., Intervirology 44:98-114 (2001)), WO 01/98333), and are preferred embodiments of the invention. Naturally occurring variants of HBcAg with deletions in the MIR have also been described (Pumpens, P.
- HBcAg contains a long arginine tail (Pumpens, P. and Grens, E., Intervirology 44:98-114 (2001))which is dispensable for capsid assembly and capable of binding nucleic acids (Pumpens, P. and Grens, E., Intervirology 44:98-114 (2001)).
- HBcAg either comprising or lacking this arginine tail are both embodiments of the invention.
- the VLP is a VLP of a RNA phage.
- the major coat proteins of RNA phages spontaneously assemble into VLPs upon expression in bacteria, and in particular in E. coli.
- Specific examples of bacteriophage coat proteins which can be used to prepare compositions of the invention include the coat proteins of RNA bacteriophages such as bacteriophage Q ⁇ (SEQ ID NO:10; PIR Database, Accession No. VCBPQ ⁇ . referring to Q ⁇ CP and SEQ ID NO: 11; Accession No. AAA16663 referring to Q ⁇ Al protein) and bacteriophage fr (SEQ ID NO: 13; PIR Accession No. VCBPFR).
- the at least one HIV polypeptide is fused to a Q ⁇ coat protein.
- Fusion protein constructs wherein epitopes have been fused to the C- terminus of a truncated form of the Al protein of Q ⁇ , or inserted within the Al protein have been described (Kozlovska, T. M., et al., Intervirology, 39:9-15 (1996)).
- the Al protein is generated by suppression at the UGA stop codon and has a length of 329 aa, or 328 aa, if the cleavage of the N-terminal methionine is taken into account.
- mosaic particles As described by Kozlovska et al. (Intervirology, 39: 9-15 (1996)), assembly of the particles displaying the fused epitopes typically requires the presence of both the Al protein-HIV-polypeptide fusion and the wt CP to form a mosaic particle.
- embodiments comprising virus-like particles, and hereby in particular the VLPs of the RNA phage Q ⁇ coat protein, which are exclusively composed of VLP subunits having at least one HIV polypeptide fused thereto, are also within the scope of the present invention.
- the production of mosaic particles may be effected in a number of ways.
- the CP gene stop codon is modified into UAA, and a second plasmid expressing the Al protein-antigen fusion is cotransformed.
- the second plasmid encodes a different antibiotic resistance and the origin of replication is compatible with the first plasmid (Kozlovska, T. M., et al, Intervirology 39:9-15 (1996)).
- CP and the Al protein-antigen fusion are encoded in a bicistronic manner, operatively linked to a promoter such as the Trp promoter, as described in Figure 1 of Kozlovska et al., Intervirology, 39:9-15 (1996).
- the HIV polypeptide is inserted between amino acid 2 and 3 (numbering of the cleaved CP, that is wherein the N-terminal methionine is cleaved) of the fr CP, thus leading to an HIV polypeptide -fr CP fusion protein.
- Vectors and expression systems for construction and expression of fr CP fusion proteins self- assembling to VLP and useful in the practice of the invention have been described (Pushko P. et al, Prot. Eng. 6:883-891 (1993)).
- the HIV polypeptide sequence is inserted into a deletion variant of the fr CP after amino acid 2, wherein residues 3 and 4 of the fr CP have been deleted (Pushko P. et al., Prot. Eng. 6:883-891 (1993)). Fusion of epitopes in the N-terminal protuberant ⁇ -hairpin of the coat protein of
- RNA phage MS-2 and subsequent presentation of the fused epitope on the self-assembled VLP of RNA phage MS-2 has also been described (WO 92/13081), and fusion of an HIV polypeptide by insertion or substitution into the coat protein of MS-2 RNA phage is also falling under the scope of the invention.
- the HIV polypeptide is fused to a capsid protein of papillomavirus.
- the HIV polypeptide is fused to the major capsid protein LI of bovine papillomavirus type 1 (BPV-1).
- the HIV polypeptide is fused to a Ty protein capable of being incorporated into a Ty VLP.
- the HIV polypeptide is fused to the pi or capsid protein encoded by the TYA gene (Roth, J.F., Yeast 16:785-795 (2000)).
- the yeast retrotransposons Tyl, 2, 3 and 4 have been isolated from Saccharomyces Serevisiae, while the retrotransposon Tfl has been isolated from Schizosaccharomyces Pombae (Boeke, J.D. and Sandmeyer, S.B., "Yeast Transposable elements," in The molecular and Cellular Biology of the Yeast
- the retrotransposons Tyl and 2 are related to the copia class of plant and animal elements, while Ty3 belongs to the gypsy family of retrotransposons, which is related to plants and animal retroviruses.
- the pi protein also referred to as Gag or capsid protein, has a length of 440 amino acids.
- PI is cleaved during maturation of the VLP at position 408, leading to the p2 protein, the essential component of the VLP. Fusion proteins to pi and vectors for the expression of said fusion proteins in
- an HIV polypeptide may be fused to pi by inserting a sequence coding for the HIV polypeptide into the BamHl site of the pMA5620 plasmid.
- the cloning of sequences coding for foreign epitopes into the pMA5620 vector leads to expression of fusion proteins comprising amino acids 1-381 of pi of Tyl-15, fused C-terminally to the N-terminus of the foreign epitope.
- N-terminal fusion of an HIV polypeptide, or internal insertion into the pi sequence, or substitution of part of the pi sequence are also meant to fall within the scope of the invention.
- VLPs suitable for fusion of antigens or antigenic determinants are, for example, Retrovirus-like-particles (WO9630523), HIV2 Gag (Kang, Y.C., et al, Biol. Chem. 380:353-364 (1999)), Cowpea Mosaic Virus (Taylor, K.M.et al., Biol. Chem. 380:387-392 (1999)), parvovirus VP2 VLP (Rueda, P. et al., Virology 263:89-99 (1999)), HBsAg (US 4,722,840, EP0201416B1).
- VLPs suitable for the practice of the invention are also those described in Intervirology 39:1 (1996). Further examples of VLPs contemplated for use in the invention are: HPV-1, HPV-6, HPV-11, HPV-16, HPV-18, HPV-33, HPV-45, CRPV, COPV, HIV GAG, Tobacco Mosaic Virus. Virus-like particles of S V-40, Polyomavirus, Adenovirus, Herpes Simplex Virus, Rotavirus and Norwalk virus have also been made, and chimeric VLPs of those VLPs comprising an HIV polypeptide are also within the scope of the present invention.
- antigens fused to the virus-like particle by insertion within the sequence of the virus-like particle building monomer are also within the scope of the present invention.
- antigens can be inserted in a form of the virus-like particle building monomer containing deletions.
- the viruslike particle building monomer may not be able to form virus-like structures in the absence of the inserted antigen.
- recombinant DNA technology can be utilized to fuse a heterologous protein to a VLP protein (Kratz, P.A., et al., Proc. Natl. Acad. Sci. USA 96:1915 (1999)).
- the present invention encompasses VLPs recombinantly fused or chemically conjugated (including both covalently and non covalently conjugations) to an antigen (or portion thereof, preferably at least 10, 20 or 50 amino acids) of the present invention to generate fusion proteins or conjugates.
- the fusion does not necessarily need to be direct, but can occur through linker sequences.
- linker sequences are typically added at one or both ends of the epitopes.
- linker sequences preferably comprise sequences recognized by the proteasome, proteases of the endosomes or other vesicular compartment of the cell.
- a peptide bond in which the conjugate can be a contiguous polypeptide, i.e. a fusion protein.
- a fusion protein according to the present invention, different peptides or polypeptides are linked in frame to each other to form a contiguous polypeptide.
- a first portion of the fusion protein comprises an antigen or immunogen and a second portion of the fusion protein, either N-terminal or C-terminal to the first portion, comprises a VLP.
- internal insertion into the VLP with optional linking sequences on both ends of the antigen, can also be used in accordance with the present invention.
- HBcAg When HBcAg is used as the VLP, it is preferred that the antigen is linked to the C- terminal end of the HBcAg particle.
- the hepatitis B core antigen (HBcAg) exhibiting a C- terminal fusion of the MHC class I restricted peptide p33 derived from lymphocytic choriomeningitis virus (LCMV) glycoprotein can be and was typically used as a model antigen (HBcAg-p33).
- the 185 amino acids long wild type HBc protein assembles into highly structured particles composed of 180 subunits assuming icosahedral geometry.
- a flexible linker sequence e.g. a polyglycine/polyserine-containing sequence such as [Gly4 Ser]2 (Huston et al., Meth. Enzymol 203:46-88 (1991)) can be inserted into the fusion protein between the antigen and ligand.
- the fusion protein can be constructed to contain an "epitope tag", which allows the fusion protein to bind an antibody (e.g. monoclonal antibody) for example for labeling or purification purposes.
- An example of an epitope tag is a Glu-Glu-Phe tripeptide which is recognized by the monoclonal antibody YL 1/2.
- the invention also relates to the chimeric DNA which contains a sequence coding for the VLP and a sequence coding for the HIV polypeptide.
- the DNA can be expressed, for example, in insect cells transformed with Baculoviruses, in yeast or in bacteria. There are no restrictions regarding the expression system, of which a large selection is available for routine use. Preferably, a system is used which allows expression of the proteins in large amounts. In general, bacterial expression systems are preferred on account of their efficiency.
- a bacterial expression system suitable for use within the scope of the present invention is the one described by Clarke et al., J. Gen. Virol. 71 : 1109-1117 (1990); Borisova et al., J. Virol.
- yeast expression system An example of a suitable yeast expression system is the one described by Emr, Methods Enzymol. 185:231-3 (1990); Baculovirus systems, which have previously been used for preparing capsid proteins, are also suitable. Constitutive or inducible expression systems can be used. By the choice and possible modification of available expression systems it is possible to control the form in which the proteins are obtained.
- the antigen to which an enhanced immune response is desired is coupled, fused or otherwise attached in frame to the Hepatitis B virus capsid (core) protein (HBcAg).
- core Hepatitis B virus capsid
- the at least one HIV polypeptide is bound to the virus-like particle by at least one covalent bond.
- the least one HIV polypeptide is bound to the virus-like particle by at least one covalent bond, said covalent bond being a non-peptide bond leading to an HIV polypeptide array and HIV polypeptide -VLP conjugate, respectively.
- This HIV polypeptide array and conjugate, respectively has typically and preferably a repetitive and ordered structure since the at least one HIV polypeptide is bound to the VLP in an oriented manner.
- HIV-peptides of the invention are bound to the VLP.
- the formation of a repetitive and ordered HIV polypeptide -VLP array and conjugate, respectively, is ensured by an oriented and directed as well as defined binding and attachment, respectively, of the at least one HIV polypeptide to the VLP as will become apparent in the following.
- the typical inherent highly repetitive and organized structure of the VLPs advantageously contributes to the display of the HIV polypeptide in a highly ordered and repetitive fashion leading to a highly organized and repetitive HIV polypeptide -VLP array and conjugate, respectively.
- the preferred inventive conjugates and arrays differ from prior art conjugates in their highly organized structure, dimensions, and in the repetitiveness of the antigen on the surface of the array.
- the preferred embodiment of this invention furthermore, allows expression of the particle in an expression host guaranteeing proper folding and assembly of the VLP, to which the HIV polypeptide is then further coupled.
- the present invention discloses methods of binding or associationof HIV polypeptide to VLPs.
- the at least one HIV polypeptide is bound to the VLP by way of chemical cross-linking, typically and preferably by using a heterobifunctional cross-linker.
- Several hetero-bifunctional cross- linkers are known to the art.
- the hetero-bifunctional cross- linker contains a functional group which can react with preferred first attachment sites, i.e. with the side-chain amino group of lysine residues of the VLP or at least one VLP subunit, and a further functional group which can react with a preferred second attachment site, i.e. a cysteine residue fused to the HIV polypeptide and optionally also made available for reaction by reduction.
- the first step of the procedure typically called the derivatization, is the reaction of the VLP with the cross-linker.
- the product of this reaction is an activated VLP, also called activated carrier.
- unreacted cross-linker is removed using usual methods such as gel filtration or dialysis.
- the HIV polypeptide is reacted with the activated VLP, and this step is typically called the coupling step.
- Unreacted HIV polypeptide may be optionally removed in a fourth step, for example by dialysis.
- Several hetero-bifunctional cross-linkers are known to the art. These include the preferred cross-linkers SMPH (Pierce), Sulfo-MBS, Sulfo- EMCS, Sulfo-GMBS, Sulfo-SIAB, Sulfo-SMPB, Sulfo-SMCC, SVSB, SIA and other cross-linkers available for example from the Pierce Chemical Company (Rockford, IL, USA) , and having one functional group reactive towards amino groups and one functional group reactive towards cysteine residues.
- SMPH Fastce
- Sulfo-MBS Sulfo-EMCS
- Sulfo-GMBS Sulfo-SIAB
- Sulfo-SMPB Sulfo-SMCC
- SVSB SIA and other
- cross-linkers all lead to formation of a thioether linkage.
- Another class of cross-linkers suitable in the practice of the invention is characterized by the introduction of a disulfide linkage between the HIV polypeptide and the VLP upon coupling.
- Preferred cross-linkers belonging to this class include for example SPDP and Sulfo-LC-SPDP (Pierce).
- the extent of derivatization of the VLP with cross-linker can be influenced by varying experimental conditions such as the concentration of each of the reaction partners, the excess of one reagent over the other, the pH, the temperature and the ionic strength.
- the degree of coupling, i.e. the amount of antigens or antigenic determinants per subunits of the VLP can be adjusted by varying the experimental conditions described above to match the requirements of the vaccine.
- a particularly favored method of binding of antigens or antigenic determinants to the VLP is the linking of a lysine residue on the surface of the VLP with a cysteine residue on the HIV polypeptide.
- fusion, coupling, attachment or binding of an amino acid linker containing a cysteine residue, as a second attachment site or as a part thereof, to the HIV polypeptide for coupling to the VLP may be required.
- Such constructs comprising said amino acid linker may also be obtained by simple peptide syntheses known in the art. Therefore, in a further preferred embodiment of the present invention, the antigen or antigenic determinant further comprises an amino acid linker, wherein preferably said amino acid linker comprises, or alternatively consists of, a second attachment site.
- amino acid linkers are favored.
- amino acid linkers are the hinge region of Immunoglobulins, glycine serine linkers (GGGGS)n (SEQ ID NO: 53), and glycine linkers (G)n all further containing a cysteine residue as second attachment site and optionally further glycine residues.
- amino acid linkers are N-terminal gammal : CGDKTHTSPP (SEQ ID NO: 54); C-terminal gamma 1: DKTHTSPPCG (SEQ ID NO: 55); N-terminal gamma 3: CGGPKPSTPPGSSGGAP (SEQ ID NO: 56); C-terminal gamma 3 : PKPSTPPGSSGGAPGGCG (SEQ ID NO: 57); N-terminal glycine linker: GCGGGG (SEQ ID NO: 58); C-terminal glycine linker: GGGGCG (SEQ ID NO: 58); C- terminal glycine-lysine linker: GGKKGC (SEQ ID NO: 60); N-terminal glycine-lysine linker: CGKKGG (SEQ ID NO: 61).
- amino acid linkers particularly suitable in the practice of the invention are CGKKGG (SEQ ID NO: 62), or CGDEGG (SEQ ID NO: 63) for N-terminal linkers, or GGKKGC (SEQ ID NO: 64) and GGEDGC (SEQ ID NO: 65), for the C-terminal linkers.
- CGKKGG SEQ ID NO: 62
- CGDEGG SEQ ID NO: 63
- GGKKGC SEQ ID NO: 64
- GGEDGC SEQ ID NO: 65
- the terminal cysteine is optionally C-terminally amidated.
- linkers useful for this invention are amino acid sequences that allow the release of the antigenic peptide, i.e. the HIV polypeptide, from the VLP. Examples for these linkers are described in Toes RE et al. J Exp Med. 2001 Jul 2;194(1):1-12. Moreover, the PAProC- a prediction algorithm for proteasomal cleavages might be used (Nussbaum AK, et. al. Immunogenetics. 2001 Mar;53(2):87-94) for prediction of aforementioned amino acid sequences that allow the release of the antigenic peptide, i.e. the HIV polypeptide, from the VLP.
- GGCG SEQ ID NO: 66
- GGC GGC or GGC-NH2
- NH2 stands for amidation
- linkers at the C-terminus of the peptide or CGG at its N-terminus are preferred as amino acid linkers.
- glycine residues will be inserted between bulky amino acids and the cysteine to be used as second attachment site, to avoid potential steric hindrance of the bulkier amino acid in the coupling reaction.
- the amino acid linker GGC-NH2 is fused to the C-terminus of the HIV polypeptide.
- the cysteine residue present on the HIV polypeptide has to be in its reduced state to react with the hetero-bifunctional cross-linker on the activated VLP, that is a free cysteine or a cysteine residue with a free sulfhydryl group has to be available.
- the cysteine residue to function as binding site is in an oxidized form, for example if it is forming a disulfide bridge, reduction of this disulfide bridge with e.g. DTT, TCEP or ⁇ -mercaptoethanol is required.
- Other methods of binding the HIV polypeptide to the VLP include methods wherein the HIV polypeptide is cross-linked to the VLP using the carbodiimide EDC, and NHS.
- the HIV polypeptide is attached to the VLP using a homo-bifunctional cross-linker such as glutaraldehyde,
- DSG DSG, BM[PEO]4, BS3, (Pierce Chemical Company, Rockford, IL, USA) or other known homo-bifunctional cross-linkers whith functional groups reactive towards amine groups or carboxyl groups of the VLP.
- VLP binding methods include methods where the VLP is biotinylated, and the HIV polypeptide expressed as a streptavidin-fusion protein, or methods wherein both the HIV polypeptide and the VLP are biotinylated, for example as described in WO 00/23955.
- the HIV polypeptide may be first bound to streptavidin or avidin by adjusting the ratio of HIV polypeptide to streptavidin such that free binding sites are still available for binding of the VLP, which is added in the next step.
- all components may be mixed in a "one pot" reaction.
- ligand-receptor pairs where a soluble form of the receptor and of the ligand is available, and are capable of being cross-linked to the VLP or the HIV polypeptide, may be used as binding agents for binding HIV polypeptide to the VLP.
- either the ligand or the receptor may be fused to the HIV polypeptide, and so mediate binding to the VLP chemically bound or fused either to the receptor, or the ligand respectively. Fusion may also be effected by insertion or substitution.
- the VLP is the VLP of a RNA phage, and in a more preferred embodiment, the VLP is the VLP of RNA phage Q ⁇ coat protein.
- One or several antigen molecules i.e. one or several antigens or antigenic determinants, can be attached to one subunit of the capsid or VLP of RNA phages coat proteins, preferably through the exposed lysine residues of the VLP of RNA phages, if sterically allowable.
- a specific feature of the VLP of the coat protein of RNA phages and in particular of the Q ⁇ coat protein VLP is thus the possibility to couple several antigens per subunit. This allows for the generation of a dense antigen array.
- the binding and attachment, respectively, of the at least one HIV polypeptide to the virus-like particle is by way of interaction and association, respectively, between at least one first attachment site of the virus-like particle and at least one second attachment of the HIV polypeptide.
- VLPs or capsids of Q ⁇ coat protein display a defined number of lysine residues on their surface, with a defined topology with three lysine residues pointing towards the interior of the capsid and interacting with the RNA, and four other lysine residues exposed to the exterior of the capsid. These defined properties favor the attachment of antigens to the exterior of the particle, rather than to the interior of the particle where the lysine residues interact with RNA.
- VLPs of other RNA phage coat proteins also have a defined number of lysine residues on their surface and a defined topology of these lysine residues.
- the first attachment site is a lysine residue and/or the second attachment comprises sulfhydryl group or a cysteine residue.
- the first attachment site is a lysine residue and the second attachment is a cysteine residue.
- the HIV polypeptide is bound via a cysteine residue, to lysine residues of the VLP of RNA phage coat protein, and in particular to the VLP of Q ⁇ coat protein.
- VLPs derived from RNA phages are their high expression yield in bacteria that allows production of large quantities of material at affordable cost.
- inventive conjugates and arrays differ from prior art conjugates in their highly organized structure, dimensions, and in the repetitiveness of the antigen on the surface of the array.
- use of the VLPs as carriers allow the formation of robust antigen arrays and conjugates, respectively, with variable antigen density.
- the use of VLPs of RNA phages, and hereby in particular the use of the VLP of RNA phage Q ⁇ coat protein allows to achieve very high epitope density.
- a density of more than 1.5 epitopes per subunit has been reached by coupling a peptide to the VLP of Q ⁇ coat protein (e.g. the human A ⁇ 1-6 peptide as described in WO 2004/016282).
- Q ⁇ coat protein e.g. the human A ⁇ 1-6 peptide as described in WO 2004/016282.
- the preparation of compositions of VLPs of RNA phage coat proteins with a high epitope density can be effected using the teaching of this application.
- an average number of HIV polypeptide per subunit of 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6 , 1.7, 1.8, 1.9, 2.0, 2.1, 2.2, 2.3, 2.4 2.5, 2.6, 2.7, 2.8, 2.9, or higher is preferred.
- the second attachment site as defined herein, may be either naturally or non- naturally present with the HIV polypeptide. In the case of the absence of a suitable natural occurring second attachment site on the HIV polypeptide, such a, then non-natural second attachment has to be engineered to the antigen.
- lysine residues are exposed on the surface of the VLP of Q ⁇ coat protein. Typically these residues are derivatized upon reaction with a cross-linker molecule. In the instance where not all of the exposed lysine residues can be coupled to an antigen, the lysine residues which have reacted with the cross-linker are left with a cross- linker molecule attached to the ⁇ -amino group after the derivatization step. This leads to disappearance of one or several positive charges, which may be detrimental to the solubility and stability of the VLP.
- arginines By replacing some of the lysine residues with arginines, as in the disclosed Q ⁇ coat protein mutants described below, we prevent the excessive disappearance of positive charges since the arginine residues do not react with the cross-linker. Moreover, replacement of lysine residues by arginines may lead to more defined antigen arrays, as fewer sites are available for reaction to the antigen. Accordingly, exposed lysine residues were replaced by arginines in the following
- Q ⁇ coat protein mutants and mutant Q ⁇ VLPs disclosed in this application Q ⁇ -240 (Lysl3-Arg; SEQ ID NO:20), Q ⁇ -250 (Lys 2-Arg, Lysl3-Arg; SEQ ID NO: 22) and Q ⁇ - 259 (Lys 2-Arg, Lysl6-Arg; SEQ ID NO:24).
- the constructs were cloned, the proteins expressed, the VLPs purified and used for coupling to HIV polypeptides.
- Q ⁇ -251 (SEQ ID NO: 23) was also constructed, and guidance on how to express, purify and couple the VLP of Q ⁇ -251 coat protein can be found throughout the application.
- a Q ⁇ mutant coat protein with one additional lysine residue suitable for obtaining even higher density arrays of antigens.
- This mutant Q ⁇ coat protein, Q ⁇ -243 (Asn 10-Lys; SEQ ID NO: 21) was cloned, the protein expressed, and the capsid or VLP isolated and purified, showing that introduction of the additional lysine residue is compatible with self-assembly of the subunits to a capsid or VLP.
- HIV polypeptide arrays and conjugates, respectively may be prepared using VLP of Q ⁇ coat protein mutants.
- a particularly favored method of attachment of antigens to VLPs, and in particular to VLPs of RNA phage coat proteins is the linking of a lysine residue present on the surface of the VLP of RNA phage coat proteins with a cysteine residue added to the antigen.
- a cysteine residue In order for a cysteine residue to be effective as second attachment site, a sulfhydryl group must be available for coupling. Thus, a cysteine residue has to be in its reduced state, that is, a free cysteine or a cysteine residue with a free sulfhydryl group has to be available.
- cysteine residue to function as second attachment site is in an oxidized form, for example if it is forming a disulfide bridge
- reduction of this disulfide bridge with e.g. DTT, TCEP or ⁇ - mercaptoethanol is required.
- concentration of reductand, and the molar excess of reductand over antigen has to be adjusted for each antigen.
- a titration range, starting from concentrations as low as 10 ⁇ M or lower, up to 10 to 20 mM or higher reductand if required is tested, and coupling of the antigen to the carrier assessed.
- the pH of the dialysis or equilibration buffer is lower than 7, preferably 6.
- the compatibility of the low pH buffer with antigen activity or stability has to be tested.
- Epitope density on the VLP of RNA phage coat proteins can be modulated by the choice of cross-linker and other reaction conditions.
- the cross-linkers Sulfo- GMBS and SMPH typically allow reaching high epitope density.
- Derivatization is positively influenced by high concentration of reactands, and manipulation of the reaction conditions can be used to control the number of antigens coupled to VLPs of RNA phage coat proteins, and in particular to VLPs of Q ⁇ coat protein.
- the position at which it should be fused, inserted or generally engineered has to be chosen.
- the selection of the position of the second attachment site may, by way of example, be based on a crystal structure of the antigen.
- Such a crystal structure of the antigen may provide information on the availability of the C- or N-termini of the molecule (determined for example from their accessibility to solvent), or on the exposure to solvent of residues suitable for use as second attachment sites, such as cysteine residues.
- Exposed disulfide bridges as is the case for Fab fragments, may also be a source of a second attachment site, since they can be generally converted to single cysteine residues through mild reduction, with e.g.
- the second attachment site will be added such that it allows generation of antibodies against the site of interaction with the natural ligands.
- the location of the second attachment site will be selected such that steric hindrance from the second attachment site or any amino acid linker containing the same is avoided.
- an antibody response directed at a site distinct from the interaction site of the self-antigen with its natural ligand is desired.
- the second attachment site may be selected such that it prevents generation of antibodies against the interaction site of the self-antigen with its natural ligands.
- Other criteria in selecting the position of the second attachment site include the oligomerization state of the antigen, the site of oligomerization, the presence of a cofactor, and the availability of experimental evidence disclosing sites in the antigen structure and sequence where modification of the antigen is compatible with the function of the self- antigen, or with the generation of antibodies recognizing the self-antigen.
- the HIV polypeptide comprises a single second attachment site or a single reactive attachment site capable of association with the first attachment sites on the core particle and the VLPs or VLP subunits, respectively.
- This further ensures a defined and uniform binding and association, respectively, of the at least one, but typically more than one, preferably more than 10, 20, 40, 80, 120, 150, 180, 210, 240, 270, 300, 360, 400, 450 HIV polypeptides to the core particle and VLP, respectively.
- the provision of a single second attachment site or a single reactive attachment site on the antigen thus, ensures a single and uniform type of binding and association, respectively leading to a very highly ordered and repetitive array.
- binding and association is effected by way of a lysine- (as the first attachment site) and cysteine- (as a second attachment site) interaction
- engineering of a second attachment site onto the HIV polypeptide require the fusion of an amino acid linker containing an amino acid suitable as second attachment site according to the disclosures of this invention.
- an amino acid linker is bound to the HIV polypeptide by way of at least one covalent bond.
- the amino acid linker comprises, or alternatively consists of, the second attachment site.
- the amino acid linker comprises a sulfhydryl group or a cysteine residue.
- the amino acid linker is cysteine.
- the attachment site is selected to be a lysine or cysteine residue that is fused in frame to the HBcAg.
- the antigen is fused to the C-terminus of HBcAg via a three leucine linker.
- an HIV polypeptide is linked to the VLP through a lysine residue, it may be advantageous to either substitute or delete one or more of the naturally resident lysine residues, as well as other lysine residues present in HBcAg variants.
- lysine residues when the naturally resident lysine residues are eliminated, another lysine will be introduced into the HBcAg as an attachment site for an HIV polypeptide. Methods for inserting such a lysine residue are known in the art. Lysine residues may also be added without removing existing lysine residues.
- the C terminus of the HBcAg has been shown to direct nuclear localization of this protein. (Eckhardt et al, J. Virol. 65:575 582 (1991)). Further, this region of the protein is also believed to confer upon the HBcAg the ability to bind nucleic acids.
- HBcAgs suitable for use in the practice of the present invention also include N terminal truncation mutants.
- Suitable truncation mutants include modified HBcAgs where 1, 2, 5, 7, 9, 10, 12, 14, 15, or 17 amino acids have been removed from the N terminus.
- variants of virus-like particles containing internal deletions within the sequence of the subunit composing the virus-like particle are also suitable in accordance with the present invention, provided their compatibility with the ordered or particulate structure of the virus-like particle.
- internal deletions within the sequence of the HBcAg are suitable (Preikschat, P., et al., J. Gen. Virol. 80:1777-1788 (1999)).
- HBcAgs suitable for use in the practice of the present invention include N- and C terminal truncation mutants.
- Suitable truncation mutants include HBcAgs where 1, 2, 5, 7, 9, 10, 12, 14, 15, and 17 amino acids have been removed from the N terminus and 1, 5, 10, 15, 20, 25, 30, 34, 35, 36, 37, 38, 3940, 41, 42 or 48 amino acids have been removed from the C terminus.
- Vaccine compositions of the invention can comprise mixtures of different HBcAgs.
- these vaccine compositions can be composed of HBcAgs which differ in amino acid sequence.
- vaccine compositions could be prepared comprising a "wild type" HBcAg and a modified HBcAg in which one or more amino acid residues have been altered (e.g., deleted, inserted or substituted). In most applications, however, only one type of a HBcAg will be used.
- the virus-like particle comprises at least one first attachment site and the antigen or antigenic determinant comprises at least one second attachment site.
- the first attachment site comprises, or preferably consists of, an amino group or a lysine residue.
- the second attachment site is preferably selected from the group consisting of (a) an attachment site not naturally occurring with said antigen or antigenic determinant; and (b) an attachment site naturally occurring with said antigen or antigenic determinant.
- the second attachment site comprises, or preferably consists of, a sulfhydryl group or a cysteine residue.
- the binding of the antigen or antigenic determinant to the virus-like particle is effected through association between the first attachment site and the second attachment site, wherein preferably the association is through at least one non-peptide bond, and wherein preferably the antigen or antigenic determinant and the virus-like particle interact through said association to form an ordered and repetitive antigen array.
- the first attachment site is a lysine residue and the second attachment site is a cysteine residue.
- the first attachment site is an amino group and the second attachment site is a sulfhydryl group.
- the antigen, and herein in particular, the polypeptide, polyprotein, peptide, epitope or polyepitope of HIV comprises one or more cytotoxic T cell epitopes, Th cell epitopes, or a combination of the two epitopes.
- the antigen or antigenic determinant comprises one, two, or more cytotoxic T cell epitopes.
- the antigen or antigenic determinant comprises one, two, or more Th cell epitopes.
- the antigen or antigenic determinant comprises one, two or more cytotoxic T cell epitopes and one, two or more Th cell epitopes.
- the antigen or antigenic determinant is a polypeptide, a polyprotein, a peptide, an epitope or a polyepitope of HIV.
- Said polypeptide, polyprotein, peptide, epitope or polyepitope of HIV is fused, coupled, bound or otherwise attached to the VLP or packaged VLP as set out throughout the present application, and leading to preferred embodiments of the invention.
- a further aspect of the present invention and a preferred embodiment of the present invention is to provide a composition for enhancing an immune response in an animal comprising: (a) a virus-like particle; (b) an immunostimulatory substance; and (c) at least one antigen or antigenic determinant; wherein said immunostimulatory substance is bound to said virus-like particle, and wherein said antigen comprises, alternatively consists essentially of, or alternatively consists of at least one HIV polypeptide, and wherein said at least one antigen or antigenic determinant is bound to said virus-like particle.
- the antigen comprises, or alternatively consists essentially of, or alternatively consists of a polyepitope, wherein the polyepitope is a combination of at least two HIV polypeptides, wherein said at least two HIV polypeptides are bound directly or by way of a linking sequence.
- VLPs bound, coupled, or otherwise fused to HIV antigens are particularly suited as a safe, non-infectious and non-replicative vaccine to induce T-cells and in particular CTLs against HIV. VLPs are particularly effective when they are packaged with immunostimulatory substances and sequences, respectively.
- VLPs target preferentially dendritic cells and macrophages (Ruedl, C. et al., Eur. J. Immunol. 32: 818-825 (2002)), ensuring antigen delivery to the most relevant antigen presenting cells.
- VLP based vaccines have therefore a much higher specificity than viral-vector or DNA based vaccines.
- Suitable HIV antigens and poylpetides, respectively, for preparation of the compositions of the invention include the following HIV protein subunits: pl7-GAG, p24-GAG, pl5-GAG, Protease, reverse transcriptase (RT), Integrase, Vif, Vpr, Vpu, Tat, Rev, gp-41-Env, g ⁇ -120-Env and Nef (Addo, M.M. et al, J. Virol. 77: 2081-2092 (2003)). Both the whole protein subunits and fragments thereof are suitable in preparing the compositions of the invention. In particular, chemically synthesized peptides having the sequence of fragments of these subunits are also included.
- Polyepitopes which may be obtained as recombinant polypeptides or as chemically synthesized long peptides, are used in a favored embodiment of the invention for binding, coupling or otherwise attachment to the VLP and preferably packaged VLP.
- the DNA sequence encoding a polyepitope may also be fused in frame to the sequence of a VLP subunit, leading to VLPs or packaged VLPs fused to the polyepitope.
- the HIV antigen is coupled to the VLP using a cross-linker containing a maleimide moiety
- the HIV antigen, a peptide or recombinant polypeptide is modified according to the disclosures of the invention to include a cysteine residue for reaction with the maleimide moiety introduced in the VLP after the derivatization step of the cross-linking procedure.
- a prominent feature of HIV infection is the ability of the virus to escape from immune control, through accumulation of mutations which are selected for by the strong CTL response elicited in the host (McMichael, A.J. & Rowland- Jones, S.L. Nature 410: 980-987 (2001)). It is therefore advantageous to immunize and induce T-cells against a diversity of epitopes, in order to limit the effect of mutations in single epitopes.
- a composition of the invention suitable for eliciting a T-cell response against a plurality of epitope will for example be prepared by coupling at least two, or alternatively a plurality of epitopes, in the form of chemically synthesized peptides modified accordingly for cross-linking, to a VLP or packaged VLP.
- VLPs or packaged VLPs each coupled to at least two, or alternatively several different HIV polypeptides and therefore epitopes are obtained.
- a preferred polyepitope of HIV for the present invention is coupled, bound, fused or otherwise attached to a VLP or packaged VLP.
- at least two, or alternatively several different polyepitopes may also be coupled, fused or otherwise attached to one VLP or packaged VLP.
- at least two, or alternatively several different HIV antigens, in the form of recombinant polypeptides are coupled or bound to one VLP or packaged VLP.
- a polyprotein, that is a fusion protein comprising two or more HIV polypeptides, modified according to the disclosures of the present invention for coupling, binding or fusion to a VLP is used as antigen or antigenic determinant.
- combination of peptides, polyepitopes and recombinant polypeptides are coupled, bound or otherwise attached to one VLP or packaged VLP.
- the HIV antigens are fused to one VLP or packaged VLP.
- the antigens or antigenic determinant of the composition of the present invention comprise, alternatively consist essentially of, or alternatively consist of a combination of at least two HIV polypeptides, wherein the at least two HIV polypeptides are selected from the at least one HIV polypeptide, and wherein the at least two HIV polypeptides are the same or different, and wherein the HIV polypeptides are bound directly or by way of a linking sequence to each other.
- Immunisation of an animal or subject with a plurality of HIV antigens is also achieved in one further embodiment of the invention by mixing different particles, each coupled, bound, fused or otherwise attached to one, two or more HIV antigens, said HIV antigens being a peptide, an epitope a recombinant polypeptide or a polyepitope.
- sequences of epitopes to be coupled, fused, bound or otherwise attached to a VLP or packaged VLP as peptide, polyepitope or included in a recombinant polypeptide or polyprotein are therefore preferably consensus sequences, obtained from the database (see above reference, or website: http://hiv-web.lanl.gov/seq-db.html) or obtained by aligning all sequences of a given antigen from the database.
- sequences from one clade of virus are selected, in function of the most prevalent clade in the geographical region where the compositions of the invention or vaccines are intended to be injected.
- Aligning sequences of the database would be known to one skilled in the art.
- the program Blast Altschul, S.F et al., J. Mol. Biol. 215:403-410 (1990); Altschul, S.F. et al, Nature Genet. 6:119-129 (1994)
- FASTA Pearson, W.R. Methods Enzymol. 183:63- 98 (1990)
- the HIV antigens p24-GAG and Nef have been found to have the highest epitope density (Addo, M.M. et al, J. Virol. 77: 2081-2092 (2003)).
- the HIV polypeptide comprises therefore p24-GAG-CTL and/or NEF-CTL and/or Th cell epitopes.
- Th cell epitopes are believed to contribute to the induction and maintenance of CTL responses, and therefore, in preferred embodiments of the invention, Th cell epitopes are included in the composition of the invention.
- Th cell epitopes may be included in a polyepitope or polyprotein.
- peptides comprising Th cell epitopes may be coupled to VLPs or packaged VLPs, or the composition of the invention may be a mixture of particles, each coupled to an individual peptide, and one or more of said peptides may comprise one or more Th cell epitopes.
- the HIV polypeptide with the second attachment site is selected from the group of the GAG polyepitopes gag-G50 (SEQ ID NO: 71), gag-G68n (SEQ ID NO: 73) and of the Nef polyepitope nef-N56 (SEQ ID NO: 72).
- Gag-50, gag-68n and nef-N56 comprise polyepitopes derived from the Clade B consensus sequences of gag and nef (The Identification of Optimal HIV-Derived CTL Epitopes in Diverse Populations Using HIV Clade-Specific Consensus, pp. 1-1-20 in HIV Molecular Immunology 2001. Edited by: Korber BTK, Brander C, Haynes BF, Koup R, Kuiken C, Moore JP, Walker BD, and Watkins D.
- the nef-N56 polyepitope starting with the aminoacid number 66 of the Nef- protein consensus sequence (SEQ ID NO: 75), comprises amino acids 66-99 (VGFPVRPQVPLRPMTYKAAVDLSHFLKEKGGLEG, (SEQ ID NO: 77), followed by amino acids 131-150 (PGIRYPLTFGWCFKLVPVEP, (SEQ ID NO: 78) of the HIV-1 clade B Nef-protein consensus sequence (SEQ ID NO: 75).
- the resulting polypeptide i.e. the combination of SEQ ID NO: 77 and SEQ ID NO: 78, has the amino acid sequence of SEQ ID NO: 83.
- the nef-N56 polyepitope additionally comprises an N-terminal Cysteine and Glycine for coupling (SEQ ID NO: 72).
- the gag-G50 polyepitope starts at the N-terminus of p24-GAG, from position 139 of the HIV-1 clade B GAG-protein consensus sequence (SEQ ID NO: 76).
- the sequence "KVVEE” ((SEQ ID NO: 79) which represents the amino acids 157-161 from the GAG consensus sequence (SEQ ID NO: 76)), and where the density of epitopes is lowest, is deleted.
- gag-G50 comprises amino acids 139-156 (QGQMVHQAISPRTLNAWV, (SEQ ID NO: 80)), followed by amino acids 162-191
- the gag-G50 polyepitope comprises an N-terminal Cysteine for coupling (SEQ ID NO: 85).
- the gag-G50 polyepitope additionally comprises a C-terminal lysine residue (SEQ ID NO: 71).
- gag-G68n epitope (SEQ ID NO: 73) is based on G50 epitope, with the addition of the more C-terminal "GEIYKRWIILGLNKIVRMY" sequence, corresponding to aminoacids 259-277 (SEQ ID NO: 82) from GAG-protein consensus sequence (SEQ ID NO: 76) to the N-terminus of the sequence of gag-G50 (excluding the N-terminal cysteine). Therefore, the resulting HIV polypeptide, i.e. the combination of SEQ ID NO: 82, SEQ ID NO: 80 and SEQ ID NO: 81, has the amino acid sequence of SEQ ID NO: 86.
- the gag-G68n epitope comprises an N-terminal Cysteine for coupling (SEQ ID NO: 87).
- the gag-G68n epitope additionally comprises a C-terminal lysine residue (SEQ ID NO: 73).
- the polyepitopes of the invention comprise a cysteine residue at the N-terminus for coupling, rather than a C-terminal cysteine, since there are more protecting strategies for N-terminal cysteines, and peptides may be further trimmed at their N-terminus for proper presentation by aminopeptidases (Goldberg A.L. et al., Mol. Immunol. 39: 147-164 (2002)). Introduction of the cysteine residue for coupling to the C-terminus rather than the N-terminus however also leads to an embodiment of this invention.
- the polyepitopes gag-G50 (SEQ ID NO: 71), nef-N56 (SEQ ID NO: 72) or gag-G68n (SEQ ID NO: 73) are coupled to the RNA phage VLPs or packaged VLPs Q ⁇ , AP205, GA, MS-2 and fr, or to HBcAg VLPs or packaged VLPs modified to harbour an additional lysine residue in their immunodominant region, i.e. HBcAgl-1851ys described in WO 02/56905 which is incorporated hereby in its entirety by way of reference.
- the two polyepitopes gag-G50 and nef-N56 are coupled both on a single VLP.
- the VLP is the VLP of RNA phages
- gag-G50 and gag-G68n, and the nef- N56 epitopes are fused to the N-terminus of the VLP of phage fr, or to the C-terminus of phage Q ⁇ .
- compositions of the invention comprising a polypeptide, a polyprotein, a peptide, an epitope or a polyepitope of HIV and optionally a further adjuvant, are useful as vaccines for induction of HIV specific T-cells in humans.
- the vaccine comprises a Q ⁇ or AP205 VLP packaged with the G8-8 or
- T-cell response induced upon vaccination is assessed in proliferation assays (for Th cell response, Belshe
- gag-G50, gag-G68n and nef-N56 devoid of the N- terminal cysteine are inserted between amino acid 2 and 3 (numbering of the cleaved CP, that is wherein the N-terminal methionine is cleaved) of the fr CP.
- gag-G50, gag-G68n and nef-N56 devoid of the N-terminal cysteine are fused to the Al protein of Q ⁇ VLP, as described above.
- the antigen being coupled, fused or otherwise attached to the virus-like particle, is a T cell epitope, either a cytotoxic or a
- the antigen is a combination of at least two, preferably different, epitopes, wherein the at least two epitopes are linked directly or by way of a linking sequence.
- These epitopes are preferably selected from the group consisting of cytotoxic and Th cell epitopes.
- a mosaic virus-like particle e.g. a virus-like particle composed of subunits attached to different antigens and epitopes, respectively, is within the scope of the present invention.
- Such a composition of the present invention can be, for example, obtained by transforming E.coli with two compatible plasmids encoding the subunits composing the virus-like particle fused to different antigens and epitopes, respectively.
- the mosaic virus-like particle is assembled either directly in the cell or after cell lysis.
- such an inventive composition can also be obtained by attaching a mixture of different antigens and epitopes, respectively, to the isolated virus-like particle.
- the HIV polypeptide of the present invention, and in particular the indicated epitope or epitopes can be synthesized or recombinantly expressed and coupled to the virus-like particle, or fused to the virus-like particle using recombinant DNA techniques. Exemplary procedures describing the attachment of antigens to virus-like particles are disclosed in WO 00/32227, in WO 01/85208 and in WO 02/056905, the disclosures of which are herewith incorporated by reference in its entirety.
- the invention also provides a method of producing a composition for enhancing an immune response in an animal comprising a VLP and an immunostimulatory substance, preferably an unmethylated CpG-containing oligonucleotide bound to the VLP which comprises incubating the VLP with the immunostimulatory substance and oligonucleotide, respectively, adding RNase and purifying said composition.
- the method further comprises the step of binding an antigen or antigenic determinant to said virus-like particle, wherein said antigen comprises, alternatively consists essentially of, or alternatively consists of an HIV polypeptide.
- the anigen or antigenic determinant is bound to the virus-like particle before incubating the virus-like particle with the immunostimulatory substance.
- the anigen or antigenic determinant is bound to the virus-like particle after purifying the composition.
- the method comprises incubating the VLP with RNase, adding the immunostimulatory substance and oligonucleotide, respectively, and purifying the composition.
- the method further comprises the step of binding an antigen or antigenic determinant to said virus-like particle, wherein said antigen comprises, alternatively consists essentially of, or alternatively consists of an HIV polypeptide.
- the anigen or antigenic determinant is bound to the virus-like particle before incubating the virus-like particle with the RNase.
- the anigen or antigenic determinant is bound to the virus-like particle after purifying the composition.
- the VLP is produced in a bacterial expression system.
- the RNase is RNase A.
- the invention further provides a method of producing a composition for enhancing an immune response in an animal comprising a VLP bound to an immunostimulatory substance, preferably to an unmethylated CpG-containing oligonucleotide which comprises disassembling the VLP, adding the immunostimulatory substance and oligonucleotide, respectively, and reassembling the VLP.
- the method can further comprise removing nucleic acids of the disassembled VLP and/or purifying the composition after reassembly.
- the method further comprises the step of binding an antigen or antigenic determinant to the virus-like particle, wherein said antigen comprises, alternatively consists essentially of, or alternatively consists of an HIV polypeptide.
- the anigen or antigenic determinant is bound to the virus-like particle before disassembling the virus-like particle. In another preferred embodiment, the anigen or antigenic determinant is bound to the virus-like particle after reassembling the virus-like particle, and preferably after purifying the composition.
- the invention also provides vaccine compositions which can be used for preventing and/or attenuating diseases or conditions.
- Vaccine compositions of the invention comprise, or alternatively consist of, an immunologically effective amount of the inventive immune enhancing composition together with a pharmaceutically acceptable diluent, carrier or excipient.
- the vaccine can also optionally comprise an adjuvant.
- the invention provides a vaccine comprising an immunologically effective amount of the inventive immune response enhancing composition together with a pharmaceutically acceptable diluent, carrier or excipient, wherein the composition comprises, (a) a virus-like particle; (b) at least one immunostimulatory substance; and (c) at least one antigen or antigenic determinant; wherein the antigen or antigenic determinant is bound to the virus-like particle, and wherein the immunostimulatory substance is bound to the virus-like particle, and wherein the antigen comprises, alternatively consists essentially of, or alternatively consists of a polypeptide, a polyprotein, a peptide, an epitope or a polyepitope of HIV.
- the vaccine further comprises an adjuvant.
- the invention further provides vaccination methods for preventing and/or attenuating diseases or conditions in animals.
- the invention provides vaccines for the prevention of infectious diseases in a wide range of animal species, particularly mammalian species such as human, mouse, or monkey, wherein the antigenic determinant is from the relevant virus infecting said species or is an antigenic determinant relevant to the particular animal model of the disease.
- Vaccines can be designed to treat infections of viral etiology such as HIV.
- RNA-phage derived VLPs in particular the VLP derived from Q ⁇ , do very efficiently induce a memory CD8 T cell response in a homologous prime-boost vaccination scheme.
- live vaccinia virus immunizations are very ineffective for the induction of a primary CD8 + T cell response and homologous boosting with vaccinia does hardly lead to an expansion of memory CD8 + T cells.
- the invention provides a method of immunizing or treating an animal comprising priming a T cell response in the animal by administering an immunologically effective amount of the inventive vaccine.
- the method further comprises the step of boosting the immune response in the animal, wherein preferably the boosting is effected by administering an immunologically effective amount of a vaccine of the invention or an immunologically effective amount of a heterologous vaccine, wherein even more preferably the heterologous vaccine is a DNA vaccine, peptide vaccine, recombinant virus or a dendritic cell vaccine.
- the invention further provides a method of immunizing or treating an animal comprising the steps of priming a T cell response in the animal, and boosting a T cell response in the animal, wherein the boosting is effected by administering an immunologically effective amount of the vaccine of the invention.
- the primimg is effected by administering an immunologically effective amount of a vaccine of the invention or an immunologically effective amount of a heterologous vaccine, wherein even more preferably said heterologous vaccine is a DNA vaccine, peptide vaccine, recombinant virus or a dendritic cell vaccine.
- the invention further provides for a composition
- a composition comprising a virus-like particle, at least one immunostimulatory substance, and at least one antigen or antigenic determinant; wherein said antigen or antigenic determinant is bound to said virus-like particle, and wherein said immunostimulatory substance is bound to said virus-like particle, and wherein said antigen comprises a cytotoxic T cell epitope, a Th cell epitope or a combination of at least two of said epitopes, wherein said at least two epitopes are bound directly or by way of a linking sequence, and wherein preferably said cytotoxic T cell epitope is a viral or a tumor cytotoxic T cell epitope.
- the present invention provides a composition, typically and preferably for enhancing an immune response in an animal comprising: (a) a viruslike particle; (b) an immunostimulatory substance; wherein said immunostimulatory substance (b) is bound to said virus-like particle (a); and (c) an antigen, wherein said antigen is mixed with said virus-like particle (a), and wherein said antigen comprises, alternatively consists essentially of, or alternatively consists of an HIV polypeptide of the invention.
- the term “mixed” refers to the combination of two or more substances, ingredients, or elements that are added together, are not chemically combined with each other and are capable of being separated. Methods of mixing antigens with virus-like particles are described in WO 04/000351 , which is incorporated herein by reference in its entirety.
- compositions of the invention when administered to an animal, they can be in a composition which contains salts, buffers, adjuvants or other substances which are desirable for improving the efficacy of the composition.
- materials suitable for use in preparing pharmaceutical compositions are provided in numerous sources including REMINGTON'S PHARMACEUTICAL SCIENCES (Osol, A, ed., Mack Publishing Co., (1990)).
- adjuvants can be used to increase the immunological response, depending on the host species, and include but are not limited to, Freund's (complete and incomplete), mineral gels such as aluminum hydroxide, surface active substances such as lysolecithin, pluronic polyols, polyanions, peptides, oil emulsions, keyhole limpet hemocyanins, dinitrophenol, and potentially useful human adjuvants such as BCG (bacille Calmette Guerin) and Corynebacterium parvum. Such adjuvants are also well known in the art.
- compositions of the invention include, but are not limited to, Monophosphoryl lipid immunomodulator, AdjuVax 100a, QS 21, QS 18, CRL1005, Aluminum salts, MF 59, and Virosomal adjuvant technology.
- the adjuvants can also comprise a mixture of these substances.
- Compositions of the invention are said to be "pharmacologically acceptable” if their administration can be tolerated by a recipient individual. Further, the compositions of the invention will be administered in a "therapeutically eff ctive amount" (i.e., an amount that produces a desired physiological effect).
- the compositions of the present invention can be administered by various methods known in the art.
- the particular mode selected will depend of course, upon the particular composition selected, the severity of the condition being treated and the dosage required for therapeutic efficacy.
- the methods of the invention can be practiced using any mode of administration that is medically acceptable, meaning any mode that produces effective levels of the active compounds without causing clinically unacceptable adverse effects.
- modes of administration include oral, rectal, parenteral, intracistemal, intravaginal, intraperitoneal, topical as by powders, ointments, drops or transdermal patch), bucal, or as an oral or nasal spray.
- parenteral refers to modes of administration which include intravenous, intramuscular, intraperitoneal, intrastemal, subcutaneous and intraarticular inj ection and infusion.
- the composition of the invention can also be injected directly in a lymph node.
- compositions for administration include sterile aqueous (e.g., physiological saline) or non-aqueous solutions and suspensions.
- non- aqueous solvents are propylene glycol, polyethylene glycol, vegetable oils such as olive oil, and injectable organic esters such as ethyl oleate.
- Carriers or occlusive dressings can be used to increase skin permeability and enhance antigen absorption.
- Combinations can be administered either concomitantly, e.g., as an admixture, separately but simultaneously or concurrently; or sequentially.
- Administration "in combination” further includes the separate administration of one of the compounds or agents given first, followed by the second.
- Dosage levels depend on the mode of administration, tr e nature of the subject, and the quality of the carrier/adjuvant formulation. Typical amounts are in the range of about 0.1 ⁇ g to about 20 mg per subject. Preferred amounts are at least about 1 ⁇ g to about 1 mg, more preferably 10 to 400 ⁇ g per subject. Multiple administration to immunize the subject is preferred, and protocols are those standard in the art adapted to the subject in question.
- compositions can conveniently be presented in unit dosage form and can be prepared by any of the methods well-known in the art of pharmacy. Methods include the step of bringing the compositions of the invention into association with a carrier which constitutes one or more accessory ingredients. In general, the compositions are prepared by uniformly and intimately bringing the compositions of the invention into association with a liquid carrier, a finely divided solid carrier, or both, and then, if necessary, shaping the product.
- Compositions suitable for oral administration can be presented as discrete units, such as capsules, tablets or lozenges, each containing a predetermined amount of the compositions of the invention. Other compositions include suspensions in aqueous liquids or non-aqueous liquids such as a syrup, an elixir or an emulsion.
- Other delivery systems can include time-release, delayed release or sustained release delivery systems. Such systems can avoid repeated administrations of the compositions of the invention described above, increasing convenience to the subject and the physician. Many types of release delivery systems are available and known to those of ordinary skill in the art.
- compositions of the invention include processes for the production of the compositions of the invention and methods of medical treatment for cancer and allergies using said compositions.
- the present invention provides an isolated polypeptide comprises, alternatively consists essentially of, or alternatively consists of an amino acid sequence selected from (a) the amino acid sequence of SEQ ID NO: 77; (b) the amino acid sequence of SEQ ID NO: 78; (c) the amino acid sequence of SEQ ID NO: 80; (d) the amino acid sequence of SEQ ID NO: 81; (e) the amino acid sequence of SEQ ID NO: 82; and (f) an amino acid sequence having at least 90% sequence identity to any of the amino acid sequences of (a) - (e) and being capable of being presented in a MHC complex.
- the present invention provides an isolated polypeptide which comprises, alternatively consists essentially of, or alternatively consists of an amino acid sequence selected from (a) the amino acid sequence of SEQ ID NO: 83; (b) the amino acid sequence of SEQ ID NO: 84; (c) the amino acid sequence of SEQ ID NO: 86; (d) an amino acid sequence having at least 90% sequence identity to any of the amino acid sequences of (a) - (c) and being capable of being presented in a MHC complex.
- the present invention provides an isolated polypeptide comprises, alternatively consists essentially of, or alternatively consists of an amino acid sequence selected from (a) the amino acid sequence of SEQ ID NO: 72; (b) the amino acid sequence of SEQ ID NO: 85; (c) the amino acid sequence of SEQ ID NO: 87; (d) an amino acid sequence having at least 90% sequence identity to any of the amino acid sequences of (a) - (c) and being capable of being presented in a MHC complex.
- the present invention provides an isolated polypeptide comprises, alternatively consists essentially of, or alternatively consists of an amino acid sequence selected from (a) the amino acid sequence of SEQ ID NO: 71; (b) the amino acid sequence of SEQ ID NO: 73; (c) an amino acid sequence having at least 90% sequence identity to any of the amino acid sequences of (a) - (b) and being capable of being presented in a MHC complex.
- the isolated polypeptides are synthesized by classical chemical synthesis known by the person skilled in the art. In a further embodiment, however, known recombinant methods for producing these inventive polypeptides could also be used for their production, as examplified in the example section of the present application.
- Preferred recombinantly produced polypeptides used for the composition of the present invention may be selected from the group consisting of without limitation GAGorig (SEQ ID NO: 100), 81GAG (SEQ ID NO: 102), GagC (SEQ ID NO: 114), or Nef74 (SEQ ID NO: 116).
- nucleic acid molecules encoding these inventive polypeptides are within the knowledge of the person skilled in the art as well as their expression in suitable host cells.
- HBcAg containing peptide p33 from LCMV is given in SEQ ID NO: 15.
- the p33-HBcAg VLPs were generated as follows: Hepatitis B clone pEco63 containing the complete viral genome of Hepatitis B virus was purchased from ATCC. The gene encoding HBcAg was introduced into the EcoRI/Hindlll restriction sites of expression vector pkk223.3 (Pharmacia) under the control of a strong tac promoter.
- the p33 peptide (KAVYNFATM) (SEQ ID NO: 67) derived from lymphocytic choriomeningitis virus (LCMV) was fused to the C-terminus of HBcAg (1-185) via a three leucine-linker by standard PCR methods.
- a clone of E. coli K802 selected for good expression was transfected with the plasmid, and cells were grown and resuspended in 5 ml lysis buffer (10 mM Na2HPO4, 30 mM NaCI, 10 mM EDTA, 0.25 % Tween-20, pH 7.0). 200 ⁇ l of lysozyme solution (20 mg/ml) was added.
- Flow through (which contains purified p33-VLP capsids) was collected and loaded onto a reducing SDS-PAGE gel for monomer molecular weight analysis. Electron microscopy was performed according to standard protocols. Thus, the structure of the p33-VLPs was assessed by electron microscopy and SDS
- p33-HBcAg VLP HBcAg-p33 VLP
- ⁇ 33- VLPs HBc33
- the cDNA of GA phage coat protein was amplified from GA phage by reverse transcription followed by a PCR amplification step, using the RevertAid First strand cDNA synthesis Kit (Fermentas).
- the cDNA was cut with the enzymes Ncol and Hindlll, and cloned in vector pQ ⁇ l85 previously cut with the same enzymes, leading to plasmid 355.24, harboring GA cDNA.
- the sequence of the inserted cDNA was checked by DNA sequencing.
- Plasmid 355.24 was transformed in E. coli JM109. Expression was performed essentially as described for Q ⁇ VLP. A single colony was inoculated in LB medium containing 20 mg/L Ampicillin overnight without shaking. This inoculum was transferred the next day into a larger flask containing M9 medium supplemented with 1% casaminoacids, 0.2% glucose and 20 mg/L Ampicillin, and incubated under shaking for 14-20 h.
- GA VLP was isolated essentially as described for Q ⁇ VLP. Cells were lysed, and the cleared lysate was loaded onto a Sepharose CL-4B column (Amersham Pharmacia). The eluate was concentrated by ammonium sulphate precipitation, and rechromatographed onto a Sepharose CL-6B column (Amersham Pharmacia). The final step was either an ultracentrifugation on sucrose gradient (20-50% w/v), or on CsCl. The isolated VLPs were subsequently dialysed against 20 mM Tris, 150 mM NaCI, pH 8.0. EXAMPLE 3
- VLPs produced in yeast contain small amounts of RNA which can be easily digested and so eliminated by incubating the VLPs with RNase A.
- RNase A enzyme has a molecular weight of about 14 kDa and is small enough to enter the VLPs to eliminate the undesired ribonucleic acids.
- Recombinantly produced BKV VLPs (SEQ ID NO: 12) were concentrated to 1 mg/ml in PBS buffer pH7.2 and incubated in the absence or presence of RNase A (200 ⁇ g/ml, Roche Diagnostics Ltd, Switzerland) for 3 h at 37°C.
- BKV VLPs were supplemented with 75 nmol/ml 5'- fluorescein labeled phosphorothioate CpG-FAM oligonucleotide (oligonucleotide from SEQ ID NO: 34) and incubated for 3 h at 37°C. Subsequently BKV VLPs were subjected to DNasel digestion for 3 h at 37°C (40 u/ml AMPD1, Sigma, Division of Fluka AG, Switzerland) or loaded without DNasel digestion.
- the samples were complemented with 6-fold concentrated DNA-loading buffer (10 mM Tris ⁇ H7.5, 10% v/v glycerol, 0.4% orange G) and run for 1 h at 65 volts in a 0.8% native tris-acetate pH 7.5 agarose gel.
- 6-fold concentrated DNA-loading buffer (10 mM Tris ⁇ H7.5, 10% v/v glycerol, 0.4% orange G)
- run for 1 h at 65 volts in a 0.8% native tris-acetate pH 7.5 agarose gel Upon staining with ethidium bromide nucleic acids are detected, while in the absence of ethidium bromide UV excitation leads to fluorescence of the fluorescein-label in the CpG-FAM.
- BKV VLPs (15 ⁇ g) was analyzed by a native 0.8% agarose gel electrophoresis after control incubation or after digestion with RNase A and subsequent incubation with double stranded (ds) DNA (246 bp) (SEQ ID NO: 17), upon staining with ethidium bromide or Coomassie Blue.
- the following samples were loaded on the gel: 1 : BKV VLPs untreated; 2: BKV VLPs RNase A treated; 3: BKV VLPs treated with RNase A and incubated with dsDNA; lane M: Gene Ruler 1 kb DNA ladder (MBI Fermentas GmbH, Heidelberg, Germany).
- BKV VLPs (15 ⁇ g) was analyzed by a native 0.8% agarose gel electrophoresis after control incubation or after digestion with RNase A and subsequent incubation with CpG-oligonucleotides (with phosphate- or with phosphorothioate (pt) backbone) upon staining with ethidium bromide or Coomassie Blue.
- the RNase A treated recombinant BKV VLPs (Example 3) were supplemented with 50 ⁇ g/ml (ds) DNA fragments (246 bp in length, dsDNA, SEQ ID NO: 17) and incubated for 3 h at 37°C.
- the samples were complemented with 6-fold concentrated DNA-loading buffer (10 mM Tris pH8.0, 10% v/v glycerol, 0.4% orange G) and run for 1 h at 65 volts in a 0.8% native tris- acetate pH8.0 agarose gel.
- BKV VLPs (15 ⁇ g) were loaded on a native 0.8% agarose gel electrophoresis and analyzed after control incubation or after digestion with RNase A and subsequent incubation with (ds) DNA upon staining with ethidium bromide or Coomassie Blue in order to assess the presence of RNA/DNA or protein.
- Packaged DNA molecules are visible in the presence of ethidium bromide as a band with the same migration as the VLP band visualized with Coomassie Blue.
- CpG-containing oligonucleotides can be packaged into BKV VLPs.
- the RNase A treated recombinant BKV VLPs (Example 3) were supplemented with 150 nmol/ml CpG- oligonucleotides CyCpG with phosphodiester backbone or CyCpGpt with phosphorothioate backbone and incubated for 3 h at 37°C.
- VLP preparations for mouse immunization were extensively dialysed (10,000-fold diluted) for 24 h against PBS pH7.2 with a 300 kDa MWCO dialysis membrane (Spectrum Medical industries Inc., Houston, USA) to eliminate RNase A and the excess of CpG-oligonucleotides.
- the samples were complemented with 6-fold concentrated DNA-loading buffer (10 mM Tris pH7.5, 10% v/v glycerol, 0.4% orange G) and run for 1 h at 65 volts in a 0.8% native tris-acetate pH7.5 agarose gel.
- BKV VLPs (15 ⁇ g) were loaded on a native 0.8%> agarose gel electrophoresis and analyzed after control incubation or after digestion with RNase A and subsequent incubation with CpG-oligonucleotides (with phosphodiester- or with phosphorothioate backbone) upon staining with ethidium bromide or Coomassie Blue in order to assess the presence of RNA/DNA or protein and the reduction of unbound CpG- oligonucleotides after dialysis. Unbound CpG-oligonucleotides are visible as a low molecular weight ethidium bromide stained band.
- VLPs containing CpG-oligonucleotides induce enhanced Thl directed immune response.
- mice Female BALB/c mice (three mice per group) were subcutaneously injected with 10 ⁇ g BKV VLPs containing phosphorothioate CpG-oligonucleotide CyCpGpt (SEQ ID NO: 34). As controls mice were subcutaneously injected with either 10 ⁇ g of RNase treated BKV VLPs alone or BKV VLPs mixed with 0.3 nmol or 20 nmol phosphorothioate CpG-oligonucleotides in 200 ⁇ l PBS pH7.2 or were left untreated. BKV VLPs were prepared as described in Example 5 and before immunization extensively purified from unbound CpG-oligonucleotide by dialysis. On day 14 after immunization blood was taken and IgGl and IgG2a antibody response to BKV VLPs was determined (see Table 1).
- Table 1 Mouse IgGl and IgG2a OD50% antibody titers to BKV VLPs on day 14 after immunization with BKV VLPs and phosphorothioate (pt) CpG-oligonucleotides.
- Immunostimulatory nucleic acids can be packaged into HBcAg VLPs comprising fusion proteins with antigens.
- HBcAg VLPs when produced in E. coli by expressing the Hepatitis B core antigen fusion protein p33-HBcAg (HBc33) (see Example 1) or the fusion protein to the peptide PI A (HBcPlA), contain RNA which can be digested and so eliminated by incubating the VLPs with RNase A.
- the gene PI A codes for a protein that is expressed by the mastocytoma tumor cell line P815.
- the dominant CTL epitope termed PI A peptide
- PI A peptide binds to MHC class I (Ld) and the complex is recognized by specific CTL clones (Brandle et al., 1998, Eur. J. Immunol. 28: 4010-4019).
- Fusion of peptide PI A-l (LPYLGWLVF) (SEQ ID NO: 74) to the C-terminus of HBcAg (aa 185, see Example 1) was performed by PCR using appropriate primers using standard molecular biology techniques.
- a three leucine linker was cloned between the HBcAg and the peptide sequence. Expression was performed as described in Example 1.
- the fusion protein of HBcAg with PI A termed HBcPl A, formed capsids when expressed in E. coli which could be purified similar to the procedure described in
- Enzymatic RNA hydrolysis Recombinantly produced HBcAg-p33 (HBc33) and HBcAg-Pl A (HBcPl A) VLPs at a concentration of 1.0 mg/ml in 1 x PBS buffer (KCl 0.2g/L, KH2PO4 0.2g/L, NaCI 8 g/L, Na2HPO4 1.15 g/L) pH 7.4, were incubated in the presence of 300 ⁇ g/ml RNase A (Qiagen AG, Switzerland) for 3 h at 37°C in a thermomixer at 650 rpm.
- RNase A Qiagen AG, Switzerland
- RNAse A HBcAg-p33 VLPs were supplemented with 130 nmol/ml CpG-oligonucleotides B- CpG, NKCpG, G10-PO (Table 2).
- the 150mer single-stranded Cyl50-1 and 253mer double stranded dsCyCpG-253, both containing multiple copies of CpG motifs were added at 130 nmol/ml or 1.2 nmol/ml, respectively, and incubated in a thermomixer for 3 h at 37°C.
- Double stranded CyCpG-253 DNA was produced by cloning a double stranded multimer of CyCpG into the EcoRV site of pBluescript KS-.
- the resulting plasmid produced in E. coli XL 1 -blue and isolated using the Qiagen Endofree plasmid Giga Kit, was digested with restriction endonucleases Xhol and Xbal and resulting restriction products were separated by agarose electrophoresis.
- the 253 bp insert was isolated by electro-elution and ethanol precipitation. Sequence was verified by sequencing of both strands.
- DNAse I treatment Packaged HBcAg-p33 VLPs were subsequently subjected to DNasel digestion (5 U/ml) for 3 h at 37°C (DNasel, RNase free Fluka AG, Switzerland) and were extensively dialysed (2 x against 200-fold volume) for 24 h against PBS pH 7.4 with a 300 kDa MWCO dialysis membrane (Spectrum Medical industries Inc., Houston, USA) to eliminate RNAse A and the excess of CpG-oligonucleotides.
- Benzonase treatment Since some single stranded oligodeoxynucleotides were partially resistant to DNasel treatment, Benzonase treatment was used to eliminate free oligonucleotides from the preparation. 100-120 U/ml Benzonase (Merck KGaA,
- VLP preparations packaged with immunostimulatroy nucleic acids used in mouse immunization experiments were extensively dialysed (2x against 200fold volume) for 24 h against PBS pH 7.4 with a 300 kDa MWCO dialysis membrane
- capsid 2-30 ⁇ g of capsid were mixed with 1 volume of 2x loading buffer (lxTBE, 42% w/v urea, 12% w/v Ficoll, 0.01 % Bromphenolblue), heated for 3 min at 95°C and loaded on a 10% (for oligonucleotides of about 20 nt length) or 15% (for > than 40 mer nucleic acids) TBE/urea polyacrylamid gel (Invitrogen). Alternatively samples were loaded on a 1% agarose gel with 6x loading dye (10 mM Tris pH 7.5, 50 mM EDTA, 10% v/v glycerol, 0.4 % orange G).
- 2x loading buffer lxTBE, 42% w/v urea, 12% w/v Ficoll, 0.01 % Bromphenolblue
- TBE/urea gels were stained with SYBRGold and agarose gels with stained with ethidium bromide.
- the oligonucleotides B-CpG, NKCpG and G10-PO were packaged into HBc33.
- the analysis of B-CpG packaged into HBc33 VLPs was done on a 1% agarose gel stained with ethidium bromide and Coomassie Blue. Loaded on the gel were 50 ⁇ g of the following samples: 1. HBc33 VLP untreated; 2. HBc33 VLP treated with RNase A; 3. HBc33 VLP treated with RNase A and packaged with B-CpG; 4.
- the amount of packaged B-CpG extracted from the VLP was analyzed on a 1.5% agarose gel stained with ethidium bromide: Loaded on gel were the following samples: 1. 0.5 nmol B-CpG control; 2. 0.5 nmol B-CpG control; 3. B-CpG oligo content HBc33 after phenol / chloroform extraction; 4.
- the analysis of NKCpG packaged into HBc33 VLPs was done on a 1% agarose gel stained with ethidium bromide and Coomassie Blue. Loaded on the gel were 15 ⁇ g of the following samples: 1. HBc33 VLP untreated; 2. HBc33 VLP treated with RNase A; 3. HBc33 VLP treated with RNase A and packaged with NKCpG; 4.
- gl Ogacga-PO packaged into HBc33 VLPs was done on a 1 % agarose gel stained with ethidium bromide and Coomassie Blue. Loaded on the gel were 15 ⁇ g of the following samples: 1. 1 kb MBI Fermentas DNA ladder; 2. HBc33 VLP untreated; 3. HBc33 VLP treated with RNase A; 4. HBc33 VLP treated with RNase A and packaged with glOgacga-PO; 5. HBc33 VLP treated with RNase A, packaged with gl Ogacga-PO, treated with Benzonase and dialysed.
- RNA content in the VLPs was strongly reduced after RNaseA treatment while most of the capsid migrated as a a slow migrating smear presumably due to the removal of the negatively charged RNA.
- the capsids contained a higher amount of nucleic acid than the RNAseA treated capsids and therefore migrated at similar velocity as the untreated capsids. Additional treatment with DNAse I or Benzonase degraded the free oligonucleotides while oligonucleotides packaged in the capsids did not degrade, clearly showing packaging of oligonucleotides.
- oligonucleotides released from the capsid with the procedure described above were of the same size than the oligonucleotide used for packaging clearly demonstrated packaging of oligonucleotides.
- Large single-stranded oligonucleotide Cy 150-1 was packaged into HBc33.
- Cy 150-1 packaged into HBc33 VLPs was analyzed on a 1% agarose gel stained with ethidium bromide and Coomassie Blue. Loaded on the gel were 15 ⁇ g of the following samples: 1. 1 kb MBI Fermentas DNA ladder; 2. HBc33 VLP untreated; 3. HBc33 VLP treated with RNase A; 4. HBc33 VLP treated with RNase A and packaged with Cyl50-1; 5. HBc33 VLP treated with RNase A, packaged with Cy 150-1, treated with DNasel and dialysed; 6.
- HBc33 VLP treated with RNase A, packaged with Cy 150-1, treated with DNasel and dialysed.
- the analysis of the amount of packaged Cyl50-1 extracted from the VLP was analyzed on a 10 % TBE/urea gel stained with SYBR Gold. Loaded on gel are the following samples: 1. 20 pmol Cyl50-1 control; 2. 10 pmol Cyl50-1 control; 3. 4 pmol Cyl50-1 control; 4. Cy 150-1 oligo content of 4 ⁇ g HBc33 after 3 min at 95°C with 1 volume TBE/urea sample buffer. RNA content in the capsid was strongly reduced after RNaseA treatment while most of the capsid migrated as a slow migrating smear.
- Capsid were diluted with 4 volumes of water and concentrated to 1 mg/ml. After incubation with an excess of Cy 150- 1 the capsid contained a bigger amount of nucleic acid and thus migrated at similar velocity as the untreated capsids. Additional treatment with DNAsel degraded the free, not packaged oligonucleotides while oligonucleotides in capsids were not degraded. Release of the DNAsel-resistant nucleic acid from the packaged VLPs by heating for 3 min at 95°C in TBE/urea loading buffer revealed the presence of the 150 mer.
- the oligonucleotide NKCpGpt was also packaged into HBcPl A.
- the analysis of NKCpGpt packaged into HBcPl A VLPs was done on a 1% agarose gel stained with ethidium bromide and Coomassie Blue. Loaded on the gel were 15 ⁇ g of the following samples: 1. 1 kb MBI Fermentas DNA ladder; 2. HBcPlA VLP untreated; 3. HBcPlA VLP treated with RNase A; 4. HBcPlA VLP treated with RNase A and packaged with NKCpGpt. Treatment with RNAse reduced nucleic acid content and slowed migration of the capsids. Addition of NKCpGpt restored nucleic acid content in capsids and fast migration.
- Immunostimulatory nucleic acids can be packaged in HBcAg-wt coupled with antigens.
- HBcAg-wt VLPs were packaged after coupling with peptide p33 (CGG-KAV YNF ATM) (SEQ ID NO : 68), derived from lymphocytic choriomeningitis virus (LCMV).
- CGG-KAV YNF ATM CGG-KAV YNF ATM
- LCMV lymphocytic choriomeningitis virus
- VLPs were dialyzed to Mes buffer (2-(N-rnorpholino) ethanesulphonic acid) pH 7.4 for 2 x 2 h using MWCO 10.000 kD dialysis membranes at 4°C.
- VLPs (50 ⁇ M) were subsequently coupled to the N-terminal cysteine of the p33 peptide (250 ⁇ M) during a 2 h incubation in a thermomixer at 25°C.
- Samples were dialyzed (MWCO 300.000) extensively to lx PBS pH 7.4 to eliminate undesired free peptide.
- HBcAg-wt VLPs derivatization with SMPH and coupling to p33 peptide was analyzed on SDS-PAGE. Samples were analysed by 16% SDS PAGE and stained with Coomassie Blue. Loaded on the gel were the following samples: l.NEB Prestained Protein Marker, Broad Range (# 7708S), 10 ⁇ l; 2. p33 peptide; 3. HBcAg-wt VLP derivatized with SMPH, before dialysis; 4. HBcAg-wt VLP derivatized with SMPH, after dialysis; 5. HBcAg-wt VLP coupled with p33, supernatant; 6.
- HBcAg-wt was visible as a 21 kD protein band. Due to the low molecular weigth of SMPH is the derivatised product only slightly larger and can not be distinguished by SDS-PAGE. Peptide alone was visible as a 3 kD band and coupled product, termed HBx33, showed a strong secondary band at approximately 24 kD accounting for more than 50% of total HBcAg-wt.
- Enzymatic RNA hydrolysis HBx33 VLPs (0.5-1.0 mg/ml, lxPBS buffer pH7.4) in the presence of RNase A (300 ⁇ g/ml, Qiagen AG, Switzerland) were diluted with 4 volumes H2O to decrease salt concentration to a final 0.2xPBS concentration and incubated for 3 h at 37°C in a thermomixer at 650 rpm.
- VLP preparations for mouse immunization were extensively dialysed (2x against 200-fold volume) for 24 h against PBS pH 7.4 with a 300 kDa MWCO dialysis membrane (Spectrum Medical industries Inc., Houston, USA) to eliminate RNase A and the excess of CpG-oligonucleotides.
- the analysis of B-CpGpt packaged into HBx33 VLPs was done on a 1% agarose gel stained with ethidium bromide and Coomassie Blue. Loaded on the gel were 50 ⁇ g of the following samples: 1. HBx33 VLP untreated; 2. HBx33 VLP treated with RNase A; 3. HBx33 VLP treated with RNase A and packaged with B-CpGpt; 4.
- HBx33 VLP treated with RNase A, packaged with B-CpGpt and treated with DNasel; 5. HBx33 VLP treated with RNase A, packaged with B-CpGpt, treated with DNasel and dialysed; 6. 1 kb MBI Fermentas DNA ladder. It could be shown that RNAse treatment reduced the nucleic acid content of the capsids and slowed their migration. Addition of B-CpGpt restored nucleic acid content and fast migration of capsids. DNAse I only digested the free oligonucleotides while the packaged oligonucleotides remained in the VLP also after dialysis.
- Immunostimulatory nucleic acids can be packaged into Q ⁇ VLPs coupled with antigens.
- Recombinantly produced virus-like particles of the RNA-bacteriophage Qb were used untreated or after coupling to p33 peptides containing an N-terminal CGG or and C-terminal GGC extension (CGG-KAVYNFATM (SEQ ID NO: 68) and KAVYNFATM-GGC (SEQ ID NO: 69)).
- Recombinantly produced Q ⁇ VLPs were derivatized with a 10 molar excess of SMPH (Pierce) for 0.5 h at 25°C, followed by dialysis against 20 mM HEPES, 150 mM NaCI, pH 7.2 at 4°C to remove unreacted SMPH.
- Q ⁇ VLPs when produced in E. coli by expressing the bacteriophage Q ⁇ capsid protein, contain RNA which can be digested and so eliminated by incubating the VLPs with RNase A.
- Q ⁇ VLPs at a concentration of 1.0 mg/ml in 20mM Hepes/150mM NaCI buffer (HBS) pH 7.4 were either digested directly by addition of RNase A (300 ⁇ g/ml, Qiagen AG, Switzerland) or were diluted with 4 volumes H2O to a final 0.2 x HBS concentration and then incubated with RNase A (60 ⁇ g/ml, Qiagen AG, Switzerland). Incubation was allowed for 3 h at 37°C in a thermomixer at 650 rpm.
- RNA hydrolysis from Qb VLPs by RNase A under low and high ionic strength was analyzed on a 1% agarose gel stained with ethidium bromide and Coomassie Blue.
- Q ⁇ VLPs were concentrated to 1 mg/ml using Millipore Microcon or Centriplus concentrators and aliquots were dialysed against lx HBS or 0.2 x HBS.
- Q ⁇ VLPs were supplemented with 130 nmol/ml CpG- oligonucleotide B-CpG and incubated in a thermomixer for 3 h at 37°C.
- Q ⁇ VLPs were subjected to Benzonase digestion (100 U/ml) for 3 h at 37°C. Samples were analysed on 1% agarose gels after staining with ethidium bromide or Coomassie Blue.
- Loaded on the gel were the following samples: 1. Qb VLP untreated; 2. Qb VLP treated with RNase A; 3. Qb VLP treated with RNase A and packaged with B-CpG in 0.2x HBS buffer pH7.2 and treated with Benzonase; 4. HBx33 VLP (see example 12) treated with RNase A, packaged with B-CpG in lx HBS buffer pH7.2 and treated with Benzonase.
- lx HBS only a very low amount of oligonucleotides could be packaged, while in 0.2 x HBS a strong ethidium bromide stained band was detectable, which colocalized with the Coomassie blue stain of the capsids.
- the Q ⁇ VLPs or Qbx33 VLPs were concentrated to 1 mg/ml using Millipore Microcon or Centriplus concentrators and supplemented with 130 nmol/ml CpG-oligonucleotides B-CpGpt, glOgacga and the 253 mer dsCyCpG-253 (Table 2) and incubated in a thermomixer for 3 h at 37°C.
- Q ⁇ VLPs or Qbx33 VLPs were subjected to DNAse I digestion (5 U/ml) or Benzonase digestion (100 U/ml) for 3 h at 37°C.
- Samples were analysed on 1% agarose gels after staining with ethidium bromide or Coomassie Blue. Loaded on the gel were 50 ⁇ g of the following samples: 1. Qbx33 VLP untreated; 2. Qbx33 VLP treated with RNase A; 3. Qbx33 VLP treated with RNase A and packaged with B-CpGpt; 4. Qbx33 VLP treated with RNase A, packaged with B-CpGpt, treated with DNasel and dialysed; 5.
- (C) depicts the analysis of the amount of packaged oligo extracted from the VLP on a 15% TBE/urea stained with SYBR Gold.
- Loaded on gel are the following samples: 1. BCpGpt oligo content of 2 ⁇ g Qbx33 VLP after proteinase K digestion and RNase A treatment; 2. 20 pmol B-CpGpt control; 3. 10 pmol B-CpGpt control; 4. 5 pmol B-CpGpt control.
- Loaded on another gel were 15 ⁇ g of the following samples: 1. MBI Fermentas 1 kb DNA ladder; 2. Qbx33 VLP untreated; 3. Qbx33 VLP treated with RNase A; 4.
- the different nucleic acids B-CpGpt, glOgacga and the 253mer dsDNA could be packaged into Qbx33.
- Packaged nucleic acids were resistant to DNAse I digestion and remained packaged during dialysis.
- Packaging of B-CpGpt was confirmed by release of the nucleic acid by proteinase K digestion followed by agarose electrophoresis and ethidium bromide staining.
- the pellet was resolubilized in 7 M urea, while the supernatant was dialyzed 3 days against NET buffer (20 mM Tris-HCl, pH 7.8 with 5mM EDTA and 150 mM NaCI) with 3 changes of buffer. Alternatively, dialysis was conducted in continuous mode over 4 days. The dialyzed solution was centrifuged at 8000 rpm for 20 minutes, and the pellet was resolubilized in 7 M urea, while the supernatant was pelletted with ammonium sulphate (60% saturation), and resolubilized in a 7 M urea buffer containing 10 mM DTT.
- NET buffer 20 mM Tris-HCl, pH 7.8 with 5mM EDTA and 150 mM NaCI
- the pellet was isolated by centrifiigation at 8000 rpm, for 20 minutes. It was resolubilized in 7 M urea, 10 mM DTT, and loaded on a short Sepharose 4B column (1.5 X 27 cm Sepharose 4B, 2 ml/h, 7 M urea, 10 mM DTT as elution buffer). Mainly one peak, with a small shoulder eluted from the column. The fractions containing the AP205 coat protein were identified by SDS-PAGE, and pooled, excluding the shoulder. This yielded a sample of 10.3 ml.
- the protein concentration was estimated spectrophotometrically by measuring an aliquot of protein diluted 25 -fold for the measurement, using the following formula: (1.55 x OD280 - 0.76 x OD260) x volume. The average concentration was of 1 nmol/ml of VLP (2.6 mg/ml). The ratio of absorbance at 280 mn vs. 260 nm was of 0.12/0.105. Reassembly: 1.1 ml beta-mercaptoethanol was added to the sample, and the following reassembly reactions were set up:
- the VLP reassembled in the presence of the CyCpG was purified over a Sepharose 4B column (1 X 50 cm), eluted with NET buffer (1 ml/h). The fractions were analyzed by Ouchterlony assay, and the fractions containing VLP were pooled. This resulted in a sample of 8 ml, which was desalted against water by dialysis, and dried. The yield of capsid was of 10 mg. Analysis of resolubilized material in a 0.6% agarose gel stained with ethidium-bromide showed that the capsids were empty of nucleic acids.
- the AP205 VLPs could also be reassembled in the absence of detectable oligodeoxynucleotide, as measured by agarose gel electrophoresis using ethidium bromide staining. Oligodeoxynucleotides could thus be successfully bound to AP205 VLP after initial disassembly of the VLP, purification of the disassembled coat protein from nucleic acids and subsequent reassembly of the VLP in the presence of oligodeoxynucleotide.
- Reassembly The protein preparation resulting from the Sephadex G-75 gel filtration purification step was precipitated with ammonium sulphate at 60% saturation, and the resulting pellet solubilized in 2 ml 7 M urea, 10 mM DTT. The sample was diluted with 8 ml of 10% 2-mercaptoethanol in NET buffer, and dialyzed for 1 hour against 40 ml of 10% 2-mercaptoethanol in NET buffer. Reassembly was initiated by adding 0.4 ml of a CyCpG solution (109 nmol/ml) to the protein sample in the dialysis bag. Dialysis in continous mode was set up, and NET buffer used as eluting buffer.
- Dialysis was pursued for two days and a sample was taken for EM analysis after completion of this dialysis step.
- the dialyzed reassembly solution was subsequently dialyzed against 50% v/v Glycerol in NET buffer, to achieve concentration.
- One change of buffer was effected after one day of dialysis.
- the dialysis was pursued over a total of three days.
- the dialyzed and concentrated reassembly solution was purified by gel filtration over a Sepharose 4-B column (1X60 cm) at a flow rate of 1 ml/hour, in NET buffer. Fractions were tested in an Ouchterlony assay, and fractions containing capsids were dried, resuspended in water, and rechromatographed on the 4-B column equilibrated in 20 mM Hepes pH 7.6. Using each of the following three formula:
- the reassembled AP205 VLPs were analyzed by EM as described above, agarose gel electrophoresis and SDS-PAGE under non-reducing conditions.
- the EM analysis of disassembled material shows that the treatment of AP205 VLP with guanidinium-chloride essentially disrupts the capsid assembly of the VLP.
- Reassembly of this disassembled material with an oligonucleotide yielded capsids, which were purified and further enriched by gel filtration. Two sizes of particles were obtained; particles of about 25 nm diameter and smaller particles are visible in the electron micrograph. No reassembly was obtained in the absence of oligonucleotides. Loading of the reassembled particles on agarose electrophoresis showed that the reassembled particles contained nucleic acids.
- the derivatized AP205 was reacted at a concentration of 1 mg/ml with either a 2.5-fold, or with a 5-fold excess of peptide, diluted from a 20 mM stock in DMSO, for 2 hours at 15 °C. The sample was subsequently flash frozen in liquid nitrogen for storage.
- the coupling reaction was analyzed on an SDS-PAGE. Loaded on a gel were the following samples: protein marker; derivatized AP205 VLP (d); AP205 VLP coupled with a 2.5-fold excess of peptide, supernatant (s); AP205 VLP coupled with a 2.5-fold excess of peptide, pellet (p); AP205 VLP coupled with a 5-fold excess of peptide, supernatant (s); AP205 VLP coupled with a 5-fold excess of peptide, pellet (p).
- the result of the coupling reaction revealed that a higher degree of coupling could be achieved by using a 5-fold excess of peptide rather than with a 2.5 fold excess of peptide in the coupling reaction.
- Non-enzymatic hydrolysis of the RNA content of VLPs and packaging of immunostimulatory nucleic acids are included in the RNA content of VLPs and packaging of immunostimulatory nucleic acids.
- 150 mM NaCI was dialysed either against 2000 ml of 50 mM TrisHCl pH 8.0, 50 mM NaCI, 5% glycerol, 10 mM MgC12 or 2000 ml of 4 mM HEPES, pH 7.4, 30 mM NaCI for 2 h at 4°C in SnakeSkinTM pleated dialysis tubing (Pierce, Cat. No. 68035). Each of the dialysis buffers was exchanged once and dialysis was allowed to continue for another 16 h at 4°C. The dialysed solution was clarified for 10 minutes at 14 000 rpm (Eppendorf 5417 R, in fixed angle rotor F45-30-11, used in all the following steps) and proteinconcentration was again determined by Bradford analysis.
- Q ⁇ VLPs in 50 mM TrisHCl pH 8.0, 50 mM NaCI, 5% glycerol, 10 mM MgC12 were diluted with the corresponding buffer to a final protein concentration of 1 mg/ml whereas Q ⁇ VLPs in 4 mM HEPES pH 7.4, 30 mM NaCI were diluted with the corresponding buffer to a final protein concentration of 0.5 mg/ml.
- This capsid-containing solutions were centrifuged again for 10 minutes at 14 000 rpm at 4°C.
- the supematants were than incubated with ZnSO4 which was added to a final concentration of 2.5 mM for 24 h at 60°C in an Eppendorf Thermomixer comfort at 550 rpm.
- ZnSO4-treated Q ⁇ VLPs was analyzed by agarose gelelectrophoresis: Q ⁇ VLPs which had been purified from E.coli and dialysed either against buffer 1 (50 mM TrisHCl pH 8.0, 50 mM NaCI, 5% glycerol, 10 mM MgC12) or buffer 2 (4 mM HEPES, pH 7.4, 30 mM NaCI) were incubated either without or in the presence of 2.5 mM zinc sulfate (ZnSO4) for 24 hrs at 60°C.
- buffer 1 50 mM TrisHCl pH 8.0, 50 mM NaCI, 5% glycerol, 10 mM MgC12
- buffer 2 4 mM HEPES, pH 7.4, 30 mM NaCI
- oligodeoxynucleotides into ZnSO4-treated VLPs ZnSO4-treated and dialysed Q ⁇ capsids with a protein concentration (as determined by Bradford analysis) beween 0.4 mg/ml and 0.9 mg/ml (which corresponds to a concentration of capsids of 159 nM and 357.5 nM, respectively) were used for the packaging of the oligodeoxynucleotides.
- the oligodeoxynucleotides were added at a 300- fold molar excess to the of Q ⁇ -VLP capsids and incubated for 3 h at 37°C in an Eppendorf Thermomixer comfort at 550 rpm .
- the nucleic acid content of ZnSO4- and oligodeoxynucleotide treated Q ⁇ VLPs was analyzed by Benzonase and proteinase K digestion and polyacrylamide TBE/Urea gelelectrophoresis: Oligodeoxynucleotides were packaged into ZnSO4-treated Q ⁇ VLPs as described above. 25 ⁇ g of these VLPs were digested with 25 ⁇ l Benzonase (Merck, Cat. No. 1.01694.0001) according to the manufactures instructions. After heat- inactivation of the nuclease (30 minutes at 80°C) the VLPs were treated with Proteinase K (final enzyme concentration was 0.5 mg/ml) according to the manufactures instructions.
- Intact Q ⁇ VLPs (which had been purified from E.coli) did not contain nucleic acids of similar size than those which had been extracted from ZnSO4- and oligodeoxynucleotide treated Q ⁇ capsids.
- nucleic acids isolated from the latter VLPs were comigrating with the oligodeoxynucleotides which had been used in the reassembly reaction. This results confirmed that the used oligodeoxynucleotides were packaged into ZnSO4-treated Q ⁇ capsids.
- Q ⁇ VLPs were treated with RNaseA as described in Example 9 under low ionic strength conditions (20 mM Hepes pH 7.4 or 4 mM Hepes, 30 mM NaCI, pH 7.4 ).
- VLPs such as described in Examples 2, 3, 7, and 10, i.e. GA, BKV,
- HBcAg, and AP205 are treated.
- Q ⁇ VLPs and AP205 VLPs were treated with ZnSO4 under low ionic strength conditions (20 mM Hepes pH 7.4 or 4 mM Hepes,
- Example 11 30 mM NaCI pH 7.4) as described in Example 11.
- AP205 VLP (1 mg/ml) in either 20 mM Hepes pH 7.4 or 20 mM Hepes, 1 mM Tris, pH 7.4 was treated for 48 h with 2.5 mM ZnSO4 at 50°C in an Eppendorf Thermomixer comfort at 550 rpm.
- Q ⁇ and AP205 VLP samples were clarified as described in Example 11 and supematants were dialysed in 10.000 MWCO Spectra/Por® dialysis tubing (Spectrum, Cat. nr. 128 118) against first 2 1 20 mM Hepes, pH 7.4 for 2 h at 4°C and, after buffer exchange, overnight.
- Samples were clarified after dialysis as described in Example 11 and protein concentration in the supematants was determined by Bradford analysis.
- samples were treated with 125 U Benzonase/ml VLPs (Merck KGaA, Darmstadt, Germany) in the presence of 2 mM MgC12 and incubated for 3 h at 37°C before dialysis.
- Samples were dialysed in 300.000 MWCO Spectra/Por® dialysis tubing (Spectrum, Cat. nr. 131 447) against 20 mM Hepes, pH 7.4 for 2 h at 4°C, and after buffer exchange overnight against the same buffer. After dialysis samples were clarified as described in Example 11 and protein concentration in the supematants were determined by Bradford analysis.
- Q ⁇ VLPs, packaged with ISS were coupled to p33 peptides containing a C- terminal GGC extension (KAVYNFATM-GGC) (SEQ ID NO: 69), resulting in Qb VLPs termed Qb-ISS-33 VLPs.
- Qb-ISS-33 VLPs a C- terminal GGC extension
- Packaged Q ⁇ VLPs in 20 mM Hepes, pH 7.4 were derivatized with a 10-fold molar excess of SMPH (Pierce) for 0.5 h at 25°C, followed by two dialysis steps of 2 hours each against 20 mM HEPES pH 7.4 at 4°C to remove unreacted SMPH.
- AP205 VLPs (1.24 mg/ml) packaged with G8-8 oligonucleotide as described above were derivatized and coupled to HIVpl7 (71-85) containing a N-terminal GGC extension (CGG-GSEEIRSLYNTVATL) (SEQ ID NO: 70), resulting in AP205-G8-8- HIVpl7 VLPs.
- AP205 VLPs (packaged with G8-8), in 20 mM Hepes pH 7.4, were derivatized with a 20-fold molar excess of SMPH for 0.5 h at 25°C, and subsequently dialysed two times against 20 mM HEPES, pH 7.4 at 4°C to remove unreacted SMPH.
- Peptide was added to the dialyzed derivatization mixture in a 10-fold molar excess and allowed to react for 2 h in a thermomixer at 25°C.
- Samples were dialysed in 10.000 MWCO dialysis tubing against 20 mM Hepes pH 7.4 for 2 h at 4°C, and after buffer exchange, overnight against the same buffer. After dialysis, samples were clarified as described in Example 11 and protein concentration in the supematants were determined by Bradford analysis. Coupling efficiency of peptide HIVpl7 to AP205 was analysed by SDS-PAGE on 16% PAGE Tris-Glycine gels.
- G8-8 oligonucleotide packaging in AP205 was analysed on 1% agarose gels and, after proteinase K digestion, G8-8 oligonucleotide amount in AP205-G8-8-HIVpl7 was analysed on TBE/urea gels as described in Example 7.
- Q ⁇ VLPs containing NKCpG oligonucleotides and subsequently coupled to p33 peptide were termed Qb-NKCpG-33 VLPs.
- Qb-NKCpG-33 VLPs On a 1 % agarose gel, the fluorescent band visible on the ethidium bromide stained gel co-migrates with the protein band visible on the Coomassie Blue stained gel demonstrating packaging.
- both RNaseA and ZnSO4 treated Q ⁇ VLPs contain NKCpG oligonucleotides before as well as after coupling to p33 peptide.
- Coupling efficiency of the p33 peptide is maintained as can be judged from the multiple coupling products visible after SDS-PAGE analysis on a 16 % PAGE Tris-Glycine gel, as bands migrating slower than residual Q ⁇ VLP subunit monomers which have not reacted with peptide.
- the packaging efficiency can be estimated from the analysis of the TBE/urea gel by comparison of the signal of the oligonucleotide from the packaged Qb-NKCpG-33 lane with the signal of the oligonucleotide standard loaded on the same gel.
- Packaged amounts of NKCPG were between 1 and 4 nmol/ 100 ⁇ g Qb-NKCpG-33 VLPs.
- Q ⁇ VLPs containing G8-8 oligonucleotides and subsequently coupled to p33 peptide were termed Qb-G8-8-33 VLPs.
- Ethidium bromide staining of G8-8 packaged Q ⁇ VLPs can be seen on a 1 % agarose gel stained with ethidium bromide. Comigration of the ethidium bromide fluorescent band with the Q ⁇ VLP protein band visible on the same gel subsequently stained with Coomassie Blue demonstrates packaging.
- Coupling efficiency can be estimated to be 30% by SDS-PAGE analysis on a 16 % PAGE Tris-Glycine gel.
- Analysis of the G8-8 content of Qb-G8-8-33 VLPs was done on a 1% agarose gel, where the amount of oligonucleotide packaged was of approximately 1 nmol/100 ⁇ g Qb-G8-8-33 VLPs.
- Packaging of G8-8 oligonucleotides into AP205 VLPs was analyzed by gelelectrophoresis. Staining of G8-8 packaged AP205 VLPs can be seen on a 1 % agarose gel stained with ethidium bromide.
- Coupling efficiency with the HIVpl7 peptide could be estimated from the SDS-PAGE analysis on a 16 % PAGE Tris-Glycine gel where multiple coupling bands migrating slower than the residual AP205 VLP monomer subunits, which did not react with peptide, are visible. Coupling efficiency was comparable to the coupling efficiency obtained for the Qb-G8-8-33 VLPs.
- Qbx33 VLPs (Q ⁇ VLPs coupled to peptide p33, see Example 9) were treated with RNaseA under low ionic conditions (20 mM Hepes pH 7.4) as described in Example 9 to hydrolyse RNA content of the Qbx33 VLP.
- RNaseA under low ionic conditions (20 mM Hepes pH 7.4
- Qbx33 VLPs were mixed with guanosine flanked oligonucleotides (Table 3: G3-6, G7-7, G8-8, G9-9, G6, G10-PO, from a 1 mM oligonucleotide stock in 10 mM Tris pH 8) and incubated as described in Example 12.
- Qbx33 VLPs were treated with Benzonase and dialysed in 300.000 MWCO tubing.
- Samples with oligos G7-7, G8-8 and G9-9 were extensively dialysed over 3 days with 4 buffer exchanges to remove free oligo.
- Packaging was analysed on 1% agarose gels and, after proteinase K digestion, on TBE/urea gels as described in Example 7.
- oligonucleotide packaging was analyzed by agarose gelelectrophoresis. Upon oligonucleotide packaging, a fluorescent band migrating slightly slower than reference untreated Q ⁇ VLP becomes visible on the 1 % agarose gel stained with ethidium bromide indicating the presence of oligonucleotides. The signal is maintained after treatment with Benzonase, indicating packaging of the oligonucleotides within the Qbx33 VLPs. The packaging efficiency can be estimated from the TBE/urea gel electrophoresis.
- the amount of the G3-6 oligonucleotide (approximately 4 nmol/100 ⁇ g Qbx33 VLPs) packaged is much higher than the amount of packaged G8-8 oligonucleotide (approximately 1 nmol/100 ⁇ g Qbx33 VLPs). This indicates a dependence of packaging ability on the length of the guanosine nucleotides tail flanking the CpG motif.
- Q ⁇ VLPs were treated with ZnSO4 under low ionic strength conditions (20 mM Hepes pH 7.4 or 4 mM Hepes, 30 mM NaCI, pH 7.4) as described in Example 11.
- AP205 VLPs (1 mg/ml) in either 20 mM Hepes pH 7.4 or 20 mM Hepes, 1 mM Tris, pH 7.4 were treated for 48 h with 2.5 mM ZnSO4 at 50°C in an Eppendorf Thermomixer comfort at 550 rpm.
- Q ⁇ and AP205 VLP samples were clarified as in Example 11 and dialysed against 20 mM Hepes, pH 7.4 as in Example 12.
- Incubated poly (I:C) was added in a 10-fold molar excess to either ZnSO4-treated Q ⁇ or AP205 VLPs (1-1.5 mg/ml) and the mixtures were incubated for 3 h at 37°C in a thermomixer at 650 rpm. Subsequently, excess of free poly (I:C) was enzymatically hydrolysed by incubation with 125 U Benzonase per ml VLP mixture in the presence of 2 mM MgC12 for 3 h at 37°C in a thermomixer at 300 rpm.
- Q ⁇ VLPs (1 mg/ml) packaged with poly (I:C) were derivatized and coupled to p33 peptide (KAVYNFATM-GGC) (SEQ ID NO: 69) as described in Example 12, resulting in Qb-pIC-33.
- the packaged Q ⁇ VLP was derivatized with a 2.1 -fold molar excess of SMPH (Pierce) for 0.5 h at 25°C, followed by two dialysis steps against 20 mM HEPES, pH 7.4 at 4°C to remove unreacted SMPH. Peptides were added in a 2.1 -fold molar excess and allowed to react for 1.5 h in a thermomixer at 25°C.
- HIVpl7 (71-85) containing a N-terminal GGC extension (CGG-GSEEIRSLYNTVATL) (SEQ ID NO: 70), resulting in AP205-pIC-HIVpl7 VLPs.
- AP205 VLPs in 20 mM Hepes, pH 7.4 were derivatized with a 20-fold molar excess of SMPH for 0.5 h at 25°C, and subsequently dialysed two times against 20 mM HEPES, pH 7.4 at 4°C to remove unreacted SMPH.
- Peptide was added to the dialyzed derivatization mixture in a 10-fold molar excess and allowed to react for 2 h in a thermomixer at 25°C.
- Packaging of poly (I:C) into ZnSO4 treated AP205 VLPs and the coupling product AP205-pIC-HIV ⁇ l7 after coupling to HIVpl7 was analyzed by agarose gelelectrophoresis.
- Coupling efficiency of the HIVpl7 peptide is estimated from the appearance of multiple coupling products visible as bands migrating slower than AP205 VLP subunit monomer, which did not react with peptide, after SDS-PAGE analysis on a 16 % PAGE Tris-Glycine gel electrophoresis. Coupling efficiency was overall comparable to the coupling efficiency obtained for the Qb-G8-8-33 VLPs and the AP205-G8-8-HIVpl7 VLPs (Example 12). The packaging efficiency could be estimated from the TBE gel, which showed that the packaged amounts of poly (I:C) in the AP205-pIC-HIVpl7 VLP is approximately 10 pmol/100 ⁇ g VLP.
- HBcAg VLPs are treated with RNaseA under low ionic strength conditions (20 mM Hepes pH 7.4) as described in Example 9 to hydrolyse RNA content of the VLP.
- VLPs are mixed with guanosine flanked oligonucleotides (Table 3; G3-6, G7-7, G8-8, G9-9, G10-PO or G6, 1 mM stock in 10 mM Tris pH 8) and incubated as described in Example 12.
- VLPs are treated with Benzonase and dialysed in 300.000 MWCO tubing. Packaging is analysed on 1% agarose gels and on TBE/urea gels after proteinase K digestion as described in Example 7.
- GA VLPs are treated with RNaseA under low ionic conditions (20 mM Hepes pH 7.4) as described in Example 9 to hydrolyse RNA content of the VLP.
- VLPs are mixed with guanosine flanked oligonucleotides (Table 3; G3-6, G7-7, G8-8, G9-9, G10-PO or G6, 1 mM stock in 10 mM Tris pH8) and incubated as described in Example 12.
- VLPs are treated with Benzonase and dialysed in 300.000 MWCO tubing. Packaging is analysed on 1% agarose gels and on TBE/urea gels after proteinase K digestion as described in Example 7.
- HBcAg VLPs are treated with ZnSO4 under low ionic strength conditions (20 mM Hepes pH 7.4 or 4 mM Hepes, 30 mM NaCI, pH 7.4 ) as described in Example 11 and are dialysed against 20 mM Hepes pH 7.4 as in Example 12.
- Poly (I:C) is added in a 10-fold molar excess to HBcAg VLPs (1-1.5 mg/ml) and incubated for 3 h at 37°C in a thermomixer at 650 rpm as described in Example 14.
- GA VLPs are treated with ZnSO4 under low ionic strength conditions (20 mM Hepes pH 7.4 or 4 mM Hepes, 30 mM NaCI, pH 7.4 ) as described in Example 11 and are dialysed against 20 mM Hepes, pH 7.4 as in Example 12.
- Poly (I:C) is added in a 10-fold molecular excess to GA VLPs (1-1.5 mg/ml) and incubated for 3 h at 37°C in a thermomixer at 650 rpm as described in Example 14.
- the column was equilibrated with 20 mM sodium phosphate buffer pH 7 and the sample was diluted 1 :15 in water to adjust a conductivity below 10 mS/cm in order to achieve proper binding of the coat protein to the column.
- the elution of the bound coat protein was accomplished by a step gradient to 20 mM sodium phosphate / 500 mM sodium chloride and the protein was collected in a fraction volume of approx. 25 ml.
- the column was regenerated with 0.5 M NaOH.
- the isolated Q ⁇ coat protein dimer (the eluted fraction from the cation exchange column) was applied (in two runs) onto a Sephacryl S-100 HR column (xk26/60, 320 ml, Amersham Bioscience) equilibrated with 20 mM sodium phosphate / 250 mM sodium chloride; pH 6.5. Chromatography was performed at RT with a flow rate of 2.5 ml/min. Absorbance was monitored at 260 nm and 280 nm. Fractions of 5 ml were collected. The column was regenerated with 0.5 M NaOH.
- Reassembly by dialysis A stock solution of purified Q ⁇ coat protein dimer at a concentration of 2 mg/ml was used for the reassembly of Q ⁇ VLP in the presence of the oligodeoxynucleotide G8-8 or G10-PO.
- the concentration of oligodeoxynucleotide in the reassembly mixture was 10 ⁇ M.
- the concentration of coat protein dimer in the reassembly mixture was 40 ⁇ M (approx. 1.13 mg/ml).
- Stock solutions of urea and DTT were added to the solution to give final concentrations of 1 M urea and 5 mM DTT respectively.
- the oligodeoxynucleotide was added as last component, together with H 2 O, giving a final volume of the reassembly reaction of 3 ml.
- This solution was dialysed at 4 °C for 72 h against 1500 ml buffer containing 20 mM TrisHCl, 150 mM NaCI, pH 8.0.
- the dialysed reassembly mixture was centrifuged at 14 000 rpm for 10 minutes at 4 °C. A negligible sediment was discarded while the supernatant contained the reassembled and packaged VLPs.
- Reassembled and packaged VLPs were concentrated with centrifugal filter devices (Millipore, UFV4BCC25, 5K NMWL) to a final protein concentration of 3 mg/ml. Protein concentration was determined by Bradford analysis.
- Reassembly by diafiltration 20 ml of a stock solution of purified coat protein (1.5 mg/ml) was mixed with stock solutions of urea, DTT, oligodeoxynucleotide G10-PO and water. The oligodeoxynucleotide was added as last component. The volume of the mixture is 30 ml and the final concentrations of the components are 35 ⁇ M dimeric coat protein (reflecting 1 mg/ml), 35 ⁇ M oligodeoxynucleotide, 1 M urea and 2.5 mM DTT.
- the mixture was then diafiltrated against 300 ml of 20 mM sodium phosphate / 250 mM sodium chloride, pH 7.2, in a tangential flow filtration apparatus at RT, using a Pellicon XL membrane cartridge (Biomax 5K, Millipore).
- the total flow rate was set to 10 ml/min and the permeate flow rate set to 2.5 ml/min.
- H 2 O 2 was added to the solution to a final concentration of 7 mM and the solution was further incubated at RT for 60 min, to accelerate the formation of the structural disulfide bonds in the formed VLPs.
- A) Hydrodynamic size of reassembled capsids Q ⁇ capsids, which had been reassembled in the presence of oligodeoxynucleotide G8-8 or G10-PO, were analyzed by dynamic light scattering (DLS) and compared to intact Q ⁇ VLPs, which had been purified from E.coli. Reassembled capsids showed the same hydrodynamic size (which depends both on mass and conformation) as the intact Q ⁇ VLPs.
- DLS dynamic light scattering
- the reactions were then mixed with a TBE-Urea sample buffer and loaded on a 15 % polyacrylamide TBE-Urea gel (Novex ® , Invitrogen Cat. No. EC6885).
- a qualitative as well as quantitative standard 1 pmol, 5 pmol and 10 pmol of the oligodeoxynucleotide which was used for the reassembling reaction, was loaded on the same gel. This gel was stained with SYBR ® -Gold (Molecular Probes Cat. No. S- 11494).
- the SYBR ® -Gold stain showed that the reassembled Q ⁇ capsids contained nucleic acid co-migrating with the oligodeoxynucleotides which were used in the reassembly reaction.
- resistance to benzonase digestion of the nucleic acid content of the Q ⁇ VLPs which had been reassembled in the presence of oligodeoxynucleotides and isolation of the oligodeoxynucleotide from purified particles by proteinase K digestion demonstrate packaging of the oligodeoxynucleotide.
- Human PBMC are isolated from buffy coats and treated with the indicated ISS in RPMI medium containing 10% FCS for 18h. IFN alpha in the supematants is measured by ELISA, using an antibody set provided by PBL Biomedical Laboratories. PBMC are stained with mouse anti-human CD8-FITC, mouse anti-human CD19-PE and anti-human CD69-APC and analyzed by flow cytometry. Decreasing the number of flanking Gs in the other oligonucleotides results in lower IFN alpha secretion.
- G10-PO, G9-9 and G8-8 upregulate CD69 on the cell membrane of CD8 T cells to a nearly similar extend.
- G10-PO, G9-9 and G8-8 have comparable high activity on human cells, therefore they can be used as ISS in Qb-HIV vaccine.
- Qbx33 VLPs loaded with G3-6, G6, G10-PO or poly (I:C) induces protection against p33 -recombinant Vaccinia virus challenge
- B6 mice were subcutaneously immunized with Qbx33 alone or loaded with G3-6 or G6 or poly (I:C) (see Examples 12 and 14). Eight days later, mice were challenged with 1.5 x 106 pfu of recombinant Vaccinia virus, expressing the LCMV-p33 antigen. After 4 days, mice were sacrificed and the viral titers in ovaries were measured as previously described (Bachmann et al, Eur. J. Imunol. 1994, 24:2228). As depicted in FIG.
- mice receiving the Qbx33 vaccine loaded with either G3-6 or G6 or poly (I:C) were protected from viral challenge.
- na ⁇ ve mice and mice immunized with Qbx33 alone did not eliminate the virus from the ovaries.
- VLP alone is not sufficient to induce protective CTL immune response
- VLP loaded with CpG or poly (I:C) are very efficient in priming na ⁇ ve CTL.
- immunization of mice with Qbx33 loaded with G10-PO was priming p33-specific CTL (6.2% +/- 1.4% vs 0.2% +/-0.1% in na ⁇ ve mice), as well as inducing protection from recombinant Vaccinia virus challenge.
- gag-G50 Coupling of gag-G50, nef-N56 and gag-G68n peptide antigen to Q ⁇ VLP
- gag-G50 sequence: CQGQMVHQAISPRTLNAWVKA
- FSPEVIPMFSALSEGATPQDLNTMLNTVK (SEQ ID NO: 71) and nef-N56 (sequence: CGVGFPVRPQVPLRPMTYKAAVDLSHFLKEKGGLE GPGIRYPLTFGWCFKLVPVEP) (SEQ ID NO: 72) and gag-G68n (sequence: CGEIYKRWIILGLNKIVRMYQGQMVHQAISPRTLNAWVK AFSPEVIPMFSALSEGATPQDLNTMLNTVK) (SEQ ID NO: 73) were chemically synthesized. The peptides were ordered from the company SynPep, P.O. Box 2999, Dublin, CA 94568, USA.
- Q ⁇ VLP (Seq-ID No. 10) was then reacted at a concentration of 1.2 mg/ml (determined in a Bradford assay), with 0.85 mM SMPH (Pierce) for 30 minutes at room temperature (RT).
- the reaction mixture was then diafiltrated against 20 mM phosphate buffer pH 7.2 and 50 mM MES pH 6.0 was added for gag-G50 coupling reactions, and 50 mM Tris pH 8.5 for nef-N56 coupling reactions.
- a 5 mM stock of peptide was dissolved in DMSO and an equimolar amount TCEP was added to the peptide in order to have reducing reaction conditions.
- the derivatised Q ⁇ particles reacted at a concentration of 1 mg/ml with 0.214 mM gag-G50, 0.214 mM nef-N56 or 0.535 mM gag-G68n. Both peptides, gag-G50 and nef-N56, were also coupled under the same conditions, but for the buffer, which was 50 mM Tris pH 8.5. The coupling reaction was left to proceed for 2 hours at 25°C; samples were taken for SDS-PAGE analysis, and the reaction mixtures dialyzed 2 X 2 hours against a 1000-fold volume 20 mM phosphate, 0.05% Tween, pH 7.2.
- the dialyzed samples were flash frozen in liquid nitrogen in aliquots for storage at -80°C until further use. An aliquot was thawed, and coupling of the antigen to a Q ⁇ subunit assessed by SDS-PAGE. The results of the coupling reactions analyzed before the dialysis are shown in FIG. 2 and FIG. 3. Analysis of the dialyzed coupling reaction showed a similar picture.
- Q ⁇ VLP packaged with G8-8 oligonucleotide made as described in Example 12 is coupled to HIV peptides as described in Example 22.
- the sequences of the coupled peptides are gag-G50 (sequence:
- gag-G68n sequence: CGEIYKRWIILGLNKIVRMYQGQMVHQAISPRTLNAWVKAFSPEVIPMFS ALSEG
- Q ⁇ VLP is coupled to HIV peptides gag-G50, gag-G68n, or nef-N56 as described in Example 22.
- Q ⁇ VLP coupled either to gag-G50, gag-G68n, or nef-N56 is packaged with G8-8 oligonucleotide and analysed as described in Example 9.
- GAGorig A protein called GAGorig was PCR amplified from primers (gaglnhefo (SEQ ID NO: 1)
- gag2fo (SEQ ID NO: 89), gag3fo (SEQ ID NO: 90), i-gag4ba (SEQ ID NO: 91), i-gag5ba (SEQ ID NO: 92), gag6fo-b (SEQ ID NO: 93), gag7fo (SEQ ID NO: 94), i- gag8ba (SEQ ID NO: 95), i-gag9-b (SEQ ID NO: 96), i-gaglOb-Notba (SEQ ID NO: 97)) using a gene synthesis approach.
- the resulting fragment was cloned at the restriction sites Nhel/Notl into the vector pMOD-GST/El (SEQ ID NO: 98).
- GST-GAGorig was digested with enterokinase (Invitrogen, Basel, Switzerland).
- the GAGorig peptide (SEQ ID NO: 100) was purified on a reversed phase column (15RPC ST 4.66/100; Amersham, Otelfingen, Switzerland) and coupled to Q ⁇ to create Q ⁇ -GAGorig particles.
- a gene called 81GAG was PCR amplified from the template GST-GAGorig.
- a first fragment was generated using the primers 80gaglnhe (SEQ ID NO: 103) and i-80gag2 (SEQ ID NO: 104), and a second one with the primers 80gag3 (SEQ ID NO: 105) and i-81gag4 (SEQ ID NO: 106). These two fragments were used as templates in a second, so called assembly PCR using the primers 80gaglnhe and i-
- the resulting PCR fragment was cloned at the restriction sites Nhel/Notl into the vector pMOD-GST/El ((SEQ ID NO: 98)).
- the cells were lysed by sonication and the protein GST-81 GAG could be purified using glutathione- sepharose 4B beads (Amersham, Otelfingen, Switzerland) according to the manufacturer's instructions.
- the primers gagClfo (SEQ ID NO: 107), i-gagC2ba (SEQ ID NO: 108) and the template GAGorig (SEQ ID NO: 99) were used to create a first N-terminal GagC fragment.
- the second PCR fragment was created using the oligos Gag3Cfo (SEQ ID NO: 109), i-gag6Cba (SEQ ID NO: 112) as primers and the oligos gagC4fo (SEQ ID NO: 110), i-gagC5ba (SEQ ID NO: 111) as templates. These two fragments were PCR assembled using them as templates and the oligos i-gag6Cba, gagClfo as primers.
- the created PCR fragment was cloned at the restriction sites Nhel/Notl into pMOD-GST/El (SEQ ID NO: 98).
- the cells were lysed by sonication and the protein GST-GagC could be purified using glutathione-sepharose 4B beads (Amersham, Otelfingen) according to the manufacturer's instructions.
- the purified fusion protein GST- GagC was digested with enterokinase (Invitrogen, Basel, Switzerland).
- the GagC peptide (SEQ ID NO: 114) was purified on a reversed phase column (15RPC ST 4.66/100; Amersham, Otelfingen) and coupled to Q ⁇ to create soluble Q ⁇ -GagC particles.
- the inclusion bodies were resuspended in 0.12 1 wash buffer (20 mM Tris pH8, 23% sucrose, 0.5% Triton X- 100, 1 mM EDTA) and sonicated three times for 30 s. That washing procedure was performed three times.
- the purified inclusion bodies showed a band of the expected size (36 kD) for GST-Nef74 (SEQ ID NO: 116) on a SDS-PAGE satained with coomassie blue. Then, the inclusion bodies were resuspended and incubated over night in 20 ml 6 M guanidine, 0.1 M Tris ⁇ H8, 0.1 M DTT.
- This suspension was diluted to 225 ml with 6 M guanidine, 20 mM Tris pH8 to a protein concentration of approximately 0.1 g/1 and then dialysed at 4°C over night against 4.5 1 of 400 mM arginine, 0.1 M Tris pH8. This dialysis procedure was repeated once for 4 hours. The dialysed sample was centrifuged for 30 minutes at 20O00 rpm in a sorvall SS-34 rotor and then dialysed twice against 4.5 1 20 mM Tris pH8, 5 % glycerol, 0.1 mM DTT.
- the refolded GST-Nef74 was centrifuged for 30 minutes at 20O00 rpm in a sorvall SS-34 rotor and concentrated in a Millipore filter unit (5000 Da cut-off membrane) to 10 ml.
- Q ⁇ VLP (SEQ ID NO: 10) was reacted at a concentration of 3.06 g/1 (determined in a Bradford assay), with 1.09 mM SMPH (Pierce; Perbio Science, Lausanne, Switzerland) for 30 minutes at room temperature. The reaction mixture was then dialysed twice against 0.5 120 mM hepes buffer pH 7.4. The protein Nef74 (SEQ ID NO: 116) was dissolved in DMSO containing reducing 2 mM TCEP (Pierce; Perbio Science, Lausanne) and incubated for 1 hour at room temperature.
- Nef74 peptide SEQ ID NO: 116
- 2 mM TCEP Pieris; Perbio Science, Lausanne
- the peptide was incubated for 1 hour at room temperature.
- the derivatised Q ⁇ particles reacted at a concentration of 0.7 g/1 with 50, or 25, or 12.5 ⁇ M Nef74.
- the coupling reaction was left to proceed for 2 hours at 25°C and samples were taken for SDS-PAGE analysis. Soluble Q ⁇ -Nef74 has been identified (23 kD). The coupling efficiency of the insoluble fraction of the vaccine was higher than that of the soluble fraction.
Abstract
Description



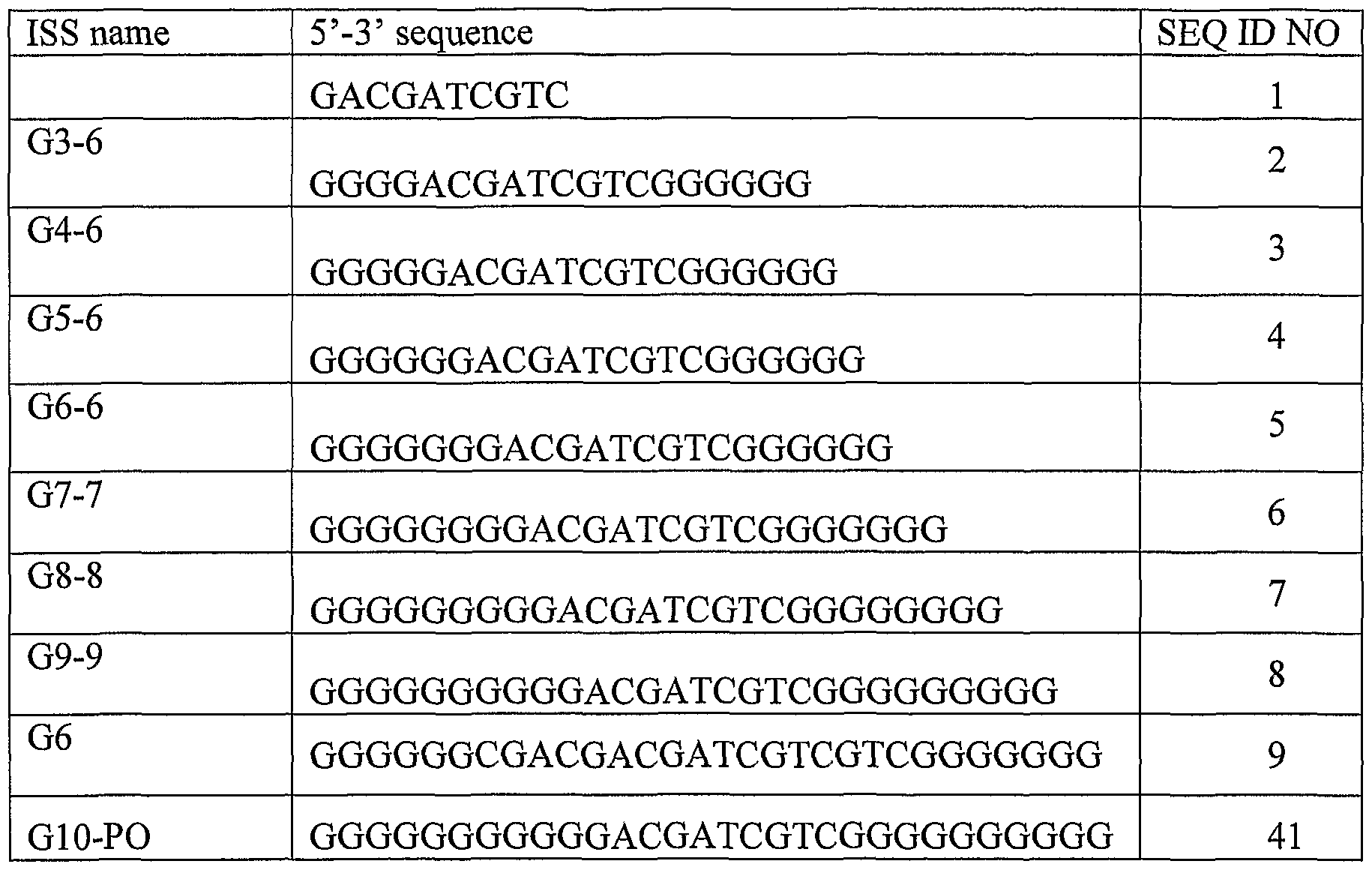
Claims
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002519165A CA2519165A1 (en) | 2003-03-26 | 2004-03-25 | Hiv-peptide-carrier-conjugates |
| EP04723204A EP1605972A2 (en) | 2003-03-26 | 2004-03-25 | Hiv-peptide-carrier-conjugates |
| AU2004224761A AU2004224761A1 (en) | 2003-03-26 | 2004-03-25 | HIV-peptide-carrier-conjugates |
| US10/550,580 US20060210588A1 (en) | 2003-03-26 | 2004-03-25 | Hiv-peptide-carrier-conjugates |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US45734803P | 2003-03-26 | 2003-03-26 | |
| US60/457,348 | 2003-03-26 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| WO2004084939A2 true WO2004084939A2 (en) | 2004-10-07 |
| WO2004084939A3 WO2004084939A3 (en) | 2005-03-31 |
| WO2004084939A8 WO2004084939A8 (en) | 2005-06-09 |
Family
ID=33098221
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2004/003163 WO2004084939A2 (en) | 2003-03-26 | 2004-03-25 | Hiv-peptide-carrier-conjugates |
| PCT/EP2004/003165 WO2004084940A1 (en) | 2003-03-26 | 2004-03-25 | Packaging of immunostimulatory oligonucleotides into virus-like particles: method of preparation and use |
| PCT/EP2004/003164 WO2004085635A1 (en) | 2003-03-26 | 2004-03-25 | Melan-a peptide analogue-virus-like-particle conjugates |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2004/003165 WO2004084940A1 (en) | 2003-03-26 | 2004-03-25 | Packaging of immunostimulatory oligonucleotides into virus-like particles: method of preparation and use |
| PCT/EP2004/003164 WO2004085635A1 (en) | 2003-03-26 | 2004-03-25 | Melan-a peptide analogue-virus-like-particle conjugates |
Country Status (13)
| Country | Link |
|---|---|
| US (2) | US7517520B2 (en) |
| EP (3) | EP1606398A1 (en) |
| JP (1) | JP5022028B2 (en) |
| KR (1) | KR20050115913A (en) |
| CN (2) | CN100560719C (en) |
| AU (3) | AU2004224762B2 (en) |
| BR (1) | BRPI0408623A (en) |
| CA (3) | CA2517675A1 (en) |
| MX (1) | MXPA05009289A (en) |
| NZ (1) | NZ542323A (en) |
| RU (1) | RU2351362C2 (en) |
| WO (3) | WO2004084939A2 (en) |
| ZA (3) | ZA200507063B (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006037787A3 (en) * | 2004-10-05 | 2006-07-06 | Cytos Biotechnology Ag | Vlp-antigen conjugates and their uses as vaccines |
| WO2006083706A2 (en) * | 2005-01-31 | 2006-08-10 | Vaxinnate Corporation | Method to identify polypeptide toll-like receptor (tlr) ligands |
| WO2006134125A1 (en) * | 2005-06-14 | 2006-12-21 | Cytos Biotechnology Ag | Antigen conjugates and uses thereof |
| FR2887457A1 (en) * | 2005-06-23 | 2006-12-29 | Fond Bettencourt Schueller | TRANSCUTANE TARGETING VACCINATION |
| EP2407534A1 (en) | 2010-07-14 | 2012-01-18 | Neo Virnatech, S.L. | Methods and reagents for obtaining transcriptionally active virus-like particles and recombinant virions |
| US8338582B2 (en) * | 2005-09-05 | 2012-12-25 | Fundacao De Amparo A Pesquisa Do Estado De Sao Paulo | Anti-HIV immunogens and methods for inducing an immune response |
| WO2013092720A1 (en) | 2011-12-22 | 2013-06-27 | F. Hoffmann-La Roche Ag | Full length antibody display system for eukaryotic cells and its use |
| US10086056B2 (en) | 2015-01-15 | 2018-10-02 | University Of Copenhagen | Virus-like particle with efficient epitope display |
| US11129882B2 (en) | 2015-10-30 | 2021-09-28 | University Of Copenhagen | Virus like particle with efficient epitope display |
Families Citing this family (115)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20030026782A1 (en) * | 1995-02-07 | 2003-02-06 | Arthur M. Krieg | Immunomodulatory oligonucleotides |
| US6207646B1 (en) * | 1994-07-15 | 2001-03-27 | University Of Iowa Research Foundation | Immunostimulatory nucleic acid molecules |
| US20030022854A1 (en) | 1998-06-25 | 2003-01-30 | Dow Steven W. | Vaccines using nucleic acid-lipid complexes |
| US7320793B2 (en) * | 2001-01-19 | 2008-01-22 | Cytos Biotechnology Ag | Molecular antigen array |
| ES2335979T3 (en) * | 2001-09-14 | 2010-04-07 | Cytos Biotechnology Ag | IMMUNOSTIMULATOR CPG PACKAGING IN VIRUS SIMILAR PARTICLES: PREPARATION METHOD AND ITS USE. |
| EP1513552B1 (en) * | 2002-06-20 | 2010-12-01 | Cytos Biotechnology AG | Packaged virus-like particles in combination with cpg for use as adjuvants with allergens : method of preparation and use |
| US7138252B2 (en) * | 2002-07-17 | 2006-11-21 | Cytos Biotechnology Ag | Molecular antigen arrays |
| DE60336902D1 (en) * | 2002-07-19 | 2011-06-09 | Cytos Biotechnology Ag | VACCINE COMPOSITIONS CONTAINING AMYLOID BETA 1-6 ANTIGENARRAYS |
| EP2241325B1 (en) | 2002-10-29 | 2012-02-08 | Coley Pharmaceutical Group, Inc. | Use of CPG oligonucleotides in the treatment of hepatitis C virus infection |
| AU2004224762B2 (en) * | 2003-03-26 | 2009-12-24 | Kuros Us Llc | Packaging of immunostimulatory oligonucleotides into virus-like particles: method of preparation and use |
| US7537767B2 (en) * | 2003-03-26 | 2009-05-26 | Cytis Biotechnology Ag | Melan-A- carrier conjugates |
| US9090673B2 (en) | 2003-12-12 | 2015-07-28 | City Of Hope | Synthetic conjugate of CpG DNA and T-help/CTL peptide |
| US8153426B2 (en) | 2004-08-19 | 2012-04-10 | University College Cardiff Consultants Limited | Preparation of antigen-presenting human gamma-delta T cells and use in immunotherapy |
| KR100721928B1 (en) * | 2004-11-05 | 2007-05-28 | 주식회사 바이오씨에스 | Pharmaceutical composition for treating or preventing dermatitis comprising CpG oligodeoxynucleotide |
| ATE521621T1 (en) * | 2004-11-29 | 2011-09-15 | Changchun Huapu Biotechnology Co Ltd | CPG SINGLE STRAND DESOXYNUCLEOTIDES FOR USE AS ADJUVANTS |
| BRPI0608455A2 (en) * | 2005-03-18 | 2010-01-05 | Cytos Biotechnology Ag | a composition comprising cat allergen conjugates as well as vaccines, pharmaceutical compositions and medicaments containing them, and a process for their production |
| US20110008831A1 (en) * | 2005-05-26 | 2011-01-13 | Cytos Biotechnology Ag | Scalable fermentation process |
| EP1736538A1 (en) * | 2005-06-21 | 2006-12-27 | Cytos Biotechnology AG | Process for the preparative purification of virus-like-particles (VLPs) |
| PT2179737E (en) * | 2005-07-01 | 2013-12-05 | Index Pharmaceuticals Ab | Modulating responsiveness to steroids |
| PL2269622T3 (en) * | 2005-07-01 | 2014-05-30 | Index Pharmaceuticals Ab | CpG oligonucleotides used for enhancing steroid activity in a steroid dependent patient |
| US7468186B2 (en) * | 2005-07-22 | 2008-12-23 | City Of Hope | Polyomavirus cellular epitopes and uses therefor |
| US8338173B2 (en) * | 2005-08-11 | 2012-12-25 | University College Cardiff Consultants Limited | Preparation of antigen-presenting human γδ T cells and use in immunotherapy |
| WO2007039458A2 (en) * | 2005-09-21 | 2007-04-12 | Cytos Biotechnology Ag | Hiv peptide conjugates and uses thereof |
| CA2625969A1 (en) * | 2005-10-28 | 2007-05-03 | Index Pharmaceuticals Ab | Composition and method for the prevention, treatment and/or alleviation of an inflammatory disease |
| US20080044438A1 (en) * | 2006-03-17 | 2008-02-21 | Ostroff Gary R | Yeast Cell Particles As Oral Delivery Vehicles For Antigens |
| SG172696A1 (en) | 2006-06-12 | 2011-07-28 | Cytos Biotechnology Ag | Processes for packaging oligonucleotides into virus-like particles of rna bacteriophages |
| AU2013204383B2 (en) * | 2006-06-12 | 2016-09-22 | Kuros Us Llc | Processes for packaging oligonucleotides into virus-like particles of RNA bacteriophages |
| WO2008024427A2 (en) * | 2006-08-23 | 2008-02-28 | Science & Technology Corporation @ Unm | A virus-like platform for rapid vaccine discovery |
| US8586728B2 (en) | 2006-12-12 | 2013-11-19 | Cytos Biotechnology Ag | Oligonucleotides containing high concentrations of guanine monomers |
| WO2008071774A1 (en) * | 2006-12-14 | 2008-06-19 | Cytos Biotechnology Ag | Purification process for coat protein of rna bacteriophages |
| CN104001170B (en) | 2008-06-27 | 2016-08-24 | 硕腾有限责任公司 | Novel adjunvant composition |
| BRPI0923225A2 (en) | 2008-12-02 | 2016-10-04 | Chiralgen Ltd | Phosphorus-modified nucleic acid synthesis method |
| EP2376108B1 (en) * | 2008-12-09 | 2017-02-22 | Pfizer Vaccines LLC | IgE CH3 PEPTIDE VACCINE |
| WO2010122164A1 (en) | 2009-04-23 | 2010-10-28 | Cytos Biotechnology Ag | VIRUS-LIKE PARTICLES OF BACTERIOPHAGE φCB5 |
| EA022699B1 (en) | 2009-05-27 | 2016-02-29 | Селекта Байосайенсиз, Инк. | Targeted synthetic nanocarriers with ph sensitive release of immunomodulatory agents |
| US8431530B2 (en) | 2009-06-12 | 2013-04-30 | Morehouse School Of Medicine | Compositions and methods for treating aids or cancer by inhibiting the secretion of microparticles |
| MX342945B (en) | 2009-07-06 | 2016-10-18 | Ontorii Inc * | Novel nucleic acid prodrugs and methods use thereof. |
| DE102009034779A1 (en) | 2009-07-25 | 2011-02-03 | Emc Microcollections Gmbh | Synthetic analogues of bacterial lipopeptides and their application for the therapy and prophylaxis of allergic diseases |
| KR20210149203A (en) * | 2009-11-02 | 2021-12-08 | 더 트러스티스 오브 더 유니버시티 오브 펜실바니아 | Foot and mouth disease virus (fmdv) consensus proteins, coding sequences therefor and vaccines made therefrom |
| WO2011109422A2 (en) | 2010-03-02 | 2011-09-09 | The United States Of America, Represented By The Secretary, Department Of Health And Human Services | Compositions and methods for the treatment of cancer |
| JP6199036B2 (en) | 2010-03-16 | 2017-09-20 | バイオエヌテック アーゲーBioNTech AG | Tumor vaccination involved in humoral immune response to self-protein |
| EP2366709A1 (en) * | 2010-03-16 | 2011-09-21 | BioNTech AG | Tumor vaccination involving a humoral immune response against self-proteins |
| CN107029223A (en) | 2010-05-26 | 2017-08-11 | 西莱克塔生物科技公司 | Synthesize nano-carrier combined vaccine |
| US9511135B2 (en) | 2010-09-14 | 2016-12-06 | Stc.Unm | Immunogenic respiratory syncytial virus glycoprotein-containing VLPs and related compositions, constructs, and therapeutic methods |
| JP5868324B2 (en) | 2010-09-24 | 2016-02-24 | 株式会社Wave Life Sciences Japan | Asymmetric auxiliary group |
| US9994443B2 (en) | 2010-11-05 | 2018-06-12 | Selecta Biosciences, Inc. | Modified nicotinic compounds and related methods |
| GB201021867D0 (en) * | 2010-12-23 | 2011-02-02 | Mologen Ag | Non-coding immunomodulatory DNA construct |
| US20120301498A1 (en) | 2011-04-29 | 2012-11-29 | Selecta Biosciences, Inc. | Controlled release of immunosuppressants from synthetic nanocarriers |
| WO2013012758A1 (en) | 2011-07-19 | 2013-01-24 | Ontorii, Inc. | Methods for the synthesis of functionalized nucleic acids |
| KR20140050698A (en) | 2011-07-29 | 2014-04-29 | 셀렉타 바이오사이언시즈, 인크. | Synthetic nanocarriers that generate humoral and cytotoxic t lymphocyte (ctl) immune responses |
| CN102336821B (en) * | 2011-08-11 | 2014-10-15 | 北京永泰免疫应用科技有限公司 | New Melan-A epitope peptide and application thereof in preventing and/or treating tumours |
| JP6081483B2 (en) * | 2011-12-12 | 2017-02-15 | セル・メディカ・リミテッド | The process of proliferating T cells |
| RU2485973C1 (en) | 2012-02-28 | 2013-06-27 | Общество с ограниченной ответственностью "НТфарма" | Recombinant trivalent influenza vaccine |
| WO2013177440A2 (en) * | 2012-05-23 | 2013-11-28 | The Board Of Trustees Of The Leland Stanford Junior University | Ordered flagellin array as an immunostimulant |
| CN104684893B (en) | 2012-07-13 | 2016-10-26 | 日本波涛生命科学公司 | Asymmetric auxiliary group |
| KR102450907B1 (en) | 2012-07-13 | 2022-10-04 | 웨이브 라이프 사이언시스 리미티드 | Chiral control |
| RU2677639C2 (en) | 2012-07-13 | 2019-01-18 | Шин Ниппон Биомедикал Лэбораториз, Лтд. | Chiral nucleic acid adjuvant |
| CA2936092A1 (en) | 2013-01-23 | 2014-07-31 | The Board Of Trustees Of The Leland Stanford Junior University | Stabilized hepatitis b core polypeptide |
| WO2014145205A2 (en) | 2013-03-15 | 2014-09-18 | St. Jude Children's Research Hospital | Methods and compositions of p27kip1 transcription modulators |
| MX2015011487A (en) | 2013-03-15 | 2016-02-03 | Univ Pennsylvania | Foot and mouth disease virus (fmdv) consensus proteins, coding sequences therefor and vaccines made therefrom. |
| BR122020023215B1 (en) | 2013-05-03 | 2022-11-22 | Selecta Biosciences, Inc | COMPOSITION AND KIT OF TOLEROGENIC SYNTHETIC NANOCARRIERS TO REDUCE OR PREVENT ANAPHYLAXIS IN RESPONSE TO A NON-ALLERGEN ANTIGEN |
| MX2015016691A (en) | 2013-06-04 | 2016-04-04 | Selecta Biosciences Inc | Repeated administration of non-immunosupressive antigen specific immunotherapeutics. |
| NZ757210A (en) | 2013-09-19 | 2022-12-23 | Zoetis Services Llc | Oil-based adjuvants |
| JP6545177B2 (en) | 2013-09-29 | 2019-07-17 | セント ジュード チルドレンズ リサーチ ホスピタル インコーポレイテッド | Aryl-substituted aminomethyl spectinomycin analogues as antimicrobial agents |
| US20170035864A1 (en) * | 2013-12-09 | 2017-02-09 | Bullet Biotechnology, Inc. | SPECIFIC VIRUS-LIKE PARTICLE-CpG OLIGONUCLEOTIDE VACCINES AND USES THEREOF |
| CN104761644A (en) * | 2014-01-03 | 2015-07-08 | 百奇生物科技(苏州)有限公司 | Fusion protein MBP-MART-1 expressed by Escherichia coli, and preparation method and application thereof |
| JPWO2015108047A1 (en) | 2014-01-15 | 2017-03-23 | 株式会社新日本科学 | Chiral nucleic acid adjuvant having immunity induction activity and immunity induction activator |
| US10322173B2 (en) | 2014-01-15 | 2019-06-18 | Shin Nippon Biomedical Laboratories, Ltd. | Chiral nucleic acid adjuvant having anti-allergic activity, and anti-allergic agent |
| US10149905B2 (en) | 2014-01-15 | 2018-12-11 | Shin Nippon Biomedical Laboratories, Ltd. | Chiral nucleic acid adjuvant having antitumor effect and antitumor agent |
| KR20230152178A (en) | 2014-01-16 | 2023-11-02 | 웨이브 라이프 사이언시스 리미티드 | Chiral design |
| GB2523187A (en) * | 2014-02-18 | 2015-08-19 | Mologen Ag | Covalently closed non-coding immunomodulatory DNA construct |
| EP3160453A1 (en) | 2014-06-25 | 2017-05-03 | Selecta Biosciences, Inc. | Methods and compositions for treatment with synthetic nanocarriers and immune checkpoint inhibitors |
| IL292574A (en) | 2014-09-07 | 2022-06-01 | Selecta Biosciences Inc | Methods and compositions for attenuating anti-viral transfer vector immune responses |
| JP6715775B2 (en) * | 2014-12-25 | 2020-07-01 | 国立研究開発法人医薬基盤・健康・栄養研究所 | Non-aggregating immunostimulatory oligonucleotide |
| EP3240801B1 (en) | 2014-12-31 | 2021-01-20 | Checkmate Pharmaceuticals, Inc. | Combination tumor immunotherapy |
| LT3244920T (en) | 2015-01-16 | 2023-08-25 | Zoetis Services Llc | Foot-and-mouth disease vaccine |
| US20180161340A1 (en) | 2015-06-18 | 2018-06-14 | St. Jude Children's Research Hospital | Methods and compositions for the prevention and treatment of hearing loss |
| LU92821B1 (en) | 2015-09-09 | 2017-03-20 | Mologen Ag | Combination comprising immunostimulatory oligonucleotides |
| WO2017044890A1 (en) * | 2015-09-10 | 2017-03-16 | Academia Sinica | Bird flu vaccine combination comprising virus-like particles and novel adjuvants |
| GB2542425A (en) | 2015-09-21 | 2017-03-22 | Mologen Ag | Means for the treatment of HIV |
| US20170157215A1 (en) | 2015-12-04 | 2017-06-08 | Jomoco, Corp. | Compositions and methods to mitigate or prevent an immune response to an immunogenic therapeutic molecule in non-human primates |
| US20190046638A1 (en) | 2016-04-01 | 2019-02-14 | Checkmate Pharmaceuticals, Inc. | Fc RECEPTOR-MEDIATED DRUG DELIVERY |
| PT3448364T (en) | 2016-04-29 | 2022-05-04 | Icahn School Med Mount Sinai | Targeting the innate immune system to induce long-term tolerance and to resolve macrophage accumulation in atherosclerosis |
| CA3038089A1 (en) | 2016-09-27 | 2018-04-05 | Selecta Biosciences, Inc. | Recombinant immunotoxins for use in the treatment of cancer |
| GB201616365D0 (en) * | 2016-09-27 | 2016-11-09 | Helsingin Yliopisto | Non-genetic modification of enveloped viruses |
| JP2020506890A (en) | 2017-01-07 | 2020-03-05 | セレクタ バイオサイエンシーズ インコーポレーテッドSelecta Biosciences,Inc. | Patterned administration of immunosuppressants coupled to synthetic nanocarriers |
| JP2020510687A (en) | 2017-03-11 | 2020-04-09 | セレクタ バイオサイエンシーズ インコーポレーテッドSelecta Biosciences,Inc. | Methods and compositions relating to combination treatment with synthetic nanocarriers including anti-inflammatory and immunosuppressive agents |
| GB2571696B (en) | 2017-10-09 | 2020-05-27 | Compass Pathways Ltd | Large scale method for the preparation of Psilocybin and formulations of Psilocybin so produced |
| EP3694543A1 (en) | 2017-10-13 | 2020-08-19 | Selecta Biosciences, Inc. | Methods and compositions for attenuating anti-viral transfer vector igm responses |
| CN112236522A (en) | 2018-04-09 | 2021-01-15 | 查克美特制药公司 | Packaging of oligonucleotides into virus-like particles |
| CN112771070A (en) | 2018-07-16 | 2021-05-07 | 西莱克塔生物科技公司 | Methods and compositions of OTC constructs and vectors |
| WO2020018587A1 (en) | 2018-07-16 | 2020-01-23 | Selecta Biosciences, Inc. | Methods and compositions of mma constructs and vectors |
| CN112639077B (en) * | 2018-08-07 | 2023-06-20 | 中国科学院生物物理研究所 | Method for activating CD4+ T cells |
| EP3902530A4 (en) * | 2018-12-28 | 2023-01-25 | Ramot at Tel-Aviv University Ltd. | Polymeric nanovaccines and uses thereof |
| KR20220008824A (en) | 2019-04-17 | 2022-01-21 | 컴퍼스 패쓰파인더 리미티드 | How to treat anxiety disorders, headache disorders and eating disorders with psilocybin |
| KR20220004121A (en) | 2019-04-28 | 2022-01-11 | 셀렉타 바이오사이언시즈, 인크. | Methods of treating a subject with pre-existing immunity to a viral transfer vector |
| BR112021023594A2 (en) | 2019-05-28 | 2022-02-08 | Selecta Biosciences Inc | Methods and compositions for attenuated antiviral transfer vector immune response |
| MX2022004825A (en) | 2019-10-23 | 2022-10-10 | Regeneron Pharma | Synthetic rig-i-like receptor agonists. |
| DE102020113731B4 (en) * | 2020-05-20 | 2024-02-08 | FiberBridge Photonics GmbH | Fiberglass and fiberglass product |
| CA3182002A1 (en) * | 2020-06-09 | 2021-12-16 | Charles Richardson | Method of making virus-like particle |
| WO2022098901A1 (en) | 2020-11-04 | 2022-05-12 | Selecta Biosciences, Inc. | Compositions for reducing immune responses against immunoglobulin proteases |
| AU2022206197A1 (en) | 2021-01-05 | 2023-07-13 | Selecta Biosciences, Inc. | Viral vector dosing protocols |
| JP2024511067A (en) | 2021-03-19 | 2024-03-12 | トレインド セラピューティクス ディスカバリー,インコーポレーテッド | Compounds and methods of use for modulating trained immunity |
| JP2024514577A (en) | 2021-04-09 | 2024-04-02 | セレクタ バイオサイエンシーズ インコーポレーテッド | Synthetic nanocarriers containing immunosuppressants in combination with high affinity IL-2 receptor agonists to enhance immune tolerance |
| US20230141563A1 (en) | 2021-10-12 | 2023-05-11 | Selecta Biosciences, Inc. | Methods and compositions for attenuating anti-viral transfer vector igm responses |
| US20230140196A1 (en) | 2021-10-12 | 2023-05-04 | Selecta Biosciences, Inc. | Viral vector dosing protocols |
| WO2023086615A1 (en) | 2021-11-14 | 2023-05-19 | Selecta Biosciences, Inc. | Multiple dosing with viral vectors |
| US20230263906A1 (en) | 2022-01-10 | 2023-08-24 | Selecta Biosciences, Inc. | High affinity il-2 receptor agonists and synthetic nanocarrier dose sparing |
| WO2023172624A1 (en) | 2022-03-09 | 2023-09-14 | Selecta Biosciences, Inc. | Immunosuppressants in combination with anti-igm agents and related dosing |
| WO2023172628A1 (en) | 2022-03-09 | 2023-09-14 | Selecta Biosciences, Inc. | Immunosuppressant in combination with high affinity il-2 receptor agonists and related dosing |
| WO2023183568A1 (en) | 2022-03-25 | 2023-09-28 | Selecta Biosciences, Inc. | Synthetic nanocarriers comprising an immunosuppressant in combination with high affinity il-2 receptor agonists and anti-igm agents |
| US20230381277A1 (en) | 2022-04-08 | 2023-11-30 | Selecta Biosciences, Inc. | High affinity il-2 receptor agonists and immunosuppressants to enhance immune tolerance |
| WO2023242155A1 (en) * | 2022-06-14 | 2023-12-21 | Janssen Vaccines & Prevention B.V. | Compositions and methods for the diagnosis of hiv infection |
| WO2024036324A1 (en) | 2022-08-11 | 2024-02-15 | Selecta Biosciences, Inc. | Compositions and methods related to immunoglobulin proteases and fusions thereof |
| WO2024047091A2 (en) | 2022-08-30 | 2024-03-07 | Saiba Animal Health Ag | Veterinary compositions of modified virus-like particles of cmv and ngf antigens |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5939598A (en) | 1990-01-12 | 1999-08-17 | Abgenix, Inc. | Method of making transgenic mice lacking endogenous heavy chains |
| WO2000039304A2 (en) | 1998-12-31 | 2000-07-06 | Chiron Corporation | Polynucleotides encoding antigenic hiv type c polypeptides, polypeptides and uses thereof |
| WO2001022972A2 (en) | 1999-09-25 | 2001-04-05 | University Of Iowa Research Foundation | Immunostimulatory nucleic acids |
Family Cites Families (96)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3160926D1 (en) | 1980-04-15 | 1983-10-27 | Beecham Group Plc | Allergens modified with polysarcosines |
| US5204096A (en) | 1984-03-07 | 1993-04-20 | New York Blood Center, Inc. | Pre-S gene coded peptide hepatitis B immunogens, vaccines, diagnostics, and synthetic lipid vesicle carriers |
| US4722840A (en) | 1984-09-12 | 1988-02-02 | Chiron Corporation | Hybrid particle immunogens |
| US4959314A (en) | 1984-11-09 | 1990-09-25 | Cetus Corporation | Cysteine-depleted muteins of biologically active proteins |
| FR2581394B1 (en) | 1985-05-02 | 1988-08-05 | Grp Genie Genetique | PARTICLES HAVING IMMUNOGENIC PROPERTIES OF THE HBS ANTIGEN AND CARRYING A FOREIGN ANTIGENIC SITE TO THE EPITOPES CARRIED BY THE HBS ANTIGEN, ANIMAL VECTORS AND CELLS FOR THE PRODUCTION OF SUCH PARTICLES AND COMPOSITIONS CONTAINING SUCH PARTICLES FOR THE PRODUCTION |
| US5374426A (en) | 1986-09-03 | 1994-12-20 | University Of Saskatchewan | Rotavirus nucleocapsid protein VP6 in vaccine compositions |
| US5143726A (en) | 1986-12-09 | 1992-09-01 | The Scripps Research Institute | T cell epitopes of the hepatitis B virus nucleocapsid protein |
| US4918166A (en) | 1987-04-10 | 1990-04-17 | Oxford Gene Systems Limited | Particulate hybrid HIV antigens |
| US5057540A (en) | 1987-05-29 | 1991-10-15 | Cambridge Biotech Corporation | Saponin adjuvant |
| GB8903313D0 (en) | 1989-02-14 | 1989-04-05 | Wellcome Found | Conjugates |
| NZ235315A (en) | 1989-09-19 | 1991-09-25 | Wellcome Found | Chimaeric hepadnavirus core antigen proteins and their construction |
| US5334394A (en) | 1990-06-22 | 1994-08-02 | The Regents Of The University Of California | Human immunodeficiency virus decoy |
| EP0468520A3 (en) | 1990-07-27 | 1992-07-01 | Mitsui Toatsu Chemicals, Inc. | Immunostimulatory remedies containing palindromic dna sequences |
| GB9101550D0 (en) | 1991-01-24 | 1991-03-06 | Mastico Robert A | Antigen-presenting chimaeric protein |
| GB9114003D0 (en) | 1991-06-28 | 1991-08-14 | Mastico Robert A | Chimaeric protein |
| ZA934199B (en) | 1992-06-18 | 1994-01-10 | Akzo Nv | Carrier system against gnrh |
| GB9213601D0 (en) * | 1992-06-26 | 1992-08-12 | Mastico Robert A | Protein based delivery system |
| EP0652890B1 (en) | 1992-07-27 | 1998-01-14 | HYBRIDON, Inc. | Oligonucleotide alkylphosphonothioates |
| FR2695563B1 (en) | 1992-09-11 | 1994-12-02 | Pasteur Institut | Microparticles carrying antigens and their use for the induction of humoral or cellular responses. |
| GB9227068D0 (en) | 1992-12-29 | 1993-02-24 | British Bio Technology | Novel proteinaceous particles |
| WO1995026204A1 (en) | 1994-03-25 | 1995-10-05 | Isis Pharmaceuticals, Inc. | Immune stimulation by phosphorothioate oligonucleotide analogs |
| US6727230B1 (en) * | 1994-03-25 | 2004-04-27 | Coley Pharmaceutical Group, Inc. | Immune stimulation by phosphorothioate oligonucleotide analogs |
| US5874560A (en) | 1994-04-22 | 1999-02-23 | The United States Of America As Represented By The Department Of Health And Human Services | Melanoma antigens and their use in diagnostic and therapeutic methods |
| WO1995029193A2 (en) | 1994-04-22 | 1995-11-02 | The Government Of The United States Of America Represented By The Secretary, Department Of Health And Human Services | Melanoma antigens |
| PT772619E (en) | 1994-07-15 | 2006-10-31 | Univ Iowa Res Found | OLIGONUCLEOTIDOS IMUNOMODULADORES |
| US6207646B1 (en) | 1994-07-15 | 2001-03-27 | University Of Iowa Research Foundation | Immunostimulatory nucleic acid molecules |
| US6429199B1 (en) * | 1994-07-15 | 2002-08-06 | University Of Iowa Research Foundation | Immunostimulatory nucleic acid molecules for activating dendritic cells |
| US7935675B1 (en) | 1994-07-15 | 2011-05-03 | University Of Iowa Research Foundation | Immunostimulatory nucleic acid molecules |
| US20030050263A1 (en) | 1994-07-15 | 2003-03-13 | The University Of Iowa Research Foundation | Methods and products for treating HIV infection |
| US6239116B1 (en) * | 1994-07-15 | 2001-05-29 | University Of Iowa Research Foundation | Immunostimulatory nucleic acid molecules |
| US5935821A (en) | 1995-01-17 | 1999-08-10 | Board Of Trustees Of The University Of Kentucky | Polynucleotides related to monoclonal antibody 1A7 and use for the treatment of melanoma and small cell carcinoma |
| WO1996030523A2 (en) | 1995-03-31 | 1996-10-03 | Hans Wolf | Antigen presentation system based on retrovirus-like particles |
| US5780448A (en) | 1995-11-07 | 1998-07-14 | Ottawa Civic Hospital Loeb Research | DNA-based vaccination of fish |
| EP0879284B1 (en) | 1996-01-30 | 2009-07-29 | The Regents of The University of California | Gene expression vectors which generate an antigen specific immune response and methods of using the same |
| EP0855184A1 (en) | 1997-01-23 | 1998-07-29 | Grayson B. Dr. Lipford | Pharmaceutical composition comprising a polynucleotide and an antigen especially for vaccination |
| GB9702021D0 (en) | 1997-01-31 | 1997-03-19 | Imperial College | Medicaments |
| DK1005368T3 (en) | 1997-03-10 | 2010-01-04 | Ottawa Hospital Res Inst | Use of nucleic acids containing non-methylated CpG dinucleotide in combination with alum as adjuvants |
| US6406705B1 (en) * | 1997-03-10 | 2002-06-18 | University Of Iowa Research Foundation | Use of nucleic acids containing unmethylated CpG dinucleotide as an adjuvant |
| AU7350798A (en) | 1997-04-29 | 1998-11-24 | Universiteit Utrecht | Corona virus-like particles as tools for vaccination and therapy |
| JP2002508748A (en) | 1997-05-01 | 2002-03-19 | カイロン コーポレイション | Use of virus-like particles as adjuvants |
| AU7690898A (en) | 1997-05-20 | 1998-12-11 | Ottawa Civic Hospital Loeb Research Institute | Vectors and methods for immunization or therapeutic protocols |
| JP4101888B2 (en) | 1997-06-06 | 2008-06-18 | ダイナバックス テクノロジーズ コーポレイション | Immunostimulatory oligonucleotides, compositions thereof and methods of use thereof |
| AU732468B2 (en) | 1997-06-23 | 2001-04-26 | Ludwig Institute For Cancer Research | Isolated nona- and decapeptides which bind to HLA molecules, and the use thereof |
| US6025470A (en) | 1997-06-23 | 2000-02-15 | Ludwig Institute For Cancer Research | Isolated nona- and decapeptides which bind to HLA molecules, and the use thereof |
| FR2764826B1 (en) | 1997-06-24 | 1999-09-03 | Tempora O | METHOD FOR CLEANING POROUS SURFACE, PARTICULARLY STONE, AND SUITABLE COMPOSITION |
| US6169175B1 (en) | 1997-08-06 | 2001-01-02 | Centers For Disease Control And Prevention | Preparation and use of recombinant influenza A virus M2 construct vaccines |
| JP4663113B2 (en) | 1997-09-05 | 2011-03-30 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | Use of immunostimulatory oligonucleotides to prevent or reduce antigen-stimulated granulocyte-mediated inflammation |
| US5989868A (en) | 1997-09-12 | 1999-11-23 | The Board Of Regents Of The University Of Oklahoma | Fusion protein systems designed to increase soluble cytoplasmic expression of heterologous proteins in esherichia coli |
| US6171591B1 (en) | 1997-12-08 | 2001-01-09 | Pentamer Pharmaceuticals, Inc. | Recombinant nodavirus compositions and methods |
| ID26669A (en) | 1998-02-12 | 2001-01-25 | Immune Complex Corp | INTI HEPATITIS B PROTEINS MODIFIED BY STRATEGY AND ITS DECREASES |
| ATE356630T1 (en) | 1998-04-03 | 2007-04-15 | Univ Iowa Res Found | METHOD AND PRODUCTS FOR STIMULATING THE IMMUNE SYSTEM USING IMMUNOTHERAPEUTIC OLIGONUCLEOTIDES AND CYTOKINE |
| US6541438B1 (en) * | 1998-05-01 | 2003-04-01 | The Procter & Gamble Company | Laundry detergent and/or fabric care compositions comprising a modified cellulase |
| US5990085A (en) | 1998-05-04 | 1999-11-23 | Michigan State University | Inhibin-HBc fusion protein |
| JP2002514397A (en) * | 1998-05-14 | 2002-05-21 | コーリー ファーマシューティカル ゲーエムベーハー | Methods for hematopoietic regulation using CpG oligonucleotides |
| AU761396B2 (en) | 1998-06-30 | 2003-06-05 | Om Pharma | Novel acyl pseudodipeptides, preparation method and pharmaceutical compositions containing same |
| WO2000006588A1 (en) | 1998-07-27 | 2000-02-10 | University Of Iowa Research Foundation | STEREOISOMERS OF CpG OLIGONUCLEOTIDES AND RELATED METHODS |
| US5962636A (en) | 1998-08-12 | 1999-10-05 | Amgen Canada Inc. | Peptides capable of modulating inflammatory heart disease |
| AU777225B2 (en) | 1998-09-03 | 2004-10-07 | Coley Pharmaceutical Gmbh | G-motif oligonucleotides and uses thereof |
| JP4486257B2 (en) | 1998-10-21 | 2010-06-23 | アメリカ合衆国 | Virus-like particles for induction of autoantibodies |
| US6380364B1 (en) | 1998-11-23 | 2002-04-30 | Loyola University Of Chicago | Chimeric biotin-binding papillomavirus protein |
| WO2000032227A2 (en) | 1998-11-30 | 2000-06-08 | Cytos Biotechnology Ag | Ordered molecular presentation of antigens, method of preparation and use |
| NZ512056A (en) | 1998-12-04 | 2004-01-30 | Biogen Inc | HBV core antigen particles with multiple immunogenic components attached via peptide ligands |
| BR9916522A (en) | 1998-12-23 | 2002-12-24 | Thompson Boyce Plant Res | Vector of expression of plants, cell, plant seed, polynucleotide, immunogenic composition, methods to elicit an immune response in a mammal and to isolate polypeptide, and transgenic plant or plant cell |
| TR200102493T2 (en) | 1999-02-25 | 2002-02-21 | Smithkline Beecham Biologicals S.A. | Epitopes or mimotopes derived from the C-epsilon-3 or C-epsilon-4 domains of IgE. |
| US6977245B2 (en) | 1999-04-12 | 2005-12-20 | The United States Of America As Represented By The Department Of Health And Human Services | Oligodeoxynucleotide and its use to induce an immune response |
| ES2228497T3 (en) | 1999-04-19 | 2005-04-16 | Glaxosmithkline Biologicals S.A. | ADJUTIVE COMPOSITION INCLUDING SAPONINA AND AN IMMUNO STIMULANT OLIGONUCLEOTIDE. |
| WO2001016320A1 (en) * | 1999-08-30 | 2001-03-08 | Ludwig Institute For Cancer Research | Isolated nona and decapeptides which bind to hla molecules, and the use thereof |
| WO2001022990A2 (en) * | 1999-09-27 | 2001-04-05 | Coley Pharmaceutical Group, Inc. | Methods related to immunostimulatory nucleic acid-induced interferon |
| US6949520B1 (en) | 1999-09-27 | 2005-09-27 | Coley Pharmaceutical Group, Inc. | Methods related to immunostimulatory nucleic acid-induced interferon |
| WO2001095935A1 (en) | 2000-01-20 | 2001-12-20 | Ottawa Health Research Institute | Immunostimulatory nucleic acids for inducing a th2 immune response |
| EP1253942A4 (en) | 2000-02-01 | 2004-06-16 | Tanox Inc | Cd40-binding apc-activating molecules |
| US7585847B2 (en) | 2000-02-03 | 2009-09-08 | Coley Pharmaceutical Group, Inc. | Immunostimulatory nucleic acids for the treatment of asthma and allergy |
| US6756044B1 (en) | 2000-02-09 | 2004-06-29 | Genvec, Inc. | Antigenic complexes and methods |
| MXPA02009895A (en) | 2000-04-07 | 2004-09-06 | Univ Leeds Innovations Ltd | Hepatitis b core antigen fusion proteins. |
| WO2001085208A2 (en) | 2000-05-05 | 2001-11-15 | Cytos Biotechnology Ag | Molecular antigen arrays and vaccines |
| AU6616301A (en) | 2000-06-22 | 2002-01-02 | Celltech Pharmaceuticals Ltd | Modification of hepatitis b core antigen |
| AUPQ912000A0 (en) | 2000-07-31 | 2000-08-24 | Crown In The Right Of The Queensland Department Of Health, The | Improved virus like particles |
| US20030138769A1 (en) | 2000-08-16 | 2003-07-24 | Birkett Ashley J. | Immunogenic HBc chimer particles having enhanced stability |
| WO2002053141A2 (en) | 2000-12-14 | 2002-07-11 | Coley Pharmaceutical Group, Inc. | Inhibition of angiogenesis by nucleic acids |
| US7094409B2 (en) * | 2001-01-19 | 2006-08-22 | Cytos Biotechnology Ag | Antigen arrays for treatment of allergic eosinophilic diseases |
| US7128911B2 (en) * | 2001-01-19 | 2006-10-31 | Cytos Biotechnology Ag | Antigen arrays for treatment of bone disease |
| US7320793B2 (en) | 2001-01-19 | 2008-01-22 | Cytos Biotechnology Ag | Molecular antigen array |
| US20030050268A1 (en) * | 2001-03-29 | 2003-03-13 | Krieg Arthur M. | Immunostimulatory nucleic acid for treatment of non-allergic inflammatory diseases |
| ES2335979T3 (en) * | 2001-09-14 | 2010-04-07 | Cytos Biotechnology Ag | IMMUNOSTIMULATOR CPG PACKAGING IN VIRUS SIMILAR PARTICLES: PREPARATION METHOD AND ITS USE. |
| AU2002347404A1 (en) * | 2001-09-14 | 2003-04-01 | Cytos Biotechnology Ag | In vivo activation of antigen presenting cells for enhancement of immune responses induced by virus like particles |
| US7115266B2 (en) * | 2001-10-05 | 2006-10-03 | Cytos Biotechnology Ag | Angiotensin peptide-carrier conjugates and uses thereof |
| EP1432443B1 (en) | 2001-10-05 | 2009-01-21 | Cytos Biotechnology AG | Angiotensin peptide-carrier conjugates and uses thereof |
| DK1455593T3 (en) | 2001-10-06 | 2013-08-26 | Merial Ltd | PROCEDURES AND COMPOSITIONS FOR PROMOTING GROWTH AND INJURY IMMUNITY OF YOUNG ANIMALS |
| US20030219459A1 (en) * | 2002-01-18 | 2003-11-27 | Cytos Biotechnology Ag | Prion protein carrier-conjugates |
| EP1513552B1 (en) | 2002-06-20 | 2010-12-01 | Cytos Biotechnology AG | Packaged virus-like particles in combination with cpg for use as adjuvants with allergens : method of preparation and use |
| AU2004224762B2 (en) * | 2003-03-26 | 2009-12-24 | Kuros Us Llc | Packaging of immunostimulatory oligonucleotides into virus-like particles: method of preparation and use |
| US7537767B2 (en) * | 2003-03-26 | 2009-05-26 | Cytis Biotechnology Ag | Melan-A- carrier conjugates |
| US20060210588A1 (en) * | 2003-03-26 | 2006-09-21 | Cytos Biotechnology Ag | Hiv-peptide-carrier-conjugates |
| US20060251623A1 (en) * | 2003-07-10 | 2006-11-09 | Caytos Biotechnology Ag | Packaged virus-like particles |
| RU2409667C2 (en) * | 2004-09-21 | 2011-01-20 | Цитос Биотехнологи Аг | Virus-like particles including hybrid protein of ap205 bacteriophage coat protein and antigen polypeptide |
| EP1973608A1 (en) * | 2005-12-14 | 2008-10-01 | Cytos Biotechnology AG | Immunostimulatory nucleic acid packaged particles for the treatment of hypersensitivity |
-
2004
- 2004-03-25 AU AU2004224762A patent/AU2004224762B2/en not_active Expired
- 2004-03-25 WO PCT/EP2004/003163 patent/WO2004084939A2/en active Application Filing
- 2004-03-25 ZA ZA200507063A patent/ZA200507063B/en unknown
- 2004-03-25 AU AU2004224761A patent/AU2004224761A1/en not_active Abandoned
- 2004-03-25 WO PCT/EP2004/003165 patent/WO2004084940A1/en active Application Filing
- 2004-03-25 CA CA002517675A patent/CA2517675A1/en not_active Abandoned
- 2004-03-25 NZ NZ542323A patent/NZ542323A/en unknown
- 2004-03-25 EP EP04723203A patent/EP1606398A1/en not_active Withdrawn
- 2004-03-25 RU RU2005129727/13A patent/RU2351362C2/en not_active IP Right Cessation
- 2004-03-25 WO PCT/EP2004/003164 patent/WO2004085635A1/en active Application Filing
- 2004-03-25 CA CA002519165A patent/CA2519165A1/en not_active Abandoned
- 2004-03-25 CN CNB2004800079060A patent/CN100560719C/en not_active Expired - Fee Related
- 2004-03-25 EP EP04723204A patent/EP1605972A2/en not_active Withdrawn
- 2004-03-25 CN CN200910175527A patent/CN101675993A/en active Pending
- 2004-03-25 JP JP2006504862A patent/JP5022028B2/en not_active Expired - Fee Related
- 2004-03-25 US US10/550,518 patent/US7517520B2/en active Active - Reinstated
- 2004-03-25 BR BRPI0408623-6A patent/BRPI0408623A/en not_active IP Right Cessation
- 2004-03-25 CA CA002517839A patent/CA2517839A1/en not_active Abandoned
- 2004-03-25 ZA ZA200507562A patent/ZA200507562B/en unknown
- 2004-03-25 KR KR1020057017197A patent/KR20050115913A/en not_active Application Discontinuation
- 2004-03-25 EP EP04723207A patent/EP1605973B1/en not_active Expired - Lifetime
- 2004-03-25 MX MXPA05009289A patent/MXPA05009289A/en active IP Right Grant
- 2004-03-25 AU AU2004223736A patent/AU2004223736B2/en not_active Ceased
-
2005
- 2005-08-16 ZA ZA200506541A patent/ZA200506541B/en unknown
-
2009
- 2009-03-24 US US12/410,085 patent/US20100098722A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5939598A (en) | 1990-01-12 | 1999-08-17 | Abgenix, Inc. | Method of making transgenic mice lacking endogenous heavy chains |
| WO2000039304A2 (en) | 1998-12-31 | 2000-07-06 | Chiron Corporation | Polynucleotides encoding antigenic hiv type c polypeptides, polypeptides and uses thereof |
| WO2001022972A2 (en) | 1999-09-25 | 2001-04-05 | University Of Iowa Research Foundation | Immunostimulatory nucleic acids |
Non-Patent Citations (24)
| Title |
|---|
| ALLEN, T.M. ET AL., J. IMMUNOL., vol. 164, 2000, pages 4968 - 4978 |
| BACHMANN; ZINKERNAGEL, ANN. REV. IMMUNOL., vol. 15, 1997, pages 235 - 270 |
| BEAUCAGE, S. L.; CARUTHERS, M. H., TET. LET., vol. 22, 1981, pages 1859 |
| BELSHE R.B. ET AL., J. INF. DIS., vol. 183, 2001, pages 1343 - 1352 |
| ENGLEMAN, E. G., CYTOTECHNOLOGY, vol. 25, 1997, pages 1 |
| FROEHLER ET AL., NUCL. ACID. RES., vol. 14, 1986, pages 5399 - 5407 |
| GAFFNEY ET AL., TET. LET., vol. 29, 1988, pages 2619 - 2622 |
| GAREGG ET AL., TET. LET., vol. 27, 1986, pages 4051 - 4054 |
| GAREGG ET AL., TET. LET., vol. 27, 1986, pages 4055 - 4058 |
| GERBER ET AL., J VIROL, vol. 75, 2001, pages 4752 - 4760 |
| GLUCKMAN, J. C., CYTOKINES, CELLULAR AND MOLECULAR THERAPY, vol. 3, 1997, pages 187 |
| GOLMOHAMMADI, R. ET AL., STRUCTURE, vol. 4, 1996, pages 543 - 554 |
| HANKE, T. ET AL., NAT. MED., vol. 6, 2000, pages 951 - 955 |
| J. MOL. BIOL., vol. 285, 1999, pages 1 - 32 |
| JIN X. ET AL., J. EXP. MED., vol. 189, 1999, pages 1365 - 1372 |
| KLINMAN ET AL., DRUG NEWS PERSPECT, vol. 13, 2000, pages 289 - 296 |
| MOINGEON P. ET AL., J.BIOTECHNOL., vol. 98, 2002, pages 189 - 198 |
| OXENIUS, A. ET AL., PROC. NATL. ACAD. SCI. USA, vol. 99, 2002, pages 13747 - 13752 |
| SAMBROOK, T. ET AL.: "Molecular Cloning: A Laboratory Manual", 1989, COLD SPRING HARBOR LABORATORY PRESS |
| SHIVER, J.W. ET AL., NATURE, vol. 415, 2002, pages 331 - 335 |
| STEINMAN, R. M., EXPERIMENTAL HEMATOLOGY, vol. 24, 1996, pages 849 |
| STORNI ET AL., J IMMUNOL, vol. 168, 2002, pages 2880 - 2886 |
| VAN SCHOOTEN, W. ET AL., MOLECULAR MEDICINE TODAY, vol. 255, June 1997 (1997-06-01) |
| YUAN ET AL., J. VIROL., vol. 73, 1999, pages 10122 - 10128 |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7959928B2 (en) | 2004-10-05 | 2011-06-14 | Cytos Biotechnology Ag | VLP-antigen conjugates and their uses as vaccines |
| WO2006037787A3 (en) * | 2004-10-05 | 2006-07-06 | Cytos Biotechnology Ag | Vlp-antigen conjugates and their uses as vaccines |
| WO2006083706A2 (en) * | 2005-01-31 | 2006-08-10 | Vaxinnate Corporation | Method to identify polypeptide toll-like receptor (tlr) ligands |
| WO2006083706A3 (en) * | 2005-01-31 | 2007-04-26 | Vaxinnate Corp | Method to identify polypeptide toll-like receptor (tlr) ligands |
| WO2006134125A1 (en) * | 2005-06-14 | 2006-12-21 | Cytos Biotechnology Ag | Antigen conjugates and uses thereof |
| FR2887457A1 (en) * | 2005-06-23 | 2006-12-29 | Fond Bettencourt Schueller | TRANSCUTANE TARGETING VACCINATION |
| US8956617B2 (en) | 2005-06-23 | 2015-02-17 | Fondation Bettencourt-Schueller | Vaccination by transcutaneous targeting |
| US8338582B2 (en) * | 2005-09-05 | 2012-12-25 | Fundacao De Amparo A Pesquisa Do Estado De Sao Paulo | Anti-HIV immunogens and methods for inducing an immune response |
| EP2407534A1 (en) | 2010-07-14 | 2012-01-18 | Neo Virnatech, S.L. | Methods and reagents for obtaining transcriptionally active virus-like particles and recombinant virions |
| WO2012007557A1 (en) | 2010-07-14 | 2012-01-19 | Neo Virnatech, S.L. | Methods and reagents for obtaining transcriptionally active virus-like particles and recombinant virions |
| WO2013092720A1 (en) | 2011-12-22 | 2013-06-27 | F. Hoffmann-La Roche Ag | Full length antibody display system for eukaryotic cells and its use |
| US10086056B2 (en) | 2015-01-15 | 2018-10-02 | University Of Copenhagen | Virus-like particle with efficient epitope display |
| US10526376B2 (en) | 2015-01-15 | 2020-01-07 | University Of Copenhagen | Virus-like particle with efficient epitope display |
| US11497800B2 (en) | 2015-01-15 | 2022-11-15 | University Of Copenhagen | Virus-like particle with efficient epitope display |
| US11129882B2 (en) | 2015-10-30 | 2021-09-28 | University Of Copenhagen | Virus like particle with efficient epitope display |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2004223736B2 (en) | Melan-A peptide analogue-virus-like-particle conjugates | |
| US20110097417A1 (en) | Melan-a-carrier conjugates | |
| AU2009200115B2 (en) | Packaging of immunostimulatory substances into virus-like particles: method of preparation and use | |
| AU2002339224A1 (en) | Packaging of immunostimulatory substances into virus-like particles: method of preparation and use | |
| EP1425040A2 (en) | In vivo activation of antigen presenting cells for enhancement of immune responses induced by virus like particles | |
| US20060210588A1 (en) | Hiv-peptide-carrier-conjugates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| CFP | Corrected version of a pamphlet front page | ||
| CR1 | Correction of entry in section i |
Free format text: IN PCT GAZETTE 41/2004 UNDER (71) THE ADDRESS SHOULD READ "WAGISTRASSE" |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4109/DELNP/2005 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2519165 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004224761 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200507562 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10550580 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2004224761 Country of ref document: AU Date of ref document: 20040325 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004224761 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004723204 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004723204 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10550580 Country of ref document: US |