WO2007001975A1 - Piperidine derivatives useful as histamine h3 antagonists - Google Patents
Piperidine derivatives useful as histamine h3 antagonists Download PDFInfo
- Publication number
- WO2007001975A1 WO2007001975A1 PCT/US2006/023800 US2006023800W WO2007001975A1 WO 2007001975 A1 WO2007001975 A1 WO 2007001975A1 US 2006023800 W US2006023800 W US 2006023800W WO 2007001975 A1 WO2007001975 A1 WO 2007001975A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- alkyl
- group
- aryl
- mmol
- Prior art date
Links
- 0 CC*CCC(C1)N(*)CCC1N Chemical compound CC*CCC(C1)N(*)CCC1N 0.000 description 5
- NGRAZFZFUIQXCR-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCC1C(N(CC1)CCC1Oc1c(C(OC)=O)[s]cc1Br)=O)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1C(N(CC1)CCC1Oc1c(C(OC)=O)[s]cc1Br)=O)=O NGRAZFZFUIQXCR-UHFFFAOYSA-N 0.000 description 1
- RLGBRERZUYUHRC-UHFFFAOYSA-N CCC(C)c1cc(Cl)ccc1NC(CC1)CCN1C(OC(C)(C)C)=O Chemical compound CCC(C)c1cc(Cl)ccc1NC(CC1)CCN1C(OC(C)(C)C)=O RLGBRERZUYUHRC-UHFFFAOYSA-N 0.000 description 1
- GPVFLUBOWYOABY-UHFFFAOYSA-N CCOC(C1CCN(Cc2ccc(CN(C)C)[o]2)CC1)=O Chemical compound CCOC(C1CCN(Cc2ccc(CN(C)C)[o]2)CC1)=O GPVFLUBOWYOABY-UHFFFAOYSA-N 0.000 description 1
- PIZRVCOLKVUARQ-UHFFFAOYSA-N CN1CC(CN(CC2)CCC2C(N(CC2)CCC2Oc(c(F)cc(F)c2)c2F)=O)CC1 Chemical compound CN1CC(CN(CC2)CCC2C(N(CC2)CCC2Oc(c(F)cc(F)c2)c2F)=O)CC1 PIZRVCOLKVUARQ-UHFFFAOYSA-N 0.000 description 1
- NKIGMVAVKJSMES-UHFFFAOYSA-O CN1NC=CC(CN(Cc(c(F)ccc2)c2F)C(CC2)CCN2C(C2(CCN(CC(C=C[NH3+])=C[NH2+2])CC2)F)=O)=C1 Chemical compound CN1NC=CC(CN(Cc(c(F)ccc2)c2F)C(CC2)CCN2C(C2(CCN(CC(C=C[NH3+])=C[NH2+2])CC2)F)=O)=C1 NKIGMVAVKJSMES-UHFFFAOYSA-O 0.000 description 1
- PCWRXIRLSXVNNJ-UHFFFAOYSA-N Fc(cc1F)cc(F)c1OC1CCNCC1 Chemical compound Fc(cc1F)cc(F)c1OC1CCNCC1 PCWRXIRLSXVNNJ-UHFFFAOYSA-N 0.000 description 1
- IBLMYGXJKQIGSN-UHFFFAOYSA-N Fc1ccc(CBr)c(F)c1 Chemical compound Fc1ccc(CBr)c(F)c1 IBLMYGXJKQIGSN-UHFFFAOYSA-N 0.000 description 1
- QHDKGQLEHAOVHJ-UHFFFAOYSA-O Fc1ccc(COC2CC[NH2+]CC2)c(F)c1 Chemical compound Fc1ccc(COC2CC[NH2+]CC2)c(F)c1 QHDKGQLEHAOVHJ-UHFFFAOYSA-O 0.000 description 1
- LGUCFVGCLQSJDU-UHFFFAOYSA-N Nc1cc(CN(CC2)CCC2C(N(CC2)CCC2OCc(c(F)c2)ccc2F)=O)ccn1 Chemical compound Nc1cc(CN(CC2)CCC2C(N(CC2)CCC2OCc(c(F)c2)ccc2F)=O)ccn1 LGUCFVGCLQSJDU-UHFFFAOYSA-N 0.000 description 1
- OBEGNVNUXQKIOW-UHFFFAOYSA-N O=C(C(F)(F)F)N(Cc(c(F)c1)ncc1F)C1CCNCC1 Chemical compound O=C(C(F)(F)F)N(Cc(c(F)c1)ncc1F)C1CCNCC1 OBEGNVNUXQKIOW-UHFFFAOYSA-N 0.000 description 1
- SSHAZVIQAIKSFI-UHFFFAOYSA-O O=C(C1(CCN(CC2=C[NH2+]NC=C2)CC1)F)N(CC1)CCC1NCc(c(F)ccc1)c1F Chemical compound O=C(C1(CCN(CC2=C[NH2+]NC=C2)CC1)F)N(CC1)CCC1NCc(c(F)ccc1)c1F SSHAZVIQAIKSFI-UHFFFAOYSA-O 0.000 description 1
- HBTODJVTRUUOQW-UHFFFAOYSA-N O=C(C1(CCNCC1)F)N(CC1)CCC1NCc(c(F)ccc1)c1F Chemical compound O=C(C1(CCNCC1)F)N(CC1)CCC1NCc(c(F)ccc1)c1F HBTODJVTRUUOQW-UHFFFAOYSA-N 0.000 description 1
- JHCKFFYUSIXFOL-UHFFFAOYSA-N O=C(C1CCN(CC2CNCC2)CC1)N(CC1)CCC1Oc(c(F)cc(F)c1)c1F Chemical compound O=C(C1CCN(CC2CNCC2)CC1)N(CC1)CCC1Oc(c(F)cc(F)c1)c1F JHCKFFYUSIXFOL-UHFFFAOYSA-N 0.000 description 1
- CLKAMJFVHJNYTG-UHFFFAOYSA-N O=C(C1CCN(Cc2ccncc2)CC1)N(CC1)CCC1NC(NCc(cc1)ccc1Cl)=O Chemical compound O=C(C1CCN(Cc2ccncc2)CC1)N(CC1)CCC1NC(NCc(cc1)ccc1Cl)=O CLKAMJFVHJNYTG-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/18—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carboxylic acids, or sulfur or nitrogen analogues thereof
- C07D295/182—Radicals derived from carboxylic acids
- C07D295/185—Radicals derived from carboxylic acids from aliphatic carboxylic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/08—Bronchodilators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/02—Non-specific cardiovascular stimulants, e.g. drugs for syncope, antihypotensives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Definitions
- the present invention relates to heteroatom-linked substituted piperidines and derivatives thereof useful as histamine H 3 antagonists.
- the invention also relates to pharmaceutical compositions comprising said compounds and their use in treating inflammatory diseases, allergic conditions, obesity, metabolic syndrome, cognition deficit disorders, cardiovascular and central nervous system disorders.
- the invention also relates to the use of a combination of histamine H 3 antagonists of this invention with histamine Hi compounds for the treatment of inflammatory diseases and allergic conditions, as well to the use of a combination of an histamine H 3 antagonist of this invention with other actives useful for treating obesity, metabolic syndrome or cognition deficit disorders.
- Pharmaceutical compositions comprising a combination of one or more novel histamine H 3 antagonist compounds of the invention with one or more histamine H-i compounds or one or more compounds useful for treating obesity, metabolic syndrome or cognition deficit disorders are also contemplated.
- H 1 , H 2 , H 3 and H 4 have been characterized by their pharmacological behavior.
- the H 1 receptors are those that mediate the response antagonized by conventional antihistamines.
- H 1 receptors are present, for example, in the ileum, the skin, and the bronchial smooth muscle of humans and other mammals.
- the most prominent H 2 receptor-mediated responses are the secretion of gastric acid in mammals and the chronotropic effect in isolated mammalian atria.
- H 4 receptors are expressed primarily on eosinophils and mast cells and have been shown to be involved in the chemotaxis of both cell types.
- H 3 receptor sites are found on sympathetic nerves, where they modulate sympathetic neurotransmission and attenuate a variety of end organ responses under control of the sympathetic nervous system. Specifically, H 3 receptor activation by histamine attenuates norepinephrine outflow to resistance and capacitance vessels, causing vasodilation. In addition, in rodents, peripheral H 3 receptors are expressed in brown adipose tissue, suggesting that they may be involved in thermogenesis regulation. H 3 receptors are also present in the CNS. H 3 receptor expression is observed in cerebral cortex, hippocampal formation, hypothalamus and other parts of the human and animal brain.
- H 3 receptors are expressed on histaminergic neurons and, as heteroreceptors, on neurons involved in other neurotransmitter systems, where H 3 receptor activation results in presynaptic inhibition of neurotransmitter release.
- H 3 receptors have been implicated in the regulation of histamine hypothalamic tone, which in turn has been associated with the modulation of sleeping, feeding and cognitive processes in the human brain (see, for example, Leurs et al., Nature Reviews, Drug Discovery, 4, (2005), 107). It is also known and has been described in the literature that histamine is involved in regulation of cognitive and memory processes in the human brain (see, for example, Life Sciences, 72, (2002), 409-414).
- H 3 receptor antagonists may be useful in treating various neuropsychiatric conditions, where cognitive deficits are an integral part of the disease, specifically ADHD, schizophrenia and Alzheimer's disease (see, for example, Hancock, A.; Fox, G. in Drug Therapy ⁇ e ⁇ . Buccafusco, J.J.). (Birkhauser, Basel, 2003).
- Imidazole H3 receptor antagonists are well known in the art. More recently, non-imidazole H3 receptor antagonists have been disclosed in US Patents 6,720,328 and 6,849,621 and in and 2004/0019099. WO 2003/103669 and WO 2003/088967 (US Published Applications 2004/0097483 and 2004/0048843) disclose 1-(4- piperidinyl)-benzimidazolone and 1-(4-piperidinyl)benzimidazole derivatives. All these patents or publications are incorporated by reference.
- the present invention provides novel compounds of formula I: or a pharmaceutically acceptable salt thereof, wherein: a is O, 1 or 2; b is 0, 1 or 2; n is 1 , 2 or 3; p is 1 , 2 or 3;
- M 1 is CH or N
- M 2 is CH, CF or N
- M 3 is CH or N with the proviso that when M 2 and M 3 are each N, p is 2 or 3;
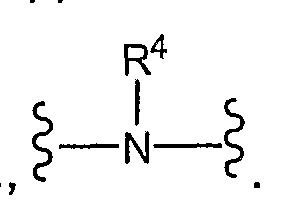
- X is -N(R 4 )-, -N(R 4 )-CH(R 19 )-, -CH(R 19 )-N(R 4 )-, -(CH 2 )rC(O)-N(R 4 )-, -O-(CH 2 ) 2 -C(O)-N(R 4 )-, -CH 2 -O-(CH 2 ) 3 -C(O)-N(R 4 )-, -CH(R 19 )CH(R 19 )N(R 4 )-, -(CH 2 ) t - N(R 4 )-C(O)-, -C(O)-N(R 4 )-CH 2 -, -(CH 2 ) r -N(R 19 )C(O)N(R 19 )-, -N(R 19 )C(O)N(R 19 )-(CH 2 ) r , -(CH 2 VOC(O)
- R 1 is H, R 10 -alkyl, R 10 -cycloalkyl, R 10 -aryl, R 10 -monoheteroaryl, R 10 -heterocycloalkyl or a group of the formula: - A -
- ring A is a monoheteroaryl ring
- said monoheteroaryl or monoheteroaryl ring is a monocyclic ring having 1 to 4 heteroatoms selected from O, S, and N, said heteroatoms interrupting an aromatic carbocyclic ring structure having from 1 to 6 carbon atoms, provided that there are no adjacent oxygen and/or sulfur atoms present with monoheteroaryl rings, such as isothiazole, isoxazole, oxazole, triazole, tetrazole, thiazole, thiophene, furane, pyrrole, pyrazole, pyrane, pyrimidine, pyrazine, furaranyl, pyridazine, and pyridine (incuding pyridine N-oxide) being preferred;
- R 2 is R 16 -alkyl, R 16 -alkenyl, R 16 -aryl, R 16 -heteroaryl, R 16 -cycloalkyl or R 16 -heterocycloalkyl;
- R 3 is H, alkyl, R 21 -aryl, R 22 -cycloalkyl, R 22 -heterocycloalkyl, R 21 -heteroaryl or -C(O)NH 2 ;
- R 4 is H, alkyl, haloalkyl, R 18 -aryl, R 18 -heteroaryl, R 18 -arylalkyl, -C(O)R 12 or -SO 2 R 13 ;
- R 7 is H, alkyl, haloalkyl, R 10 -aryl or R 10 -heteroaryl;
- R 8 and R 8 independently are 1 , 2 or 3 substituents independently selected from the group consisting of H, R 10 -cycloalkyl, R 10 -heterocycloalkyl, R 10 -aryl, R 10 -heteroaryl and haloalkyl, preferably -CF 3 ; each R 9 is independently selected from the group consisting of H and alkyl;
- R 12 is alkyl, R 18 -cycloalkyl, R 18 -aryl, R 18 -heteroaryl or R 18 -heterocycloalkyl;
- R 13 is alkyl, aryl or alkylsulfonylalkyl
- R 16 is 1 , 2 or 3 substituents independently selected from the group consisting of H, halo, alkyl, R 10 -cycloalkyl, -OH, alkoxy, hydroxyalkyl, R 10 -aryl, R 10 -heteroaryl, R 10 - heterocycloalkyl, R 10 -aryloxy, haloalkyl, preferably -CF 3 , haloalkoxy, preferably -OCF 3 , -NO 2 , -CO 2 R 17 , -N(R 17 ) 2 , -alkylene-N(R 17 ) 2 , -CON(R 17 ) 2 , -NHC(O)R 17 , -NHC(O)OR 17 , - NHSO 2 R 17 , -SO 2 N(R 17 ) 2 and -CN; each R 17 is independently selected from the group consisting of H, alkyl, haloalkyl, aryl, heteroaryl, cycl
- R 18 is 1 , 2 or 3 substituents independently selected from the group consisting of H, alkyl, halo, alkoxy, haloalkyl, preferably -CF 3 , -NO 2 , -CN and -alkylene-N(R 17' ) 2 ; each R 17 is independently selected from the group consisting of H, alkyl, haloalkyl, aryl, heteroaryl, cycloalkyl and heterocycloalkyl R 19 is independently selected from the group consisting of H and alkyl;
- R 20 is independently selected from the group consisting of H and alkyl
- R 21 is 1 , 2, 3 or 4 substituents independently selected from the group consisting of H, halo, alkyl, -OH, alkoxy, haloalkyl, preferably -CF 3 , or -CHF 2 , haloalkoxy, preferably -OCF 3 , -NO 2 , -CN, -C(O)N(R 19 ) 2 and -N(R 19 ) 2 ;
- R 22 is 1 , 2 or 3 substituents independently selected from the group consisting of halo, alkyl, -OH, alkoxy, haloalkyl, preferably -CF 3 , -NO 2 and -CN; and
- R 23 is independently 1 , 2 or 3 substituents independently selected from the group consisting of H, R 10 -cycloalkyl, R 10 -heterocycloalkyl, R 10 -aryl, heteroaryl, haloalkyl, preferably -CF 3 , halo, -CN, -OH, alkoxy, haloalkoxy, preferably -OCF 3 , -NO 2 , and -N(R 9 ) 2 .
- R 23' is independently 1 , 2 or 3 substituents independently selected from the group consisting of H, R 10 -cycloalkyl, R 10 -heterocycloalkyl, R 10 -aryl, heteroaryl, haloalkyl, preferably -CF 3 , -OH, alkoxy, haloalkoxy, preferably -OCF 3 , -NO 2 , and - N(R 9 ) 2 .
- This invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising an effective amount of at least one compound of formula I and a pharmaceutically acceptable carrier.
- This invention further provides a method of treating: allergy, allergy-induced airway (e.g., upper airway) responses (e.g., pruritis, sneezing, rhinorrhea, mucosal inflammation; see, for example, McLeod, JPET, 305 (2003) 1037), congestion (e.g., nasal congestion), hypotension, cardiovascular disease, diseases of the Gl tract, hyper- and hypo- motility and acidic secretion of the gastro-intestinal tract, metabolic syndrome, obesity, sleeping disorders (e.g., hypersomnia, somnolence, and narcolepsy), hypo- and hyperactivity of the central nervous system (for example, agitation and depression), cognition deficit disorders (such as attention deficit hyperactivity disorder (ADHD), Alzheimer's Disease (AD) and schizophrenia) nonalcoholic fatty liver disease (NAFLD), hepatic steatosis, nonalcoholic steatohepatitis (NASH), cirrhosis, hepatocellular carcinoma and/or other CNS
- Patient means a mammal, typically a human, although veterinary use is also contemplated.
- compounds of this invention are useful for treating congestion, metabolic syndrome, obesity and cognition deficit disorders.
- This invention further provides a pharmaceutical composition
- a pharmaceutical composition comprising an effective amount of a combination of at least one compound of formula I and at least one H 1 receptor antagonist in combination with a pharmaceutically acceptable carrier.
- This invention further provides a method of treating allergy, allergy-induced airway responses, and/or congestion comprising administering to a patient in need of such treatment an effective amount of a combination of at least one compound of formula I and at least one Hi receptor antagonist.
- This invention further provides a pharmaceutical composition comprising an effective amount of a combination of at least one compound of formula I and at least one other compound useful in treating obesity, metabolic syndrome, cognition deficit disorders, NAFLD, hepatic steatosis, NASH, cirrhosis, or hepatacellular carcinoma in combination with a pharmaceutically acceptable carrier.
- This invention further provides a method of treating obesity, metabolic syndrome or cognition deficit disorders comprising administering to a patient in need of such treatment an effective amount of a combination of at least one compound of formula I and at least one other compound useful in treating obesity, metabolic syndrome, cognition deficit disorders, NAFLD, hepatic steatosis, NASH, cirrhosis, or hepatacellular carcinoma.
- Kits comprising a compound of formula I in a pharmaceutical composition, and a separate Hi receptor antagonist in a pharmaceutical composition in a single package are also contemplated, as are kits comprising a compound of formula I in a pharmaceutical composition, and at least one separate compound useful in treating obesity, metabolic syndrome, cognition deficit disorders, NAFLD, hepatic steatosis, NASH, cirrhosis or hepatacellular carcinoma in a pharmaceutical composition in a single package.
- R 1 is preferably R 10 -aryl or R 10 -heteroaryl. More preferably, R 1 is R 10 -phenyl or R 10 -heteroaryl wherein heteroaryl is a 6-membered ring, especially R 10 -pyridyl.
- R 10 is preferably 1 , 2 or 3 substituents independently selected from H, alkyl, halo, -CF 3 , -
- M 1 is preferably N.
- M 2 is preferably CH or CF, more preferably CF.
- M 3 is preferably N.
- Variables n and p are preferably each 2.
- Variables a and b are preferably each independently 0 or 1 , more preferably 0.
- R 2 is preferably R 16 -heteroaryl or R 16 -heterocycloalkyl, more preferably a 5 or 6 membered R 16 -heteroaryl or a 4, 5 or 6-membered R 16 -heterocycloalkyl. More preferred R 2 groups are R 16 -pyridyl, R 16 -pyrimidyl, R 16 -pyradazinyl,
- R 16 -tetrahydropyranyl, R 16 -azetidinyl, R 16 -oxazolyl and R 16 -thiazolyl When R 2 is R 16 - pyridyl, R 16 -pyrimidyl, R 16 -pyradazinyl, R 16 -oxazolyl or R 16 -thiazolyl, R 16 is preferably 1 or 2 substituents independently selected from H, -CH 3 , -NH 2 and -NHCH 3 . When R 2 is R 16 -tetrahydropyranyl or R 16 -azetidinyl, R 16 is preferably 1 or 2 substituents independently selected from H and -CH 3 .
- 2-amino pyridyl 2- amino oxazolyl, 2-amino thiazolyl, 1-methyl-azetidinyl and tetrahydropyranyl, with 2- amino pyridyl being most preferred.
- R 3 is preferably H, R 22 -aryl, wherein R 22 is H, Or -C(O)NH 2 , most preferably H.
- X is preferably -NR 4 -, -NR 4 CH 2 -, -O-, -OCH 2 -, -NR 4 C(O)-, -C(O)NR 4 -, -S- or -SO 2 -, more preferably -O-, -NR 4 - or -S-.
- R 4 is preferably H, alkyl or -C(O)R 12 , more preferably H or alkyl.
- R 12 is preferably alkyl or aryl, more preferably alkyl or phenyl.
- R 5 and R 6 are independently preferably selected from H, alkyl, OH or fluoro.
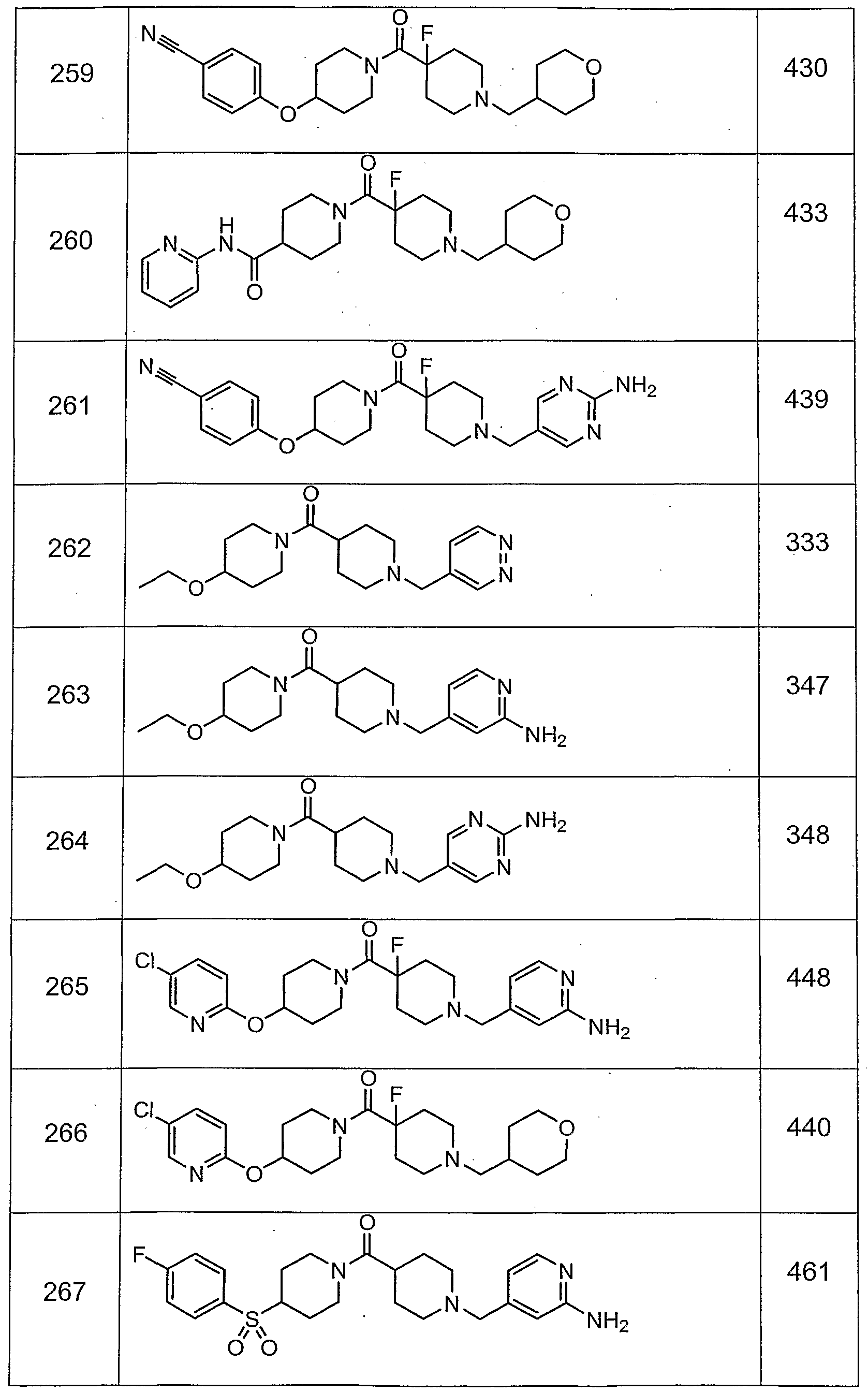
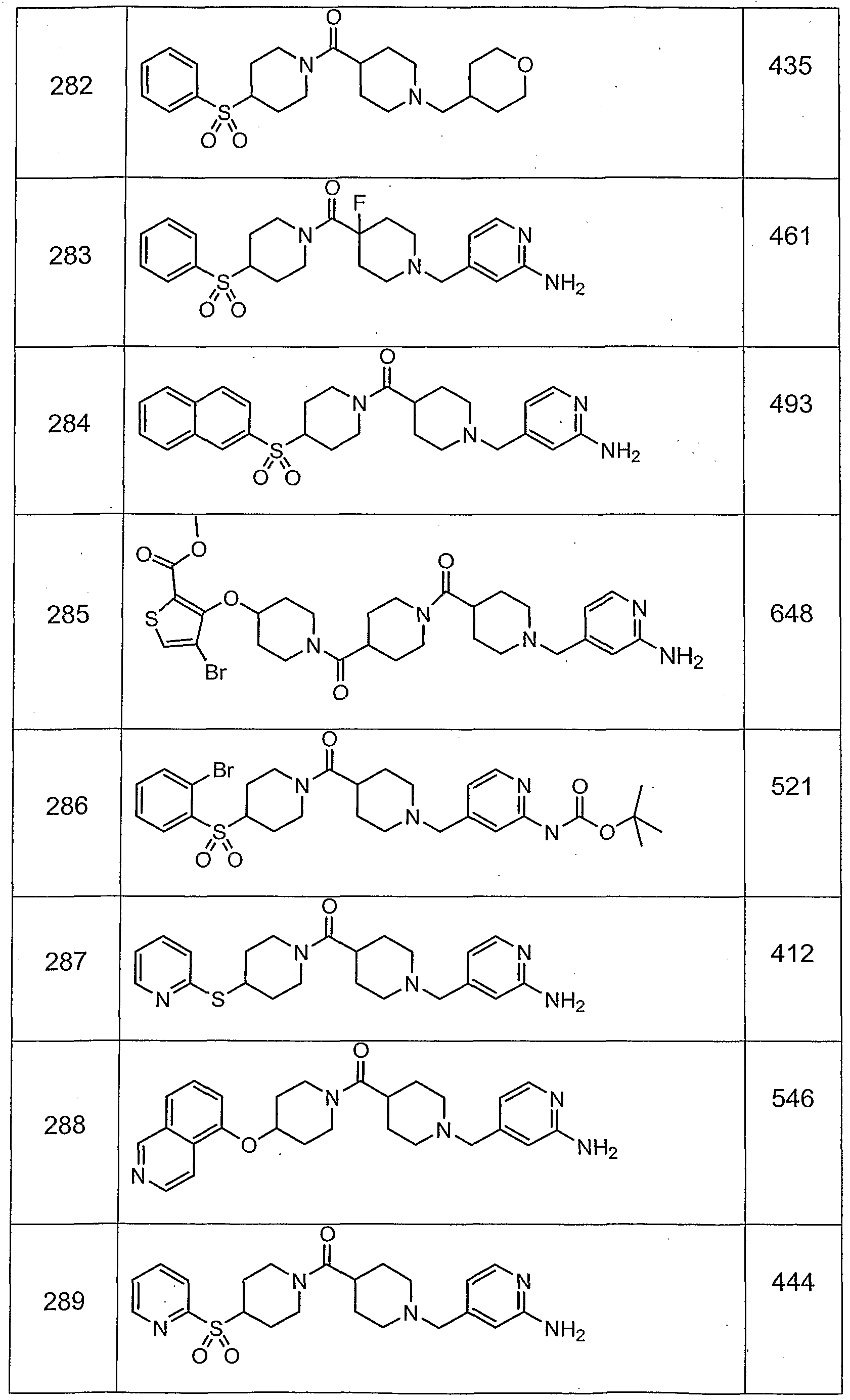
- Preferred compounds among those exemplified below are examples 11 , 15, 24, 26N, 30, 31 , 33, 34, 35, 36, 80, 81 , 88, 195, 202, 219, 220, 225, 227, 230, 237, 256, 257, 259, 260, 265, 274, 280, 290, 294, 295, 298, 299, 300, 301 , 308, 309 and 310.
- More preferred compounds of this invention are selected from examples 15, 26N, 33, 34, 35, 36, 80, 81 , 219, 237, 257, 265, 274 298, 299, 300 and 301.
- the left side of all divalent radicals is attached to the left portion of formula I and the right side of all divalent radicals is attached the the right side of the formula based upon where the variable is in formula I; e.g; when X is -CH 2 -0-(CH 2 ) 3 -C(O)- N(R 4 )-, R 1 is attached to the left or "-CH 2 -O" side of the variable and the right or "N(R 4 )-" side is attached to the left hand ring along with the R 3 substituent.
- alkyl represents straight and branched carbon chains and contains from one to six carbon atoms
- alkylene represents a divalent straight or branched alkyl chain, e.g., ethylene (-CH 2 -) or propylene (-CH 2 CH 2 CH 2 -);
- haloalkyl or haloalkoxy represent alkyl or alkoxy chains as defined above wherein one or more hydrogen atoms are replaced by halogen atoms, e.g., -CF 3 , CF 3 CH 2 CH 2 -, CF 3 CF 2 - or CF 3 O-;
- aryl (including the aryl portion of arylalkyl) represents a monocyclic or multicyclic carbocyclic group containing from 6 to 15 carbon atoms and having at least one aromatic ring (e.g., aryl is a phenyl or naphthyl ring), with all available substitutable
- Preferred cycloalkyl rings contain about 3 to about 7 ring atoms.
- suitable monocyclic cycloalkyls include cyclopropyl, cyclopentyl, cyclohexyl, cycloheptyl and the like.
- suitable multicyclic cycloalkyls include 1-decalinyl, norbomyl, adamantyl and the like.
- halogen represents fluoro, chloro, bromo and iodo
- heteroaryl represents cyclic groups having 1 to 4 heteroatoms selected from O, S or N, said heteroatom interrupting a carbocyclic ring structure and having a sufficient number of delocalized pi electrons to provide aromatic character, with the aromatic heterocyclic groups preferably containing from 2 to 14 carbon atoms.
- the rings do not contain adjacent oxygen and/or sulfur atoms.
- Examples include but are not limited to isothiazolyl, isoxazolyl, oxazolyl, furazanyl, triazolyl, tetrazolyl, thiazolyl, thienyl, furanyl (furyl), pyrrolyl, pyrazolyl, pyranyl, pyrimidinyl, pyrazinyl, pyridazinyl, pyridyl (e.g., 2-, 3-, or 4-pyridyl), pyridyl N-oxide (e.g., 2-, 3-, or 4-pyridyl N-oxide), triazinyl, pteridinyl, indolyl (benzopyrrolyl), pyridopyrazinyl, isoquinolinyl, quinolinyl, naphthyridinyl; all available substitutable carbon and nitrogen atoms can be substituted as defined.
- heterocycloalkyl represents a saturated carbocylic ring containing from 3 to 15 carbon atoms, preferably from 4 to 6 carbon atoms, which carbocyclic ring is interrupted by 1 to 3 hetero atoms selected from -O-, -S-, -SO-, -SO 2 or -NR 40 - wherein R 40 represents H 1 C 1 to C 6 alkyl, arylalkyl, -C(O)R 20 , -C(O)OR 20 , or
- each R is independently selected from the group consisting of H, alkyl, phenyl and benzyl
- examples include but are not limited to 2- or 3- tetrahydrofuranyl, 2- or 3- tetrahydrothienyl, 2-, 3- or 4-piperidinyl, 2- or 3-pyrrolidinyl, 2- or 3-piperizinyl, 2- or 4-dioxanyl, 1 ,3-dioxolanyl, 1 ,3,5-trithianyl, pentamethylene sulfide, perhydroisoquinolinyl, decahydroquinolinyl, trimethylene oxide, azetidinyl, 1- azacycloheptanyl, 1 ,3-dithianyl, 1 ,3,5-trioxanyl, morpholinyl, thiomorpholinyl, 1 ,4- thioxanyl, and 1 ,3,5-hexahydr
- Effective amount or “therapeutically effective amount” is meant to describe an amount of compound or a composition of the present invention effective in inhibiting the above-noted diseases and thus producing the desired therapeutic, ameliorative, inhibitory or preventative effect.
- a line drawn into a ring means that the indicated bond may be attached to any of the substitutable ring carbon atoms.
- substituted means that one or more hydrogens on the designated atom is replaced with a selection from the indicated group, provided that the designated atom's normal valency under the existing circumstances is not exceeded, and that the substitution results in a stable compound. Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- stable compound' or “stable structure” is meant a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent.
- Claim 1 does not include compounds known by the skilled artisan to be unstable.
- the term “purified”, “in purified form” or “in isolated and purified form” for a compound refers to the physical state of said compound after being isolated from a synthetic process or natural source or combination thereof.
- the term “purified”, “in purified form” or “in isolated and purified form” for a compound refers to the physical state of said compound after being obtained from a purification process or processes described herein or well known to the skilled artisan, in sufficient purity to be characterizable by standard analytical techniques described herein or well known to the skilled artisan.
- protecting groups When a functional group in a compound is termed "protected", this means that the group is in modified form to preclude undesired side reactions at the protected site when the compound is subjected to a reaction. Suitable protecting groups will be recognized by those with ordinary skill in the art as well as by reference to standard textbooks such as, for example, T. W. Greene et al, Protective Groups in organic Synthesis (1991), Wiley, New York.
- composition is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- Prodrugs and solvates of the compounds of the invention are also contemplated herein.
- a discussion of prodrugs is provided in T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems (1987) 14 of the A.C.S. Symposium Series, and in Bioreversible Carriers in Drug Design, (1987) Edward B. Roche, ed., American Pharmaceutical Association and Pergamon Press.
- the term "prodrug” means a compound (e.g, a drug precursor) that is transformed in vivo to yield a compound of Formula (I) or a pharmaceutically acceptable salt, hydrate or solvate of the compound. The transformation may occur by various mechanisms (e.g., by metabolic or chemical processes), such as, for example, through hydrolysis in blood.
- a prodrug can comprise an ester formed by the replacement of the hydrogen atom of the acid group with a group such as, for example, (Ci-C 8 )alkyl, (C 2 -C-
- a prodrug can be formed by the replacement of the hydrogen atom of the alcohol group with a group such as, for example, (C 1 -C 6 )alkanoyloxymethyl, 1-((C 1 -C 6 )alkanoyloxy)- ethyl, 1-methyl-1-((C 1 -C 6 )alkanoyloxy)ethyl, (C 1 -C 6 )alkoxycarbonyloxymethyl, N-(Cr C 6 )alkoxycarbonylaminomethyl, succinoyl, (C 1 -C 6 )alkanoyl, ⁇ -amino(C 1 -C 4 )alkanyl, arylacyl and ⁇ -aminoacyl, or ⁇ -aminoacyl- ⁇ -aminoacyl, where each ⁇ -aminoacyl group is independently selected from the naturally occurring L-amino acids, P(O)(
- a prodrug can be formed by the replacement of a hydrogen atom in the amine group with a group such as, for example, R"-carbonyl, R"O-carbonyl, NR"R"'-carbonyl where R" and R"' are each independently (CrC 10 )alkyl, (C 3 -C 7 ) cycloalkyl, benzyl, or R"- carbonyl is a natural ⁇ -aminoacyl or natural ⁇ -aminoacyl, — C(OH)C(O)OY 1 wherein Y 1 is H, (C 1 -C 6 )alkyl or benzyl, — C(OY 2 )Y 3 wherein Y 2 is (C 1 -C 4 ) alkyl and Y 3 is (C 1 - C 6 )alkyl, carboxy (C r C 6 )alkyl, amino(Ci-C 4 )alky
- C 6 )alkylaminoalkyl — C(Y 4 )Y 5 wherein Y 4 is H or methyl and Y 5 is mono-N — or di- N,N-(C 1 -C 6 )alkylamino morpholino, piperidin-1-yl or pyrrol id in-1-yl, and the like.
- Solvate means a physical association of a compound of this invention with one or more solvent molecules. This physical association involves varying degrees of ionic and covalent bonding, including hydrogen bonding. In certain instances the solvate will be capable of isolation, for example when one or more solvent molecules are incorporated in the crystal lattice of the crystalline solid.
- Solvate encompasses both solution-phase and isolatable solvates. Non-limiting examples of suitable solvates include ethanolates, methanolates, and the like.
- “Hydrate” is a solvate wherein the solvent molecule is H 2 O.
- the compounds of Formula I can form salts which are also within the scope of this invention.
- Reference to a compound of Formula I herein is understood to include reference to salts thereof, unless otherwise indicated.
- the term "salt(s)", as employed herein, denotes acidic salts formed with inorganic and/or organic acids, as well as basic salts formed with inorganic and/or organic bases.
- zwitterions inner salts may be formed and are included within the term "salt(s)" as used herein.
- Salts of the compounds of the Formula I may be formed, for example, by reacting a compound of Formula I with an amount of acid or base, such as an equivalent amount, in a medium such as one in which the salt precipitates or in an aqueous medium followed by lyophilization.
- Exemplary acid addition salts include acetates, ascorbates, benzoates, benzenesulfonates, bisulfates, borates, butyrates, citrates, camphorates, camphorsulfonates, fumarates, hydrochlorides, hydrobromides, hydroiodides, lactates, maleates, methanesulfonates, naphthalenesulfonates, nitrates, oxalates, phosphates, propionates, salicylates, succinates, sulfates, tartarates, thiocyanates, toluenesulfonates (also known as tosylates,) and the like.
- Exemplary basic salts include ammonium salts, alkali metal salts such as sodium, lithium, and potassium salts, alkaline earth metal salts such as calcium and magnesium salts, salts with organic bases (for example, organic amines) such as dicyclohexylamines, t-butyl amines, and salts with amino acids such as arginine, lysine and the like.
- Basic nitrogen-containing groups may be quarternized with agents such as lower alkyl halides (e.g. methyl, ethyl, and butyl chlorides, bromides and iodides), dialkyl sulfates (e.g.
- dimethyl, diethyl, and dibutyl sulfates dimethyl, diethyl, and dibutyl sulfates
- long chain halides e.g. decyl, lauryl, and stearyl chlorides, bromides and iodides
- aralkyl halides e.g. benzyl and phenethyl bromides
- esters of the present compounds include the following groups: (1 ) carboxylic acid esters obtained by esterification of the hydroxy groups, in which the non-carbonyl moiety of the carboxylic acid portion of the ester grouping is selected from straight or branched chain alkyl (for example, acetyl, n- propyl, t-butyl, or n-butyl), alkoxyalkyl (for example, methoxymethyl), aralkyl (for example, benzyl), aryloxyalkyl (for example, phenoxymethyl), aryl (for example, phenyl optionally substituted with, for example, halogen, C- ⁇ _ 4 alkyl, or C 1-4 alkoxy or amino); (2) sulfonate esters, such as alkyl- or aralkylsulfonyl (for example, methanesulfonyl); (3) amino acid esters (for example, L-valyl or L-isoleucyl);
- One or more compounds of the invention may also exist as, or optionally converted to, a solvate.
- Preparation of solvates is generally known.
- M. Caira et al, J. Pharmaceutical ScL, 93(3), 601-611 (2004) describe the preparation of the solvates of the antifungal fluconazole in ethyl acetate as well as from water.
- Similar preparations of solvates, hemisolvate, hydrates and the like are described by E. C. van Tonder et al, AAPS PharmSciTech., 5(1), article 12 (2004); and A. L. Bingham et al, Chem. Commun., 603-604 (2001 ).
- a typical, non-limiting, process involves dissolving the inventive compound in desired amounts of the desired solvent (organic or water or mixtures thereof) at a higher than ambient temperature, and cooling the solution at a rate sufficient to form crystals which are then isolated by standard methods.
- Analytical techniques such as, for example I. R. spectroscopy, show the presence of the solvent (or water) in the crystals as a solvate (or hydrate).
- Compounds of Formula I, and salts, solvates, esters and prodrugs thereof may exist in their tautomeric form (for example, as an amide or imino ether). All such tautomeric forms are contemplated herein as part of the present invention.
- All stereoisomers for example, geometric isomers, optical isomers and the like
- of the present compounds including those of the salts, solvates, esters and prodrugs of the compounds as well as the salts, solvates and esters of the prodrugs
- those which may exist due to asymmetric carbons on various substituents including enantiomeric forms (which may exist even in the absence of asymmetric carbons), rotameric forms, atropisomers, and diastereomeric forms, are contemplated within the scope of this invention, as are positional isomers (such as, for example, 4- pyridyl and 3-pyridyl).
- Individual stereoisomers of the compounds of the invention may, for example, be substantially free of other isomers, or may be admixed, for example, as racemates or with all other, or other selected, stereoisomers.
- the chiral centers of the present invention can have the S or R configuration as defined by the IUPAC 1974 Recommendations.
- the use of the terms "salt”, “solvate”, “ester”, “prodrug” and the like, is intended to equally apply to the salt, solvate, ester and prodrug of enantiomers, stereoisomers, rotamers, tautomers, positional isomers, racemates or prodrugs of the inventive compounds.
- Polymorphic forms of the compounds of Formula I, and of the salts, solvates, esters and prodrugs of the compounds of Formula I are intended to be included in the present invention.
- the compounds of this invention can be combined with an Hi receptor antagonist (i.e., the compounds of this invention can be combined with an Hi receptor antagonist in a pharmaceutical composition, or the compounds of this invention can be administered with an Hi receptor antagonist).
- Hi receptor antagonists include, without limitation: astemizole, azatadine, azelastine, acrivastine, brompheniramine, cetirizine, chlorpheniramine, clemastine, cyclizine, carebastine, cyproheptadine, carbinoxamine, descarboethoxyloratadine, diphenhydramine, doxylamine, dimethindene, ebastine, epinastine, efletirizine, fexofenadine, hydroxyzine, ketotifen, loratadine, levocabastine, meclizine, mizolastine, mequitazine, mianserin, noberastine, norastemizole, picumast, pyrilamine, promethazine, terfenadine, tripelennamine,
- the H 1 receptor antagonist is used at its known therapeutically effective dose, or the H 1 receptor antagonist is used at its normally prescribed dosage.
- said Hi receptor antagonist is selected from: azatadine, brompheniramine, cetirizine, chlorpheniramine, carebastine, descarboethoxyloratadine, diphenhydramine, ebastine, fexofenadine, loratadine, or norastemizole. More preferably, said Hi antagonist is selected from loratadine, descarboethoxyloratadine, fexofenadine or cetirizine.
- nasal congestion is treated.
- metabolic syndrome refers to a combination of risk factors for cardiovascular disease (CVD) identified in the National Cholesterol Education Program's Adult Treatment Panel III report. See for example the discussion by Grundy et al in Circulation, 109 (2004), 433-438.
- CVD cardiovascular disease
- the components of metabolic syndrome are: 1 ) abdominal obesity; 2) atherogenic dyslipidemia; 3) raised blood pressure; 4) insulin resistance; 5) proinflammatory state; and 6) prothrombotic state.
- Weight loss drugs include appetite suppressants, metabolic rate enhancers and nutrient absorption inhibitors.
- Appetite suppressant agents useful for treating obesity or metabolic syndrome include cannabinoid receptor 1 (CBi) antagonists or inverse agonists (e.g., rimonabant); Neuropeptide Y (NPY1 , NPY2, NPY4 and NPY5) antagonists; metabotropic glutamate subtype 5 receptor (mGluR ⁇ ) antagonists (e.g., 2-methyl-6-(phenylethynyl)-pyridine and 3[(2-methyl-1 ,4-thiazol-4-yl)ethynyl]pyridine); melanin-concentrating hormone receptor (MCH1 R and MCH2R) antagonists; melanocortin receptor agonists (e.g., Melanotan-ll and Mc4r agonists); serotonin uptake inhibitors (e.g., dexfenfluramine and fluoxetine); serotonin (5HT) transport inhibitors
- Metabolic rate enhancers include acetyl-CoA carboxylase-2 (ACC2) inhibitors; beta adrenergic receptor 3 ( ⁇ 3) agonists; diacylglycerol acyltransferase inhibitors (DGAT1 and DGAT2); fatty acid synthase (FAS) inhibitors (e.g., Cerulenin); phosphodiesterase (PDE) inhibitors (e.g., theophylline, pentoxifylline, zaprinast, sildenafil, amrinone, milrinone, cilostamide, rolipram and cilomilast); thyroid hormone ⁇ agonists; uncoupling protein activators (UCP-1 ,2 or 3) (e.g., phytanic acid, 4-[(E)-2-(5, 6,7,8- tetramethyl-2-naphthalenyl)-1-propenyl]benzoic acid and retinoic acid); acyl-estrogen
- Nutrient absorption inhibitors include lipase inhibitors (e.g., orlistat, lipstatin, tetrahydrolipstatin, teasaponin and diethylumbelliferyl phosphate); fatty acid transporter inhibitors; dicarboxylate transporter inhibitors; glucose transporter inhibitors; and phosphate transporter inhibitors.
- lipase inhibitors e.g., orlistat, lipstatin, tetrahydrolipstatin, teasaponin and diethylumbelliferyl phosphate
- fatty acid transporter inhibitors e.g., orlistat, lipstatin, tetrahydrolipstatin, teasaponin and diethylumbelliferyl phosphate
- dicarboxylate transporter inhibitors e.g., dicarboxylate transporter inhibitors
- glucose transporter inhibitors e.
- Specific compounds for use in the combination for treating obesity and metabolic syndrome include rimonabant, 2-methyl-6-(phenylethynyl)-pyridine, 3[(2- methyl-1 ,4-thiazol-4-yl)ethynyl]pyridine, Melanotan-ll, dexfenfluramine, fluoxetine, paroxetine, fluoxetine, fenfluramine, fluvoxamine, sertaline, imipramine, desipramine, talsupram, nomifensine, leptin, nalmefene, 3-methoxynaltrexone, naloxone, nalterxone, butabindide, axokine, sibutramine, topiramate, phytopharm compound 57, Cerulenin, theophylline, pentoxifylline, zaprinast, sildenafil, amrinone, milrinone, cilostamide, rolipram, cilomilast
- Preferred compounds for use in the combination for treating obesity and metabolic syndrome include rimonabant, dexfenfluramine, fenfluramine, phentermine, leptin, nalmefene, axokine, sibutramine, topiramate, phytopharm compound 57, oleoyl-estrone and orlistat.
- NAFLD nonalcoholic fatty liver disease
- NASH nonalcoholic steatohepatitis
- cirrhosis hepatacellular carcinoma or obesity.
- Typical HMG-CoA reductase inhibitors include statins such as lovastatin, simvastatin, pravastatin, atorvastatin, fluvastatin, resuvastatin, cerivastatin, rivastatin and pitavastatin.
- statins such as lovastatin, simvastatin, pravastatin, atorvastatin, fluvastatin, resuvastatin, cerivastatin, rivastatin and pitavastatin.
- sterol absorption inhibitor means a compound capable of inhibiting the absorption of one or more sterols, including but not limited to cholesterol, phytosterols (such as sitosterol, campesterol, stigmasterol and avenosterol), 5 ⁇ -stanols (such as cholestanol, 5 ⁇ -campestanol, 5 ⁇ -sitostanol), and/or mixtures thereof, when administered in a therapeutically effective (sterol and/or 5 ⁇ - stanol absorption inhibiting) amount to a mammal or human.
- suitable substituted azetidinones and methods of making the same include those disclosed in U.S. Patents Nos.
- Typical compounds for use in combination with an H 3 antagonist of this invention for the treatment of cognition deficit disorders are atomoxetine and dexmethylphenidate for the treatment of ADHD, olanzapine, risperidone or aripiprazole for treatment of schizophrenia, and donepezil, heptylphysostigmine, tacrine, rivastigmine or galantamine for the treatment of Alzheimer's Disease.
- the H 3 antagonist and other compound can be administered simultaneously (at the same time, in a single dosage form or in separate dosage forms) or sequentially (first one and then the other over a period of time).
- M 1 -Y constitutes an amide fragment
- specific examples of these approaches wherein M 1 -Y constitutes an amide fragment include amide coupling between a secondary amine, representative of the AB fragment, and carboxylic acid lithium salt, representative of the CD or C fragment, respectively.
- a carboxylic acid lithium salt is converted into the corresponding acid chloride and then coupled with the amine.
- PG is a protecting group.
- Conversion of the ABC intermediate into a compound of formula I is accomplished, for example, in the particular case when M 3 represents N, through deprotection of the secondary amine, followed by its reaction with an electrophilic D fragment, which most typically is an aldehyde (reductive amination) or a halide (alkylation) (Scheme 2), but also can be represented by an epoxide or other electrophile.
- an electrophilic D fragment which most typically is an aldehyde (reductive amination) or a halide (alkylation) (Scheme 2), but also can be represented by an epoxide or other electrophile.
- the last step in either synthetic sequence involves cleavage of the protecting groups present in the molecule to yield final compounds.
- M 1 -Y-M 2 constitutes a ketone (i.e., M 1 is CH, Y is -C(O)- and
- connection between B and C fragments may be established through the reaction of appropriate B ring-based carbon nucleophile (e.g., Grignard reagent or a transition metal-based reagent (Zn, Pd, Sn)), derived from the corresponding B-ring cycloalkyl halide, with a C ring-based Weinreb amide or aldehyde, generated from an acid (see Scheme 2), or any other suitable electrophile, such as, for example, a vinyl halide.
- B ring-based carbon nucleophile e.g., Grignard reagent or a transition metal-based reagent (Zn, Pd, Sn)
- Zn, Pd, Sn transition metal-based reagent
- D-electrophiles are a one-carbon aldehyde or alkyl halide attached to an R 2 group (Z' is a bond).
- Longer-chain D-electrophiles are synthesized through chain extension of one-carbon starting D-aldehydes (previously described or commercially available) by various methods known to those skilled in the art.
- Those methods include, but are not limited to, the reactions of starting aldehydes with alkylmetal reagents, carbon-phosphorus reagents (known as Wittig reactions and Horner-Emmons reactions), and reactions with other carbon nucleophiles, followed by appropriate functional elaboration, to obtain compounds where Z is an appropriately substituted Ci to C 5 alkyl or alkenyl group.
- the corresponding D fragment with the elongated Z is prepared by coupling an aryl halide with an appropriate alkyl or alkenyl metal (e.g., Li or MgHaI) reagent, optionally in the presence of an appropriate transition metal catalyst (e.g, Cu, Ni).
- an appropriate alkyl or alkenyl metal e.g., Li or MgHaI
- an appropriate transition metal catalyst e.g, Cu, Ni
- the A fragment contains a nucleophilic functional group, which is reacted with an electrophilic functional group that is part of the B fragment.
- Examples of this approach include, but are not limited to those illustrated in Scheme 3; in particular, reactions comprise reductive amination of an aldehyde or a ketone, nucleophilic aliphatic substitution or an amide coupling or an esterification.
- fragment A contains an electrophilic functionality, which reacts with a nucleophilic functional group on fragment B.
- compounds are synthesized by assembling BCD part of the molecule first, and then adding on the A fragment.
- the connection between A and BCD is established using the methods described above for the assembly of the AB portion.
- compounds are synthesized by preparing the BC portion of the molecule first, then adding on the A or D fragment, then following the approaches described above.
- One skilled in the art will recognize that most fragments will contain more than one functional group and that selective protection-deprotection may be an integral part of the above mentioned transformations. Examples of actual procedures employed in the syntheses of the corresponding compounds are described below.
- the starting materials and reagents used in preparing compounds described are either available from commercial suppliers such as Aldrich Chemical Co.
- Ki Dissociation Constant for substrate/receptor complex
- pA2 -logEC 5 o, as defined by J. Hey, Eur. J. Pharmacol., (1995), Vol. 294, 329-335
- Ci/mmol Curie/mmol (a measure of specific activity)
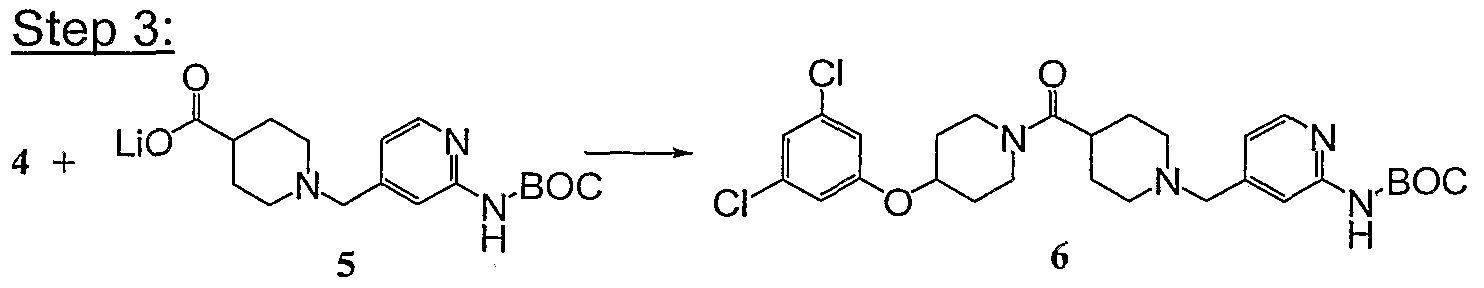
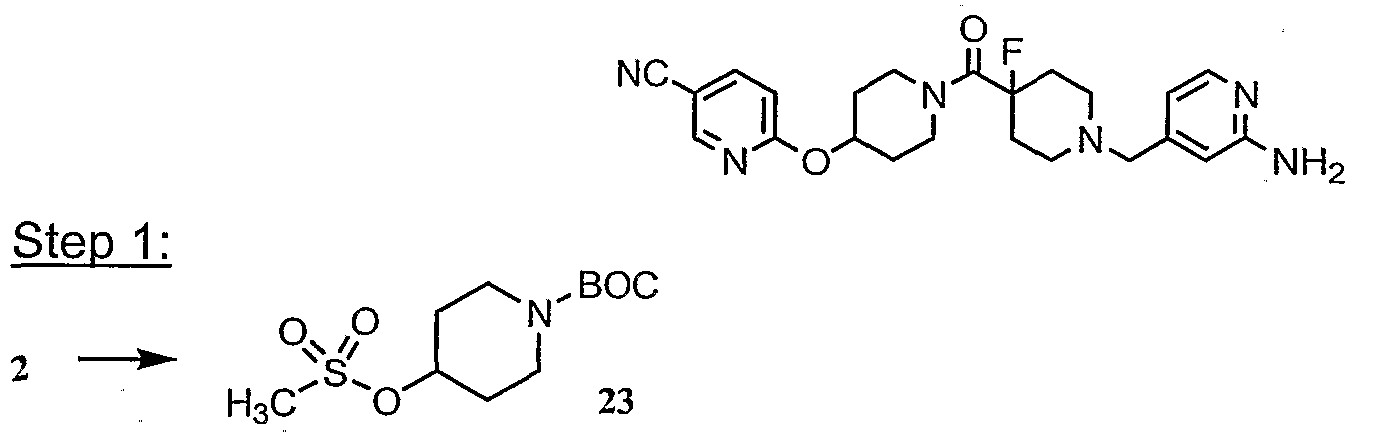
- Steps 3-4 Compound 8c was converted into the title compound using the procedures described in Steps 3 and 4 of Example 1. MS: (M+1 ) 402.
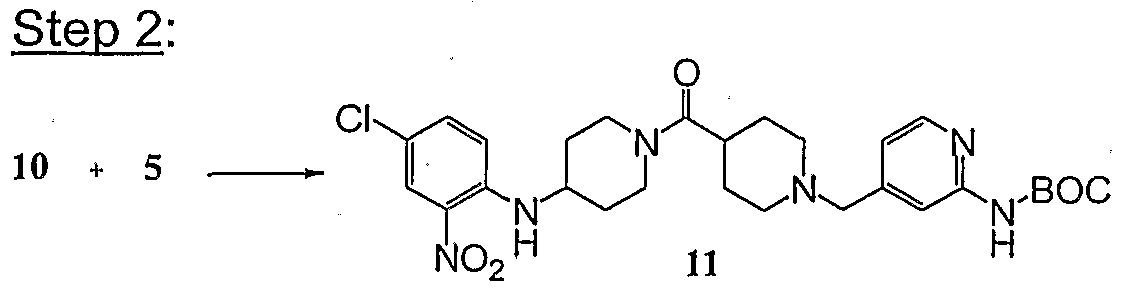
- Step 2 Compound 13 was converted into Example 7 and compound 13a into Example 7A using a procedure similar to that in Step 3 of Example 5.
- Ex. 7 MS: (M+1) 601 ;
- Ex. 7A MS: (M+1) 759.
- Step 3 A suspension of 15 (6.2 g, 10 mmol) in EtOH (20 ml) was treated with SnCI 2 (9.5 g, 50 mmol), heated to reflux for 3 h, and then stirred overnight at 20 0 C. The reaction mixture was poured on saturated aqueous NaHCO 3 , extracted with CH 2 CI 2 , dried over Na 2 SO 4 , and concentrated. Chromatography (5% 1 N NH 3 -MeOH/EtOAc) provided the title compound (5.2 g, 52%) as a yellow solid. MS: (M+1 ) 487.
- Example 9 was converted into the title compound as described in Example 8, Step L MS: (M+1 ) 594.
- Step 4 Compound 58 was converted into the title compound using procedures similar to those in Steps 3 and 4 of Example 1. MS: (M+1 ) 533.
- Steps 2-3 Compound 59 was converted into the title compound using procedures similar to those described in Steps 3 and 4 of Example 11. MS: (M+1 ) 462.
- Oxalyl chloride (1.9 ml, 21.78 mmol) was added dropwise to a stirred solution of 1-Boc-isonipecotic acid 16 (4.5 g, 19.63 mmol) in CH 2 CI 2 (150 ml) at RT. A catalytic amount of DMF (40 ⁇ l) was added. The resulting mixture was stirred for 1 h 15 min. Et 3 N (6.8 ml, 48.79 mmol) was added slowly to the mixture, followed by 4- hydroxypiperidine (2.68 g, 26.50 mmol). Reaction was continued for 2.5 d. The mixture was washed with H 2 O (50 ml), a 1.0 M HCI aq.
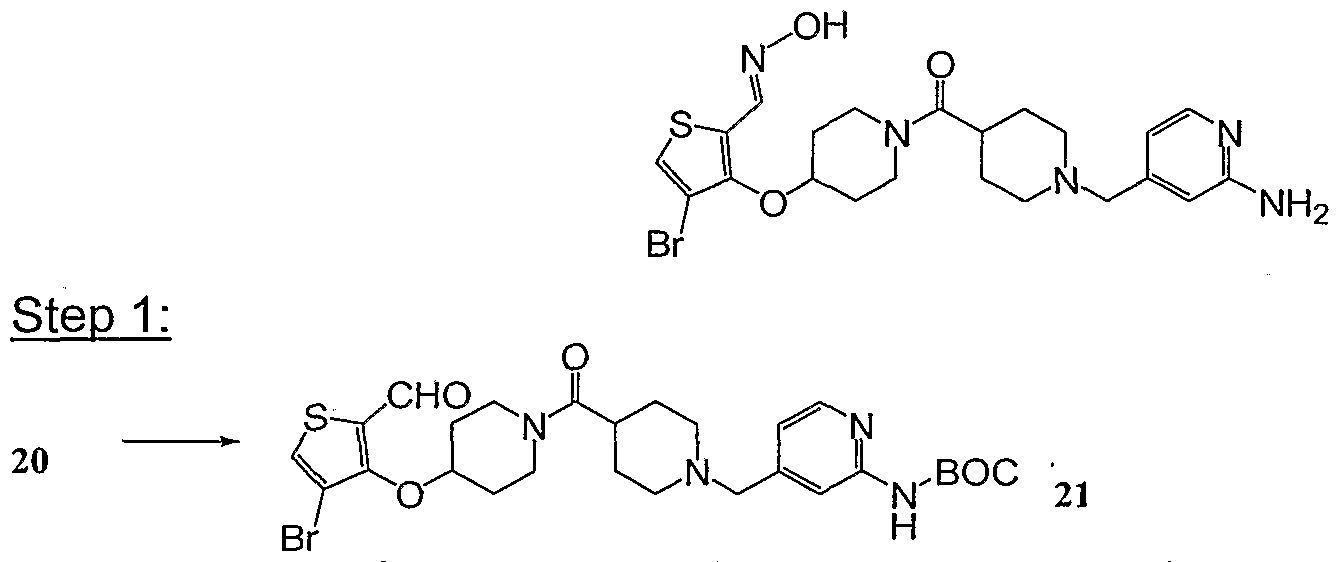
- the thiophene ester 20 (305 mg, 0.48 mmol) was dissolved in THF (5 ml) and Et 2 O (10 ml), cooled in an ice bath, and treated with a 1.0 M solution of LiALH 4 in ether (0.95 ml, 0.95 mmol). The mixture was stirred for 1.5h at O 0 C and 10 min at RT. A small volume of H 2 O and a 1.0 M NaOH aqueous solution were added alternately to the mixture. The mixture was diluted with EtOAc, stirred for an additional 1 h, and filtered through a 1-in silica gel pad, rinsing with EtOAc. The filtrate was concentrated in vacuo to an oil (200 mg).
- the oil from above was dissolved in CH 2 CI 2 (5 ml), and N-methylmorpholino oxide (NMO) (96 mg, 0.82 mmol) was added, followed by a catalytic amount of tetrapropylamonium perruthenate (TPAP) (12 mg, 0.034 mmol). After 2 h, the mixture was quenched with H 2 O and extracted with CH 2 CI 2 . The organic extracts were washed with a 10% Na 2 S 2 O 3 aq. solution and brine, dried with Na 2 SO 4 , and filtered through a 1-in Celite pad.
- NMO N-methylmorpholino oxide
- TPAP tetrapropylamonium perruthenate
- Ethyl isonipecotate (3.62 g.; 23 mmol) and 31 (1.73 g, 17.8 mmol) were stirred at RT in 15 ml of CH 2 CI 2 for 90 min.
- NaBH(OAc) 3 (4.24 g, 20 mmol) was then added, and the mixture stirred under N 2 for 20 h.
- the mixture was diluted with CH 2 CI 2 , washed with saturated aqueous NaHCO 3 , then with brine, and dried with anhydrous Na 2 SO 4 .
- Compound 40 was prepared as described for compound 37 in Example 20.
- Step 2 Compound 41 was converted into the title compound using the procedures of Steps 2 to 4 of Example 1. MS: (M+1 ) 531.
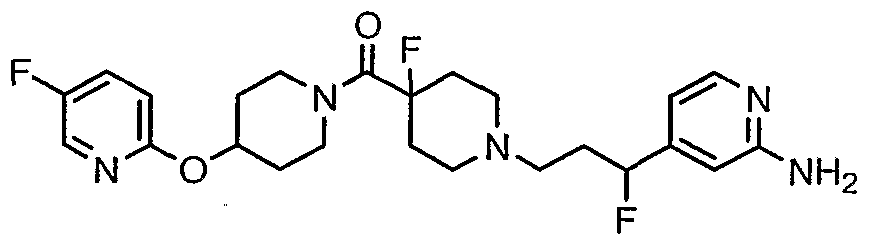
- Compound 42 was prepared using the procedure described for preparing compound 37, followed by BOC deprotection as described in the conversion of 3 into 4 in Example 1 , followed by EDC mediated coupling with N-BOC- ⁇ -fluoroisonipecotic acid, followed by BOC deprotection.
- Compound 42 (0.41 g; 1.22 mmol) and pyridazine-4-carbaldehyde (0.2 g; 1.82 mmol) (prepared as described in US 6,720,328) were stirred at RT in dry CH 2 CI 2 (14 ml) containing activated molecular sieves (5.0 g).
- Example 23 MS: (M+1 ) 430; 23A: MS: (M+1 ) 522.
- Step 2 Compound 46 was converted into the title compound using the procedures described in Steps 2 to 4 of Example 1. MS: (M+1 ) 444.
- the microtiter plate was then sealed and shaken at 25°C for 20 h.
- the solutions were filtered through a polypropylene frit into a second microtiter plate containing MP-TsOH resin (-100 mg). After the top plate was washed with MeCN (0.5 ml), the plate was removed, the bottom microtiter plate sealed and shaken at 25°C for 2 h.
- the solutions were filtered through a polypropylene frit and the resin was washed with CH 2 CI 2 (3X), then MeOH (3X) to remove unreacted reagents.
- 2,2-Dimethylcyclopentanone (0.10 g, 2.0 eq) was added to a 0.02 M solution of 47 in CH 2 CI 2 .
- Titanium tetraisopropoxide (0.15 ml, 1.2 eq) was added and the resulting mixture was stirred at RT for 2.5 h, NaBH(OAc) 3 (0.18 g, 2.0 eq) was added and the stirring was continued for 15 h.
- the mixture was diluted with CH 2 CI 2 (50 ml) and washed with aq sat K 2 CO 3 (50 ml). The layers were separated and the aq layer extracted with CH 2 CI 2 (2 x 50 ml).
- Example 28A was prepared from 47 using procedure of Example 23, followed by BOC-deprotection. MS: (M+1 ) 426.
- Step 2 A solution of Example 28A (0.107 g), a mixture of 37% aq formaldehyde (2 ml) and formic acid (1 ml) was heated at reflux for 16 h, then diluted with CH 2 CI 2 (30 ml) and washed with aq sat K 2 CO 3 (30 ml). The aq. layer was extracted with CH 2 CI 2 (2 x 30 ml), and the combined organic phase dried. The solvent was evaporated under vacuo and the residue purified by column chromatography (SiO 2 , CH 2 CI 2 : 0.4 N NH 3 in MeOH 3:1 ) to give Example 28 (0.052 g, 47%) as colorless crystals. MS: (M+1 ) 440.
- the suspension was sealed and heated at 60 0 C for 12 h.
- the resin was then washed with MeOH (3X), THF, then CH 2 CI 2 and dried in vacuo.
- the resin was then treated multiple times (8-10) with 0.1 N TFA in CH 2 CI 2 containing 20% triethylsilane for 5-10 mins each.
- the resin was then washed with CH 2 CI 2 (4x) and dried in vacuo to provide resin 50.
- a CH 2 CI 2 solution of 50 (70 mg, 0.0546 mmol) and DIEA (0.19 ml, 20 equiv) was treated with a CH 2 CI 2 solution of the acid chloride of FMOC-isonipecotic acid (2 equiv).
- the suspension was shaken at RT for 20 h, then the resin was washed with MeOH (3X), then CH 2 CI 2 , and dried in vacuo.
- the resin was then treated three times with 20% piperdine in DMF for 20 min each, then washed (3X) with DMF, THF and CH 2 CI 2 , then dried in vacuo to provide 51.
- Step 2 Compound 54 was converted into the title compound using procedures analogous to Steps 2 to 4 in Example 1. MS: (M+1 ) 440.
- Step 2 Compound 62 was converted into the title compound using procedures analogous to Steps 2 to 4 in Example 1. MS: (M+1 ) 462.
- Compound 47b was prepared using the procedures analogous to those of steps 1 and 2 from example 1 and converted into 47a by using the procedure analogous to step 5 of example 296, followed by procedure analogous to step 3 of example 13. Step 5:
- Compound 72 was prepared in two steps from compound 69a using procedures analogous to Steps 3 and 4 of Example 1. Conversion of 72 into 73 was accomplished using procedure of Step 3 in Example 1. Step 3: Compound 73 was converted into the title compound using a procedure analogous to Step 4 of Example 1. MS: (M+1 ) 458.
- Step 2 Compound 74 was converted into the title compound using a procedure analogous to Step 4 of Example 1. MS: (M+1 ) 460.
- Step i
- Ozone was passed through a -78 C solution of 76 (1.0 g, 3.76 mmol) in a 1 :1 mixture of MeOH-CH 2 CI 2 (20 ml) until the solution turned blue.
- the solution was purged for 5 min. with oxygen, after which NaBH 4 (0.7 g, 18.8 mmol) was added in portions, and reaction mixture was allowed to warm to RT.
- the reaction mixture was diluted with aqueous NH 4 CI and extracted with CH 2 CI 2 .
- the organic phase was separated, dried (Na 2 SO 4 ) and concentrated to produce 1.0 g of crude alcohol 77, which was used without purification.
- Step 6 Compound 79 was converted into the title compound using a procedure analogous to Step 4 of Example 1. MS: (M+1 ) 478.
- Compound 84a was prepared from 84 using procedure analogous to step 3 of example 13.
- This intermediate was dissolved in CH 2 CI 2 (2 ml) and treated with formalin (0.1 ml, 1.34 mmol) and NaBH(OAc) 3 (60 mg, 0.28 mmol). Two drops of AcOH were added. The reaction was continued overnight, and quenched with a saturated NaHCO 3 aqueous solution (30 ml). The aqueous mixture was extracted with CH 2 CI 2 (30 ml x3).
- Boc-protected amine 96 was stirred in 20% TFA/CH 2 CI 2 overnight at room temperature. TFA was removed under vacuum, and the residue was subjected to aqueous NaHCO3 work-up - CH 2 CI 2 extraction to produce 426 mg of amine 97 as a white solid.
- the source of the H 3 receptors in this experiment was guinea pig brain.
- the source of H 3 receptors was recombinant human receptor, expressed in HEK-293 (human embryonic kidney) cells.
- the animals weighed 400-600 g.
- the brain tissue was homogenized with a solution of 50 mM Tris, pH 7.5.
- the final concentration of tissue in the homogenization buffer was 10% w/v.
- the homogenates were centrifuged at 1 ,000 x g for 10 min. in order to remove clumps of tissue and debris.
- the resulting supernatants were then centrifuged at 50,000 x g for 20 min. in order to sediment the membranes, which were next washed three times in homogenization buffer (50,000 x g for 20 min. each).
- the membranes were frozen and stored at -70 0 C until needed.
- Bound ligand was separated from unbound ligand by filtration, and the amount of radioactive ligand bound to the membranes was quantitated by liquid scintillation spectrometry. All incubations were performed in duplicate and the standard error was always less than 10%.
- Compounds that inhibited more than 70% of the specific binding of radioactive ligand to the receptor were serially diluted to determine a Kj (nM).
- Compounds of formula I have a Kj within the range of about 1 to about 10000 nM.
- Preferred compounds of formula I have a Kj within the range of about 0.2 to about 100 nM. More preferred compounds of formula I have a Kj within the range of about 0.2 to about 20 nM.
- the compound of Example 11 has a Ki of 0.2 nM in the guinea pig receptor assay, while the compound of Example 256 has a K 1 of 0.7 nM in the recombinant human receptor assay.
- the term "at least one compound of formula I” means that one to three different compounds of formula I may be used in a pharmaceutical composition or method of treatment. Preferably one compound of formula I is used.
- "at least one H-i receptor antagonist " or "at least one other compound (or agent) for treating obesity, metabolic syndrome or cognition deficit disorders” means that one to three different Hi antagonists or other compounds may be used in a pharmaceutical composition or method of treatment. Preferably, one Hi antagonist or one other compound for treating obesity, metabolic syndrome or cognition deficit disorders is used in the combinations.
- inert, pharmaceutically acceptable carriers can be either solid or liquid.
- Solid form preparations include powders, tablets, dispersible granules, capsules, cachets and suppositories.
- the powders and tablets may be comprised of from about 5 to about 95 percent active ingredient.
- Suitable solid carriers are known in the art, e.g. magnesium carbonate, magnesium stearate, talc, sugar or lactose. Tablets, powders, cachets and capsules can be used as solid dosage forms suitable for oral administration. Examples of pharmaceutically acceptable carriers and methods of manufacture for various compositions may be found in A. Gennaro (ed.), The Science and Practice of Pharmacy, 20 th Edition, (2000), Lippincott Williams & Wilkins, Baltimore, MD.
- Liquid form preparations include solutions, suspensions and emulsions. As an example may be mentioned water or water-propylene glycol solutions for parenteral injection or addition of sweeteners and opacifiers for oral solutions, suspensions and emulsions. Liquid form preparations may also include solutions for intranasal administration.
- Aerosol preparations suitable for inhalation may include solutions and solids in powder form, which may be in combination with a pharmaceutically acceptable carrier, such as an inert compressed gas, e.g. nitrogen.
- a pharmaceutically acceptable carrier such as an inert compressed gas, e.g. nitrogen.
- solid form preparations which are intended to be converted, shortly before use, to liquid form preparations for either oral or parenteral administration.
- liquid forms include solutions, suspensions and emulsions.
- the compounds of the invention may also be deliverable transdermally.
- the transdermal compositions can take the form of creams, lotions, aerosols and/or emulsions and can be included in a transdermal patch of the matrix or reservoir type as are conventional in the art for this purpose.
- the compound is administered orally.
- the pharmaceutical preparation is in a unit dosage form.
- the preparation is subdivided into suitably sized unit doses containing appropriate quantities of the active component, e.g., an effective amount to achieve the desired purpose.
- the quantity of active compound in a unit dose of preparation may be varied or adjusted from about 1 mg to about 150 mg, preferably from about 1 mg to about 75 mg, more preferably from about 1 mg to about 50 mg, according to the particular application.
- the actual dosage employed may be varied depending upon the requirements of the patient and the severity of the condition being treated. Determination of the proper dosage regimen for a particular situation is within the skill of the art. For convenience, the total daily dosage may be divided and administered in portions during the day as required.
- a typical recommended daily dosage regimen for oral administration can range from about 1 mg/day to about 300 mg/day, preferably 1 mg/day to 75 mg/day, in two to four divided doses.
- the two active components may be co-administered simultaneously or sequentially, or a single pharmaceutical composition comprising a H 3 antagonist and an H 1 antagonist in a pharmaceutically acceptable carrier can be administered.
- the components of the combination can be administered individually or together in any conventional dosage form such as capsule, tablet, powder, cachet, suspension, solution, suppository, nasal spray, etc.
- the dosage of the Hi antagonist can be determined from published material, and may range from 1 to 1000 mg per dose. When used in combination, the dosage levels of the individual components are preferably lower than the recommended individual dosages because of the advantageous effect of the combination.
- kits comprising in a single package, one container comprising an H 3 antagonist in a pharmaceutically acceptable carrier, and a separate container comprising an Hi antagonist in a pharmaceutically acceptable carrier, with the H 3 and Hi antagonists being present in amounts such that the combination is therapeutically effective.
- a kit is advantageous for administering a combination when, for example, the components must be administered at different time intervals or when they are in different dosage forms.
- the invention comprises a combination of H 3 antagonist and at least one other compound for treating obesity, metabolic syndrome, cognition deficit disorders, NAFLD, hepatic steatosis, NASH, cirrhosis, or hepatacellular carcinoma
- the two active components may be co-administered simultaneously or sequentially, or a single pharmaceutical composition comprising a H 3 antagonist and another compound in a pharmaceutically acceptable carrier can be administered.
- the components of the combination can be administered individually or together in any conventional dosage form such as capsule, tablet, powder, cachet, suspension, solution, suppository, nasal spray, etc.
- the dosage of the other compound for treating obesity, metabolic syndrome or cognition deficit disorders can be determined from published material, and may range from 1 to 1000 mg per dose.
- kits comprising in a single package, one container comprising an H 3 antagonist in a pharmaceutically acceptable carrier, and a separate container or containers comprising at least one other compound for treating obesity, metabolic syndrome, cognition deficit disorders NAFLD, hepatic steatosis, NASH, cirrhosis, or hepatacellular carcinoma in a pharmaceutically acceptable carrier, with the H 3 antagonists and other compounds being present in amounts such that the combination is therapeutically effective.
- a kit is advantageous for administering a combination when, for example, the components must be administered at different time intervals or when they are in different dosage forms.
Abstract
Description








































































































































Claims



Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002610959A CA2610959A1 (en) | 2005-06-20 | 2006-06-19 | Piperidine derivatives useful as histamine h3 antagonists |
| JP2008518276A JP2008546784A (en) | 2005-06-20 | 2006-06-19 | Piperidine derivatives useful as histamine H3 antagonists |
| DE602006010870T DE602006010870D1 (en) | 2005-06-20 | 2006-06-19 | PIPERIDINE DERIVATIVES SUITED AS ANTAGONISTS OF HISTAMINE H3 |
| EP06773528A EP1902046B1 (en) | 2005-06-20 | 2006-06-19 | Piperidine derivatives useful as histamine h3 antagonists |
| MX2008000115A MX2008000115A (en) | 2005-06-20 | 2006-06-19 | Piperidine derivatives useful as histamine h3 antagonists. |
| AU2006262441A AU2006262441A1 (en) | 2005-06-20 | 2006-06-19 | Piperidine derivatives useful as histamine H3 antagonists |
| AT06773528T ATE450526T1 (en) | 2005-06-20 | 2006-06-19 | PIPERIDINE DERIVATIVES SUITABLE AS ANTAGONISTS OF HISTAMINE H3 |
| IL187984A IL187984A0 (en) | 2005-06-20 | 2007-12-06 | Piperidine derivatives useful as histamine h3 antagonists |
| HK08110560.1A HK1121737A1 (en) | 2005-06-20 | 2008-09-23 | Piperidine derivatives useful as histamine h3 antagonists |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US69211005P | 2005-06-20 | 2005-06-20 | |
| US60/692,110 | 2005-06-20 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007001975A1 true WO2007001975A1 (en) | 2007-01-04 |
| WO2007001975A8 WO2007001975A8 (en) | 2008-01-24 |
Family
ID=36968725
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2006/023800 WO2007001975A1 (en) | 2005-06-20 | 2006-06-19 | Piperidine derivatives useful as histamine h3 antagonists |
Country Status (17)
| Country | Link |
|---|---|
| US (2) | US7635705B2 (en) |
| EP (1) | EP1902046B1 (en) |
| JP (1) | JP2008546784A (en) |
| KR (1) | KR20080021082A (en) |
| CN (1) | CN101243072A (en) |
| AR (1) | AR054787A1 (en) |
| AT (1) | ATE450526T1 (en) |
| AU (1) | AU2006262441A1 (en) |
| CA (1) | CA2610959A1 (en) |
| DE (1) | DE602006010870D1 (en) |
| ES (1) | ES2337727T3 (en) |
| HK (1) | HK1121737A1 (en) |
| IL (1) | IL187984A0 (en) |
| MX (1) | MX2008000115A (en) |
| TW (1) | TW200738676A (en) |
| WO (1) | WO2007001975A1 (en) |
| ZA (1) | ZA200710968B (en) |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007075555A2 (en) * | 2005-12-21 | 2007-07-05 | Schering Corporation | Combination of an h3 antagonist/inverse agonist and an appetite suppressant |
| WO2008051873A2 (en) * | 2006-10-20 | 2008-05-02 | Arete Therapeutics, Inc. | Phenylurea compounds as soluble epoxide hydrolase inhibitors |
| WO2008108957A2 (en) * | 2007-03-02 | 2008-09-12 | Schering Corporation | Piperidinyl-piperidine and piperazinyl-piperidine for use in the treatment of diabetes or pain |
| WO2008112022A1 (en) * | 2007-03-13 | 2008-09-18 | Arete Therapeutics, Inc. | 4 -pi peridinylurea compounds as soluble epoxide hydrolase inhibitors |
| WO2009010454A2 (en) * | 2007-07-13 | 2009-01-22 | Addex Pharma S.A. | Amido derivatives and their use as positive allosteric modulators of metabotropic glutamate receptors |
| US20120322719A1 (en) * | 2011-06-16 | 2012-12-20 | The Feinstein Institute For Medical Research | Methods of treatment of fatty liver disease by pharmacological activation of cholinergic pathways |
| US8466153B2 (en) | 2006-12-08 | 2013-06-18 | Janssen Pharmaceutica N.V. | Piperidinylamino-pyridazines and their use as fast dissociating dopamine 2 receptor antagonists |
| US8530474B2 (en) | 2008-07-03 | 2013-09-10 | Janssen Pharmaceutica Nv | Substituted 6-(1-piperazinyl)-pyridazines as 5-HT6 receptor antagonists |
| WO2013151982A1 (en) | 2012-04-03 | 2013-10-10 | Arena Pharmaceuticals, Inc. | Methods and compounds useful in treating pruritus, and methods for identifying such compounds |
| US8765750B2 (en) | 2010-01-22 | 2014-07-01 | Taiho Pharmaceutical Co., Ltd. | Piperazine compound having a PGDS inhibitory effect |
| US8791120B2 (en) | 2007-02-13 | 2014-07-29 | Janssen Pharmaceutica Nv | Fast-dissociating dopamine 2 receptor antagonists |
| US8889674B2 (en) | 2009-03-05 | 2014-11-18 | Shionogi & Co., Ltd. | Piperidine and pyrrolidine derivatives having NPY Y5 receptor antagonism |
| US8895562B2 (en) | 2008-07-31 | 2014-11-25 | Janssen Pharmaceutica Nv | Piperazin-1-yl-trifluoromethyl-substituted-pyridines as fast dissociating dopamine 2 receptor antagonists |
| US8906921B2 (en) | 2007-04-23 | 2014-12-09 | Janssen Pharmaceutica Nv | 4-alkoxypyridazine derivatives as fast dissociating dopamine 2 receptor antagonists |
| US8933101B2 (en) | 2007-04-23 | 2015-01-13 | Janssen Pharmaceutica Nv | Thia(dia)zoles as fast dissociating dopamine 2 receptor antagonists |
| US8940743B2 (en) | 2005-10-26 | 2015-01-27 | Janssen Pharmaceutica Nv | Piperidin-4-yl-pyridazin-3-ylamine derivatives as fast dissociating dopamine 2 receptor antagonists |
| CN109082465A (en) * | 2018-08-20 | 2018-12-25 | 苏州市广济医院 | The molecular marker and application thereof of Olanzapine induction carbohydrate metabolism disturbance related disease |
| WO2021187605A1 (en) * | 2020-03-19 | 2021-09-23 | 田辺三菱製薬株式会社 | NITROGEN-CONTAINING HETEROCYCLIC α-CYANO CARBONYL COMPOUND |
| US11198699B2 (en) | 2019-04-02 | 2021-12-14 | Aligos Therapeutics, Inc. | Compounds targeting PRMT5 |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BRPI0807829A2 (en) * | 2007-01-29 | 2014-08-05 | Arete Therapeutics Inc | "Soluble EPOXIDE HYDROLASE INHIBITORS FOR TREATMENT OF METABOLIC SYNDROME AND RELATED DISORDERS". |
| EP2141994A4 (en) * | 2007-04-26 | 2011-05-18 | Avalon Pharmaceuticals | Multi-ring compounds and uses thereof |
| WO2010027567A2 (en) | 2008-07-23 | 2010-03-11 | Schering Corporation | Tricyclic spirocycle derivatives and methods of use thereof |
| US8337835B2 (en) * | 2009-04-10 | 2012-12-25 | Washington University | Use of an endogenous ligand for peroxisome proliferator activated receptor alpha to treat liver disorders |
| AU2011261375B2 (en) | 2010-06-04 | 2016-09-22 | Albany Molecular Research, Inc. | Glycine transporter-1 inhibitors, methods of making them, and uses thereof |
| CN103402971A (en) * | 2011-01-07 | 2013-11-20 | 塔加西普特公司 | Nicotinic receptor non-competitive antagonists |
| CA2848711C (en) | 2011-09-16 | 2019-08-06 | Galectin Therapeutics, Inc. | Galacto-rhamnogalacturonate compositions for the treatment of non-alcoholic steatohepatitis and non-alcoholic fatty liver disease |
| CN102657868A (en) * | 2012-05-30 | 2012-09-12 | 中国药科大学 | 5-hydroxy tryptamine 2 receptor antagonist or transporter inhibitor for treating fatty liver or hyperlipemia |
| CN104619329B (en) | 2012-06-06 | 2018-01-26 | 卡莱克汀医疗有限公司 | For treating the galactolipin rhamnose galacturonic ester composition of the disease associated with elevated nitric oxide synthase type |
| US8652527B1 (en) | 2013-03-13 | 2014-02-18 | Upsher-Smith Laboratories, Inc | Extended-release topiramate capsules |
| US9101545B2 (en) | 2013-03-15 | 2015-08-11 | Upsher-Smith Laboratories, Inc. | Extended-release topiramate capsules |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001058891A2 (en) * | 2000-02-11 | 2001-08-16 | Vertex Pharmaceuticals Incorporated | Piperazine and piperidine derivatives for treatment or prevention of neuronal damage |
| WO2002032893A2 (en) * | 2000-10-17 | 2002-04-25 | Schering Corporation | Piperidine compounds as anti-allergic |
| WO2002072570A2 (en) * | 2001-03-13 | 2002-09-19 | Schering Corporation | Non-imidazole compounds as histamine h3 antagonists |
| WO2003088967A1 (en) * | 2002-04-18 | 2003-10-30 | Schering Corporation | (1-4-piperidinyl) benzimidazole derivatives useful as histamine h3 antagonists |
| WO2003103669A1 (en) * | 2002-04-18 | 2003-12-18 | Schering Corporation | 1-(4-piperidinyl) benzimidazolones as histamine h3 antagonists |
| WO2004000831A1 (en) * | 2002-06-24 | 2003-12-31 | Schering Corporation | Indole derivatives useful as histamine h3 antagonists |
| US20040198743A1 (en) * | 2003-01-31 | 2004-10-07 | Schering Corporation | Methods for treating allergic skin and allergic ocular conditions using combinations of histamine receptor antagonists |
| WO2004101546A1 (en) * | 2003-04-23 | 2004-11-25 | Glaxo Group Limited | Piperazine derivatives and their use for the treatment of neurological and psychiatric diseases |
Family Cites Families (64)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US661145A (en) * | 1900-08-06 | 1900-11-06 | Otto Hausmann | Attachment for bicycles. |
| US856473A (en) * | 1906-05-21 | 1907-06-11 | Daniel E Krause | Drilling-machine. |
| US4983567A (en) * | 1987-03-16 | 1991-01-08 | Biomeasure, Inc. | Immunomodulators and methods of making same |
| US4983597A (en) | 1989-08-31 | 1991-01-08 | Merck & Co., Inc. | Beta-lactams as anticholesterolemic agents |
| US5688787A (en) * | 1991-07-23 | 1997-11-18 | Schering Corporation | Substituted β-lactam compounds useful as hypochlesterolemic agents and processes for the preparation thereof |
| HUT67341A (en) * | 1991-07-23 | 1995-03-28 | Schering Corp | Substituted beta-lactam compounds useful as hypocholesterolemic agents, pharmaceutical compositions containing the same and process for the production thereof |
| US5561227A (en) * | 1991-07-23 | 1996-10-01 | Schering Corporation | Process for the stereospecific synthesis of azetidinones |
| US5688785A (en) * | 1991-07-23 | 1997-11-18 | Schering Corporation | Substituted azetidinone compounds useful as hypocholesterolemic agents |
| LT3300B (en) | 1992-12-23 | 1995-06-26 | Schering Corp | Combination of a cholesterol biosynhtesis inhibitor and a beta- lactam cholesterol absorbtion inhibitor |
| LT3595B (en) * | 1993-01-21 | 1995-12-27 | Schering Corp | Spirocycloalkyl-substituted azetidinones useful as hypocholesterolemic agents |
| NZ268798A (en) * | 1993-07-09 | 1997-01-29 | Schering Corp | Process for the preparation of substituted azetidinones |
| US5631365A (en) * | 1993-09-21 | 1997-05-20 | Schering Corporation | Hydroxy-substituted azetidinone compounds useful as hypocholesterolemic agents |
| US5627176A (en) * | 1994-03-25 | 1997-05-06 | Schering Corporation | Substituted azetidinone compounds useful as hypocholesterolemic agents |
| US5624920A (en) * | 1994-11-18 | 1997-04-29 | Schering Corporation | Sulfur-substituted azetidinone compounds useful as hypocholesterolemic agents |
| US5633246A (en) * | 1994-11-18 | 1997-05-27 | Schering Corporation | Sulfur-substituted azetidinone compounds useful as hypocholesterolemic agents |
| US5656624A (en) * | 1994-12-21 | 1997-08-12 | Schering Corporation | 4-[(heterocycloalkyl or heteroaromatic)-substituted phenyl]-2-azetidinones useful as hypolipidemic agents |
| US5618707A (en) * | 1996-01-04 | 1997-04-08 | Schering Corporation | Stereoselective microbial reduction of 5-fluorophenyl-5-oxo-pentanoic acid and a phenyloxazolidinone condensation product thereof |
| WO1997016424A1 (en) | 1995-11-02 | 1997-05-09 | Schering Corporation | Process for preparing 1-(4-fluorophenyl)-3(r)-(3(s)-hydroxy-3-([phenyl or 4-fluorophenyl])-propyl)-4(s)-(4-hydroxyphenyl)-2-azetidinone |
| US5739321A (en) * | 1996-05-31 | 1998-04-14 | Schering Corporation | 3-hydroxy γ-lactone based enantionselective synthesis of azetidinones |
| US5886171A (en) * | 1996-05-31 | 1999-03-23 | Schering Corporation | 3-hydroxy gamma-lactone based enantioselective synthesis of azetidinones |
| MY116130A (en) | 1996-08-16 | 2003-11-28 | Schering Corp | Treatment of upper airway allergic responses |
| US5756470A (en) * | 1996-10-29 | 1998-05-26 | Schering Corporation | Sugar-substituted 2-azetidinones useful as hypocholesterolemic agents |
| US5869479A (en) * | 1997-08-14 | 1999-02-09 | Schering Corporation | Treatment of upper airway allergic responses |
| US6133001A (en) * | 1998-02-23 | 2000-10-17 | Schering Corporation | Stereoselective microbial reduction for the preparation of 1-(4-fluorophenyl)-3(R)-[3(S)-Hydroxy-3-(4-fluorophenyl)propyl)]-4(S)-(4 -hydroxyphenyl)-2-azetidinone |
| JP2002518368A (en) * | 1998-06-17 | 2002-06-25 | デュポン ファーマシューティカルズ カンパニー | Cyclic hydroxamic acids as metalloproteinase inhibitors |
| US5919672A (en) * | 1998-10-02 | 1999-07-06 | Schering Corporation | Resolution of trans-2-(alkoxycarbonylethyl)-lactams useful in the synthesis of 1-(4-fluoro-phenyl)-3(R)- (S)-hydroxy-3-(4-fluorophenyl)-propyl!-4(S)-(4-hydroxyphenyl)-2-azetidinone |
| US6207822B1 (en) * | 1998-12-07 | 2001-03-27 | Schering Corporation | Process for the synthesis of azetidinones |
| US6593078B1 (en) * | 1999-04-16 | 2003-07-15 | Schering Corporation | Use of azetidinone compounds |
| US20020045686A1 (en) * | 2000-08-24 | 2002-04-18 | The Yokohama Rubber Co., Ltd | Rubber composition having improved wet skid resistance and rolling resistance |
| DE10042447A1 (en) * | 2000-08-29 | 2002-03-28 | Aventis Pharma Gmbh | Vertebrate intestinal protein that absorbs cholesterol and use of this protein to identify inhibitors of intestinal cholesterol transport |
| US6982251B2 (en) * | 2000-12-20 | 2006-01-03 | Schering Corporation | Substituted 2-azetidinones useful as hypocholesterolemic agents |
| IL156552A0 (en) * | 2000-12-21 | 2004-01-04 | Aventis Pharma Gmbh | Diphenyl azetidinone derivatives, method for the production thereof, medicaments containing these compounds, and their use |
| MXPA03005155A (en) * | 2000-12-21 | 2003-09-10 | Aventis Pharma Gmbh | Novel 1,2-diphenzylazetidinones, method for producing the same, medicaments containing said compounds, and the use thereof for treating disorders of the lipid metabolism. |
| AU2002231688A1 (en) * | 2000-12-21 | 2002-07-01 | Sanofi-Aventis Deutschland Gmbh | Diphenyl azetidinone derivatives, method for the production thereof, medicaments containing these compounds, and their use |
| TWI291957B (en) | 2001-02-23 | 2008-01-01 | Kotobuki Pharmaceutical Co Ltd | Beta-lactam compounds, process for repoducing the same and serum cholesterol-lowering agents containing the same |
| AU2002258605B2 (en) * | 2001-03-28 | 2006-01-12 | Merck Sharp & Dohme Corp. | Enantioselective synthesis of azetidinone intermediate compounds |
| US20030137689A1 (en) * | 2002-01-18 | 2003-07-24 | General Instrument Corporation. | Remote printer driver server |
| US7105505B2 (en) * | 2002-04-18 | 2006-09-12 | Schering Corporation | Benzimidazole derivatives useful as histamine H3 antagonists |
| DE10227506A1 (en) | 2002-06-19 | 2004-01-08 | Aventis Pharma Deutschland Gmbh | Ring-substituted diphenylazetidinones, processes for their preparation, pharmaceutical compositions containing them and their use |
| DE10227508A1 (en) | 2002-06-19 | 2004-01-08 | Aventis Pharma Deutschland Gmbh | Acid group-substituted diphenylazetidinones, processes for their preparation, pharmaceutical compositions containing them and their use |
| DE10227507A1 (en) | 2002-06-19 | 2004-01-08 | Aventis Pharma Deutschland Gmbh | Cationically substituted diphenylazetidinones, processes for their preparation, medicaments containing these compounds and their use |
| GB0215579D0 (en) | 2002-07-05 | 2002-08-14 | Astrazeneca Ab | Chemical compounds |
| JP4575772B2 (en) | 2002-08-06 | 2010-11-04 | サノフィ−アベンティス・ドイチュラント・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング | Method for isolating intestinal cholesterol binding protein |
| ATE406364T1 (en) * | 2003-03-07 | 2008-09-15 | Schering Corp | SUBSTITUTED AZETIDINONE DERIVATIVES, THEIR PHARMACEUTICAL FORMULATIONS AND THEIR USE IN THE TREATMENT OF HYPERCHOLESTEROLEMIA |
| DE602004018617D1 (en) * | 2003-03-07 | 2009-02-05 | Schering Corp | SUBSTITUTED AZETIDINONE DERIVATIVES, THEIR PHARMACEUTICAL FORMULATIONS AND THEIR USE FOR THE TREATMENT OF HYPERCHOLESTEROLMIA |
| MXPA05009502A (en) * | 2003-03-07 | 2005-10-18 | Schering Corp | Substituted azetidinone compounds, formulations and uses thereof for the treatment of hypercholesterolemia. |
| DE10314610A1 (en) | 2003-04-01 | 2004-11-04 | Aventis Pharma Deutschland Gmbh | New diphenylazetidinone with improved physiological properties, process for its preparation, medicaments containing these compounds and its use |
| GB0308333D0 (en) | 2003-04-10 | 2003-05-14 | Glaxo Group Ltd | Novel compounds |
| US20040224952A1 (en) * | 2003-05-07 | 2004-11-11 | Cowart Marlon D. | Fused bicyclic-substituted amines as histamine-3 receptor ligands |
| JP2005015434A (en) | 2003-06-27 | 2005-01-20 | Kotobuki Seiyaku Kk | Serum cholesterol-lowering agent or prophylactic or therapeutic agent for atherosclerosis |
| GB0315360D0 (en) | 2003-07-01 | 2003-08-06 | Burns Robert N | Anti-theft device |
| AU2003259547A1 (en) | 2003-07-31 | 2005-02-14 | Hetero Drugs Limited | Ezetimibe polymorphs |
| EP1660456A2 (en) | 2003-08-25 | 2006-05-31 | Microbia Inc. | Quaternary salt derivatives of 1,4-diphenylazetidin-2-ones |
| EP1660446A2 (en) | 2003-08-28 | 2006-05-31 | Microbia, Inc. | Tethered dimers and trimers of 1,4-diphenylazetidn-2-ones |
| GB0321228D0 (en) | 2003-09-10 | 2003-10-08 | Inpharmatica Ltd | Modulating cell activity |
| CA2540364A1 (en) | 2003-09-26 | 2005-04-07 | Forbes Medi-Tech Inc. | Method of inhibiting the expression of a multi-drug resistance genes and inhibiting the production of proteins resulting from the expression of such genes thereby enhancing the effectiveness of chemotherapeutic agents to treat cancers |
| EP1522541A1 (en) | 2003-10-07 | 2005-04-13 | Lipideon Biotechnology AG | Novel hypocholesterolemic compounds |
| US20070135357A1 (en) | 2003-10-30 | 2007-06-14 | Sings Heather L | Anti-hypercholesterolemic compounds |
| WO2005042692A2 (en) | 2003-10-31 | 2005-05-12 | Forbes Medi-Tech Inc. | A method of inhibiting the expression of genes which mediate cellular cholesterol influx in animal cells and inhibiting the production of proteins resulting from the expression of such genes using cholesterol absorption inhibitors |
| JP4839221B2 (en) | 2003-11-07 | 2011-12-21 | ジェイ ジェイ ファーマ,インコーポレイテッド | Combination therapy combination to increase HDL |
| AU2004288822A1 (en) | 2003-11-10 | 2005-05-26 | Microbia, Inc. | 4-Biarylyl-1-phenylazetidin-2-ones |
| AU2004303742B2 (en) | 2003-12-23 | 2008-06-19 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitory activity |
| GB0329778D0 (en) | 2003-12-23 | 2004-01-28 | Astrazeneca Ab | Chemical compounds |
| JP2007516287A (en) | 2003-12-23 | 2007-06-21 | メルク エンド カムパニー インコーポレーテッド | Anti-hypercholesterolemic compound |
-
2006
- 2006-06-19 JP JP2008518276A patent/JP2008546784A/en not_active Withdrawn
- 2006-06-19 EP EP06773528A patent/EP1902046B1/en active Active
- 2006-06-19 US US11/455,625 patent/US7635705B2/en not_active Expired - Fee Related
- 2006-06-19 KR KR1020077030855A patent/KR20080021082A/en not_active Application Discontinuation
- 2006-06-19 DE DE602006010870T patent/DE602006010870D1/en active Active
- 2006-06-19 MX MX2008000115A patent/MX2008000115A/en active IP Right Grant
- 2006-06-19 CN CNA2006800301178A patent/CN101243072A/en active Pending
- 2006-06-19 AT AT06773528T patent/ATE450526T1/en not_active IP Right Cessation
- 2006-06-19 AU AU2006262441A patent/AU2006262441A1/en not_active Abandoned
- 2006-06-19 ES ES06773528T patent/ES2337727T3/en active Active
- 2006-06-19 CA CA002610959A patent/CA2610959A1/en not_active Abandoned
- 2006-06-19 TW TW095121918A patent/TW200738676A/en unknown
- 2006-06-19 WO PCT/US2006/023800 patent/WO2007001975A1/en active Application Filing
- 2006-06-20 AR ARP060102617A patent/AR054787A1/en not_active Application Discontinuation
-
2007
- 2007-12-06 IL IL187984A patent/IL187984A0/en unknown
- 2007-12-18 ZA ZA200710968A patent/ZA200710968B/en unknown
-
2008
- 2008-09-23 HK HK08110560.1A patent/HK1121737A1/en not_active IP Right Cessation
-
2009
- 2009-07-22 US US12/507,563 patent/US7846946B2/en not_active Expired - Fee Related
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001058891A2 (en) * | 2000-02-11 | 2001-08-16 | Vertex Pharmaceuticals Incorporated | Piperazine and piperidine derivatives for treatment or prevention of neuronal damage |
| WO2002032893A2 (en) * | 2000-10-17 | 2002-04-25 | Schering Corporation | Piperidine compounds as anti-allergic |
| WO2002072570A2 (en) * | 2001-03-13 | 2002-09-19 | Schering Corporation | Non-imidazole compounds as histamine h3 antagonists |
| WO2003088967A1 (en) * | 2002-04-18 | 2003-10-30 | Schering Corporation | (1-4-piperidinyl) benzimidazole derivatives useful as histamine h3 antagonists |
| WO2003103669A1 (en) * | 2002-04-18 | 2003-12-18 | Schering Corporation | 1-(4-piperidinyl) benzimidazolones as histamine h3 antagonists |
| WO2004000831A1 (en) * | 2002-06-24 | 2003-12-31 | Schering Corporation | Indole derivatives useful as histamine h3 antagonists |
| US20040198743A1 (en) * | 2003-01-31 | 2004-10-07 | Schering Corporation | Methods for treating allergic skin and allergic ocular conditions using combinations of histamine receptor antagonists |
| WO2004101546A1 (en) * | 2003-04-23 | 2004-11-25 | Glaxo Group Limited | Piperazine derivatives and their use for the treatment of neurological and psychiatric diseases |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9751860B2 (en) | 2005-10-26 | 2017-09-05 | Janssen Pharmaceutica Nv | Piperidin-4YL-pyridazin-3-ylamine derivatives as fast dissociating dopamine 2 receptor antagonists |
| US9468640B2 (en) | 2005-10-26 | 2016-10-18 | Janssen Pharmaceutica Nv | Piperidin-4-yl-pyridazin-3-ylamine derivatives as fast dissociating dopamine 2 receptor antagonists |
| US8940743B2 (en) | 2005-10-26 | 2015-01-27 | Janssen Pharmaceutica Nv | Piperidin-4-yl-pyridazin-3-ylamine derivatives as fast dissociating dopamine 2 receptor antagonists |
| WO2007075555A3 (en) * | 2005-12-21 | 2007-12-21 | Schering Corp | Combination of an h3 antagonist/inverse agonist and an appetite suppressant |
| WO2007075555A2 (en) * | 2005-12-21 | 2007-07-05 | Schering Corporation | Combination of an h3 antagonist/inverse agonist and an appetite suppressant |
| WO2008051873A2 (en) * | 2006-10-20 | 2008-05-02 | Arete Therapeutics, Inc. | Phenylurea compounds as soluble epoxide hydrolase inhibitors |
| WO2008051873A3 (en) * | 2006-10-20 | 2008-06-19 | Arete Therapeutics Inc | Phenylurea compounds as soluble epoxide hydrolase inhibitors |
| US8466153B2 (en) | 2006-12-08 | 2013-06-18 | Janssen Pharmaceutica N.V. | Piperidinylamino-pyridazines and their use as fast dissociating dopamine 2 receptor antagonists |
| US9422296B2 (en) | 2007-02-13 | 2016-08-23 | Janssen Pharmaceutica Nv | Fast-dissociating dopamine 2 receptor antagonists |
| US8791120B2 (en) | 2007-02-13 | 2014-07-29 | Janssen Pharmaceutica Nv | Fast-dissociating dopamine 2 receptor antagonists |
| WO2008108957A3 (en) * | 2007-03-02 | 2009-02-05 | Schering Corp | Piperidinyl-piperidine and piperazinyl-piperidine for use in the treatment of diabetes or pain |
| WO2008108957A2 (en) * | 2007-03-02 | 2008-09-12 | Schering Corporation | Piperidinyl-piperidine and piperazinyl-piperidine for use in the treatment of diabetes or pain |
| WO2008112022A1 (en) * | 2007-03-13 | 2008-09-18 | Arete Therapeutics, Inc. | 4 -pi peridinylurea compounds as soluble epoxide hydrolase inhibitors |
| US8906921B2 (en) | 2007-04-23 | 2014-12-09 | Janssen Pharmaceutica Nv | 4-alkoxypyridazine derivatives as fast dissociating dopamine 2 receptor antagonists |
| US8933101B2 (en) | 2007-04-23 | 2015-01-13 | Janssen Pharmaceutica Nv | Thia(dia)zoles as fast dissociating dopamine 2 receptor antagonists |
| WO2009010454A2 (en) * | 2007-07-13 | 2009-01-22 | Addex Pharma S.A. | Amido derivatives and their use as positive allosteric modulators of metabotropic glutamate receptors |
| WO2009010454A3 (en) * | 2007-07-13 | 2009-03-05 | Addex Pharmaceuticals Sa | Amido derivatives and their use as positive allosteric modulators of metabotropic glutamate receptors |
| US8524726B2 (en) | 2007-07-13 | 2013-09-03 | Addex Pharma S.A. | Amido derivatives and their use as positive allosteric modulators of metabotropic glutamate receptors |
| US8530474B2 (en) | 2008-07-03 | 2013-09-10 | Janssen Pharmaceutica Nv | Substituted 6-(1-piperazinyl)-pyridazines as 5-HT6 receptor antagonists |
| US8895562B2 (en) | 2008-07-31 | 2014-11-25 | Janssen Pharmaceutica Nv | Piperazin-1-yl-trifluoromethyl-substituted-pyridines as fast dissociating dopamine 2 receptor antagonists |
| US8889674B2 (en) | 2009-03-05 | 2014-11-18 | Shionogi & Co., Ltd. | Piperidine and pyrrolidine derivatives having NPY Y5 receptor antagonism |
| US8765750B2 (en) | 2010-01-22 | 2014-07-01 | Taiho Pharmaceutical Co., Ltd. | Piperazine compound having a PGDS inhibitory effect |
| US8865641B2 (en) * | 2011-06-16 | 2014-10-21 | The Feinstein Institute For Medical Research | Methods of treatment of fatty liver disease by pharmacological activation of cholinergic pathways |
| US20120322719A1 (en) * | 2011-06-16 | 2012-12-20 | The Feinstein Institute For Medical Research | Methods of treatment of fatty liver disease by pharmacological activation of cholinergic pathways |
| WO2013151982A1 (en) | 2012-04-03 | 2013-10-10 | Arena Pharmaceuticals, Inc. | Methods and compounds useful in treating pruritus, and methods for identifying such compounds |
| CN109082465A (en) * | 2018-08-20 | 2018-12-25 | 苏州市广济医院 | The molecular marker and application thereof of Olanzapine induction carbohydrate metabolism disturbance related disease |
| US11198699B2 (en) | 2019-04-02 | 2021-12-14 | Aligos Therapeutics, Inc. | Compounds targeting PRMT5 |
| WO2021187605A1 (en) * | 2020-03-19 | 2021-09-23 | 田辺三菱製薬株式会社 | NITROGEN-CONTAINING HETEROCYCLIC α-CYANO CARBONYL COMPOUND |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2007001975A8 (en) | 2008-01-24 |
| US7846946B2 (en) | 2010-12-07 |
| US20090286830A1 (en) | 2009-11-19 |
| US7635705B2 (en) | 2009-12-22 |
| IL187984A0 (en) | 2008-03-20 |
| DE602006010870D1 (en) | 2010-01-14 |
| TW200738676A (en) | 2007-10-16 |
| EP1902046A1 (en) | 2008-03-26 |
| HK1121737A1 (en) | 2009-04-30 |
| KR20080021082A (en) | 2008-03-06 |
| JP2008546784A (en) | 2008-12-25 |
| ATE450526T1 (en) | 2009-12-15 |
| AU2006262441A1 (en) | 2007-01-04 |
| ES2337727T3 (en) | 2010-04-28 |
| US20070015807A1 (en) | 2007-01-18 |
| MX2008000115A (en) | 2008-03-18 |
| CN101243072A (en) | 2008-08-13 |
| CA2610959A1 (en) | 2007-01-04 |
| AR054787A1 (en) | 2007-07-18 |
| ZA200710968B (en) | 2009-03-25 |
| EP1902046B1 (en) | 2009-12-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7635705B2 (en) | Heteroatom-linked substituted piperidines and derivatives thereof useful as histamine H3 antagonists | |
| US7638531B2 (en) | Phenoxypiperidines and analogs thereof useful as histamine H3 antagonists | |
| US7408066B2 (en) | Carbon-linked substituted piperidines and derivatives thereof useful as histamine H3 antagonists | |
| US7879880B2 (en) | Substituted aniline derivatives useful as histamine H3 antagonists | |
| US7482468B2 (en) | Imidazole and benzimidazole derivatives useful as histamine H3 antagonists | |
| US20050267095A1 (en) | 3- or 4-monosubstituted phenol and thiophenol derivatives useful as H3 ligands | |
| EP3526203B1 (en) | N-aryl and n-heteroaryl piperidine derivatives as liver x receptor beta agonists, compositions, and their use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680030117.8 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006262441 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2610959 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 187984 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12007502774 Country of ref document: PH Ref document number: 564099 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006773528 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2008518276 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/000115 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020077030855 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2006262441 Country of ref document: AU Date of ref document: 20060619 Kind code of ref document: A |