WO2007090071A2 - 6-modified bicyclic nucleic acid analogs - Google Patents
6-modified bicyclic nucleic acid analogs Download PDFInfo
- Publication number
- WO2007090071A2 WO2007090071A2 PCT/US2007/061183 US2007061183W WO2007090071A2 WO 2007090071 A2 WO2007090071 A2 WO 2007090071A2 US 2007061183 W US2007061183 W US 2007061183W WO 2007090071 A2 WO2007090071 A2 WO 2007090071A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- oligomeric compound
- nucleoside
- mmol
- compound
- substituted
- Prior art date
Links
- 0 *[C@@]([C@]1O[C@@]2*)O[C@]2(CO*)[C@]1O* Chemical compound *[C@@]([C@]1O[C@@]2*)O[C@]2(CO*)[C@]1O* 0.000 description 8
- XOGMFHOLVGTVQM-VBFQVJHPSA-N FCC([C@@](COCc1ccccc1)(C1)O2)O[C@H]1[C@@H]2[U] Chemical compound FCC([C@@](COCc1ccccc1)(C1)O2)O[C@H]1[C@@H]2[U] XOGMFHOLVGTVQM-VBFQVJHPSA-N 0.000 description 1
- STEDEQGVMDEZHE-VBFQVJHPSA-N OCC([C@@](COCc1ccccc1)(C1)O2)O[C@H]1[C@@H]2[U] Chemical compound OCC([C@@](COCc1ccccc1)(C1)O2)O[C@H]1[C@@H]2[U] STEDEQGVMDEZHE-VBFQVJHPSA-N 0.000 description 1
- ABQOJAWNMSWYPN-IBTYICNHSA-N OC[C@@]1(CO[C@@H]2C1)O[C@H]2[U] Chemical compound OC[C@@]1(CO[C@@H]2C1)O[C@H]2[U] ABQOJAWNMSWYPN-IBTYICNHSA-N 0.000 description 1
- REVACISYLLJEDR-RVSMYKNHSA-N [U][C@@H]1O[C@@]2(COCc3ccccc3)NC3O[C@@H]1C23 Chemical compound [U][C@@H]1O[C@@]2(COCc3ccccc3)NC3O[C@@H]1C23 REVACISYLLJEDR-RVSMYKNHSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention provides 6-modified bicyclic nucleosides and oligomeric compounds and compositions prepared therefrom. More particularly, the present invention provides nucleosides having a 2'-O-C(H)(R)-4' bridge and oligomers and compositions prepared therefrom.
- R is in a particular configuration providing either the (R) or (S) isomer.
- the oligomeric compounds and compositions of the present invention hybridize to a portion of a target RNA resulting in loss of normal function of the target RNA.
- Antisense technology is an effective means for reducing the expression of one or more specific gene products and can therefore prove to be uniquely useful in a number of therapeutic, diagnostic, and research applications.
- Chemically modified nucleosides are routinely used for incorporation into antisense sequences to enhance one or more properties such as for example nuclease resistance.
- One such group of chemical modifications includes bicyclic nucleosides wherein the furanose portion of the nucleoside includes a bridge connecting two atoms on the furanose ring thereby forming a bicyclic ring system.
- Such bicyclic nucleosides have various names including BNA's and LNA's for bicyclic nucleic acids or locked nucleic acids respectively.
- LNA's are toxic. See, e.g., Swayze, E. E.; Siwkowski, A. M.; Wancewicz, E. V.; Migawa, M. T.; Wyrzykiewicz, T. K.; Hung, G.; Monia, B. P.; Bennett, C. F., Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucl. Acids Res., doi: 10.1093/nar/gkll071 (Dec. 2006, advanced online publication).
- 6-substituted BNA's and antisense compounds prepared therefrom useful for modulating gene expression pathways, including those relying on mechanisms of action such as RNaseH, RNAi and dsRNA enzymes, as well as other antisense mechanisms based on target degradation or target occupancy.
- the present invention provides a bicyclic nucleoside having the formula:
- Bx is a heterocyclic base moiety
- T 1 is H or a hydroxyl protecting group
- T 2 is H, a hydroxyl protecting group or a reactive phosphorus group
- Z is C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, substituted C 1 -C 6 alkyl, substituted C 2 -C 6 alkenyl, substituted C 2 -C 6 alkynyl, acyl, substituted acyl, or substituted amide.
- Z is CpC 6 alkyl or substituted CpC 6 alkyl. In another embodiment, Z is Ci-C 6 alkyl. In another embodiment, Z is methyl (CH 3 -). In another embodiment, Z is ethyl (CH 3 CH 2 -). In another embodiment, Z is substituted Ci-C 6 alkyl. In another embodiment, Z is substituted methyl. In another embodiment, Z is substituted ethyl.
- the substituent group is C 1 -C 6 alkoxy (e.g., Z is Ci-C 6 alkyl substituted with one or more Ci-C 6 alkoxy).
- the Ci-C 6 alkoxy substituent group is CH 3 O- (e.g., Z is CH 3 OCH 2 -).
- the Ci-C 6 alkoxy substituent group can be further substituted such as N(JiJ 2 )CH 2 O- (e.g., Z is N(JiJ 2 )CH 2 OCH 2 -).
- the substituent group is halogen (e.g., Z is Cj-C 6 alkyl substituted with one or more halogen).
- the halogen substituent group is fluoro (e.g., Z is CH 2 FCH 2 -, CHF 2 CH 2 - or CF 3 CH 2 -).
- the substituent group is hydroxyl (e.g., Z is Ci-C 6 alkyl substituted with one or more hydroxyl).
- Z is HOCH 2 -.
- Z is CH 3 -, CH 3 CH 2 -, -CH 2 OCH 3 , -CH 2 F or HOCH 2 -.
- the Z group is C 1 -C 6 alkyl substituted with one or more X x , wherein each X x is independently halo (e.g., fluoro), hydroxyl, alkoxy (e.g., CH 3 O-), substituted alkoxy or azido.
- the Z group is -CH 2 X X , wherein X x is halo (e.g., fluoro), hydroxyl, alkoxy (e.g., CH 3 O-) or azido.
- the Z group is in the (i?)-configuration:
- the Z group is in the ( ⁇ -configuration:
- each T 1 and T 2 is a hydroxyl protecting group.
- a preferred list of hydroxyl protecting groups includes benzyl, benzoyl, 2,6-dichlorobenzyl, t-butyldimethylsilyl, t- butyldiphenylsilyl, mesylate, tosylate, dimethoxytrityl (DMT), 9-phenylxanthine-9-yl (Pixyl) and 9-(p-methoxyphenyl)xanthine-9-yl (MOX).
- Ti is a hydroxyl protecting group selected from acetyl, benzyl, t-butyldimethylsilyl, t-butyldiphenylsilyl and dimethoxytrityl wherein a more preferred hydroxyl protecting group is T 1 is 4,4'-dimethoxytrityl.
- T 2 is a reactive phosphorus group wherein preferred reactive phosphorus groups include diisopropylcyanoethoxy phosphoramidite and H-phosphonate.
- T 1 is 4,4'-dimethoxytrityl and T 2 is diisopropylcyanoethoxy phosphoramidite.
- the present invention also provides oligomeric compounds having at least one monomer of the formula:
- Bx is a heterocyclic base moiety
- T 3 is H, a hydroxyl protecting group, a linked conjugate group or an internucleoside linking group attached to a nucleoside, a nucleotide, an oligonucleoside, an oligonucleotide, a monomelic subunit or an oligomeric compound;
- T 4 is H, a hydroxyl protecting group, a linked conjugate group or an internucleoside linking group attached to a nucleoside, a nucleotide, an oligonucleoside, an oligonucleotide, a monomelic subunit or an oligomeric compound; wherein at least one of T 3 and T 4 is an internucleoside linking group attached to a nucleoside, a nucleotide, an oligonucleoside, an oligonucleotide, a monomelic subunit or an oligomeric compound; and
- Z is C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, substituted C r C 6 alkyl, substituted C 2 -C 6 alkenyl, substituted C 2 -C 6 alkynyl, acyl, substituted acyl, or substituted amide.
- At least one Z is C 1 -C 6 alkyl or substituted C 1 -C 6 alkyl. Li another embodiment, each Z is, independently, Cj-C 6 alkyl or substituted CpC 6 alkyl. In another embodiment, at least one Z is C 1 -C 6 alkyl. In another embodiment, each Z is, independently, C 1 - C 6 alkyl. In another embodiment, at least one Z is methyl. In another embodiment, each Z is methyl. In another embodiment, at least one Z is ethyl. In another embodiment, each Z is ethyl. In another embodiment, at least one Z is substituted C 1 -C 6 alkyl. In another embodiment, each Z is, independently, substituted Ci-C 6 alkyl. In another embodiment, at least one Z is substituted methyl. In another embodiment, each Z is substituted methyl. In another embodiment, at least one Z is substituted ethyl, hi another embodiment, each Z is substituted ethyl.
- At least one substituent group is C 1 -C 6 alkoxy (e.g., at least one Z is C 1 -C 6 alkyl substituted with one or more C 1 -C 6 alkoxy).
- each substituent group is, independently, C 1 -C 6 alkoxy (e.g., each Z is, independently, C 1 -C 6 alkyl substituted with one or more C 1 -C 6 alkoxy).
- At least one C 1 -C 6 alkoxy substituent group is CH 3 O- (e.g., at least one Z is CH 3 OCH 2 -). In another embodiment, each C 1 -C 6 alkoxy substituent group is CH 3 O- (e.g., each Z is CH 3 OCH 2 -). In one embodiment, at least one substituent group is halogen (e.g., at least one Z is C 1 -C 6 alkyl substituted with one or more halogen). In another embodiment, each substituent group is, independently, halogen (e.g., each Z is, independently, C 1 -C 6 alkyl substituted with one or more halogen).
- At least one halogen substituent group is fluoro (e.g., at least one Z is CH 2 FCH 2 -, CHF 2 CH 2 - or CF 3 CH 2 -).
- each halo substituent group is fluoro (e.g., each Z is, independently, CH 2 FCH 2 -, CHF 2 CH 2 - or CF 3 CH 2 -).
- At least one substituent group is hydroxyl (e.g., at least one Z is C 1 - C 6 alkyl substituted with one or more hydroxyl). In another embodiment, each substituent group is, independently, hydroxyl (e.g., each Z is, independently, C 1 -C 6 alkyl substituted with one or more hydroxyl). In another embodiment, at least one Z is HOCH 2 -. In another embodiment, each Z is HOCH 2 -.
- At least one Z is CH 3 -, CH 3 CH 2 -, CH 2 OCH 3 -, CH 2 F- or HOCH 2 -.
- each Z is, independently, CH 3 -, CH 3 CH 2 -, CH 2 OCH 3 -, CH 2 F- or HOCH 2 -.
- At least one Z group is C 1 -C 6 alkyl substituted with one or more X x , wherein each X x is, independently, halo (e.g., fluoro), hydroxyl, alkoxy (e.g., CH 3 O-) or azido.
- each Z group is, independently, C 1 -C 6 alkyl substituted with one or more X x , wherein each X x is independently halo (e.g., fluoro), hydroxyl, alkoxy (e.g., CH 3 O-) or azido.
- X x is independently halo (e.g., fluoro), hydroxyl, alkoxy (e.g., CH 3 O-) or azido.
- at least one Z group is -CH 2 X X , wherein X x is halo (e.g., fluoro), hydroxyl, alkoxy (e.g., CH 3 O-) or azido.
- each Z group is, independently, -CH 2 X X , wherein each X x is, independently, halo (e.g., fluoro), hydroxyl, alkoxy (e.g., CH 3 O-) or azido.
- halo e.g., fluoro
- hydroxyl e.g., CH 3 O-
- alkoxy e.g., CH 3 O-
- azido e.g., hydroxyl
- at least one Z is CH 3 -.
- each Z is, CH 3 -.
- the Z group of at least one monomer is in the (R)- configuration represented by the formula: or the formula:
- the Z group of each monomer of the formula is in the (R)- configuration.
- the Z group of at least one monomer is in the (S)- configuration represented by the formula:
- the Z group of each monomer of the formula is in the (S)- configuration.
- T 3 is H or a hydroxyl protecting group. In another embodiment T 4 is H or a hydroxyl protecting group. In a further embodiment T 3 is an internucleoside linking group attached to a nucleoside, a nucleotide or a monomelic subunit. In another embodiment T 4 is an internucleoside linking group attached to a nucleoside, a nucleotide or a monomelic subunit. In another embodiment T 3 is an internucleoside linking group attached to an oligonucleoside or an oligonucleotide. In a further embodiment T 4 is an internucleoside linking group attached to an oligonucleoside or an oligonucleotide.
- T 3 is an internucleoside linking group attached to an oligomeric compound.
- T 4 is an internucleoside linking group attached to an oligomeric compound.
- at least one of T 3 and T 4 comprises an internucleoside linking group selected from phosphodiester or phosphorothioate.
- oligomeric compounds have at least one region of at least two contiguous monomers of the formula:
- the oligomeric compound comprises at least two regions of at least two contiguous monomers of the above formula. In a further embodiment the oligomeric compound comprises a gapped oligomeric compound. In another embodiment the oligmeric compound comprises at least one region of from about 8 to about 14 contiguous ⁇ -D-2'- deoxyribofuranosyl nucleosides. In a further embodiment the oligomeric compound comprises at least one region of from about 9 to about 12 contiguous ⁇ -D-2'-deoxyribofuranosyl nucleosides.
- the oligomeric compound comprises at least one region of from 2 to three contiguous monomers of the above formula, an optional second region of 1 or 2 contiguous monomers of the above formula and a third region of from 8 to 14 ⁇ -D-2'-deoxyribofuranosyl nucleosides wherein the third region is located between the first and the second regions.
- the oligomeric compond comprises from 8 to 10 ⁇ -D-2'-deoxyribofuranosyl nucleosides.
- oligomeric compounds are provided having from about 8 to about 40 nucleosides and/or modified nucleosides or mimetics in length. In a further embodiment oligomeric compound comprise from about 8 to about 20 nucleosides and/or modified nucleosides or mimetics in length. Ln an even further embodiment oligomeric compounds comprise from about 10 to about 16 nucleosides and/or modified nucleosides or mimetics in length. In another embodiment oligomeric compounds comprise from about 10 to about 14 nucleosides and/or modified nucleosides or mimetics in length.
- Also provided are methods of inhibiting gene expression comprising contacting one or more cells, a tissue or an animal with an oligomeric compound of the invention.
- the present invention provides 6-modified bicyclic nucleosides, oligomeric compounds and compositions prepared therefrom, novel synthetic intermediates, and methods of preparing the nucleosides, oligomeric compounds, compositions, and novel synthetic intermediates. More particularly, the present invention provides nucleosides having a bridge between the 4' and T- positions of the ribose portion having the formula: 2'-O-C(H)(Z)-4' and oligomers and compositions prepared therefrom. In a preferred embodiment, Z is in a particular configuration providing either the (R) or (S) isomer.
- the oligomeric compounds and compositions of the present invention are designed to hybridize to a portion of a target RNA.
- the oligomeric compounds of the present invention can be used in the design of aptamers which are oligomeric compounds capable of binding to aberrant proteins in an in vivo setting.
- Bicyclic nucleosides of the present invention are useful for enhancing desired properties of oligomeric compounds in which they are incorporated.
- the oligomers of the present invention may also be useful as primers and probes in diagnostic applications.
- the 6-modified bicyclic nucleosides of the present invention have the structure shown below:
- the present invention provides bicyclic nucleosides having formula I:
- Bx is a heterocyclic base moiety
- T 1 is H or a hydroxyl protecting group
- T 2 is H, a hydroxyl protecting group or a reactive phosphorus group; and Z is C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 1 -C 6 alkylidenyl, C 3 -C 6 alkenylidenyl, substituted C 1 -C 6 alkyl, substituted C 2 -C 6 alkenyl, substituted C 2 -C 6 alkynyl, substituted C 1 -C 6 alkylidenyl, substituted C 3 -C 6 alkenylidenyl, acyl, substituted acyl, substituted amide, thiol, or substituted thio.
- bicyclic nucleosides are prepared having reactive groups orthogonally protected and further comprising a reactive phosphorus group. Such bicyclic nucleosides are useful as monomers for oligomer synthesis.
- One illustrative example of such a bicyclic nucleoside monomer has the formula:
- the bicyclic nucleoside monomer shown is genetically referred to as a dimethoxytrityl phosphoramidite or more formally using IUPAC naming nomenclature as (15',3i?,4i?,6i?,75)-7-[2-cyanoethoxy(diisopropylamino)phosphinoxy]-l- (4,4 ' -dimethoxytrityloxymethyl)-3 -(uracil- 1 -yl)-6-methyl-2, 5 -dioxa-bicyclo [2.2.1 Jheptane.
- the 6-modified bicyclic nucleosides of the present invention are useful for modifying otherwise unmodified oligomeric compounds at one or more positions.
- modified oligomeric compounds can be described as having a particular motif.
- Motifs amenable to the present invention include but are not limited to a gapped motif, a hemimer motif, a blockmer motif, a fully modified motif, a positionally modified motif and an alternating motif.
- linkages can also be used including but not limited to phosphodiester and phosphorothioate linkages used uniformly or in combinations.
- the positioning of 6-modified bicyclic nucleosides and the use of linkage strategies can be easily optimized for the best activity for a particular target. Representative U.S.
- patents that teach the preparation of representative motifs include, but are not limited to, 5,013,830; 5,149,797; 5,220,007; 5,256,775; 5,366,878; 5,403,711; 5,491,133; 5,565,350; 5,623,065; 5,652,355; 5,652,356; and 5,700,922, certain of which are commonly owned with the instant application, and each of which is herein incorporated by reference in its entirety.
- stable compound and “stable structure” are meant to indicate a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent. Only stable compounds are contemplated herein.
- substituents within the compounds described herein are present to a recursive degree.
- "recursive substituent” means that a substituent may recite another instance of itself. Because of the recursive nature of such substituents, theoretically, a large number may be present in any given claim.
- One of ordinary skill in the art of medicinal chemistry and organic chemistry understands that the total number of such substituents is reasonably limited by the desired properties of the compound intended. Such properties include, by way of example and not limitation, physical properties such as molecular weight, solubility or log P, application properties such as activity against the intended target, and practical properties such as ease of synthesis.
- Recursive substituents are an intended aspect of the invention.
- One of ordinary skill in the art of medicinal and organic chemistry understands the versatility of such substituents.
- alkyl refers to a saturated straight or branched hydrocarbon radical containing up to twenty four carbon atoms.
- alkyl groups include, but are not limited to, methyl, ethyl, propyl, butyl, isopropyl, n-hexyl, octyl, decyl, dodecyl and the like.
- Alkyl groups typically include from 1 to about 24 carbon atoms, more typically from 1 to about 12 carbon atoms (Ci-Ci 2 alkyl) with from 1 to about 6 carbon atoms being more preferred.
- the term "lower alkyl” as used herein includes from 1 to about 6 carbon atoms.
- Alkyl groups as used herein may optionally include one or more further substitutent groups.
- alkenyl refers to a straight or branched hydrocarbon chain radical containing up to twenty four carbon atoms and having at least one carbon-carbon double bond.
- alkenyl groups include, but are not limited to, ethenyl, propenyl, butenyl, 1 - methyl-2-buten-l-yl, dienes such as 1,3 -butadiene and the like.
- Alkenyl groups typically include from 2 to about 24 carbon atoms, more typically from 2 to about 12 carbon atoms with from 2 to about 6 carbon atoms being more preferred.
- Alkenyl groups as used herein may optionally include one or more further substitutent groups.
- alkynyl refers to a straight or branched hydrocarbon radical containing up to twenty four carbon atoms and having at least one carbon-carbon triple bond.
- alkynyl groups include, but are not limited to, ethynyl, 1-propynyl, 1-butynyl, and the like.
- Alkynyl groups typically include from 2 to about 24 carbon atoms, more typically from 2 to about 12 carbon atoms with from 2 to about 6 carbon atoms being more preferred.
- Alkynyl groups as used herein may optionally include one or more further substitutent groups.
- aminoalkyl refers to an amino substituted alkyl radical. This term is meant to include Ci-C 12 alkyl groups having an amino substituent at any position and wherein the alkyl group attaches the aminoalkyl group to the parent molecule. The alkyl and/or amino portions of the aminoalkyl group can be further substituted with substituent groups.
- aliphatic refers to a straight or branched hydrocarbon radical containing up to twenty four carbon atoms wherein the saturation between any two carbon atoms is a single, double or triple bond.
- An aliphatic group preferably contains from 1 to about 24 carbon atoms, more typically from 1 to about 12 carbon atoms with from 1 to about 6 carbon atoms being more preferred.
- the straight or branched chain of an aliphatic group may be interupted with one or more heteroatoms that include nitrogen, oxygen, sulfur and phosphorus.
- heteroatoms include without limitation polyalkoxys, such as polyalkylene glycols, polyamines, and polyimines.
- Aliphatic groups as used herein may optionally include further substitutent groups.
- alicyclic refers to a cyclic ring system wherein the ring is aliphatic.
- the ring system can comprise one or more rings wherein at least one ring is aliphatic.
- Preferred alicyclics include rings having from about 5 to about 9 carbon atoms in the ring.
- Alicyclic as used herein may optionally include further substitutent groups.
- alkoxy refers to a radical formed between an alkyl group and an oxygen atom wherein the oxygen atom is used to attach the alkoxy group to a parent molecule.
- alkoxy groups include, but are not limited to, methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, sec-butoxy, tert-butoxy, n-pentoxy, neopentoxy, n-hexoxy and the like.
- Alkoxy groups as used herein may optionally include further substitutent groups.
- halo and "halogen,” as used herein, refer to an atom selected from fluorine, chlorine, bromine and iodine.
- aryl and aromatic refer to a mono- or polycyclic carbocyclic ring system radicals having one or more aromatic rings.
- aryl groups include, but are not limited to, phenyl, naphthyl, tetrahydronaphthyl, indanyl, idenyl and the like.
- Preferred aryl ring systems have from about 5 to about 20 carbon atoms in one or more rings.
- Aryl groups as used herein may optionally include further substitutent groups.
- aralkyl and arylalkyl refer to a radical formed between an alkyl group and an aryl group wherein the alkyl group is used to attach the aralkyl group to a parent molecule. Examples include, but are not limited to, benzyl, phenethyl and the like. Aralkyl groups as used herein may optionally include further substitutent groups attached to the alkyl, the aryl or both groups that form the radical group.
- heterocyclic radical refers to a radical mono-, or poly-cyclic ring system that includes at least one heteroatom and is unsaturated, partially saturated or fully saturated, thereby including heteroaryl groups. Heterocyclic is also meant to include fused ring systems wherein one or more of the fused rings contain at least one heteroatom and the other rings can contain one or more heteroatoms or optionally contain no heteroatoms.
- a heterocyclic group typically includes at least one atom selected from sulfur, nitrogen or oxygen.
- heterocyclic groups include, [l,3]dioxolane, pyrrolidinyl, pyrazolinyl, pyrazolidinyl, imidazolinyl, imidazolidinyl, piperidinyl, piperazinyl, oxazolidinyl, isoxazolidinyl, morpholinyl, thiazolidinyl, isothiazolidinyl, quinoxalinyl, pyridazinonyl, tetrahydrofuryl and the like.
- Heterocyclic groups as used herein may optionally include further substitutent groups.
- heteroaryl refers to a radical comprising a mono- or poly-cyclic aromatic ring, ring system or fused ring system wherein at least one of the rings is aromatic and includes one or more heteroatom. Heteroaryl is also meant to include fused ring systems including systems where one or more of the fused rings contain no heteroatoms. Heteroaryl groups typically include one ring atom selected from sulfur, nitrogen or oxygen.
- heteroaryl groups include, but are not limited to, pyridinyl, pyrazinyl, pyrimidinyl, pyrrolyl, pyrazolyl, imidazolyl, thiazolyl, oxazolyl, isooxazolyl, thiadiazolyl, oxadiazolyl, thiophenyl, furanyl, quinolinyl, isoquinolinyl, benzimidazolyl, benzooxazolyl, quinoxalinyl, and the like.
- Heteroaryl radicals can be attached to a parent molecule directly or through a linking moiety such as an aliphatic group or hetero atom.
- Heteroaryl groups as used herein may optionally include further substitutent groups.
- heteroarylalkyl refers to a heteroaryl group as previously defined having an alky radical that can attach the heteroarylalkyl group to a parent molecule.
- heteroarylalkyl groups as used herein may optionally include further substitutent groups on one or both of the heteroaryl or alkyl portions.
- mono or poly cyclic structure includes all ring systems that are single or polycyclic having rings that are fused or linked and is meant to be inclusive of single and mixed ring systems individually selected from aliphatic, alicyclic, aryl, heteroaryl, aralkyl, arylalkyl, heterocyclic, heteroaryl, heteroaromatic, heteroarylalkyl.
- Such mono and poly cyclic structures can contain rings that are uniform or have varying degrees of saturation including fully saturated, partially saturated or fully unsaturated.
- Each ring can comprise ring atoms selected from C, N, O and S to give rise to heterocyclic rings as well as rings comprising only C ring atoms which can be present in a mixed motif such as for example benzimidazole wherein one ring has only carbon ring atoms and the fused ring has two nitrogen atoms.
- mono or poly cyclic structures can be attached to a parent molecule directly through a ring atom, through a substituent group or a bifunctional linking moiety.
- acyl refers to a radical formed by removal of a hydroxyl group from an organic acid and has the general formula -C(O)-X where X is typically aliphatic, alicyclic or aromatic. Examples include aliphatic carbonyls, aromatic carbonyls, aliphatic sulfonyls, aromatic sulfinyls, aliphatic sulf ⁇ nyls, aromatic phosphates, aliphatic phosphates and the like. Acyl groups as used herein may optionally include further substitutent groups.
- hydrocarbyl includes groups comprising C, O and H. Included are straight, branched and cyclic groups having any degree of saturation. Such hydrocarbyl groups can include one or more heteroatoms selected from N, O and S and can be further mono or poly substituted with one or more substituent groups.
- substituted and substituteduent group are meant to include groups that are typically added to other groups or parent compounds to enhance desired properties or give desired effects. Substituent groups can be protected or unprotected and can be added to one available site or to many available sites in a parent compound. Substituent groups may also be further substituted with other substituent groups and may be attached directly or via a linking group such as an alkyl or hydrocarbyl group to a parent compound.
- each R 33 , R bb and R 4x is, independently, H, an optionally linked chemical functional group or a further substituent group with a preferred list including without limitation H, alkyl, alkenyl, alkynyl, aliphatic, alkoxy, acyl, aryl, aralkyl, heteroaryl, alicyclic, heterocyclic and heteroarylalkyl.
- the compounds (e.g., bicyclic nucleosides) described herein can be prepared by any of the applicable techniques of organic synthesis, as, for example, illustrated in the examples below. Many such techniques are well known in the art.
- oligomeric compounds are modified by covalent attachment of one or more conjugate groups.
- conjugate groups modify one or more properties of the attached oligomeric compound including but not limited to pharmakodynamic, pharmacokinetic, binding, absorption, cellular distribution, cellular uptake, charge and clearance.
- Conjugate groups are routinely used in the chemical arts and are linked directly or via an optional linking moiety or linking group to a parent compound such as an oligomeric compound.
- conjugate groups includes without limitation, intercalators, reporter molecules, polyamines, polyamides, polyethylene glycols, thioethers, polyethers, cholesterols, thiocholesterols, cholic acid moieties, folate, lipids, phospholipids, biotin, phenazine, phenanthridine, anthraquinone, adamantane, acridine, fluoresceins, rhodamines, coumarins and dyes.
- Linking groups or bifunctional linking moieties such as those known in the art are amenable to the present invention.
- Linking groups are useful for attachment of chemical functional groups, conjugate groups, reporter groups and other groups to selective sites in a parent compound such as for example an oligomeric compound.
- a bifunctional linking moiety comprises a hydrocarbyl moiety having two functional groups. One of the functional groups is selected to bind to a parent molecule or compound of interest and the other is selected to bind essentially any selected group such as a chemical functional group or a conjugate group.
- the linker comprises a chain structure or an oligomer of repeating units such as ethylene glyol or amino acid units.
- bifunctional linking moieties include amino, hydroxyl, carboxylic acid, thiol, unsaturations (e.g., double or triple bonds), and the like.
- bifunctional linking moieties include 8-amino-3,6-dioxaoctanoic acid (ADO), succmimidyl 4- (N-maleimidomethyl) cyclohexane-l-carboxylate (SMCC) and 6-aminohexanoic acid (AHEX or AHA).
- linking groups include, but are not limited to, substituted C 1 -C 10 alkyl, substituted or unsubstituted C 2 -C 10 alkenyl or substituted or unsubstituted C 2 -C 10 alkynyl, wherein a nonlimiting list of preferred substituent groups includes hydroxyl, amino, alkoxy, carboxy, benzyl, phenyl, nitro, thiol, thioalkoxy, halogen, alkyl, aryl, alkenyl and alkynyl.
- protecting group refers to a labile chemical moiety which is known in the art to protect reactive groups including without limitation, hydroxyl, amino and thiol groups, against undesired reactions during synthetic procedures.
- Protecting groups are typically used selectively and/or orthogonally to protect sites during reactions at other reactive sites and can then be removed to leave the unprotected group as is or available for further reactions.
- Protecting groups as known in the art are described generally in Greene and Wuts, Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons, New York (1999). Groups can be selectively incorporated into oligomeric compounds of the invention as precursors.
- an amino group can be placed into a compound of the invention as an azido group that can be chemically converted to the amino group at a desired point in the synthesis.
- groups are protected or present as precursors that will be inert to reactions that modify other areas of the parent molecule for conversion into their final groups at an appropriate time. Further representative protecting or precursor groups are discussed in
- hydroxyl protecting groups include, but are not limited to, acetyl, t-butyl, t- butoxymethyl, methoxymethyl, tetrahydropyranyl, 1-ethoxyethyl, l-(2-chloroethoxy)ethyl, p- chlorophenyl, 2,4-dinitrophenyl, benzyl, 2,6-dichlorobenzyl, diphenylmethyl, p-nitrobenzyl, bis(2-acetoxyethoxy)methyl (ACE), 2-trimethylsilylethyl, trimethylsilyl, triethylsilyl, t- butyldimethylsilyl, t-butyldiphenylsilyl, triphenylsilyl, [(triisopropylsilyl)oxy]methyl (TOM), benzoylformate, chloroacetyl, trichloroacetyl, trifluoroacetyl, pivaloyl
- hydroxyl protecting groups include, but are not limited to, benzyl, 2,6-dichlorobenzyl, t-butyldimethylsilyl, t-butyldiphenylsilyl, benzoyl, mesylate, tosylate, dimethoxytrityl (DMT), 9-phenylxanthine-9-yl (Pixyl) and 9-(p-methoxyphenyl)xanthine-9-yl (MOX).
- benzyl 2,6-dichlorobenzyl
- t-butyldimethylsilyl t-butyldiphenylsilyl
- benzoyl mesylate, tosylate, dimethoxytrityl (DMT), 9-phenylxanthine-9-yl (Pixyl) and 9-(p-methoxyphenyl)xanthine-9-yl (MOX).
- amino protecting groups include, but are not limited to, carbamate- protecting groups, such as 2-trimethylsilylethoxycarbonyl (Teoc), 1 -methyl- l-(4-biphenylyl)- ethoxycarbonyl (Bpoc), t-butoxycarbonyl (BOC), allyloxycarbonyl (Alloc), 9- fluorenylmethyloxycarbonyl (Fmoc), and benzyloxycarbonyl (Cbz); amide-protecting groups, such as formyl, acetyl, trihaloacetyl, benzoyl, and nitrophenylacetyl; sulfonamide-protecting groups, such as 2-nitrobenzenesulfonyl; and imine- and cyclic imide-protecting groups, such as phthalimido and dithiasuccinoyl.
- carbamate- protecting groups such as 2-trimethylsilylethoxycarbonyl (Teoc), 1 -methyl
- thiol protecting groups include, but are not limited to, triphenylmethyl (trityl), benzyl (Bn), and the like.
- oligomeric compounds are prepared by connecting nucleosides with optionally protected phosphorus containing internucleoside linkages.
- Representative protecting groups for phosphorus containing internucleoside linkages such as phosphodiester and phosphorothioate linkages include ⁇ -cyanoethyl, diphenylsilylethyl, ⁇ - cyanobutenyl, cyano p-xylyl (CPX), N-methyl-N-trifluoroacetyl ethyl (META), acetoxy phenoxy ethyl (APE) and butene-4-yl groups. See for example U.S. Patents Nos. 4,725,677 and Re.
- Orthogonal protection is widely used in for example automated oligonucleotide synthesis.
- a functional group is deblocked in the presence of one or more other protected functional groups which is not affected by the deblocking procedure. This deblocked functional group is reacted in some manner and at some point a further orthogonal protecting group is removed under a different set of reaction conditions. This allows for selective chemistry to arrive at a desired compound or oligomeric compound.
- the present invention provides compounds having reactive phosphorus groups useful for forming internucleoside linkages including for example phosphodiester and phosphorothioate internucleoside linkages.
- Such reactive phosphorus groups are known in the art and contain phosphorus atoms in P i ⁇ or P v valence state including, but not limited to, phosphoramidite, H- phosphonate, phosphate triesters and phosphorus containing chiral auxiliaries.
- a preferred syn- thetic solid phase synthesis utilizes phosphoramidites (P 111 chemistry) as reactive phosphites.
- the intermediate phosphite compounds are subsequently oxidized to the P v state using known methods to yield, in preferred embodiments, phosphodiester or phosphorothioate internucleotide linkages.
- oligomeric compounds useful in this invention include oligonucleotides containing modified e.g. non-naturally occurring internucleoside linkages.
- Two main classes of intemucleoside linkages are defined by the presense or absence of a phosphorus atom.
- Modified internucleoside linkages having a phosphorus atom include, but are not limited to, phosphorothioates, chiral phosphorothioates, phosphorodithioates, phosphotriesters, aminoalkylphosphotri esters, methyl and other alkyl phosphonates including 3'-alkylene phosphonates, 5'-alkylene phosphonates and chiral phosphonates, phosphinates, phosphoramidates including 3 '-amino phosphoramidate and aminoalkylphosphoramidates, thionophosphoramidates, thionoalkylphosphonates, thionoalkylphosphotriesters, selenophosphates and boranophosphates having normal 3'-5' linkages, 2'-5' linked analogs of these, and those having inverted polarity wherein one or more internucleotide linkages is a 3' to 3', 5' to 5' or 2' to 2' linkage.
- Oligonucleotides having inverted polarity can comprise a single 3' to 3' linkage at the 3'-most internucleotide linkage i.e. a single inverted nucleoside residue which may be abasic (the nucleobase is missing or has a hydroxyl group in place thereof).
- Various salts, mixed salts and free acid forms are also included.
- Modified internucleoside linkages not having a phosphorus atom include, but are not limited to, those that are formed by short chain alkyl or cycloalkyl internucleoside linkages, mixed heteroatom and alkyl or cycloalkyl internucleoside linkages, or one or more short chain heteroatomic or heterocyclic internucleoside linkages.
- siloxane backbones include those having siloxane backbones; sulfide, sulfoxide and sulfone backbones; formacetyl and thioformacetyl backbones; methylene formacetyl and thioformacetyl backbones; riboacetyl backbones; alkene containing backbones; sulfamate backbones; methyleneimino and methylenehydrazino backbones; sulfonate and sulfonamide backbones; amide backbones; and others having mixed N, O, S and CH 2 component parts.
- U.S. patents that teach the preparation of the above oligonucleosides include, but are not limited to, U.S.: 5,034,506; 5,166,315; 5,185,444; 5,214,134; 5,216,141; 5,235,033; 5,264,562; 5,264,564; 5,405,938; 5,434,257; 5,466,677; 5,470,967; 5,489,677; 5,541,307; 5,561,225; 5,596,086; 5,602,240; 5,610,289; 5,602,240; 5,608,046; 5,610,289; 5,618,704; 5,623,070; 5,663,312; 5,633,360; 5,677,437; 5,792,608; 5,646,269 and 5,677,439, certain of which are commonly owned with this application, and each of which is herein incorporated by reference.
- the compounds described herein contain one or more asymmetric centers and thus give rise to enantiomers, diastereomers, and other stereoisomeric forms that may be defined, in terms of absolute stereochemistry, as (R)- or (S)-, ⁇ or ⁇ , or as (D)- or (L)- such as for amino acids.
- the present invention is meant to include all such possible isomers, as well as their racemic and optically pure forms.
- Optical isomers may be prepared from their respective optically active precursors by the procedures described above, or by resolving the racemic mixtures. The resolution can be carried out in the presence of a resolving agent, by chromatography or by repeated crystallization or by some combination of these techniques which are known to those skilled in the art.
- any carbon-carbon double bond appearing herein is selected for convenience only and is not intended to designate a particular configuration unless the text so states; thus a carbon-carbon double bond or carbon-heteroatom double bond depicted arbitrarily herein as trans may be cis, trans, or a mixture of the two in any proportion.
- the term "oligomeric compound" refers to a polymer having at least a region that is capable of hybridizing to a nucleic acid molecule.
- oligomeric compound includes oligonucleotides, oligonucleotide analogs and oligonucleosides as well as nucleotide mimetics and/or mixed polymers comprising nucleic acid and non-nucleic acid components. Oligomeric compounds are routinely prepared linearly but can be joined or otherwise prepared to be circular and may also include branching. Oligomeric compounds can form double stranded constructs such as for example two strands hybridized to form double stranded compositions. The double stranded compositions can be linked or separate and can include overhangs on the ends.
- an oligomeric compound comprises a backbone of linked monomelic subunits where each linked monomelic subunit is directly or indirectly attached to a heterocyclic base moiety. Oligomeric compounds may also include monomelic subunits that are not linked to a heterocyclic base moiety thereby providing abasic sites.
- the linkages joining the monomelic subunits, the sugar moieties or surrogates and the heterocyclic base moieties can be independently modified.
- the linkage-sugar unit which may or may not include a heterocyclic base, may be substituted with a mimetic such as the monomers in peptide nucleic acids.
- nucleoside is a base-sugar combination.
- the base portion of the nucleoside is normally a heterocyclic base moiety.
- the two most common classes of such heterocyclic bases are purines and pyrimidines.
- Nucleotides are nucleosides that further include a phosphate group covalently linked to the sugar portion of the nucleoside. For those nucleosides that include a pentofuranosyl sugar, the phosphate group can be linked to either the 2 ⁇ 3' or 5' hydroxyl moiety of the sugar.
- the phosphate groups covalently link adjacent nucleosides to one another to form a linear polymeric compound.
- the respective ends of this linear polymeric structure can be joined to form a circular structure by hybridization or by formation of a covalent bond, however, open linear structures are generally desired.
- the phosphate groups are commonly referred to as forming the internucleoside linkages of the oligonucleotide.
- the normal internucleoside linkage of RNA and DNA is a 3' to 5' phosphodi ester linkage.
- oligonucleotide refers to an oligomer or polymer of ribonucleic acid (RNA) or deoxyribonucleic acid (DNA). This term includes oligonucleotides composed of naturally-occurring nucleobases, sugars and covalent internucleoside linkages.
- oligonucleotide analog refers to oligonucleotides that have one or more non-naturally occurring portions. Such non-naturally occurring oligonucleotides are often desired over naturally occurring forms because of desirable properties such as, for example, enhanced cellular uptake, enhanced affinity for nucleic acid target and increased stability in the presence of nucleases.
- oligonucleoside refers to a sequence of nucleosides that are joined by internucleoside linkages that do not have phosphorus atoms. Internucleoside linkages of this type include short chain alkyl, cycloalkyl, mixed heteroatom alkyl, mixed heteroatom cycloalkyl, one or more short chain heteroatomic and one or more short chain heterocyclic.
- internucleoside linkages include, but are not limited to, siloxane, sulfide, sulfoxide, sulfone, acetyl, formacetyl, thioformacetyl, methylene formacetyl, thioformacetyl, alkeneyl, sulfamate, methyleneimino, methylenehydrazino, sulfonate, sulfonamide, amide and others having mixed N, O, S and CH 2 component parts.
- U.S. patents that teach the preparation of the above oligonucleosides include, but are not limited to, U.S.: 5,034,506; 5,166,315; 5,185,444; 5,214,134; 5,216,141; 5,235,033; 5,264,562; 5,264,564; 5,405,938; 5,434,257; 5,466,677; 5,470,967; 5,489,677; 5,541,307; 5,561,225; 5,596,086; 5,602,240; 5,610,289; 5,602,240; 5,608,046; 5,610,289; 5,618,704; 5,623,070; 5,663,312; 5,633,360; 5,677,437; 5,792,608; 5,646,269 and 5,677,439, certain of which are commonly owned with this application, and each of which is herein incorporated by reference.
- nucleobase or “heterocyclic base moiety” as used herein, is intended to by synonymous with “nucleic acid base or mimetic thereof.”
- a nucleobase is any substructure that contains one or more atoms or groups of atoms capable of hydrogen bonding to a base of a nucleic acid.
- unmodified or “natural” nucleobases include the purine bases adenine
- Modified nucleobases include other synthetic and natural nucleobases such as 5-methylcytosine (5-me-C), 5-hydroxymethyl cytosine, xanthine, hypoxanthine, 2-aminoadenine, 6-methyl and other alkyl derivatives of adenine and guanine, 2-propyl and other alkyl derivatives of adenine and guanine, 2-thiouracil, 2-thiothymine and 2-thiocytosine, 5-halouracil and cytosine, 5- propynyl (-C ⁇ C-CH 3 ) uracil and cytosine and other alkynyl derivatives of pyrimidine bases, 6- azo uracil, cytosine and thymine, 5-uracil (pseudouracil), 4-thiouracil, 8-halo,
- nucleobases include tricyclic pyrimidines such as phenoxazine cytidine(lH- pyrimido[5,4-b][l,4]benzoxazin-2(3H)-one), phenothiazine cytidine (lH-pyrimido[5,4- b][l,4]benzothiazin-2(3H)-one), G-clamps such as a substituted phenoxazine cytidine (e.g.
- nucleobases may also include those in which the purine or pyrimidine base is replaced with other heterocycles, for example 7-deaza-adenine, 7-deazaguanosine, 2- aminopyridine and 2-pyridone. Further nucleobases include those disclosed in United States Patent No.
- Modified nucleobases include, but are not limited to, universal bases, hydrophobic bases, promiscuous bases, size-expanded bases, and fluorinated bases as defined herein. Certain of these nucleobases are particularly useful for increasing the binding affinity of the oligomeric compounds of the invention. These include 5-substituted pyrimidines, 6-azapyrimidines and N- 2, N-6 and O-6 substituted purines, including 2-aminopropyladenine, 5-propynyluracil and 5- propynylcytosine. 5-methylcytosine substitutions have been shown to increase nucleic acid duplex stability by 0.6-1.2 °C (Sanghvi, Y.S., Crooke, S.T.
- Oligomeric compounds of the present invention may also contain one or more nucleosides having modified sugar moieties.
- the furanosyl sugar ring can be modified in a number of ways including substitution with a substituent group, bridging to form a BNA and substitution of the 4'-0 with a heteroatom such as S or N(R).
- Some representative U.S. patents that teach the preparation of such modified sugars include, but are not limited to, U.S.:
- nucleoside mimetic is intended to include those structures used to replace the sugar or the sugar and the base not the linkage at one or more positions of an oligomeric compound such as for example nucleoside mimetics having morpholino or bicyclo[3.1.Ojhexyl sugar mimetics e.g. non furanose sugar units with a phosphodiester linkage.
- nucleoside mimetic overlaps with the slightly broader term “nucleoside mimetic” but is intended to indicate replacement of the sugar unit (furanose ring) only.
- the oligomeric compounds in accordance with the present invention can comprise from about 8 to about 80 nucleosides and/or modified nucleosides or mimetics in length.
- the invention embodies oligomeric compounds of 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, or 80 nucleosides and/or modified nucleosides or mimetics in length, or any range therewithin.
- the oligomeric compounds of the invention are 8 to 40 nucleosides and/or modified nucleosides or mimetics in length.
- this embodies oligomeric compounds of 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39 or 40 nucleosides and/or modified nucleosides or mimetics in length, or any range therewithin.
- the oligomeric compounds of the invention are 8 to 20 nucleosides and/or modified nucleosides or mimetics in length.
- this embodies oligomeric compounds of 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20 nucleosides and/or modified nucleosides or mimetics in length, or any range therewithin.
- the oligomeric compounds of the invention are 10 to 16 nucleosides and/or modified nucleosides or mimetics in length.
- the oligomeric compounds of the invention are 10 to 14 nucleosides and/or modified nucleosides or mimetics in length.
- this embodies oligomeric compounds of 10, 11, 12, 13 or 14 nucleosides and/or modified nucleosides or mimetics in length, or any range therewithin.
- Chimeric oligomeric compounds have differentially modified nucleosides at two or more positions and are generally defined as having a motif.
- Chimeric oligomeric compounds of the invention may be formed as composite structures of two or more oligonucleotides, oligonucleotide analogs, oligonucleosides and/or oligonucleotide mimetics as described above. Representative U.S.
- RNA and related analogs have been increasing as efforts in RNAi increase.
- the primary RNA synthesis strategies that are presently being used commercially include 5'-O-DMT-2'-O-t-butyldimethylsilyl (TBDMS), 5'-O-DMT-2'-O-[l(2-fluorophenyl)-4-methoxypiperidin-4-yl] (FPMP), 2'-O- [(triisopropylsilyl)oxy]methyl (2 ? -O-CH 2 -O-Si(iPr) 3 (TOM), and the 5'-O-silyl ether-2'-ACE (5 ?
- RNA synthesis activator advertised to reduce coupling times especially with TOM and TBDMS chemistries. Such an activator would also be amenable to the present invention.
- TBDMS 5'-O-DMT-2'-O-t-butyldimethylsilyl
- TOM 2'-O-[(triisopropylsilyl)oxy]methyl
- DOD/ACE (5'-O-bis(trimethylsiloxy)cyclododecyloxysilyl ether-2'-O-bis(2- acetoxyethoxy)methyl
- FPMP 5'-O-DMT-2'-O-[l(2-fluorophenyl)-4-methoxypiperidin-4-yl] .
- RNA synthesis strategies are amenable to the present invention.
- Strategies that would be a hybrid of the above e.g. using a 5 '-protecting group from one strategy with a 2'-O-protecting from another strategy is also amenable to the present invention.
- hybridization means the pairing of complementary strands of oligomeric compounds.
- one mechanism of pairing involves hydrogen bonding, which may be Watson-Crick, Hoogsteen or reversed Hoogsteen hydrogen bonding, between complementary nucleoside or nucleotide bases (nucleobases) of the strands of oligomeric compounds.
- nucleobases complementary nucleoside or nucleotide bases
- adenine and thymine are complementary nucleobases which pair through the formation of hydrogen bonds.
- Hybridization can occur under varying circumstances.
- An oligomeric compound is specifically hybridizable when binding of the compound to the target nucleic acid interferes with the normal function of the target nucleic acid to cause a loss of activity, and there is a sufficient degree of complementarity to avoid non-specific binding of the oligomeric compound to non-target nucleic acid sequences under conditions in which specific binding is desired, i.e., under physiological conditions in the case of in vivo assays or therapeutic treatment, and under conditions in which assays are performed in the case of in vitro assays.
- “Complementary,” as used herein, refers to the capacity for precise pairing of two nucleobases regardless of where the two are located.
- a nucleobase at a certain position of an oligomeric compound is capable of hydrogen bonding with a nucleobase at a certain position of a target nucleic acid, the target nucleic acid being a DNA, RNA, or oligonucleotide molecule
- the position of hydrogen bonding between the oligonucleotide and the target nucleic acid is considered to be a complementary position.
- the oligomeric compound and the further DNA, RNA, or oligonucleotide molecule are complementary to each other when a sufficient number of complementary positions in each molecule are occupied by nucleobases which can hydrogen bond with each other.
- an oligomeric compound need not be 100% complementary to that of its target nucleic acid to be specifically hybridizable.
- an oligonucleotide may hybridize over one or more segments such that intervening or adjacent segments are not involved in the hybridization event (e.g., a loop structure or hairpin structure).
- the oligomeric compounds of the present invention can comprise at least about 70%, at least about 80%, at least about 90%, at least about 95%, or at least about 99% sequence complementarity to a target region within the target nucleic acid sequence to which they are targeted.
- an oligomeric compound in which 18 of 20 nucleobases of the oligomeric compound are complementary to a target region, and would therefore specifically hybridize would represent 90 percent complementarity.
- the remaining noncomplementary nucleobases may be clustered or interspersed with complementary nucleobases and need not be contiguous to each other or to complementary nucleobases.
- an oligomeric compound which is 18 nucleobases in length having 4 (four) noncomplementary nucleobases which are flanked by two regions of complete complementarity with the target nucleic acid would have 77.8% overall complementarity with the target nucleic acid and would thus fall within the scope of the present invention.
- Percent complementarity of an oligomeric compound with a region of a target nucleic acid can be determined routinely using BLAST programs (basic local alignment search tools) and PowerBLAST programs known in the art (Altschul et al., J. MoI. Biol., 1990, 215, 403-410; Zhang and Madden, Genome Res., 1997, 7, 649-656).
- oligomeric compounds such as antisense oligomeric compounds, antisense oligonucleotides, ribozymes, external guide sequence (EGS) oligonucleotides, alternate splicers, primers, probes, and other oligomeric compounds which hybridize to at least a portion of the target nucleic acid.
- these oligomeric compounds may be introduced in the form of single-stranded, double-stranded, circular or hairpin oligomeric compounds and may contain structural elements such as internal or terminal bulges or loops.
- the oligomeric compounds of the invention may elicit the action of one or more enzymes or structural proteins to effect modification of the target nucleic acid.
- RNAse H a cellular endonuclease which cleaves the RNA strand of an RNA:DNA duplex. It is known in the art that single- stranded oligomeric compounds which are "DNA-like" elicit RNAse H. Activation of RNase H, therefore, results in cleavage of the RNA target, thereby greatly enhancing the efficiency of oligonucleotide-mediated inhibition of gene expression. Similar roles have been postulated for other ribonucleases such as those in the RNase III and ribonuclease L family of enzymes.
- oligomeric compound is a single-stranded antisense oligonucleotide
- double-stranded RNA (dsRNA) molecules has been shown to induce potent and specific antisense-mediated reduction of the function of a gene or its associated gene products. This phenomenon occurs in both plants and animals and is believed to have an evolutionary connection to viral defense and transposon silencing.
- suitable target segments may be employed in a screen for additional oligomeric compounds that modulate the expression of a selected protein.
- Modulators are those oligomeric compounds that decrease or increase the expression of a nucleic acid molecule encoding a protein and which comprise at least an 8-nucleobase portion which is complementary to a suitable target segment.
- the screening method comprises the steps of contacting a suitable target segment of a nucleic acid molecule encoding a protein with one or more candidate modulators, and selecting for one or more candidate modulators which decrease or increase the expression of a nucleic acid molecule encoding a protein. Once it is shown that the candidate modulator or modulators are capable of modulating (e.g.
- the modulator may then be employed in further investigative studies of the function of the peptide, or for use as a research, diagnostic, or therapeutic agent in accordance with the present invention.
- the suitable target segments of the present invention may also be combined with their respective complementary antisense oligomeric compounds of the present invention to form stabilized double-stranded (duplexed) oligonucleotides.
- Such double stranded oligonucleotide moieties have been shown in the art to modulate target expression and regulate translation as well as RNA processsing via an antisense mechanism.
- double-stranded moieties may be subject to chemical modifications (Fire et al., Nature, 1998, 391, 806-811; Timmons and Fire, Nature 1998, 395, 854; Timmons et al., Gene, 2001, 263, 103-112; Tabara et al., Science, 1998, 282, 430-431; Montgomery et al., Proc. Natl. Acad. Sci. USA, 1998, 95, 15502-15507; Tuschl et al., Genes Dev., 1999, 13, 3191-3197; Elbashir et al., Nature, 2001, 411, 494-498; Elbashir et al., Genes Dev. 2001, 15, 188-200).
- oligomeric compounds of the present invention can also be applied in the areas of drug discovery and target validation.
- the present invention comprehends the use of the oligomeric compounds and targets identified herein in drug discovery efforts to elucidate relationships that exist between proteins and a disease state, phenotype, or condition.
- These methods include detecting or modulating a target peptide comprising contacting a sample, tissue, cell, or organism with the oligomeric compounds of the present invention, measuring the nucleic acid or protein level of the target and/or a related phenotypic or chemical endpoint at some time after treatment, and optionally comparing the measured value to a non-treated sample or sample treated with a further oligomeric compound of the invention.
- These methods can also be performed in parallel or in combination with other experiments to determine the function of unknown genes for the process of target validation or to determine the validity of a particular gene product as a target for treatment or prevention of a particular disease, condition, or phenotype.
- RNAi activity Effect of nucleoside modifications on RNAi activity is evaluated according to existing literature (Elbashir et al., Nature (2001), 411, 494-498; Nishikura et al., Cell (2001), 107, 415- 416; and Bass et al., Cell (2000), 101, 235-238.)
- oligomeric compounds of the present invention can be utilized for diagnostics, therapeutics, prophylaxis and as research reagents and kits. Furthermore, antisense oligonucleotides, which are able to inhibit gene expression with 17, specificity, are often used by those of ordinary skill to elucidate the function of particular genes or to distinguish between functions of various members of a biological pathway.
- the oligomeric compounds of the present invention can be used as tools in differential and/or combinatorial analyses to elucidate expression patterns of a portion or the entire complement of genes expressed within cells and tissues.
- expression patterns within cells or tissues treated with one or more oligomeric compounds are compared to control cells or tissues not treated with oligomeric compounds and the patterns produced are analyzed for differential levels of gene expression as they pertain, for example, to disease association, signaling pathway, cellular localization, expression level, size, structure or function of the genes examined. These analyses can be performed on stimulated or unstimulated cells and in the presence or absence of other compounds and or oligomeric compounds which affect expression patterns.
- Examples of methods of gene expression analysis known in the art include DNA arrays or microarrays (Brazma and ViIo, FEBS Lett., 2000, 480, 17-24; Celis, et al., FEBS Lett., 2000, 480, 2-16), SAGE (serial analysis of gene expression)(Madden, et al., Drug Discov. Today, 2000, 5, 415-425), READS (restriction enzyme amplification of digested cDNAs) (Prashar and Weissman, Methods Enzymol., 1999, 303, 258-72), TOGA (total gene expression analysis) (Sutcliffe, et al., Proc. Natl. Acad. Sci. U. S.
- oligomeric compounds of the invention are useful for research and diagnostics, because these oligomeric compounds hybridize to nucleic acids encoding proteins.
- oligonucleotides that are shown to hybridize with such efficiency and under such conditions as disclosed herein as to be effective protein inhibitors will also be effective primers or probes under conditions favoring gene amplification or detection, respectively.
- These primers and probes are useful in methods requiring the specific detection of nucleic acid molecules encoding proteins and in the amplification of the nucleic acid molecules for detection or for use in further studies.
- Hybridization of the antisense oligonucleotides, particularly the primers and probes, of the invention with a nucleic acid can be detected by means known in the art.
- Such means may include conjugation of an enzyme to the oligonucleotide, radiolabelling of the oligonucleotide or any other suitable detection means. Kits using such detection means for detecting the level of selected proteins in a sample may also be prepared.
- tert-Butyldiphenylsilyl chloride (1.73 mL, 6.7 mmol) was added to a cold (O 0 C) solution of nucleoside 10 (from above), triethylamine (1.4 mL, 10.0 mmol) and 4-dimethylaminopyridine (80 mg, 0.7 mmol) in CH 2 Cl 2 (9 mL). After stirring for 16h at rt, the reaction was poured into EtOAc and the organic phase was sequentially washed with 5% aqueous HCl, saturated NaHCO 3 , dried (Na 2 SO 4 ) and concentrated under vacuum. Purification by column chromatography (SiO 2 , eluting with 50% EtOAc/hexanes) provided nucleoside 11 (2.02 g, 79% from 8) as a white solid.
- Triethylamine trihydrofluoride (2.98 mL, 18.3 mmol) was added to a solution of nucleoside 12 (1.86 g, 3.7 mmol) and triethylamine (1.03 mL, 7.3 mmol) in THF (36 mL), in a polypropylene tube. After stirring at rt for 16h, the reaction was concentrated under vacuum and the residue dissolved in EtOAc. The organic layer was sequentially washed with water, saturated NaHCO 3 , brine, dried (Na 2 SO 4 ) and concentrated under vacuum. Purification by column chromatography (SiO 2 , 15% MeOH/CHCl 3 ) provided nucleoside 13 (1.31 g, product contaminated with triethylamine) as a white solid.
- nucleoside 14 (1.85 g, 89%) as a white foam.
- 2-Cyanoethyl tetraisopropylphorodiamidite (0.69 mL, 2.2 mmol) was added to a solution of nucleoside 14 (0.83 g, 1.4 mmol), tetrazole (80 mg, 1.2 mmol) and 7V-methylimidazole (29 ⁇ L, 0.36 mmol) in DMF (7.2 mL). After stirring at rt for 8h, the reaction was poured into EtOAc and the organic layer was washed with 90% brine, brine, dried (Na 2 SO 4 ) and concentrated. The residue was dissolved in minimum amount of EtOAc and this solution was added to hexanes.
- nucleoside 16 tert-Butyldimethylsilyl chloride (0.79 g, 5.2 mmol) was added to a solution of nucleoside 14 (1.0 g, 1.7 mmol) and imidazole (0.7Og, 10.4 mmol) in DMF (3.5 mL). After stirring at rt for 16h, the reaction was poured into EtOAc and the organic phase was sequentially extracted with brine, dried (Na 2 SO 4 ) and concentrated under vacuum. Purification by column chromatpography (SiO 2 , eluting with 50% EtOAc/hexanes) provided nucleoside 16 (1.17 g, 99%) as a white solid.
- Phosphorus oxychloride (1.27 mL, 13.6 mmol) was added to a cold (O 0 C) suspension of 1 ,2,4-triazole (4.0 g, 58.0 mmol) in CH 3 CN (21 mL). After stirring for 15 min, triethylamine (9.57 mL, 68 mmol) was added to the reaction and the stirring continued for 30 min. A solution of nucleoside 16 (1.17g, 1.7 mmol) in CH 3 CN (10 mL) was added to the reaction at O 0 C. After stirring for 10 min, the ice bath was removed and the reaction was stirred at rt for 4h. The solvent was then removed under vacuum and the residue was partitioned between EtOAc and water. The organic layer was then washed with saturated NaHCO 3 , brine, dried (Na 2 SO 4 ) and concentrated under vacuum to provide crude 17, which was used without any further purification.
- Aqueous ammonia (4 mL) was added to a solution of nucleoside 17 (from above) in dioxane (20 mL). After stirring at rt for 16h, the reaction was concentrated under vacuum and dried over high vacuum for 8h to provide nucleoside 18, which was used without any further purification.
- Benzoic anhydride (0.65 g, 2.9 mmol) was added to a solution of nucleoside 18 (from above) in DMF (3 mL). After stirring at rt for 16h, the reaction was poured into EtOAc and the organic layer was extracted with saturated NaHCO 3 , brine, dried (Na 2 SO 4 ) and concentrated under vacuum. Purification by column chromatography (SiO 2 , eluting with 50% EtOAc/hexanes) provided nucleoside 19 (1.2 g, 90% from 16) as a white solid.
- Triethylamine trihydrofluoride (1.48 mL, 9.1 mmol) was added to a solution of nucleoside 19 (1.86 g, 3.7 mmol) and triethylamine (1.03 mL, 7.3 mmol) in THF (15 mL) a polypropylene tube. After stirring at rt for 16h, the reaction was concentrated under vacuum and the residue was dissolved in EtOAc and the organic layer was sequentially washed with water, saturated NaHCO 3 , brine, dried (Na 2 SO 4 ) and concentrated under vacuum.
- nucleoside 20 (0.91 g, 90%) as a white solid.
- aldehyde 24 was used without any further purification.
- the crude aldehyde 24 from above was dissolved in a mixture of THF:H 2 O (1:1, 100 mL) and the reaction was cooled in an ice bath.
- Formaldehyde (25 mL, 35%w/w) and IN NaOH (100 mL) were added to the reaction.
- formaldehyde (5 mL) was added to the reaction and the stirring was continued for an additional 32h.
- Methanesulfonyl chloride (0.11 mL, 1.4 mmol) was added to a cold (O 0 C) solution of alcohol 30 (from above), triethylamine (1.77 mL, 10.5 mmol) and 4-dimethylaminopyridine (85 mg, 0.7 mmol) in CH 2 Cl 2 (21 mL). After stirring at rt for Ih, the reaction was poured into CHCl 3 and the organic layer was sequentially washed with 5% aqueous HCl, saturated NaHCO 3 , brine, dried (Na 2 SO 4 ) and concentrated under vacuum to provide mesylate 31, which was used without any purification.
- N,O-Bis(trimethylsilyl)acetamide (3.45 mL, 14.0 mmol) was added to a suspension of diacetate 32 (3.0 g, 4.1 mmol) and uracil (0.57 g, 5.1 mmol) in CH 3 CN (20 mL). After heating at 4O 0 C for 15 min to get a clear solution, trimethylsilyl triflate (0.95 mL, 5.3 mmol) was added to the reaction. After refluxing for 2h, the reaction was cooled to rt and poured into EtOAc. The organic layer was washed with saturated NaHCO 3 , brine, dried (Na 2 SO 4 ) and concentrated under vacuum to provide crude nucleoside 33, which was used without any purification.
- nucleoside 35 (1.25 g, 80%) as a white solid.
- nucleoside (36) Triethylamine trihydroflouride (2.4 mL, 14.7 mmol) was added to a solution of nucleoside 35 (1.25 g, 2.5 mmol) and triethlyamine (1.0 mL, 7.4 mmol) in THF (25 mL) in a polypropylene tube. After stirring at rt for 24h, the reaction was concentrated under vacuum and the residue was dissolved in EtOAc. The organic layer was then washed with water, saturated NaHCO 3 , brine, dried and concentrated (Na 2 SO 4 ). Purification by column chromatography (SiO 2 , eluting with 5% to 10% MeOH/CHCl 3 ) provided nucleoside 36 (0.88 g) as a white solid (product contaminated with Et 3 N).
- Dimethoxytrityl chloride (0.91 g, 2.7 mmol) was added to a solution of nucleoside 36 (from above) in pyridine (12 mL). After stirring at rt for 16h, the reaction was poured into EtOAc and the organic layer was washed with brine, dried and concentrated. Purification by column chromatography (SiO 2 , eluting with 90% EtOAc/hexanes) provided nucleoside 37 (1.28 g, 86% from 36) as a white solid.
- 2-Cyanoethyl tetraisopropylphorodiamidite (0.46 mL, 1.5 mmol) was added to a solution of nucleoside 37 (0.59 g, 1.0 mmol), tetrazole (57 mg, 0.82 mmol) and N-methylimidazole (20 ⁇ L, 0.25 mmol) in DMF (5 mL). After stirring at rt for 8h, the reaction was poured into EtOAc and the organic layer was washed with 90% brine, brine, dried (Na 2 SO 4 ) and concentrated.
- nucleoside 39 (0.68 g, 97%) as a white solid.
- Phosphorus oxychloride (0.74 mL, 8.0 mmol) was added to a cold (O 0 C) suspension of 1 ,2,4-triazole (2.35 g, 34.0 mmol) in CH 3 CN (16 mL). After stirring for 15 min, triethylamine (5.6 mL, 40 mmol) was added to the reaction and the stirring continued for 30 min. A solution of nucleoside 39 (0.68 g, 1.0 mmol) in CH 3 CN (7 mL) was added to the reaction at O 0 C. After stirring for 10 min, the ice bath was removed and the reaction was stirred at rt for 4h. The solvent was then removed under vacuum and the residue was partitioned between EtOAc and water. The organic layer was then washed with saturated NaHCO 3 , brine, dried (Na 2 SO 4 ) and concentrated under vacuum to provide crude 40, which was used without any further purification.
- Aqueous ammonia (2.5 mL) was added to a solution of nucleoside 40 (from above) in dioxane (12 mL). After stirring at rt for 16h, the reaction was concentrated under vacuum and dried over high vacuum for 8h to provide nucleoside 41, which was used without any further purification.
- nucleoside 41 (from above) in DMF (2 mL). After stirring at rt for 16h, the reaction was poured into EtOAc and the organic layer was extracted with saturated NaHCO 3 , brine, dried (Na 2 SO 4 ) and concentrated under vacuum. Purification by column chromatography (SiO 2 , eluting with 50% EtOAc/hexanes) provided nucleoside 42 (0.72 g, 91% from 39) as a white solid.
- nucleoside 43 (0.53 g, 87%) as a white solid.
- N,0-Bis(trimethylsilyl)acetamide (1.1 mL, 4.50 mmol) was added to a suspension of diacetate 32 (1.0 g, 1.4 mmol) and 6-iV-benzoyladenine (0.48 g, 2.0 mmol) in dichloroethane (14 mL).
- the reaction mixture turned clear after refluxing 45 minutes and was cooled in an ice bath and trimethylsilyl triflate (0.49 mL, 2.7 mmol) was added. After refluxing for 8 hours the reaction was cooled to room temperature and poured into EtOAc. The organic layer was washed with saturated NaHCO 3 and brine then dried (Na 2 SO 4 ) and concentrated under vacuum to provide crude nucleoside 45, which was used without purification.
- nucleoside 45 (from above) was added to a solution of nucleoside 45 (from above) in MeOH (14 mL). After stirring at room temperature for 24 hours the reaction was concentrated under vacuum. The residue was suspended in EtOAc, extracted with water and brine then dried (Na 2 SO 4 ) and concentrated under vacuum. Purification by column chromatography (SiO 2 , eluting with 1 to 2.5% MeOH/CHCl 3 ) provided nucleoside 46 as a white solid (0.69 g, 73% from 32).
- Nucleoside 47 is prepared from nucleoside 46 by reaction with benzoic anhydride (1.5-2 eq) in dry DMF.
- Phosphoramidite 51 is prepared from nucleoside 47 using the procedures illustrated in Example 3 for the phosphoramidite 38 from nucleoside 34.
- Methanesulfonyl chloride (1.33 mL, 16.8 mmol) was added dropwise to a cold (O 0 C) solution of alcohol 28 (7.37 g, 12.0 mmol), triethylamine (2.82 mL, 20.2 mmol) and DMAP (0.20 g, 1.1 mmol) in dichloromethane (25 mL). After stirring for 2 hours at room temperature, the reaction was diluted with dichloromethane and the organic layer was washed with 5% HCl, saturated sodium bicarbonate solution, brine, dried (Na 2 SO 4 ) and concentrated. The crude mesylate 52 thus obtained was used without further purification.
- Phosphoramidite 60 is prepared from diacetate 53 using the procedures illustrated in Example 3 for the phosphoramidite 51 from diacetate 32.
- N,OBis(trimethylsilyl)acetamide (3.8 mL, 15.5 mmol) was added to a suspension of diacetate 32 (3.44 g, 4.7 mmol) and 2-amino-6-chloropurine (1.18 g, 7.0 mmol) in dichloroethane (46 mL). After refluxing 45 minutes to get a clear solution, the reaction was cooled in an ice bath and trimethylsilyl triflate (1.69 mL, 9.4 mmol) was added. After refluxing for 8 hours the reaction was cooled to room temperature and poured into chloroform. The organic layer was washed with saturated NaHCO 3 and brine then dried (Na 2 SO 4 ) and concentrated under vacuum to provide crude nucleoside 61, which was used without purification.
- nucleoside (62) 3-Hydroxypropionitrile (1.67 mL, 24.5 mmol) was added dropwise to a stirring suspension of sodium hydride (1.07 g, 27.0 mmol, 60% w/w) in dry THF (10 mL). After stirring for 20 minutes, a solution of crude nucleoside 61 (from above) in dry THF (25 mL) was added. The stirring was continued for 5 hours at room temperature after which, the reaction was carefully quenched by the addition of a solution of saturated ammonium chloride. The reaction was poured into ethyl acetate and the organic layer was extracted with brine, dried (Na 2 SO 4 ) and concentrated. Purification of the residue by column chromatography (SiO 2 , eluting with CHCl 3 to 2.5% MeOH/CHCl 3 ) provided nucleoside 62 (3.18 g, 82% from 32) as a light brown solid.
- nucleoside 63 (2.5 g, 71%) as a yellowish foam.
- nucleoside 64 (1.84 g, 91%).
- Triethylamine trihydroflouride (2.88 mL, 17.9 mmol) was added to a solution of nucleoside 64 (1.84 g, 3.0 mmol) and triethylamine (1.25 mL, 8.9 mmol) in THF (30 mL) in a polypropylene tube. After stirring at room temperature for 24 hours the reaction was concentrated under vacuum and the residue was dissolved in EtOAc. The organic layer was then washed with water, saturated NaHCO 3 and brine then dried (Na 2 SO 4 ) and concentrated.
- nucleoside 65 (1.05 g, 97%) as a white solid.
- nucleoside 65 (66) Dimethoxytrityl chloride (1.07 g, 3.2 mmol) was added to a solution of nucleoside 65
- nucleoside 66 (1.00 g, 2.7 mmol) in pyridine (13 mL). After stirring at room temperature for 16 hours the reaction was poured into EtOAc and the organic layer was washed with brine, dried and concentrated. Purification by column chromatography (SiO 2 , eluting with 2.5 to 5% MeOH/CHCl 3 ) provided nucleoside 66 (1.52 g, 85%) as a white foam.
- Scheme 8 (a) 2-amino-6-chloropurine, BSA, TMSOTf, DCE, reflux, 2h; (b) 3-Hydroxypropionitrile, NaH, THF, 4h; (c) lsobutyric anhydride, DMAP, DMF, 6OC, 24h; (d) DDQ, CH 2 CI 2 , H 2 O, rt, 16h; (e) Et 3 N.3HF, Et 3 N, THF, rt, 16h; (f) DMTCI, Pyridine, rt, 16h; (g) CNCH 2 CH 2 OP(N-iPr 2 ) 2 , Tetrazole, NMI, DMF.
- the phosphoramidite 74 is prepared from diacetate 53 using the same procedures illustrated for the phosphoramidite 67 from diacetate 32.
- Methanesulfonyl chloride (2.3 mL, 29.2 mmol) was added to a cold (0 °C) solution of alcohols 75a-b (13.38 g, 20.8 mmol) dissolved in triethylamine (5.3 mL, 37.9 mmol) and DMAP (0.36 g, 2.9 mmol) in dichloromethane (42 mL). After stirring for 2 hours additional methanesulfonyl chloride (0.5 mL) was added. Stirring was continued for 1 hour and the reaction was diluted with chloroform. The organic layer was sequentially washed with 5% HCl, a saturated solution of sodium bicarbonate and brine then dried (Na 2 SO 4 ) and concentrated. Purification by column chromatography (SiO 2 , eluting with 20% EtOAc/hexanes) provide mesylates 76a-b (12.8 g, 85%) as viscous oil
- nucleosides (80a and 80b) DDQ (20.0 mmol, 4.5 g) was added to a solution of nucleosides 79a-b (9.0 g, 13.3 mmol) in dichloromethane (130 mL) and water (6.5 mL). The biphasic reaction was stirred at room temperature for 2 hours after which additional DDQ (2.75 g was added to the reaction). After another 2 hours additional DDQ (1.1 g) was added to the reaction and the stirring was continued for another 4 hours after which the reaction was stored in a refrigerator for 16 hours.
- nucleosides 80a and 80b were obtained by column chromatography (SiO 2 , eluting 10 to 20% acetone/chloroform) (7.0 g combined yield, 98%).
- nucleoside 811 Triethylamine trihydrofluoride (12.2 mL, 74.8 mmol) was added to a solution of nucleoside 80a (6.7 g, 12.5 mmol) and triethylamine (5.2 mL, 37.4 mmol) in THF (120 mL). After stirring at room temperature for 16 hours the reaction was concentrated to dryness under vacuum. The residue was purified by column chromatography (SiO 2 , eluting with 7.5% to 12.5 % MeOH/CHCl 3 ) to provide nucleoside 81 (contaminated with triethylamine.hydrofiouride salt, yield >100%), which was used without further purification.
- DMTCl 4,4'-Dimethoxytrityl chloride
- Triethylamine.trihydro fluoride (11.6 mL, 71.5 mmol) was added to a solution of nucleoside 80b (6.43 g, 12.0 mmol) and triethylamine (5.0 mL, 35.7 mmol) in THF (125 mL). After stirring at room temperature for 16 hours the reaction was concentrated to dryness under vacuum. The residue was purified by column chromatography (SiO 2 , eluting with 7.5% to 12.5 % MeOH/CHCl 3 ) to provide nucleoside 84 (contaminated with triethylamine.hydroflouride salt, yield >100%), which was used without further purification.
- DMTCl 4,4'-Dimethoxytrityl chloride
- 2-Cyanoethyl tetraisopropylphosphordiamidite (1.58 mL, 5.0 mmol) was added to a solution of nucleoside 85 (2.0 g, 3.3 mmol), tetrazole (0.19 g, 2.7 mmol) and iV-methylimidazole (68 ⁇ L, 0.83 mmol) in DMF (17 mL). After stirring at room temperature for 8 hours the reaction was poured into EtOAc. The organic layer was washed with 90% brine then brine and dried (Na 2 SO 4 ) and concentrated.
- nucleoside 87 tert-Butyldimethylsilyl chloride (2.40 g, 15.9 mmol) was added to a solution of nucleoside 82 (3.20 g, 5.3 mmol) and imidazole (2.16 g, 31.8 mmol) in DMF (10.6 mL). After stirring at room temperature for 16 hours the reaction was poured into EtOAc. The organic phase was sequentially extracted with brine, dried (Na 2 SO 4 ) and concentrated under vacuum. Purification by column chromatography (SiO 2 , eluting with 50% EtOAc/hexanes) provided nucleoside 87 (3.70 g, 98%) as a white solid.
- nucleoside 89 (from above) was added to a solution of nucleoside 89 (from above) in DMF (10 mL). After stirring at room temperature for 16 hours the reaction was poured into EtOAc. The organic layer was extracted with saturated NaHCO 3 and brine then dried (Na 2 SO 4 ) and concentrated under vacuum. Purification by column chromatography (SiO 2 , eluting with 50% EtOAc/hexanes) provided nucleoside 90 (3.86 g, 91% from 87) as a white solid.
- Triethylamine trihydrofluoride (4.54 mL, 27.9 mmol) was added to a solution of nucleoside 90 (3.81 g, 4.7 mmol) and triethylamine (1.56 mL, 11.2 mmol) in THF (46 mL) a polypropylene tube. After stirring at room temperature for 16 hours the reaction was dried under vacuum and the residue was dissolved in EtOAc. The organic layer was sequentially washed with water, saturated NaHCO 3 and brine then dried (Na 2 SO 4 ) and concentrated under vacuum. Purification by column chromatography (SiO 2 , eluting with 5% MeOH/CHCl 3 ) provided nucleoside 91 (3.07 g, 94%) as a white solid.
- nucleoside 93) tert-Butyldimethylsilyl chloride (2.25 g, 15.0 mmol) was added to a solution of nucleoside 85 (3.0 g, 5.0 mmol) and imidazole (2.03 g, 29.9 mmol) in DMF (10 mL). After stirring at room temperature for 16 hours the reaction was poured into EtOAc. The organic phase was sequentially extracted with brine, dried (Na 2 SO 4 ) and concentrated under vacuum. Purification by column chromatography (SiO 2 , eluting with 50% EtOAc/hexanes) provided nucleoside 93 (3.45 g, 97%) as a white solid.
- Phosphorus oxychloride (3.59 mL, 38.5 mmol) was added to a cold (0 °C) suspension of 1,2,4-triazole (11.3 g, 163.9 mmol) in CH 3 CN (80 mL). After stirring for 15 minutes triethylamine (27.0 mL, 192.8 mmol) was added to the reaction and the stirring continued for 30 minutes. A solution of nucleoside 93 (3.45 g, 4.82 mmol) in CH 3 CN (20 mL) was added to the reaction at 0 °C. After stirring for 10 minutes the ice bath was removed and the reaction was stirred at room temperature for 4 hours. The solvent was then removed under vacuum and the residue was partitioned between EtOAc and water. The organic layer was then washed with a saturated solution OfNaHCO 3 and brine then dried (Na 2 SO 4 ) and concentrated under vacuum to provide crude 94, which was used without further purification.
- Aqueous ammonia (10 mL) was added to a solution of nucleoside 94 (from above) in dioxane (50 mL). After stirring at room temperature for 16 hours the reaction was concentrated under vacuum and dried over high vacuum for 8 hours to provide nucleoside 95, which was used without further purification.
- nucleoside 95 from above was added to a solution of nucleoside 95 (from above) in DMF (9 mL). After stirring at room temperature for 16 hours the reaction was poured into EtOAc. The organic layer was extracted with saturated NaHCO 3 and brine then dried (Na 2 SO 4 ) and concentrated under vacuum. Purification by column chromatography (SiO 2 , eluting with 50% EtOAc/hexanes) provided nucleoside 96 (3.53 g, 89% from 93) as a white solid.
- nucleoside 97 Triethylamine trihydrofluoride (4.20 mL, 25.8 mmol) was added to a solution of nucleoside 96 (3.53 g, 4.3 mmol) and triethylamine (1.43 mL, 10.3 mmol) in THF (43 mL) in a polypropylene tube. After stirring at room temperature for 16 hours the reaction was dried under vacuum and the residue was dissolved in EtOAc. The organic layer was sequentially washed with water, saturated NaHCO 3 and brine then dried (Na 2 SO 4 ) and concentrated under vacuum. Purification by column chromatography (SiO 2 , eluting with 25% to 40% acetone/CHCl 3 ) provided nucleoside 97 (2.87 g, 95%) as a white solid.
- R TBDPS 102b
- R TBDPS
- Phosphoramidite 105 is prepared from diacetate 77a-b using the procedures illustrated for the synthesis of phosphoramidite 83 from diacetate mixture 77a-b.
- Phosphoramidite 108 is prepared from nucleoside 102b using the procedures illustrated for the synthesis of phosphoramidite 86 from 80b.
- Phosphoramidite 114 is prepared from diacetate 77a-b using the procedures illustrated for the synthesis of phosphoramidite 83 from diacetate mixture 77a-b.
- Phosphoramidite 117 is prepared from nucleoside 111b using the procedures illustrated for the synthesis of phosphoramidite 86 from 80b.
- Tetrabutylammonium fluoride (IM in THF, 10.00 mL, 10.0 mmol) was added to a solution of olefin 118 (4.83 g, 8.1 mmol) in THF (35 mL). The reaction was stirred at room temperature for 16 hours after which the solvent was removed under vacuum and the residue was dissolved in EtOAc. The organic layer was washed with water, brine, dried (Na 2 SO 4 ) and concentrated. Purification by column chromatography (SiO 2 , eluting with 40 % EtOAc in hexanes) provided alcohol 119 (2.79 g, 97%) as a colorless oil.
- Osmium Tetroxide (OsO 4 , 25% solution in iPrOH, ImL) was added to a solution of olefin 120 (1.80 g, 4.0 mmol) and N-methylmorpholine-JV-oxide (NMO, 0.94 g, 8.0 mmol) in 95% acetone/water (25 mL). After stirring for 16h at room temperature, additional OsO 4 solution (0.5 mL) and NMO (0.40 g) were added to the reaction. After stirring for a total 48 hours, the reaction was diluted with EtOAc and washed with 10% NaHSO 3 , brine, dried (Na 2 SO 4 ) and concentrated. Purification by column chromatography (SiO 2 , eluting with 40 to 50% EtOAc in hexanes) provided diol 121 (1.68 g, 87%, ca. 1:1 mixture of isomers) as a colorless oil.
- TBSCl (0. 66 g, 4.4 mmol) was added to a cold (0 0 C) solution of diol 121 (1.63 g, 3.4 mmol) in pyridine (17 mL). After stirring for 4 h at 0 0 C, the reaction was diluted with EtOAc and the organic layer was washed with water, brine, dried and concentrated. Purification by column chromatography (SiO 2 , eluting with 10 to 20% EtOAc in hexanes) provided alcohols 122 and 123 (0.90 g and 1.17 g, absolute stereochemistry not assigned) as colorless oils.
- Triethylamine trihydroflouride (0.64 mL, 4.0 mmol) was added to a solution of mesylate 124 (0.44 g, 0.6 mmol) and triethylamine (0.23 mL, 1.7 mmol) in THF (7 mL). After stirring for 16 hours at room temperature, the reaction was diluted with EtOAc and the organic phase was washed with saturated NaHCO 3 , brine, dried (Na 2 SO 4 ) and concentrated. Purification by column chromatography (SiO 2 , eluting with 50% EtOAc in hexanes) provided alcohol 125 (0.40 g, quantitative).
- N,C>-Bis(trimethylsilyl)acetamide (0.8 mL, 3.3 mmol) was added to a suspension of diacetate 127 (0.45 g, 0.65 mmol) and uracil (0.15 g, 1.3 mmol) in CH 3 CN (3.5 mL). After heating at 4O 0 C for 15 min to get a clear solution, trimethylsilyl trifiate (0.24 mL, 1.3 mmol) was added to the reaction. After refiuxing for 2 hours, the reaction was cooled to room temperature and poured into EtOAc.
- Bx ⁇ /-Bz-Cytosine 130c
- Bx ⁇ /-Bz-Adenine 13Od
- Bx ⁇ /-lsobu-Guanine
- Nucleosides 128b, 128c and 128d are prepared from sugar precursor 127 by a Vorbrugen reaction using 7V-Bz-cytosine, 6-7V-Bz-adenine and 2-amino-6-chloropurine respectively (Scheme 18).
- Treatment of 128b and 128c with K 2 CO 3 and MeOH provides nucleosides 129b and 129c respectively.
- Treatment of 128d with sodium hydride and 3-hydroxypropionitrile provides nucleoside 129d.
- Transient protection of the hydroxyl group with TMSCl followed by reaction with benzoyl chloride provides nucleosides 130b and 130c respectively.
- nucleosides 129b and 129c can also be accomplished by reacting nucleosides 129b and 129c with benzoic anhydride using DMF as the solvent.
- Nucleoside 13Od is prepared by transient protection with excess TMSCl in pyridine followed by reaction with isobutyryl chloride.
- R, R 1 and R 2 are each independently H, alkyl, alkenyl, alkynyl, substituted alkyl, substituted alkenyl, substituted alkynyl, or a protecting group
- Nucleoside 131 is prepared from nucleoside 130 by treatment with a fluorinating agent such as DAST using dichloromethane as the solvent.
- Nucleoside 132 is prepared from 130 by first oxidizing the primary hydroxyl group with Dess-Martin periodinane or under Swern conditions followed by treatment of the resulting aldehyde with DAST.
- Nucleoside 133 is prepared from 130 by first oxidizing the primary hydroxyl group with Dess-Martin periodinane or under Swern conditions followed by reductive amination of the resulting aldehyde with a primary or a secondary amine in the presence of glacial acetic acid and a reducing agent such as sodium cyanoborohydride.
- Nucleoside 134 is prepared from 130 by converting the hydroxyl group to a leaving group (mesylate, tosylate, halide) followed by heating with excess sodium azide.
- Nucleoside 135 is prepared from 130 by oxidation of the primary alcohol to a carboxylic acid followed by reaction with a amine in the presence of HATU or any other peptide coupling reagent.
- Nucleoside 136 is prepared from 130 by activating the hydroxyl group with carbonyl dimimdazole followed by reaction with a amine.
- Nucleoside 137 is prepared from 130 by deprotonating the hydroxyl group with an appropriate base followed by quenching the anion with an alkylating reagent.
- Nucleoside 138 is prepared from 130 by converting the hydroxyl group to a leaving group followed by displacement with a thiol nucleophile.
- Nucleoside 139 is prepared from 134 by reduction of the azide group followed by reaction with an isocyanate or an isothiocyanate.
- Nucleoside 140 is prepared from 134 by reduction of the azido group and reaction with FmocNCS to provide an activated thiourea. Further reaction of the frnoc activated thiourea with an amine in the presence of EDC provides the substituted guanidine. Removal of the frnoc protecting group liberates nucleoside 140.
- Nucleoside 142 is prepared from nucleoside 130-140 by catalytic hydrogenation to remove the 3'- and 5'-O protecting groups.
- 142 can be prepared from 130-140 by first removing the 3'0-Nap group with DDQ followed by a catalytic hydrogenation to remove the 5'0-benzyl group. Subsequent protection of the 5' hydroxyl group as the dimethoxytrityl ether followed by a phosphitilation reaction provides phosphoramidite 143.
- nucleoside 131a 51 mg, 52%, contaminated with 15-20% of a ring opened impurity as a mixture of isomers. ).
- nucleoside 141a (41 mg, quantitative as a mixture of isomers. ).
- nucleoside 141a 41 mg, from above
- 10% palladium on charcoal (10 mg) in methanol (2 mL) was hydrogenated using a hydrogen balloon. After 3 hours, all starting nucleoside 141a was consumed (as indicated by LCMS analysis of the reaction mixture).
- the reaction was filtered through celite and the filtrate concentrated under reduced pressure. Purification by column chromatography (SiO 2 , 10 to 20% methanol in chloroform) provided nucleoside 142a (14 mg, 50%) as a mixture of isomers.
- LCMS retention time 1.72 min; M+Na calcd. 311.08, found 311.0.
- DMTCl (24 mg, 0.07 mmol) was added to a solution of nucleoside 142a (14 mg, 0.049 mmol) in pyridine (0.25 mL). After stirring at room temperature for 3 hours, the reaction was concentrated under reduced pressure. Purification by column chromatography (SiO 2 , 20 to 30% acetone in chloroform) provided nucleoside 142aa (16 mg, 55%) as a mixture of isomers. 19 F NMR (CDCl 3 ): ⁇ -228.6 (t) and -230.91 (dt). LCMS: retention time 3.56 min; M+Na calcd. 613.21, found 613.1.
- Amidite (143a) is prepared from nucleoside 142aa using a phosphitilation reaction as described in example 1.
- nucleoside Phosphoramidites The preparation of nucleoside phosphoramidites is performed following procedures that are illustrated herein and in the art such as but not limited to US Patent 6,426,220 and published PCT WO 02/36743.
- oligomeric compounds used in accordance with this invention may be conveniently and routinely made through the well-known technique of solid phase synthesis.
- Equipment for such synthesis is sold by several vendors including, for example, Applied Biosystems (Foster City, CA). Any other means for such synthesis known in the art may additionally or altematively be employed. It is well known to use similar techniques to prepare oligonucleotides such as the phosphorothioates and alkylated derivatives.
- Alkyl phosphonate oligonucleotides can be prepared as described in U.S. Patent 4,469,863.
- 3 '-Deoxy-3 '-methylene phosphonate oligonucleotides can be prepared as described in U.S. Patents 5,610,289 or 5,625,050.
- Phosphoramidite oligonucleotides can be prepared as described in U.S. Patent, 5,256,775 or U.S. Patent 5,366,878.
- Alkylphosphonothioate oligonucleotides can be prepared as described in published PCT applications PCT/US94/00902 and PCT/US93/06976 (published as WO 94/17093 and WO 94/02499, respectively).
- 3 '-Deoxy-3 '-amino phosphoramidate oligonucleotides can be prepared as described in U.S. Patent 5,476,925.
- Phosphotriester oligonucleotides can be prepared as described in U.S. Patent 5,023,243.
- Borano phosphate oligonucleotides can be prepared as described in U.S. Patents 5,130,302 and 5,177,198.
- Formacetal and thioformacetal linked oligonucleosides can be prepared as described in U.S. Patents 5,264,562 and 5,264,564.
- Ethylene oxide linked oligonucleosides can be prepared as described in U.S. Patents
- Example 24 Oligonucleotide Isolation After cleavage from the controlled pore glass solid support and deblocking in concentrated ammonium hydroxide at 55 0 C for 12-16 hours, the oligonucleotides or oligonucleosides are recovered by precipitation out of 1 M NH 4 OAc with >3 volumes of ethanol. Synthesized oligonucleotides are analyzed by electrospray mass spectroscopy (molecular weight determination) and by capillary gel electrophoresis. The relative amounts of phosphorothioate and phosphodiester linkages obtained in the synthesis is determined by the ratio of correct molecular weight relative to the -16 amu product (+/-32 +/-48).
- oligonucleotides are purified by HPLC, as described by Chiang et al., J. Biol. Chem. 1991, 266, 18162- 18171. Results obtained with HPLC-purified material are generally similar to those obtained with non-HPLC purified material.
- Oligonucleotides can be synthesized via solid phase P(III) phosphoramidite chemistry on an automated synthesizer capable of assembling 96 sequences simultaneously in a 96-well format.
- Phosphodiester internucleotide linkages are afforded by oxidation with aqueous iodine.
- Phosphorothioate internucleotide linkages are generated by sulfurization utilizing 3, H- 1,2 benzodithiole-3-one 1,1 dioxide (Beaucage Reagent) in anhydrous acetonitrile.
- Standard base- protected beta-cyanoethyl-diiso-propyl phosphoramidites are purchased from commercial vendors (e.g.
- Non-standard nucleosides are synthesized as per standard or patented methods. They are utilized as base protected beta-cyanoethyldiisopropyl phosphoramidites.
- Oligonucleotides are cleaved from support and deprotected with concentrated NH 4 OH at elevated temperature (55-60°C) for 12-16 hours and the released product then dried in vacuo. The dried product is then re-suspended in sterile water to afford a master plate from which all analytical and test plate samples are then diluted utilizing robotic pipettors.
- the concentration of oligonucleotide in each well is assessed by dilution of samples and UV absorption spectroscopy.
- the full-length integrity of the individual products is evaluated by capillary electrophoresis (CE) in either the 96- well format (Beckman P/ ACETM MDQ) or, for individually prepared samples, on a commercial CE apparatus (e.g., Beckman P/ACETM 5000, ABI 270).
- Base and backbone composition is confirmed by mass analysis of the oligomeric compounds utilizing electrospray-mass spectroscopy. All assay test plates are diluted from the master plate using single and multi-channel robotic pipettors. Plates are judged to be acceptable if at least 85% of the oligomeric compounds on the plate are at least 85% full length.
- oligomeric compounds on target nucleic acid expression can be tested in any of a variety of cell types provided that the target nucleic acid is present at measurable levels. This can be routinely determined using, for example, PCR or Northern blot analysis. Cell lines derived from multiple tissues and species can be obtained from American Type Culture Collection (ATCC, Manassas, VA).
- b.END cells The mouse brain endothelial cell line b.END was obtained from Dr. Werner Risau at the Max Plank Institute (Bad Nauheim, Germany). b.END cells were routinely cultured in DMEM, high glucose (Invitrogen Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen Life Technologies, Carlsbad, CA).
- Cells were routinely passaged by trypsinization and dilution when they reached approximately 90% confluence. Cells were seeded into 96- well plates (Falcon-Primaria #353872, BD Biosciences, Bedford, MA) at a density of approximately 3000 cells/well for uses including but not limited to oligomeric compound transfection experiments.
- Oligonucleotide When cells reached 65-75% confluency, they are treated with oligonucleotide. Oligonucleotide is mixed with LIPOFECTINTM Invitrogen Life Technologies, Carlsbad, CA) in Opti-MEMTM-1 reduced serum medium (Invitrogen Life Technologies, Carlsbad, CA) to achieve the desired concentration of oligonucleotide and a LIPOFECTINTM concentration of 2.5 or 3 ⁇ g/mL per 100 nM oligonucleotide. This transfection mixture is incubated at room temperature for approximately 0.5 hours. For cells grown in 96-well plates, wells are washed once with 100 ⁇ L OPTI-MEMTM- 1 and then treated with 130 ⁇ L of the transfection mixture.
- Cells grown in 24- well plates or other standard tissue culture plates are treated similarly, using appropriate volumes of medium and oligonucleotide. Cells are treated and data are obtained in duplicate or triplicate. After approximately 4-7 hours of treatment at 37°C, the medium containing the transfection mixture is replaced with fresh culture medium. Cells are harvested 16-24 hours after oligonucleotide treatment.
- transfection reagents known in the art include, but are not limited to, CYTOFECTINTM, LIPOFECTAMINETM, OLIGOFECTAMINETM, and FUGENETM.
- Other suitable transfection methods known in the art include, but are not limited to, electroporation.
- Antisense modulation of a target expression can be assayed in a variety of ways known in the art.
- a target mRNA levels can be quantitated by, e.g., Northern blot analysis, competitive polymerase chain reaction (PCR), or real-time PCR.
- Real-time quantitative PCR is presently desired.
- RNA analysis can be performed on total cellular RNA or poly(A)+ mRNA.
- One method of RNA analysis of the present invention is the use of total cellular RNA as described in other examples herein. Methods of RNA isolation are well known in the art.
- Northern blot analysis is also routine in the art.
- Real-time quantitative (PCR) can be conveniently accomplished using the commercially available ABI PRISMTM 7600, 7700, or 7900 Sequence Detection System, available from PE- Applied Biosystems, Foster City, CA and used according to manufacturer's instructions.
- Protein levels of a target can be quantitated in a variety of ways well known in the art, such as immunoprecipitation, Western blot analysis (immunoblotting), enzyme-linked immunosorbent assay (ELISA) or fluorescence-activated cell sorting (FACS).
- Antibodies directed to a target can be identified and obtained from a variety of sources, such as the MSRS catalog of antibodies (Aerie Corporation, Birmingham, MI), or can be prepared via conventional monoclonal or polyclonal antibody generation methods well known in the art. Methods for preparation of polyclonal antisera are taught in, for example, Ausubel, F.M. et al., Current Protocols in Molecular Biology, Volume 2, pp.
- Enzyme-linked immunosorbent assays are standard in the art and can be found at, for example, Ausubel, F.M. et al., Current Protocols in Molecular Biology, Volume 2, pp. 11.2.1-11.2.22, John Wiley & Sons, Inc., 1991.
- Phenotypic assays Once target inhibitors have been identified by the methods disclosed herein, the oligomeric compounds are further investigated in one or more phenotypic assays, each having measurable endpoints predictive of efficacy in the treatment of a particular disease state or condition.
- Phenotypic assays, kits and reagents for their use are well known to those skilled in the art and are herein used to investigate the role and/or association of a target in health and disease.
- Representative phenotypic assays which can be purchased from any one of several commercial vendors, include those for determining cell viability, cytotoxicity, proliferation or cell survival (Molecular Probes, Eugene, OR; PerkinElmer, Boston, MA), protein-based assays including enzymatic assays (Panvera, LLC, Madison, WI; BD Biosciences, Franklin Lakes, NJ; Oncogene Research Products, San Diego, CA), cell regulation, signal transduction, inflammation, oxidative processes and apoptosis (Assay Designs Inc., Ann Arbor, MI), triglyceride accumulation (Sigma- Aldrich, St.
- angiogenesis assays i.e., MCF-7 cells selected for breast cancer studies; adipocytes for obesity studies
- a target inhibitors identified from the in vitro studies as well as control compounds at optimal concentrations which are determined by the methods described above.
- treated and untreated cells are analyzed by one or more methods specific for the assay to determine phenotypic outcomes and endpoints.
- Phenotypic endpoints include changes in cell morphology over time or treatment dose as well as changes in levels of cellular components such as proteins, lipids, nucleic acids, hormones, saccharides or metals. Measurements of cellular status which include pH, stage of the cell cycle, intake or excretion of biological indicators by the cell, are also endpoints of interest.
- Measurement of the expression of one or more of the genes of the cell after treatment is also used as an indicator of the efficacy or potency of the a target inhibitors.
- Hallmark genes or those genes suspected to be associated with a specific disease state, condition, or phenotype, are measured in both treated and untreated cells. In vivo studies
- the individual subjects of the in vivo studies described herein are warm-blooded vertebrate animals, which includes humans.
- PoIy(A)+ mRNA is isolated according to Miura et al., (Clin. Chem., 1996, 42, 1758- 1764). Other methods for poly(A)+ mRNA isolation are routine in the art. Briefly, for cells grown on 96-well plates, growth medium is removed from the cells and each well is washed with 200 ⁇ L cold PBS. 60 ⁇ L lysis buffer (10 mM Tris-HCl, pH 7.6, 1 mM EDTA, 0.5 M NaCl,
- NP-40 0.5% vanadyl-ribonucleoside complex
- NP-40 0.5% vanadyl-ribonucleoside complex
- the plate is gently agitated and then incubated at room temperature for five minutes.
- 55 ⁇ L of lysate is transferred to Oligo d(T) coated 96-well plates (AGCT Inc., Irvine CA). Plates are incubated for 60 minutes at room temperature, washed 3 times with 200 ⁇ L of wash buffer (10 mM Tris-HCl pH 7.6, 1 mM EDTA, 0.3 M NaCl). After the final wash, the plate is blotted on paper towels to remove excess wash buffer and then air-dried for 5 minutes.
- wash buffer 10 mM Tris-HCl pH 7.6, 1 mM EDTA, 0.3 M NaCl
- RNA Isolation 60 ⁇ L of elution buffer (5 mM Tris-HCl pH 7.6), preheated to 70°C, is added to each well, the plate is incubated on a 90°C hot plate for 5 minutes, and the eluate is then transferred to a fresh 96-well plate. Cells grown on 100 mm or other standard plates may be treated similarly, using appropriate volumes of all solutions.
- Total RNA is isolated using an RNEASY 96TM kit and buffers purchased from Qiagen Inc. (Valencia, CA) following the manufacturer's recommended procedures. Briefly, for cells grown on 96-well plates, growth medium is removed from the cells and each well is washed with 200 ⁇ L cold PBS. 150 ⁇ L Buffer RLT is added to each well and the plate vigorously agitated for 20 seconds. 150 ⁇ L of 70% ethanol is then added to each well and the contents mixed by pipetting three times up and down. The samples are then transferred to the RNEASY 96TM well plate attached to a QIA V ACTM manifold fitted with a waste collection tray and attached to a vacuum source. Vacuum is applied for 1 minute.
- RNA is then eluted by pipetting 140 ⁇ L of RNAse free water into each well, incubating 1 minute, and then applying the vacuum for 3 minutes.
- the repetitive pipetting and elution steps may be automated using a QIAGEN Bio-Robot 9604 (Qiagen, Inc., Valencia CA). Essentially, after lysing of the cells on the culture plate, the plate is transferred to the robot deck where the pipetting, DNase treatment and elution steps are carried out.
- Quantitation of a target mRNA levels was accomplished by real-time quantitative PCR using the ABI PRISMTM 7600, 7700, or 7900 Sequence Detection System (PE- Applied Biosystems, Foster City, CA) according to manufacturer's instructions.
- ABI PRISMTM 7600, 7700, or 7900 Sequence Detection System PE- Applied Biosystems, Foster City, CA
- This is a closed-tube, non-gel-based, fluorescence detection system which allows high-throughput quantitation of polymerase chain reaction (PCR) products in real-time.
- PCR polymerase chain reaction
- products in real-time quantitative PCR are quantitated as they accumulate. This is accomplished by including in the PCR reaction an oligonucleotide probe that anneals specifically between the forward and reverse PCR primers, and contains two fluorescent dyes.
- a reporter dye e.g., FAM or JOE, obtained from either PE-Applied Biosystems, Foster City, CA, Operon Technologies Inc., Alameda, CA or Integrated DNA Technologies Inc., Coralville, IA
- a quencher dye e.g., TAMRA, obtained from either PE-Applied Biosystems, Foster City, CA, Operon Technologies Inc., Alameda, CA or Integrated DNA Technologies Inc., Coralville, IA
- TAMRA obtained from either PE-Applied Biosystems, Foster City, CA, Operon Technologies Inc., Alameda, CA or Integrated DNA Technologies Inc., Coralville, IA
- annealing of the probe to the target sequence creates a substrate that can be cleaved by the 5'-exonuclease activity of Taq polymerase.
- cleavage of the probe by Taq polymerase releases the reporter dye from the remainder of the probe (and hence from the quencher moiety) and a sequence-specific fluorescent signal is generated.
- additional reporter dye molecules are cleaved from their respective probes, and the fluorescence intensity is monitored at regular intervals by laser optics built into the ABI
- PRISMTM Sequence Detection System In each assay, a series of parallel reactions containing serial dilutions of mRNA from untreated control samples generates a standard curve that is used to quantitate the percent inhibition after antisense oligonucleotide treatment of test samples.
- primer-probe sets specific to the target gene being measured Prior to quantitative PCR analysis, primer-probe sets specific to the target gene being measured are evaluated for their ability to be "multiplexed" with a GAPDH amplification reaction. In multiplexing, both the target gene and the internal standard gene GAPDH are amplified concurrently in a single sample. In this analysis, mRNA isolated from untreated cells is serially diluted.
- Each dilution is amplified in the presence of primer-probe sets specific for GAPDH only, target gene only ("single-plexing"), or both (multiplexing).
- primer-probe sets specific for GAPDH only target gene only
- target gene only target gene only
- multiplexing target gene only
- standard curves of GAPDH and target mRNA signal as a function of dilution are generated from both the single-plexed and multiplexed samples. If both the slope and correlation coefficient of the GAPDH and target signals generated from the multiplexed samples fall within 10% of their corresponding values generated from the single-plexed samples, the primer-probe set specific for that target is deemed multiplexable.
- Other methods of PCR are also known in the art.
- RT and PCR reagents were obtained from Invitrogen Life Technologies (Carlsbad, CA).
- RT real-time PCR was carried out by adding 20 ⁇ L PCR cocktail (2.5x PCR buffer minus MgCl 2 , 6.6 mM MgCl 2 , 375 ⁇ M each of dATP, dCTP, dCTP and dGTP, 375 nM each of forward primer and reverse primer, 125 nM of probe, 4 Units RNAse inhibitor, 1.25 Units PLATINUM® Taq, 5 Units MuLV reverse transcriptase, and 2.5x ROX dye) to 96-well plates containing 30 ⁇ L total RNA solution (20-200 ng).
- PCR cocktail 2.5x PCR buffer minus MgCl 2 , 6.6 mM MgCl 2 , 375 ⁇ M each of dATP, dCTP, dCTP and dGTP, 375 nM each of forward primer and reverse primer, 125
- the RT reaction was carried out by incubation for 30 minutes at 48 0 C. Following a 10 minute incubation at 95°C to activate the PLATINUM® Taq, 40 cycles of a two-step PCR protocol were carried out: 95 0 C for 15 seconds (denaturation) followed by 60 0 C for 1.5 minutes (annealing/extension).
- Gene target quantities obtained by RT, real-time PCR are normalized using either the expression level of GAPDH, a gene whose expression is constant, or by quantifying total RNA using RIBOGREENTM (Molecular Probes, Inc. Eugene, OR).
- GAPDH expression is quantified by real time RT-PCR, by being run simultaneously with the target, multiplexing, or separately.
- Total RNA is quantified using RiboGreenTM RNA quantification reagent (Molecular Probes, Inc. Eugene, OR). Methods of RNA quantification by RIBOGREENTM are taught in Jones, L. J., et al, (Analytical Biochemistry, 1998, 265, 368-374).
- RIBOGREENTM working reagent RIBOGREENTM working reagent diluted 1:350 in 1OmM Tris-HCl, 1 mM EDTA, pH 7.5
- RIBOGREENTM reagent diluted 1:350 in 1OmM Tris-HCl, 1 mM EDTA, pH 7.5 is pipetted into a 96-well plate containing 30 ⁇ L purified, cellular RNA.
- the plate is read in a CytoFluor 4000 (PE Applied Biosystems) with excitation at 485nm and emission at 530nm.
- Probes and primers may be designed to hybridize to a target sequence, using published sequence information.
- primer-probe set was designed using published sequence information (GENBANKTM accession number U92436.1, SEQ ID NO: 1).
- FAM-TTGCAGCAATTCACTGTAAAGCTGGAAAGG-TAMRA (SEQ ID NO: 4), where FAM is the fluorescent dye and TAMRA is the quencher dye.
- Western blot analysis is carried out using standard methods.
- Cells are harvested 16-20 h after oligonucleotide treatment, washed once with PBS, suspended in Laemmli buffer (100 ⁇ l/well), boiled for 5 minutes and loaded on a 16% SDS-PAGE gel. Gels are run for 1.5 hours at 150 V, and transferred to membrane for western blotting.
- Appropriate primary antibody directed to a target is used, with a radiolabeled or fluorescently labeled secondary antibody directed against the primary antibody species. Bands are visualized using a PHOSPHORIMAGERTM (Molecular Dynamics, Sunnyvale CA).
- oligomeric compounds were synthesized and tested for their ability to reduce PTEN expression over a range of doses.
- b.END cells were treated with the 6-(R or S)-CH 3 -BNA (392748 and 392749 respectively) and 6-(R or S)-CH 2 -O- CH 3 (396004 and 396005 respectively) modified oligomers at concentrations of 0.3125, 0.0625, 1.25, 2.5, 5, 10 or 20 nM using methods described herein.
- Expression levels of PTEN were determined using real-time PCR and normalized to RIBOGREENTM as described in other examples herein. Resulting dose-response curves were used to determine the EC50 as shown below.
- Tm's were assessed in 100 mM phosphate buffer, 0.1 mM EDTA, pH 7, at 260 nm using 4 ⁇ M 6-(R or S)-CH 3 -BNA or 6-(R or S)-CH 2 -O-CH 3 modified oligomers and 4 ⁇ M complementary RNA.
- SEQ ID NO. Composition (5' to 3') EC 50 Tm 0 C
- All internucleoside linkages are phosphorothioate, bolded nucleosides are 6-(R or S)-CH 3 BNA nucleosides, underlined and bolded nucleosides are 6-(R or S)-CH 2 -O-CH 3 BNA nucleosides and subscripts R and S indicate the configuration at the 6 carbon atom. It is notable that the 6-modified BNA oligomeric compounds exhibited greater potency despite the slight decrease in Tm.
- mice Six week old Balb/c mice (Jackson Laboratory, Bar Harbor, ME) were injected twice weekly for 3 weeks with a 6-CH 3 -BNA modified oligomers (either 6-(S) or 6-(R)) targeted to PTEN at a dose of 0.5 or 2 ⁇ mol/kg. The mice were sacrificed 48 hours following the final administration. Liver tissues were homogenized and mRNA levels were quantitated using realtime PCR as described herein for comparison to untreated control levels (%UTC).
- 6-CH 3 -BNA modified oligomers targeted to PTEN at a dose of 1, 2, 4 or 8 ⁇ mol/kg.
- the mice were sacrificed 72 hrs following administration. Liver tissues were homogenized and mRNA levels were quantitated using real-time PCR as described herein for comparison to untreated control levels (%UTC).
- All internucleoside linkages are phosphorothioate, bolded nucleosides are 6-CH 3 -BNA nucleosides and subscripts S and R indicate the configuration at the 6 carbon atom.
- mice Six week old Balb/c mice (Jackson Laboratory, Bar Harbor, ME) were injected once with 6-CH 3 -BNA modified oligomers (either 6-(S) or 6-(R)) targeted to PTEN at a dose of 1 , 2, 4 or 8 ⁇ mol/kg (only the 8 ⁇ mol/kg data is shown below). The mice were sacrificed 72 hrs following administration. Liver tissues were homogenized and mRNA levels were quantitated using realtime PCR as described herein for comparison to untreated control levels (%UTC).
- AU internucleoside linkages are phosphorothioate, bolded and underlined nucleosides are 6-CH 3 -O-CH 2 -BNA nucleosides and subscripts S and R indicate the configuration at the 6 carbon atom.
- nuclease stability of 6-(R or S)-CH 3 -BNA modified oligomers treated with SVPD The nuclease stability of 6-(R or S)-CH 3 -BNA (392748 and 392749 respectively) modified oligomers was determined using snake venom phosphodiesterase (SVPD). The study included a the respective 6-unsubstituted gapmer (4'-CH 2 -O-2' bridged BNA, 392745, subscript 1) and the 2'-0-MOE gapmer (2'-O-(CH 2 ) 2 -OCH 3 , 392753, subscript e) for comparison.
- SVPD snake venom phosphodiesterase
- Each oligomer is prepared as a 500 ⁇ L mixture containing: 5 ⁇ L lOO ⁇ M oligomer, 50 ⁇ L phosphodiesterase I @ 0.5 Units/mL in SVPD buffer (50 mM Tris-HcL, pH 7.5, 8 mM MgCl 2 ) final concentration 0.05 Units/mL, 445 ⁇ L SVP buffer. Samples were incubated at 37 0 C in a water bath. Aliquats (100 ⁇ L) were taken at 0, 1, 2 and 4 days with fresh enzyme added at days 1 and 2. EDTA was added to aliquats immediately after removal to quench enzyme activity. Samples were analyzed on IP HPLC/MS. SEQ ID NO. Composition (5' to 3') % full length at day 4
- the nuclease stability of 6-CH 3 -BNA modified oligomers was determined using snake venom phosphodiesterase (SVPD). Each oligomer is prepared as a 90 ⁇ L mixture containing 5 ⁇ L oligomer (2 ⁇ L of 5 ⁇ M oligomer and 3 ⁇ L of 5' 32 P-labled oligomer) 75 ⁇ L H 2 O, and 10 ⁇ L 1OX buffer (500 mM Tris-HCl, 700 mM NaCl, and 140 mM MgCl 2 at pH 8.6).
- SVPD snake venom phosphodiesterase
- All internucleoside linkages are phosphorothioate, bolded nucleosides are modifed nucleosides, subscript R and S indicate the configuration at the 6 carbon atom for 6-CH 3 -BNA nucleosides, subscript e indicates 2'-0-MOE nucleosides and subscript 1 indicates 4'-CH 2 -O-2' modified nucleosides.
- mice Six week old Balb/c mice (Jackson Laboratory, Bar Harbor, ME) were injected twice weekly for three weeks with 6-(S)-CH 3 -BNA (2-10-2, 14-mer), 4'-CH 2 -O-2'-BNA (2-10-2, 14- mer) and 2'-0-MOE (5-10-5, 20-mer) modified oligomers targeted to PTEN at a dose of 3.2, 1.0, 0.32 and 0.1 ⁇ mol/kg (only the 3.2 and 1 ⁇ mol/kg data is shown below). The mice were sacrificed 48 hrs following last administration. Liver tissues were homogenized and mRNA levels were quantitated using real-time PCR as described herein for comparison to untreated control levels (%UTC). Plasma chemistries and liver weights were determined after sacrifice.
- liver weights for the 4'-CH 2 -O-2' BNA (392063, 3.2 ⁇ mol/Kg dose group) containing oligomers 153% relative to saline.
- the liver weights for 6-(S)-CH 3 BNA (392749, 3.2 ⁇ mol/Kg dose group) containing oligomers were 117% relative to saline.
- Liver weights for 2 O-MOE containing oligomers (116847, 1.0 ⁇ mol/Kg dose group) were 116% relative to saline.
- mice 2.5, 5, 10 and 20 ⁇ mol/kg (only 5 and 10 ⁇ mol/Kg data shown).
- the mice were sacrificed 66 hrs following administration. Liver tissues were homogenized.
- one does not include a nucleoside that is chiral at the 6 carbon atom, wherein the other four (Isis Nos. 396024, 396568, 396008 and
- those four include one such nucleoside at the 1, 2, 13 and 14 positions.
- the one that does not has a relatively higher toxicity in the liver compared to the four oligonucleosides that do.
- mice Six week old Balb/c mice (Jackson Laboratory, Bar Harbor, ME) were injected once with 6-CH 3 -BNA modified oligomers targeted to PTEN at a dose of 2 or 10 ⁇ mol/kg. The mice were sacrificed 72 hrs following administration. Liver tissues were homogenized and mRNA levels were quantitated using real-time PCR as described herein for comparison to untreated control levels (% UTC).
- AU internucleoside linkages are phosphorothioate, bolded nucleosides are modifed nucleosides, subscript R and S indicate the configuration at the 6 carbon atom for 6-CH 3 -BNA and 6-CH 2 -O-CH 3 -BNA nucleosides as indicated, subscript e indicates 2'-O-MOE nucleosides and Me C indicates a 5 '-methyl cytosine nucleoside.
Abstract
Description

























































Claims
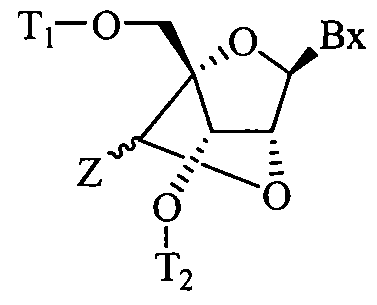











Priority Applications (78)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AT07762892T ATE482965T1 (en) | 2006-01-27 | 2007-01-27 | 6-MODIFIED BICYCLIC NUCLEIC ACID ANALOGUES |
| CN2007800105660A CN101410406B (en) | 2006-01-27 | 2007-01-27 | 6-modified bicyclic nucleic acid analogs |
| DK07762892.3T DK1984381T3 (en) | 2006-01-27 | 2007-01-27 | 6-modified bicyclic nucleic acid analogues |
| KR1020087020907A KR101304071B1 (en) | 2006-01-27 | 2007-01-27 | 6-modified bicyclic nucleic acid analogs |
| PL07762892T PL1984381T3 (en) | 2006-01-27 | 2007-01-27 | 6-modified bicyclic nucleic acid analogs |
| DE602007009487T DE602007009487D1 (en) | 2006-01-27 | 2007-01-27 | 6-MODIFIED BICYCLIC NUCLEIC ACID ANALOGUE |
| CA2640171A CA2640171C (en) | 2006-01-27 | 2007-01-27 | 6-modified bicyclic nucleic acid analogs |
| AU2007211080A AU2007211080B9 (en) | 2006-01-27 | 2007-01-27 | 6-modified bicyclic nucleic acid analogs |
| EP07762892A EP1984381B1 (en) | 2006-01-27 | 2007-01-27 | 6-modified bicyclic nucleic acid analogs |
| KR1020137007796A KR20130042043A (en) | 2006-01-27 | 2007-01-27 | 6-modified bicyclic nucleic acid analogs |
| JP2008552604A JP5342881B2 (en) | 2006-01-27 | 2007-01-27 | 6-modified bicyclic nucleic acid analogues |
| PCT/US2007/068415 WO2007136989A2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of dgat2 |
| AT07811872T ATE514777T1 (en) | 2006-05-05 | 2007-05-07 | COMPOUNDS AND METHODS FOR MODULATING GENE EXPRESSION |
| PCT/US2007/068403 WO2007131238A2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression apob |
| AU2007253909A AU2007253909B2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of GCCR |
| EP12174138A EP2505649A1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of GCGR |
| AU2007257093A AU2007257093A1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of PCSK9 |
| DK11189591.8T DK2458006T3 (en) | 2006-05-05 | 2007-05-07 | Compounds and Methods for Modulating ApoB Expression. |
| EP12174139A EP2505650A1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of PCSK9 |
| PCT/US2007/068412 WO2007134014A2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of gcgr |
| CA002651042A CA2651042A1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of sglt2 |
| CA3044969A CA3044969A1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating gene expression |
| EP07811873.4A EP2015758B1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression apob |
| EP07811878.3A EP2019692B1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of gccr |
| DK11160528.3T DK2363481T3 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating gene expression |
| EP11160528.3A EP2363481B1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating gene expression |
| EP12174131A EP2505647A1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of DGAT2 |
| JP2009510130A JP5731115B2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating gene expression |
| EP12174128A EP2505646A1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of CRP |
| US12/299,583 US8372967B2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of GCCR |
| PCT/US2007/068406 WO2007143316A2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of sglt2 |
| PT78118734T PT2015758E (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression apob |
| US12/299,764 US8673871B2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression ApoB |
| EP12174135A EP2505648A1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of PTP1B |
| ES07811874T ES2386578T3 (en) | 2006-05-05 | 2007-05-07 | Compounds and procedures to modulate PCSK9 expression |
| DK07811873.4T DK2015758T3 (en) | 2006-05-05 | 2007-05-07 | RELATIONS AND PROCEDURES FOR MODULATING APOB EXPRESSION |
| PCT/US2007/068402 WO2007131237A2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of ptp1b |
| US11/745,429 US9045754B2 (en) | 2006-05-05 | 2007-05-07 | Short antisense compounds with gapmer configuration |
| AT07811875T ATE513912T1 (en) | 2006-05-05 | 2007-05-07 | COMPOUNDS AND METHODS FOR MODULATING THE EXPRESSION OF SGLT2 |
| CN201310168425.9A CN103554205A (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of gccr |
| JP2009510131A JP5825754B2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating the expression of APOB |
| EP11176633A EP2397551A1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of PCSK9 |
| ES07811873.4T ES2471978T3 (en) | 2006-05-05 | 2007-05-07 | Compounds and procedures to modulate ApoB expression |
| BRPI0711429-0A BRPI0711429A2 (en) | 2006-05-05 | 2007-05-07 | compounds and methods for gccr expression modules |
| PCT/US2007/068404 WO2007143315A2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of pcsk9 |
| EP07811874A EP2023939B1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of pcsk9 |
| JP2009510132A JP2009536222A (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating the expression of PCSK9 |
| US12/299,605 US20090292006A1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of dgat2 |
| JP2009510134A JP5372745B2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating the expression of SGLT2 |
| EP07811872A EP2021472B1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating gene expression |
| DK07811878.3T DK2019692T3 (en) | 2006-05-05 | 2007-05-07 | RELATIONS AND METHODS FOR MODULATING EXPRESSION OF GCCR |
| JP2009510136A JP2009536039A (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating the expression of GCCR |
| EP07811875A EP2023940B1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of sglt2 |
| EP11189591.8A EP2458006B1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression APOB |
| KR1020087029617A KR101441700B1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of pcsk9 |
| MX2008014100A MX2008014100A (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of pcsk9. |
| PCT/US2007/068408 WO2007143317A2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of crp |
| US12/299,572 US8143230B2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of PCSK9 |
| CA2651309A CA2651309C (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating gene expression |
| US12/299,611 US20090326041A1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of sglt2 |
| EP12174143A EP2527442A3 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating gene expression |
| US12/299,768 US20090326042A1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of crp |
| PCT/US2007/068410 WO2007136988A2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of gccr |
| DK07811872.6T DK2021472T3 (en) | 2006-05-05 | 2007-05-07 | Compounds and Methods for Modeling Gene Expression |
| US12/299,607 US8586554B2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of PTP1B |
| EP11160534A EP2363482A1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating gene expression |
| AU2007258117A AU2007258117B2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating gene expression |
| AU2007257094A AU2007257094B2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of SGLT2 |
| PCT/US2007/068401 WO2007146511A2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating gene expression |
| NO20084738A NO20084738L (en) | 2006-05-05 | 2008-11-10 | Compounds and Methods for Modulating the Expression of PCSK9 |
| HK09107609.9A HK1128418A1 (en) | 2006-05-05 | 2009-08-18 | Compounds and methods for modulating expression of sglt2 |
| US12/883,049 US8362232B2 (en) | 2006-05-05 | 2010-09-15 | Compounds and methods for modulating expression of SGLT2 |
| US13/457,960 US20120208864A1 (en) | 2006-05-05 | 2012-04-27 | Compounds and methods for modulating expression of gcgr |
| US13/662,263 US8969316B2 (en) | 2006-05-05 | 2012-10-26 | Compounds and methods for modulating expression of DGAT2 |
| JP2013184285A JP2014033674A (en) | 2006-05-05 | 2013-09-05 | Compound and method for regulating expression of apob |
| US14/216,600 US20150057329A1 (en) | 2006-05-05 | 2014-03-17 | Compounds and methods for modulating expression apob |
| US14/698,554 US9617540B2 (en) | 2006-05-05 | 2015-04-28 | Compounds and methods for modulating gene expression |
| JP2015248195A JP6272290B2 (en) | 2006-05-05 | 2015-12-21 | Compounds and methods for modulating the expression of APOB |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US76272206P | 2006-01-27 | 2006-01-27 | |
| US60/762,722 | 2006-01-27 | ||
| US80566006P | 2006-06-23 | 2006-06-23 | |
| US60/805,660 | 2006-06-23 |
Related Child Applications (13)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/068412 Continuation WO2007134014A2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of gcgr |
| PCT/US2007/068403 Continuation-In-Part WO2007131238A2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression apob |
| PCT/US2007/068406 Continuation-In-Part WO2007143316A2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of sglt2 |
| US12/299,583 Continuation-In-Part US8372967B2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of GCCR |
| PCT/US2007/068410 Continuation-In-Part WO2007136988A2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of gccr |
| US11/745,429 Continuation-In-Part US9045754B2 (en) | 2006-05-05 | 2007-05-07 | Short antisense compounds with gapmer configuration |
| US12/299,611 Continuation-In-Part US20090326041A1 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of sglt2 |
| US12/299,607 Continuation-In-Part US8586554B2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression of PTP1B |
| US12/299,764 Continuation US8673871B2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression ApoB |
| US12/299,764 Continuation-In-Part US8673871B2 (en) | 2006-05-05 | 2007-05-07 | Compounds and methods for modulating expression ApoB |
| US29960909A Continuation | 2006-05-05 | 2009-01-29 | |
| US29958309A Continuation-In-Part | 2006-05-05 | 2009-02-03 | |
| US29961109A Continuation-In-Part | 2006-05-05 | 2009-03-11 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007090071A2 true WO2007090071A2 (en) | 2007-08-09 |
| WO2007090071A3 WO2007090071A3 (en) | 2008-01-24 |
Family
ID=38328111
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/061183 WO2007090071A2 (en) | 2006-01-27 | 2007-01-27 | 6-modified bicyclic nucleic acid analogs |
Country Status (13)
| Country | Link |
|---|---|
| US (3) | US7399845B2 (en) |
| EP (3) | EP2332951A3 (en) |
| JP (2) | JP5342881B2 (en) |
| KR (2) | KR20130042043A (en) |
| CN (2) | CN102908630B (en) |
| AT (1) | ATE482965T1 (en) |
| AU (1) | AU2007211080B9 (en) |
| CA (1) | CA2640171C (en) |
| DE (1) | DE602007009487D1 (en) |
| DK (2) | DK2314594T3 (en) |
| ES (1) | ES2516815T3 (en) |
| PL (2) | PL2314594T3 (en) |
| WO (1) | WO2007090071A2 (en) |
Cited By (209)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009124295A2 (en) * | 2008-04-04 | 2009-10-08 | Isis Pharmaceuticals, Inc. | Oligomeric compounds comprising bicyclic nucleosides and having reduced toxicity |
| WO2010036698A1 (en) * | 2008-09-24 | 2010-04-01 | Isis Pharmaceuticals, Inc. | Substituted alpha-l-bicyclic nucleosides |
| WO2010077578A1 (en) * | 2008-12-09 | 2010-07-08 | Isis Pharmaceuticals, Inc. | Bis-modified bicyclic nucleic acid analogs |
| WO2010150789A1 (en) | 2009-06-23 | 2010-12-29 | 武田薬品工業株式会社 | Method for synthesizing nucleic acid |
| US8084437B2 (en) | 2006-11-27 | 2011-12-27 | Isis Pharmaceuticals, Inc. | Methods for treating hypercholesterolemia |
| US8093222B2 (en) | 2006-11-27 | 2012-01-10 | Isis Pharmaceuticals, Inc. | Methods for treating hypercholesterolemia |
| EP2410054A1 (en) | 2006-10-18 | 2012-01-25 | Isis Pharmaceuticals, Inc. | Antisense compounds |
| US8143230B2 (en) | 2006-05-05 | 2012-03-27 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression of PCSK9 |
| EP2527442A2 (en) * | 2006-05-05 | 2012-11-28 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating gene expression |
| WO2013022967A1 (en) | 2011-08-11 | 2013-02-14 | Isis Pharmaceuticals, Inc. | Gapped oligomeric compounds comprising 5'-modified deoxyribonucleosides in the gap and uses thereof |
| WO2013068348A1 (en) | 2011-11-07 | 2013-05-16 | Santaris Pharma A/S | Lna oligomers for improvement in hepatic function |
| US8563528B2 (en) | 2009-07-21 | 2013-10-22 | Santaris Pharma A/S | Antisense oligomers targeting PCSK9 |
| USRE44760E1 (en) | 2002-11-13 | 2014-02-11 | Genzyme Corporation | Antisense modulation of apolipoprotein B-expression |
| WO2014076195A1 (en) | 2012-11-15 | 2014-05-22 | Santaris Pharma A/S | Oligonucleotide conjugates |
| WO2014118267A1 (en) | 2013-01-30 | 2014-08-07 | Santaris Pharma A/S | Lna oligonucleotide carbohydrate conjugates |
| WO2014179629A2 (en) | 2013-05-01 | 2014-11-06 | Isis Pharmaceuticals, Inc. | Compositions and methods |
| US8916694B2 (en) | 2004-05-05 | 2014-12-23 | Genzyme Corporation | SNPs of apolipoprotein B and modulation of their expression |
| EP2176280B2 (en) † | 2007-07-05 | 2015-06-24 | Isis Pharmaceuticals, Inc. | 6-disubstituted bicyclic nucleic acid analogs |
| US9107933B2 (en) | 2009-03-16 | 2015-08-18 | Isis Pharmaceuticals, Inc. | Compositions and methods of targeting apolipoprotein B for the reduction of apolipoprotein C-III |
| WO2015164693A1 (en) | 2014-04-24 | 2015-10-29 | Isis Pharmaceuticals, Inc. | Oligomeric compounds comprising alpha-beta-constrained nucleic acid |
| WO2015168589A2 (en) | 2014-05-01 | 2015-11-05 | Isis Pharmaceuticals, Inc. | Compositions and methods for modulating angiopoietin-like 3 expression |
| WO2015168618A2 (en) | 2014-05-01 | 2015-11-05 | Isis Pharmaceuticals, Inc. | Compositions and methods for modulating growth hormone receptor expression |
| EP2580228B1 (en) * | 2010-06-08 | 2016-03-23 | Ionis Pharmaceuticals, Inc. | Substituted 2'-amino and 2'-thio-bicyclic nucleosides and oligomeric compounds prepared therefrom |
| US9347061B2 (en) | 2007-03-24 | 2016-05-24 | Genzyme Corporation | Administering antisense oligonucleotides complementary to human apolipoprotein B |
| WO2017015555A1 (en) | 2015-07-22 | 2017-01-26 | Wave Life Sciences Ltd. | Oligonucleotide compositions and methods thereof |
| WO2017032726A1 (en) | 2015-08-24 | 2017-03-02 | Roche Innovation Center Copenhagen A/S | Lna-g process |
| WO2017068087A1 (en) | 2015-10-22 | 2017-04-27 | Roche Innovation Center Copenhagen A/S | Oligonucleotide detection method |
| US9695418B2 (en) | 2012-10-11 | 2017-07-04 | Ionis Pharmaceuticals, Inc. | Oligomeric compounds comprising bicyclic nucleosides and uses thereof |
| WO2017157672A1 (en) | 2016-03-18 | 2017-09-21 | Roche Innovation Center Copenhagen A/S | Acyl-protected l-lna-guanosine monomers |
| WO2017157899A1 (en) | 2016-03-14 | 2017-09-21 | F. Hoffmann-La Roche Ag | Oligonucleotides for reduction of pd-l1 expression |
| WO2017178656A1 (en) | 2016-04-14 | 2017-10-19 | Roche Innovation Center Copenhagen A/S | TRITYL-MONO-GalNAc COMPOUNDS AND THEIR USE |
| WO2017216390A1 (en) | 2016-06-17 | 2017-12-21 | F. Hoffmann-La Roche Ag | Nucleic acid molecules for reduction of papd5 or papd7 mrna for treating hepatitis b infection |
| WO2017223528A1 (en) | 2016-06-24 | 2017-12-28 | The Scripps Research Institute | Novel nucleoside triphosphate transporter and uses thereof |
| WO2018002105A1 (en) | 2016-07-01 | 2018-01-04 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides for modulating htra1 expression |
| US9879265B2 (en) | 2013-06-27 | 2018-01-30 | Roche Innovation Center Copenhagen A/S | Oligonucleotide conjugates |
| US9914922B2 (en) | 2012-04-20 | 2018-03-13 | Ionis Pharmaceuticals, Inc. | Oligomeric compounds comprising bicyclic nucleotides and uses thereof |
| US9943604B2 (en) | 2013-09-20 | 2018-04-17 | Ionis Pharmaceuticals, Inc. | Targeted therapeutic nucleosides and their use |
| WO2018087200A1 (en) | 2016-11-11 | 2018-05-17 | Roche Innovation Center Copenhagen A/S | Therapeutic oligonucleotides capture and detection |
| US9982257B2 (en) | 2012-07-13 | 2018-05-29 | Wave Life Sciences Ltd. | Chiral control |
| US10017764B2 (en) | 2011-02-08 | 2018-07-10 | Ionis Pharmaceuticals, Inc. | Oligomeric compounds comprising bicyclic nucleotides and uses thereof |
| WO2018130581A1 (en) | 2017-01-13 | 2018-07-19 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides for modulating rela expression |
| WO2018130582A1 (en) | 2017-01-13 | 2018-07-19 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides for modulating rel expression |
| WO2018130585A1 (en) | 2017-01-13 | 2018-07-19 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides for modulating relb expression |
| WO2018130584A1 (en) | 2017-01-13 | 2018-07-19 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides for modulating nfkb2 expression |
| WO2018130583A1 (en) | 2017-01-13 | 2018-07-19 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides for modulating nfkb1 expression |
| WO2018177825A1 (en) | 2017-03-29 | 2018-10-04 | Roche Innovation Center Copenhagen A/S | Orthogonal protecting groups for the preparation of stereodefined phosphorothioate oligonucleotides |
| US10144933B2 (en) | 2014-01-15 | 2018-12-04 | Shin Nippon Biomedical Laboratories, Ltd. | Chiral nucleic acid adjuvant having immunity induction activity, and immunity induction activator |
| WO2018220034A1 (en) | 2017-06-01 | 2018-12-06 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides for modulating htra1 expression |
| US10149905B2 (en) | 2014-01-15 | 2018-12-11 | Shin Nippon Biomedical Laboratories, Ltd. | Chiral nucleic acid adjuvant having antitumor effect and antitumor agent |
| US10160969B2 (en) | 2014-01-16 | 2018-12-25 | Wave Life Sciences Ltd. | Chiral design |
| US10167309B2 (en) | 2012-07-13 | 2019-01-01 | Wave Life Sciences Ltd. | Asymmetric auxiliary group |
| WO2019002237A1 (en) | 2017-06-28 | 2019-01-03 | Roche Innovation Center Copenhagen A/S | Multiple coupling & oxidation method |
| WO2019030313A2 (en) | 2017-08-11 | 2019-02-14 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for modulating ube3c expression |
| WO2019038228A1 (en) | 2017-08-22 | 2019-02-28 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for modulating tom1 expression |
| WO2019073018A1 (en) | 2017-10-13 | 2019-04-18 | Roche Innovation Center Copenhagen A/S | Methods for identifying improved stereodefined phosphorothioate oligonucleotide variants of antisense oligonucleotides utilising sub-libraries of partially stereodefined oligonucleotides |
| WO2019076842A1 (en) | 2017-10-16 | 2019-04-25 | F. Hoffmann-La Roche Ag | NUCLEIC ACID MOLECULE FOR REDUCTION OF PAPD5 AND PAPD7 mRNA FOR TREATING HEPATITIS B INFECTION |
| US10280192B2 (en) | 2011-07-19 | 2019-05-07 | Wave Life Sciences Ltd. | Methods for the synthesis of functionalized nucleic acids |
| US10280423B2 (en) | 2014-05-01 | 2019-05-07 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating complement factor B expression |
| US10307434B2 (en) | 2009-07-06 | 2019-06-04 | Wave Life Sciences Ltd. | Nucleic acid prodrugs and methods of use thereof |
| WO2019115416A2 (en) | 2017-12-11 | 2019-06-20 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for modulating fndc3b expression |
| WO2019115417A2 (en) | 2017-12-12 | 2019-06-20 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for modulating rb1 expression |
| US10329318B2 (en) | 2008-12-02 | 2019-06-25 | Wave Life Sciences Ltd. | Method for the synthesis of phosphorus atom modified nucleic acids |
| WO2019121838A1 (en) | 2017-12-21 | 2019-06-27 | F. Hoffmann-La Roche Ag | Companion diagnostic for htra1 rna antagonists |
| WO2019122279A1 (en) | 2017-12-22 | 2019-06-27 | Roche Innovation Center Copenhagen A/S | Oligonucleotides comprising a phosphorodithioate internucleoside linkage |
| WO2019122282A1 (en) | 2017-12-22 | 2019-06-27 | Roche Innovation Center Copenhagen A/S | Gapmer oligonucleotides comprising a phosphorodithioate internucleoside linkage |
| WO2019122277A1 (en) | 2017-12-22 | 2019-06-27 | Roche Innovation Center Copenhagen A/S | Novel thiophosphoramidites |
| WO2019140231A1 (en) | 2018-01-12 | 2019-07-18 | Bristol-Myers Squibb Company | Antisense oligonucleotides targeting alpha-synuclein and uses thereof |
| WO2019137883A1 (en) | 2018-01-10 | 2019-07-18 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for modulating pias4 expression |
| WO2019137974A1 (en) | 2018-01-12 | 2019-07-18 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for modulating gsk3b expression |
| WO2019138057A1 (en) | 2018-01-12 | 2019-07-18 | Roche Innovation Center Copenhagen A/S | Alpha-synuclein antisense oligonucleotides and uses thereof |
| WO2019140236A1 (en) | 2018-01-12 | 2019-07-18 | Bristol-Myers Squibb Company | Antisense oligonucleotides targeting alpha-synuclein and uses thereof |
| WO2019141723A1 (en) | 2018-01-18 | 2019-07-25 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting srebp1 |
| WO2019141656A1 (en) | 2018-01-17 | 2019-07-25 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for modulating erc1 expression |
| WO2019145386A1 (en) | 2018-01-26 | 2019-08-01 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for modulating csnk1d expression |
| WO2019154979A1 (en) | 2018-02-09 | 2019-08-15 | Genentech, Inc. | Oligonucleotides for modulating tmem106b expression |
| WO2019165453A1 (en) | 2018-02-26 | 2019-08-29 | Synthorx, Inc. | Il-15 conjugates and uses thereof |
| WO2019165067A1 (en) | 2018-02-21 | 2019-08-29 | Bristol-Myers Squibb Company | Camk2d antisense oligonucleotides and uses thereof |
| WO2019182037A1 (en) | 2018-03-20 | 2019-09-26 | 国立大学法人東京工業大学 | Antisense oligonucleotide having reduced toxicity |
| US10428019B2 (en) | 2010-09-24 | 2019-10-01 | Wave Life Sciences Ltd. | Chiral auxiliaries |
| WO2019193165A1 (en) | 2018-04-05 | 2019-10-10 | F. Hoffmann-La Roche Ag | Use of fubp1 inhibitors for treating hepatitis b virus infection |
| WO2019215065A1 (en) | 2018-05-07 | 2019-11-14 | Roche Innovation Center Copenhagen A/S | Massively parallel discovery methods for oligonucleotide therapeutics |
| WO2019215175A1 (en) | 2018-05-08 | 2019-11-14 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for modulating myh7 expression |
| WO2019219723A1 (en) | 2018-05-18 | 2019-11-21 | F. Hoffmann-La Roche Ag | Pharmaceutical compositions for treatment of microrna related diseases |
| WO2019224172A1 (en) | 2018-05-25 | 2019-11-28 | Roche Innovation Center Copenhagen A/S | Novel process for making allofuranose from glucofuranose |
| US10494633B2 (en) | 2015-11-12 | 2019-12-03 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for inducing paternal UBE3A expression |
| WO2019233922A1 (en) | 2018-06-05 | 2019-12-12 | F. Hoffmann-La Roche Ag | Oligonucleotides for modulating atxn2 expression |
| US10513706B2 (en) | 2014-04-09 | 2019-12-24 | The Scripps Research Institute | Import of unnatural or modified nucleoside triphosphates into cells via nucleic acid triphosphate transporters |
| WO2020007700A1 (en) | 2018-07-02 | 2020-01-09 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting spi1 |
| WO2020007772A1 (en) | 2018-07-02 | 2020-01-09 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting gbp-1 |
| WO2020007889A1 (en) | 2018-07-05 | 2020-01-09 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting stat1 |
| WO2020007826A1 (en) | 2018-07-05 | 2020-01-09 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting mbtps1 |
| WO2020007702A1 (en) | 2018-07-02 | 2020-01-09 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting bcl2l11 |
| WO2020007892A1 (en) | 2018-07-03 | 2020-01-09 | F. Hoffmann-La Roche Ag | Oligonucleotides for modulating tau expression |
| WO2020011653A1 (en) | 2018-07-09 | 2020-01-16 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting kynu |
| WO2020011745A2 (en) | 2018-07-11 | 2020-01-16 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting cers6 |
| WO2020011743A1 (en) | 2018-07-09 | 2020-01-16 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting mafb |
| WO2020011869A2 (en) | 2018-07-11 | 2020-01-16 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting tlr2 |
| WO2020011902A1 (en) | 2018-07-13 | 2020-01-16 | F. Hoffmann-La Roche Ag | Oligonucleotides for modulating rtel1 expression |
| WO2020011744A2 (en) | 2018-07-11 | 2020-01-16 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting cers5 |
| WO2020025563A1 (en) | 2018-07-31 | 2020-02-06 | Roche Innovation Center Copenhagen A/S | Oligonucleotides comprising a phosphorotrithioate internucleoside linkage |
| WO2020025527A1 (en) | 2018-07-31 | 2020-02-06 | Roche Innovation Center Copenhagen A/S | Oligonucleotides comprising a phosphorotrithioate internucleoside linkage |
| US10557137B2 (en) | 2015-11-06 | 2020-02-11 | Ionis Pharmaceuticals, Inc. | Modulating apolipoprotein (a) expression |
| WO2020038968A1 (en) | 2018-08-23 | 2020-02-27 | Roche Innovation Center Copenhagen A/S | Microrna-134 biomarker |
| WO2020038976A1 (en) | 2018-08-23 | 2020-02-27 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting usp8 |
| WO2020038971A1 (en) | 2018-08-23 | 2020-02-27 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting vcan |
| WO2020038973A1 (en) | 2018-08-23 | 2020-02-27 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting sptlc1 |
| WO2020043750A1 (en) | 2018-08-28 | 2020-03-05 | Roche Innovation Center Copenhagen A/S | Neoantigen engineering using splice modulating compounds |
| EP3620519A1 (en) | 2018-09-04 | 2020-03-11 | F. Hoffmann-La Roche AG | Use of isolated milk extracellular vesicles for delivering oligonucleotides orally |
| US10610571B2 (en) | 2017-08-03 | 2020-04-07 | Synthorx, Inc. | Cytokine conjugates for the treatment of proliferative and infectious diseases |
| WO2020089260A1 (en) | 2018-11-01 | 2020-05-07 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides targeting tia1 |
| WO2020104492A1 (en) | 2018-11-22 | 2020-05-28 | Roche Innovation Center Copenhagen A/S | Pyridinium salts as activators in the synthesis of stereodefined oligonucleotides |
| WO2020109344A1 (en) | 2018-11-29 | 2020-06-04 | F. Hoffmann-La Roche Ag | Occular administration device for antisense oligonucleotides |
| WO2020109343A1 (en) | 2018-11-29 | 2020-06-04 | F. Hoffmann-La Roche Ag | Combination therapy for treatment of macular degeneration |
| WO2020136125A2 (en) | 2018-12-21 | 2020-07-02 | Boehringer Ingelheim International Gmbh | Antisense oligonucleotides targeting card9 |
| WO2020152303A1 (en) | 2019-01-25 | 2020-07-30 | F. Hoffmann-La Roche Ag | Lipid vesicle for oral drug delivery |
| WO2020169695A1 (en) | 2019-02-20 | 2020-08-27 | Roche Innovation Center Copenhagen A/S | Phosphonoacetate gapmer oligonucleotides |
| WO2020169696A1 (en) | 2019-02-20 | 2020-08-27 | Roche Innovation Center Copenhagen A/S | Novel phosphoramidites |
| WO2020173845A1 (en) | 2019-02-26 | 2020-09-03 | Roche Innovation Center Copenhagen A/S | Oligonucleotide formulation method |
| WO2020178258A1 (en) | 2019-03-05 | 2020-09-10 | F. Hoffmann-La Roche Ag | Intracellular targeting of molecules |
| WO2020191377A1 (en) | 2019-03-21 | 2020-09-24 | Codiak Biosciences, Inc. | Extracellular vesicle conjugates and uses thereof |
| WO2020201339A1 (en) | 2019-04-04 | 2020-10-08 | F. Hoffmann-La Roche Ag | Oligonucleotides for modulating atxn2 expression |
| WO2020206115A2 (en) | 2019-04-03 | 2020-10-08 | Bristol-Myers Squibb Company | Angptl2 antisense oligonucleotides and uses thereof |
| WO2020212301A1 (en) | 2019-04-16 | 2020-10-22 | Roche Innovation Center Copenhagen A/S | Novel process for preparing nucleotide p(v) monomers |
| EP3730619A1 (en) | 2013-06-21 | 2020-10-28 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulation of target nucleic acids |
| WO2020221705A1 (en) | 2019-04-30 | 2020-11-05 | Roche Innovation Center Copenhagen A/S | Novel process for preparing rhenium chelated mag3 oligonucleotides |
| WO2020245233A1 (en) | 2019-06-06 | 2020-12-10 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides targeting atxn3 |
| WO2021030777A1 (en) | 2019-08-14 | 2021-02-18 | Codiak Biosciences, Inc. | Extracellular vesicle linked to molecules and uses thereof |
| WO2021030769A1 (en) | 2019-08-14 | 2021-02-18 | Codiak Biosciences, Inc. | Extracellular vesicles with nras antisense oligonucleotides |
| WO2021030776A1 (en) | 2019-08-14 | 2021-02-18 | Codiak Biosciences, Inc. | Extracellular vesicle-aso constructs targeting stat6 |
| WO2021030780A1 (en) | 2019-08-14 | 2021-02-18 | Codiak Biosciences, Inc. | Extracellular vesicle-aso constructs targeting cebp/beta |
| WO2021030773A1 (en) | 2019-08-14 | 2021-02-18 | Codiak Biosciences, Inc. | Extracellular vesicle-nlrp3 antagonist |
| WO2021030781A1 (en) | 2019-08-14 | 2021-02-18 | Codiak Biosciences, Inc. | Extracellular vesicles with antisense oligonucleotides targeting kras |
| WO2021030706A1 (en) | 2019-08-15 | 2021-02-18 | Synthorx, Inc. | Immuno oncology combination therapies with il-2 conjugates |
| WO2021041206A1 (en) | 2019-08-23 | 2021-03-04 | Synthorx, Inc. | Il-15 conjugates and uses thereof |
| WO2021050554A1 (en) | 2019-09-10 | 2021-03-18 | Synthorx, Inc. | Il-2 conjugates and methods of use to treat autoimmune diseases |
| WO2021062058A1 (en) | 2019-09-25 | 2021-04-01 | Codiak Biosciences, Inc. | Sting agonist comprising exosomes for treating neuroimmunological disorders |
| WO2021091986A1 (en) | 2019-11-04 | 2021-05-14 | Synthorx, Inc. | Interleukin 10 conjugates and uses thereof |
| WO2021122735A1 (en) | 2019-12-19 | 2021-06-24 | F. Hoffmann-La Roche Ag | Use of sept9 inhibitors for treating hepatitis b virus infection |
| WO2021122910A1 (en) | 2019-12-19 | 2021-06-24 | F. Hoffmann-La Roche Ag | Use of sbds inhibitors for treating hepatitis b virus infection |
| WO2021122921A1 (en) | 2019-12-19 | 2021-06-24 | F. Hoffmann-La Roche Ag | Use of cops3 inhibitors for treating hepatitis b virus infection |
| WO2021122869A1 (en) | 2019-12-19 | 2021-06-24 | F. Hoffmann-La Roche Ag | Use of scamp3 inhibitors for treating hepatitis b virus infection |
| WO2021122993A1 (en) | 2019-12-19 | 2021-06-24 | F. Hoffmann-La Roche Ag | Use of saraf inhibitors for treating hepatitis b virus infection |
| WO2021123086A1 (en) | 2019-12-20 | 2021-06-24 | F. Hoffmann-La Roche Ag | Enhanced oligonucleotides for inhibiting scn9a expression |
| WO2021130266A1 (en) | 2019-12-24 | 2021-07-01 | F. Hoffmann-La Roche Ag | Pharmaceutical combination of a therapeutic oligonucleotide targeting hbv and a tlr7 agonist for treatment of hbv |
| WO2021130270A1 (en) | 2019-12-24 | 2021-07-01 | F. Hoffmann-La Roche Ag | Pharmaceutical combination of antiviral agents targeting hbv and/or an immune modulator for treatment of hbv |
| US11077195B2 (en) | 2019-02-06 | 2021-08-03 | Synthorx, Inc. | IL-2 conjugates and methods of use thereof |
| EP3862362A2 (en) | 2014-05-01 | 2021-08-11 | Ionis Pharmaceuticals, Inc. | Conjugates of modified antisense oligonucleotides and their use for modulating pkk expression |
| WO2021158810A1 (en) | 2020-02-05 | 2021-08-12 | Bristol-Myers Squibb Company | Oligonucleotides for splice modulation of camk2d |
| WO2021170697A1 (en) | 2020-02-28 | 2021-09-02 | F. Hoffmann-La Roche Ag | Oligonucleotides for modulating cd73 exon 7 splicing |
| WO2021184020A1 (en) | 2020-03-13 | 2021-09-16 | Codiak Biosciences, Inc. | Methods of treating neuroinflammation |
| WO2021184021A1 (en) | 2020-03-13 | 2021-09-16 | Codiak Biosciences, Inc. | Extracellular vesicle-aso constructs targeting pmp22 |
| WO2021229036A1 (en) | 2020-05-13 | 2021-11-18 | F. Hoffmann-La Roche Ag | Oligonucleotide agonists targeting progranulin |
| WO2021231210A1 (en) | 2020-05-11 | 2021-11-18 | Genentech, Inc. | Complement component c1r inhibitors for treating a neurological disease, and related compositions, systems and methods of using same |
| WO2021231211A1 (en) | 2020-05-11 | 2021-11-18 | Genentech, Inc. | Complement component c1s inhibitors for treating a neurological disease, and related compositions, systems and methods of using same |
| WO2021231204A1 (en) | 2020-05-11 | 2021-11-18 | Genentech, Inc. | Complement component 4 inhibitors for treating neurological diseases, and related compositons, systems and methods of using same |
| WO2021233551A1 (en) | 2020-05-22 | 2021-11-25 | F.Hoffmann-La Roche Ag | Oligonucleotides for splice modulation of card9 |
| WO2021249993A1 (en) | 2020-06-09 | 2021-12-16 | Roche Innovation Center Copenhagen A/S | Guanosine analogues for use in therapeutic polynucleotides |
| WO2021263026A1 (en) | 2020-06-25 | 2021-12-30 | Synthorx, Inc. | Immuno oncology combination therapy with il-2 conjugates and anti-egfr antibodies |
| WO2021260197A1 (en) | 2020-06-26 | 2021-12-30 | F. Hoffmann-La Roche Ag | Enhanced oligonucleotides for modulating fubp1 expression |
| WO2022018155A1 (en) | 2020-07-23 | 2022-01-27 | F. Hoffmann-La Roche Ag | Lna oligonucleotides for splice modulation of stmn2 |
| WO2022018187A1 (en) | 2020-07-23 | 2022-01-27 | F. Hoffmann-La Roche Ag | Oligonucleotides targeting rna binding protein sites |
| WO2022038211A2 (en) | 2020-08-21 | 2022-02-24 | F. Hoffmann-La Roche Ag | Use of a1cf inhibitors for treating hepatitis b virus infection |
| US11261209B2 (en) | 2016-05-12 | 2022-03-01 | Roche Innovation Center Copenhagen A/S | Enhanced coupling of stereodefined oxazaphospholidine phosphoramidite monomers to nucleoside or oligonucleotide |
| US11286485B2 (en) | 2019-04-04 | 2022-03-29 | Hoffmann-La Roche Inc. | Oligonucleotides for modulating ATXN2 expression |
| WO2022076859A1 (en) | 2020-10-09 | 2022-04-14 | Synthorx, Inc. | Immuno oncology therapies with il-2 conjugates |
| WO2022076853A1 (en) | 2020-10-09 | 2022-04-14 | Synthorx, Inc. | Immuno oncology combination therapy with il-2 conjugates and pembrolizumab |
| WO2022076596A1 (en) | 2020-10-06 | 2022-04-14 | Codiak Biosciences, Inc. | Extracellular vesicle-aso constructs targeting stat6 |
| WO2022117747A2 (en) | 2020-12-03 | 2022-06-09 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides targeting atxn3 |
| WO2022117745A1 (en) | 2020-12-03 | 2022-06-09 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides targeting atxn3 |
| WO2022122613A1 (en) | 2020-12-08 | 2022-06-16 | F. Hoffmann-La Roche Ag | Novel synthesis of phosphorodithioate oligonucleotides |
| WO2022129320A1 (en) | 2020-12-18 | 2022-06-23 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides for targeting progranulin |
| WO2022136140A1 (en) | 2020-12-22 | 2022-06-30 | F. Hoffmann-La Roche Ag | Oligonucleotides targeting xbp1 |
| US11400161B2 (en) | 2016-10-06 | 2022-08-02 | Ionis Pharmaceuticals, Inc. | Method of conjugating oligomeric compounds |
| WO2022167456A1 (en) | 2021-02-02 | 2022-08-11 | F. Hoffmann-La Roche Ag | Enhanced oligonucleotides for inhibiting rtel1 expression |
| WO2022174102A1 (en) | 2021-02-12 | 2022-08-18 | Synthorx, Inc. | Lung cancer combination therapy with il-2 conjugates and an anti-pd-1 antibody or antigen-binding fragment thereof |
| WO2022174101A1 (en) | 2021-02-12 | 2022-08-18 | Synthorx, Inc. | Skin cancer combination therapy with il-2 conjugates and cemiplimab |
| WO2022178180A1 (en) | 2021-02-17 | 2022-08-25 | Codiak Biosciences, Inc. | Extracellular vesicle linked to a biologically active molecule via an optimized linker and an anchoring moiety |
| WO2022178149A2 (en) | 2021-02-17 | 2022-08-25 | Codiak Biosciences, Inc. | Extracellular vesicle-nlrp3 antagonist |
| WO2022212884A1 (en) | 2021-04-01 | 2022-10-06 | Codiak Biosciences, Inc. | Extracellular vesicle compositions |
| WO2022213118A1 (en) | 2021-03-31 | 2022-10-06 | Entrada Therapeutics, Inc. | Cyclic cell penetrating peptides |
| WO2022240760A2 (en) | 2021-05-10 | 2022-11-17 | Entrada Therapeutics, Inc. | COMPOSITIONS AND METHODS FOR MODULATING mRNA SPLICING |
| WO2022240721A1 (en) | 2021-05-10 | 2022-11-17 | Entrada Therapeutics, Inc. | Compositions and methods for modulating interferon regulatory factor-5 (irf-5) activity |
| WO2022241408A1 (en) | 2021-05-10 | 2022-11-17 | Entrada Therapeutics, Inc. | Compositions and methods for modulating tissue distribution of intracellular therapeutics |
| WO2022256538A1 (en) | 2021-06-03 | 2022-12-08 | Synthorx, Inc. | Head and neck cancer combination therapy comprising an il-2 conjugate and cetuximab |
| WO2022258555A1 (en) | 2021-06-08 | 2022-12-15 | F. Hoffmann-La Roche Ag | Oligonucleotide progranulin agonists |
| WO2022271818A1 (en) | 2021-06-23 | 2022-12-29 | Entrada Therapeutics, Inc. | Antisense compounds and methods for targeting cug repeats |
| WO2023021046A1 (en) | 2021-08-16 | 2023-02-23 | Vib Vzw | Oligonucleotides for modulating synaptogyrin-3 expression |
| WO2023034817A1 (en) | 2021-09-01 | 2023-03-09 | Entrada Therapeutics, Inc. | Compounds and methods for skipping exon 44 in duchenne muscular dystrophy |
| WO2023052317A1 (en) | 2021-09-29 | 2023-04-06 | F. Hoffmann-La Roche Ag | Rna editing |
| WO2023078883A1 (en) | 2021-11-03 | 2023-05-11 | F. Hoffmann-La Roche Ag | Oligonucleotides for modulating apolipoprotein e4 expression |
| WO2023083906A2 (en) | 2021-11-11 | 2023-05-19 | F. Hoffmann-La Roche Ag | Pharmaceutical combinations for treatment of hbv |
| WO2023104693A1 (en) | 2021-12-07 | 2023-06-15 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides targeting actl6b |
| WO2023111336A1 (en) | 2021-12-17 | 2023-06-22 | F. Hoffmann-La Roche Ag | Oligonucleotide gba agonists |
| WO2023111210A1 (en) | 2021-12-17 | 2023-06-22 | F. Hoffmann-La Roche Ag | Combination of oligonucleotides for modulating rtel1 and fubp1 |
| WO2023117738A1 (en) | 2021-12-20 | 2023-06-29 | F. Hoffmann-La Roche Ag | Threose nucleic acid antisense oligonucleotides and methods thereof |
| WO2023122750A1 (en) | 2021-12-23 | 2023-06-29 | Synthorx, Inc. | Cancer combination therapy with il-2 conjugates and cetuximab |
| WO2023122573A1 (en) | 2021-12-20 | 2023-06-29 | Synthorx, Inc. | Head and neck cancer combination therapy comprising an il-2 conjugate and pembrolizumab |
| WO2023122762A1 (en) | 2021-12-22 | 2023-06-29 | Camp4 Therapeutics Corporation | Modulation of gene transcription using antisense oligonucleotides targeting regulatory rnas |
| WO2023141507A1 (en) | 2022-01-20 | 2023-07-27 | Genentech, Inc. | Antisense oligonucleotides for modulating tmem106b expression |
| WO2023156652A1 (en) | 2022-02-21 | 2023-08-24 | F. Hoffmann-La Roche Ag | Antisense oligonucleotide |
| US11761007B2 (en) | 2015-12-18 | 2023-09-19 | The Scripps Research Institute | Production of unnatural nucleotides using a CRISPR/Cas9 system |
| WO2023217890A1 (en) | 2022-05-10 | 2023-11-16 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides targeting cfp-elk1 intergene region |
| WO2023222858A1 (en) | 2022-05-18 | 2023-11-23 | F. Hoffmann-La Roche Ag | Improved oligonucleotides targeting rna binding protein sites |
| US11834689B2 (en) | 2017-07-11 | 2023-12-05 | The Scripps Research Institute | Incorporation of unnatural nucleotides and methods thereof |
| WO2023240277A2 (en) | 2022-06-10 | 2023-12-14 | Camp4 Therapeutics Corporation | Methods of modulating progranulin expression using antisense oligonucleotides targeting regulatory rnas |
| WO2023242324A1 (en) | 2022-06-17 | 2023-12-21 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides for targeting progranulin |
| US11879145B2 (en) | 2019-06-14 | 2024-01-23 | The Scripps Research Institute | Reagents and methods for replication, transcription, and translation in semi-synthetic organisms |
| EP4332221A1 (en) | 2022-08-29 | 2024-03-06 | Roche Innovation Center Copenhagen A/S | Threose nucleic acid antisense oligonucleotides and methods thereof |
| WO2024052403A1 (en) | 2022-09-06 | 2024-03-14 | F. Hoffmann-La Roche Ag | Double-stranded rna molecule for administration to the eye |
Families Citing this family (492)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2506576C (en) * | 2002-11-18 | 2018-03-06 | Santaris Pharma A/S | Antisense gapmer oligonucleotides |
| DE102005042073B4 (en) * | 2005-08-31 | 2010-11-11 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | fiber laser |
| US8288354B2 (en) | 2005-12-28 | 2012-10-16 | The Scripps Research Institute | Natural antisense and non-coding RNA transcripts as drug targets |
| JP5342881B2 (en) * | 2006-01-27 | 2013-11-13 | アイシス ファーマシューティカルズ, インコーポレーテッド | 6-modified bicyclic nucleic acid analogues |
| ES2389737T3 (en) * | 2006-05-11 | 2012-10-31 | Isis Pharmaceuticals, Inc. | 5 'modified bicyclic nucleic acid analogs |
| AU2008286771B2 (en) | 2007-08-15 | 2013-08-15 | Isis Pharmaceuticals, Inc. | Tetrahydropyran nucleic acid analogs |
| RU2010119775A (en) | 2007-11-09 | 2011-12-27 | Айсис Фармасьютикалс,Инк. (Us) | MODULATION OF EXPRESSION OF FACTOR 7 |
| JP2011502515A (en) | 2007-11-09 | 2011-01-27 | アイシス ファーマシューティカルズ インコーポレイティッド | Regulation of factor 9 expression |
| EP2265627A2 (en) * | 2008-02-07 | 2010-12-29 | Isis Pharmaceuticals, Inc. | Bicyclic cyclohexitol nucleic acid analogs |
| WO2009117589A1 (en) * | 2008-03-21 | 2009-09-24 | Isis Pharmaceuticals, Inc. | Oligomeric compounds comprising tricyclic nucleosides and methods for their use |
| EP2285819B1 (en) * | 2008-04-04 | 2013-10-16 | Isis Pharmaceuticals, Inc. | Oligomeric compounds comprising neutrally linked terminal bicyclic nucleosides |
| WO2009143387A2 (en) * | 2008-05-22 | 2009-11-26 | Isis Pharmaceuticals, Inc. | Modulation of smrt expression |
| US20110237646A1 (en) * | 2008-08-07 | 2011-09-29 | Isis Pharmaceuticals, Inc. | Modulation of transthyretin expression for the treatment of cns related disorders |
| WO2010019270A1 (en) * | 2008-08-14 | 2010-02-18 | Isis Pharmaceuticals, Inc. | Modulation of prion expression |
| EP3081648A1 (en) | 2008-08-25 | 2016-10-19 | Excaliard Pharmaceuticals, Inc. | Antisense oligonucleotides directed against connective tissue growth factor and uses thereof |
| WO2010027832A1 (en) | 2008-08-25 | 2010-03-11 | Excaliard Pharmaceuticals, Inc. | Method for reducing scarring during wound healing using antisense compounds directed to ctgf |
| US8946172B2 (en) * | 2008-08-25 | 2015-02-03 | Excaliard Pharmaceuticals, Inc. | Method for reducing scarring during wound healing using antisense compounds directed to CTGF |
| US8604192B2 (en) * | 2008-09-24 | 2013-12-10 | Isis Pharmaceuticals, Inc. | Cyclohexenyl nucleic acids analogs |
| ES2727549T3 (en) | 2008-10-03 | 2019-10-17 | Curna Inc | Treatment of diseases related to apolipoprotein a1 by inhibition of the natural antisense transcript to apolipoprotein a1 |
| AU2009305636A1 (en) | 2008-10-15 | 2010-04-22 | Ionis Pharmaceuticals, Inc. | Modulation of Factor 11 expression |
| WO2010048585A2 (en) | 2008-10-24 | 2010-04-29 | Isis Pharmaceuticals, Inc. | Oligomeric compounds and methods |
| CA2745811C (en) | 2008-12-04 | 2021-07-13 | Joseph Collard | Treatment of tumor suppressor gene related diseases by inhibition of natural antisense transcript to the gene |
| CA2746003C (en) | 2008-12-04 | 2020-03-31 | Opko Curna, Llc | Treatment of vascular endothelial growth factor (vegf) related diseases by inhibition of natural antisense transcript to vegf |
| CN102317458B (en) | 2008-12-04 | 2018-01-02 | 库尔纳公司 | Pass through treatment of the suppression of erythropoietin(EPO) (EPO) natural antisense transcript to EPO relevant diseases |
| EP2384197B1 (en) | 2008-12-31 | 2016-04-06 | Roche Innovation Center Copenhagen A/S | Use of lna apob antisense oligomers for the treatment of acute coronary syndromes |
| WO2010090830A1 (en) * | 2009-01-20 | 2010-08-12 | Isis Pharmaceuticals, Inc. | Modulation of sirt1 expression |
| WO2010091308A2 (en) | 2009-02-06 | 2010-08-12 | Isis Pharmaceuticals, Inc. | Oligomeric compounds and methods |
| US8536320B2 (en) | 2009-02-06 | 2013-09-17 | Isis Pharmaceuticals, Inc. | Tetrahydropyran nucleic acid analogs |
| EP3009150B1 (en) | 2009-02-12 | 2019-11-13 | CuRNA, Inc. | Treatment of brain derived neurotrophic factor (bdnf) related diseases by inhibition of natural antisense transcript to bdnf |
| WO2010107733A2 (en) | 2009-03-16 | 2010-09-23 | Curna, Inc. | Treatment of nuclear factor (erythroid-derived 2)-like 2 (nrf2) related diseases by inhibition of natural antisense transcript to nrf2 |
| EP2408920B1 (en) | 2009-03-17 | 2017-03-08 | CuRNA, Inc. | Treatment of delta-like 1 homolog (dlk1) related diseases by inhibition of natural antisense transcript to dlk1 |
| EP2414520A2 (en) | 2009-03-31 | 2012-02-08 | Altair Therapeutics, Inc. | Methods of modulating an immune response to a viral infection |
| KR101722541B1 (en) | 2009-05-06 | 2017-04-04 | 큐알엔에이, 인크. | Treatment of tristetraproline(ttp) related diseases by inhibition of natural antisense transcript to ttp |
| CN103223177B (en) | 2009-05-06 | 2016-08-10 | 库尔纳公司 | By suppression therapy lipid transfer and the metabolic gene relevant disease of the natural antisense transcript for lipid transfer and metabolic gene |
| KR101742334B1 (en) | 2009-05-08 | 2017-06-01 | 큐알엔에이, 인크. | Treatment of dystrophin family related diseases by inhibition of natural antisense transcript to dmd family |
| CN102575251B (en) | 2009-05-18 | 2018-12-04 | 库尔纳公司 | The relevant disease of the reprogramming factor is treated by inhibiting the natural antisense transcript for the reprogramming factor |
| KR101703695B1 (en) | 2009-05-22 | 2017-02-08 | 큐알엔에이, 인크. | Treatment of transcription factor e3 (tfe3) and insulin receptor substrate 2 (irs2) related diseases by inhibition of natural antisense transcript to tfe3 |
| EP2435571B1 (en) | 2009-05-28 | 2016-12-14 | CuRNA, Inc. | Treatment of antiviral gene related diseases by inhibition of natural antisense transcript to an antiviral gene |
| CA2764822A1 (en) | 2009-06-12 | 2010-12-16 | Santaris Pharma A/S | New potent anti apob antisense compounds |
| EP2443238B1 (en) | 2009-06-16 | 2017-03-22 | CuRNA, Inc. | Treatment of paraoxonase 1 (pon1) related diseases by inhibition of natural antisense transcript to pon1 |
| JP5944311B2 (en) | 2009-06-16 | 2016-07-05 | クルナ・インコーポレーテッド | Treatment of collagen gene-related diseases by suppression of natural antisense transcripts against collagen genes |
| RU2566724C9 (en) | 2009-06-17 | 2019-03-12 | Байоджен Ma Инк. | Compositions and methods of modulating smn2 splicing in subject |
| CN102597238B (en) | 2009-06-24 | 2016-06-29 | 库尔纳公司 | The relevant disease of TNFR2 is treated by suppressing for the natural antisense transcript of tumor necrosis factor receptor 2 (TNFR2) |
| WO2010151674A2 (en) | 2009-06-26 | 2010-12-29 | Curna, Inc. | Treatment of down syndrome gene related diseases by inhibition of natural antisense transcript to a down syndrome gene |
| WO2011011700A2 (en) | 2009-07-24 | 2011-01-27 | Curna, Inc. | Treatment of sirtuin (sirt) related diseases by inhibition of natural antisense transcript to a sirtuin (sirt) |
| JP6128848B2 (en) | 2009-08-05 | 2017-05-17 | クルナ・インコーポレーテッド | Treatment of insulin gene (INS) -related diseases by suppression of natural antisense transcripts against the insulin gene (INS) |
| WO2011017521A2 (en) | 2009-08-06 | 2011-02-10 | Isis Pharmaceuticals, Inc. | Bicyclic cyclohexose nucleic acid analogs |
| CN102625841A (en) | 2009-08-11 | 2012-08-01 | 欧科库尔纳有限责任公司 | Treatment of Adiponectin (ADIPOQ) related diseases by inhibition of natural antisense transcript to an Adiponectin (ADIPOQ) |
| CA2771228C (en) | 2009-08-21 | 2020-12-29 | Opko Curna, Llc | Treatment of 'c terminus of hsp70-interacting protein' (chip) related diseases by inhibition of natural antisense transcript to chip |
| WO2011031482A2 (en) | 2009-08-25 | 2011-03-17 | Curna, Inc. | Treatment of 'iq motif containing gtpase activating protein' (iqgap) related diseases by inhibition of natural antisense transcript to iqgap |
| KR102175230B1 (en) | 2009-09-11 | 2020-11-06 | 아이오니스 파마수티컬즈, 인코포레이티드 | Modulation of huntingtin expression |
| JP6175236B2 (en) | 2009-09-25 | 2017-08-09 | カッパーアールエヌエー,インコーポレイテッド | Treatment of FLG-related diseases by modulating the expression and activity of filaggrin (FLG) |
| WO2011048125A1 (en) | 2009-10-20 | 2011-04-28 | Santaris Pharma A/S | Oral delivery of therapeutically effective lna oligonucleotides |
| WO2011054811A1 (en) | 2009-11-03 | 2011-05-12 | Santaris Pharma A/S | Rna antagonists targeting hsp27 combination therapy |
| WO2011084455A2 (en) | 2009-12-16 | 2011-07-14 | Opko Curna, Llc. | Treatment of membrane bound transcription factor peptidase, site 1 (mbtps1) related diseases by inhibition of natural antisense transcript to mbtps1 |
| KR101793753B1 (en) | 2009-12-23 | 2017-11-03 | 큐알엔에이, 인크. | Treatment of uncoupling protein 2 (ucp2) related diseases by inhibition of natural antisense transcript to ucp2 |
| CN102869776B (en) | 2009-12-23 | 2017-06-23 | 库尔纳公司 | HGF relevant diseases are treated by suppressing the natural antisense transcript of HGF (HGF) |
| EP2519633B1 (en) | 2009-12-29 | 2017-10-25 | CuRNA, Inc. | Treatment of nuclear respiratory factor 1 (nrf1) related diseases by inhibition of natural antisense transcript to nrf1 |
| WO2011090741A2 (en) | 2009-12-29 | 2011-07-28 | Opko Curna, Llc | TREATMENT OF TUMOR PROTEIN 63 (p63) RELATED DISEASES BY INHIBITION OF NATURAL ANTISENSE TRANSCRIPT TO p63 |
| JP6083735B2 (en) | 2009-12-31 | 2017-02-22 | カッパーアールエヌエー,インコーポレイテッド | Treatment of insulin receptor substrate 2 (IRS2) related diseases by inhibition of natural antisense transcripts against insulin receptor substrate 2 (IRS2) and transcription factor 3 (TFE3) |
| NO2521784T3 (en) | 2010-01-04 | 2018-05-05 | ||
| US8912157B2 (en) | 2010-01-06 | 2014-12-16 | Curna, Inc. | Treatment of pancreatic developmental gene related diseases by inhibition of natural antisense transcript to a pancreatic developmental gene |
| US8653047B2 (en) | 2010-01-08 | 2014-02-18 | Isis Pharmaceuticals, Inc. | Modulation of angiopoietin-like 3 expression |
| DK2524039T3 (en) | 2010-01-11 | 2018-03-12 | Curna Inc | TREATMENT OF GENDER HORMON-BINDING GLOBULIN (SHBG) RELATED DISEASES BY INHIBITION OF NATURAL ANTISENCE TRANSCRIPTS TO SHBG |
| US8779118B2 (en) | 2010-01-11 | 2014-07-15 | Isis Pharmaceuticals, Inc. | Base modified bicyclic nucleosides and oligomeric compounds prepared therefrom |
| WO2011088148A1 (en) * | 2010-01-12 | 2011-07-21 | Isis Pharmaceuticals, Inc. | Modulation of transforming growth factor-beta 1 expression |
| WO2011091390A2 (en) | 2010-01-25 | 2011-07-28 | Opko Curna, Llc | Treatment of rnase h1 related diseases by inhibition of natural antisense transcript to rnase h1 |
| EP3321361B1 (en) | 2010-02-08 | 2019-03-27 | Ionis Pharmaceuticals, Inc. | Selective reduction of allelic variants |
| AU2011213563B2 (en) | 2010-02-08 | 2015-12-24 | Ionis Pharmaceuticals, Inc. | Selective reduction of allelic variants |
| WO2011103528A2 (en) | 2010-02-22 | 2011-08-25 | Opko Curna Llc | Treatment of pyrroline-5-carboxylate reductase 1 (pycr1) related diseases by inhibition of natural antisense transcript to pycr1 |
| WO2011106689A1 (en) * | 2010-02-26 | 2011-09-01 | Isis Pharmaceuticals, Inc. | Modulation of smad3 expression |
| WO2011115817A1 (en) | 2010-03-16 | 2011-09-22 | Isis Pharmaceuticals, Inc. | Methods of preparing 2'-o-substituted purine nucleosides |
| US9193752B2 (en) | 2010-03-17 | 2015-11-24 | Isis Pharmaceuticals, Inc. | 5′-substituted bicyclic nucleosides and oligomeric compounds prepared therefrom |
| EP2553098B1 (en) | 2010-04-02 | 2017-10-11 | CuRNA, Inc. | Treatment of colony-stimulating factor 3 (csf3) related diseases by inhibition of natural antisense transcript to csf3 |
| US9044494B2 (en) | 2010-04-09 | 2015-06-02 | Curna, Inc. | Treatment of fibroblast growth factor 21 (FGF21) related diseases by inhibition of natural antisense transcript to FGF21 |
| KR101869570B1 (en) | 2010-04-28 | 2018-06-20 | 아이오니스 파마수티컬즈, 인코포레이티드 | Modified nucleosides and oligomeric compounds prepared therefrom |
| EP2625186B1 (en) | 2010-04-28 | 2016-07-27 | Ionis Pharmaceuticals, Inc. | 5' modified nucleosides and oligomeric compounds prepared therefrom |
| WO2011139695A2 (en) | 2010-04-28 | 2011-11-10 | Isis Pharmaceuticals, Inc. | Modified 5' diphosphate nucleosides and oligomeric compounds prepared therefrom |
| WO2011139911A2 (en) | 2010-04-29 | 2011-11-10 | Isis Pharmaceuticals, Inc. | Lipid formulated single stranded rna |
| WO2011139917A1 (en) | 2010-04-29 | 2011-11-10 | Isis Pharmaceuticals, Inc. | Modulation of transthyretin expression |
| EP2566966A4 (en) | 2010-05-03 | 2013-12-11 | Curna Inc | Treatment of sirtuin (sirt) related diseases by inhibition of natural antisense transcript to a sirtuin (sirt) |
| TWI586356B (en) | 2010-05-14 | 2017-06-11 | 可娜公司 | Treatment of par4 related diseases by inhibition of natural antisense transcript to par4 |
| CA2799207C (en) | 2010-05-26 | 2019-03-26 | Curna, Inc. | Treatment of atonal homolog 1 (atoh1) related diseases by inhibition of natural antisense transcript to atoh1 |
| WO2011150007A2 (en) | 2010-05-26 | 2011-12-01 | Opko Curna Llc | Treatment of methionine sulfoxide reductase a (msra) related diseases by inhibition of natural antisense transcript to msra |
| WO2011153323A2 (en) | 2010-06-02 | 2011-12-08 | Alnylam Pharmaceuticals, Inc. | Compositions and methods directed to treating liver fibrosis |
| WO2011156278A1 (en) | 2010-06-07 | 2011-12-15 | Isis Pharmaceuticals, Inc. | Bicyclic nucleosides and oligomeric compounds prepared therefrom |
| WO2011159836A2 (en) | 2010-06-15 | 2011-12-22 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating interaction between proteins and target nucleic acids |
| CA2803882C (en) | 2010-06-23 | 2022-10-18 | Opko Curna, Llc | Treatment of sodium channel, voltage-gated, alpha subunit (scna) related diseases by inhibition of natural antisense transcript to scna |
| WO2012007477A1 (en) | 2010-07-12 | 2012-01-19 | Santaris Pharma A/S | Anti hcv oligomers |
| US8980860B2 (en) | 2010-07-14 | 2015-03-17 | Curna, Inc. | Treatment of discs large homolog (DLG) related diseases by inhibition of natural antisense transcript to DLG |
| WO2012012467A2 (en) | 2010-07-19 | 2012-01-26 | Isis Pharmaceuticals, Inc. | Modulation of nuclear-retained rna |
| DK2601611T3 (en) | 2010-08-02 | 2021-02-01 | Integrated Dna Tech Inc | PROCEDURES FOR PREDICTING STABILITY AND MELTING TEMPERATURES FOR NUCLEIC ACID DUPLEXES |
| WO2012034942A1 (en) | 2010-09-13 | 2012-03-22 | Santaris Pharma A/S | Compounds for the modulation of aurora kinase b expression |
| US8993533B2 (en) | 2010-10-06 | 2015-03-31 | Curna, Inc. | Treatment of sialidase 4 (NEU4) related diseases by inhibition of natural antisense transcript to NEU4 |
| US8648053B2 (en) | 2010-10-20 | 2014-02-11 | Rosalind Franklin University Of Medicine And Science | Antisense oligonucleotides that target a cryptic splice site in Ush1c as a therapeutic for Usher syndrome |
| CA2815212A1 (en) | 2010-10-22 | 2012-04-26 | Curna, Inc. | Treatment of alpha-l-iduronidase (idua) related diseases by inhibition of natural antisense transcript to idua |
| WO2012064758A2 (en) | 2010-11-08 | 2012-05-18 | Isis Pharmaceuticals, Inc. | Methods for modulating factor 12 expression |
| EP2640853B1 (en) | 2010-11-17 | 2018-12-26 | Ionis Pharmaceuticals, Inc. | Modulation of alpha synuclein expression |
| US10000752B2 (en) | 2010-11-18 | 2018-06-19 | Curna, Inc. | Antagonat compositions and methods of use |
| WO2012066092A1 (en) | 2010-11-19 | 2012-05-24 | Santaris Pharma A/S | Compounds for the modulation of aurora kinase a expression |
| WO2012066093A1 (en) | 2010-11-19 | 2012-05-24 | Santaris Pharma A/S | Compounds for the modulation of pdz-binding kinase (pbk) expression |
| WO2012071238A2 (en) | 2010-11-23 | 2012-05-31 | Opko Curna Llc | Treatment of nanog related diseases by inhibition of natural antisense transcript to nanog |
| WO2012078967A2 (en) | 2010-12-10 | 2012-06-14 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for increasing erythropoietin (epo) production |
| WO2012079046A2 (en) | 2010-12-10 | 2012-06-14 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for inhibiting expression of klf-1 and bcl11a genes |
| SI2670411T1 (en) | 2011-02-02 | 2019-06-28 | Excaliard Pharmaceuticals, Inc. | Antisense compounds targeting connective tissue growth factor (ctgf) for use in a method of treating keloids or hypertrophic scars |
| WO2012110457A2 (en) | 2011-02-14 | 2012-08-23 | Santaris Pharma A/S | Compounds for the modulation of osteopontin expression |
| SG193923A1 (en) | 2011-03-29 | 2013-11-29 | Alnylam Pharmaceuticals Inc | Compositions and methods for inhibiting expression of tmprss6 gene |
| WO2012135736A2 (en) | 2011-04-01 | 2012-10-04 | Isis Pharmaceuticals, Inc. | Modulation of signal transducer and activator of transcription 3 (stat3) expression |
| CN103547588B (en) | 2011-04-13 | 2016-06-29 | Isis制药公司 | The antisense that PTP1B expresses regulates |
| WO2012143427A1 (en) | 2011-04-19 | 2012-10-26 | Santaris Pharma A/S | Anti polyomavirus compounds |
| MX340408B (en) | 2011-04-21 | 2016-07-07 | Ionis Pharmaceuticals Inc | Modulation of hepatitis b virus (hbv) expression. |
| CN103620035B (en) | 2011-04-25 | 2021-04-23 | 赛诺菲 | microRNA compounds and methods for modulating MIR-21 activity |
| JP6203707B2 (en) | 2011-04-27 | 2017-09-27 | アイオーニス ファーマシューティカルズ, インコーポレーテッドIonis Pharmaceuticals,Inc. | Regulation of apolipoprotein CIII (APOCIII) expression |
| WO2012151324A1 (en) | 2011-05-02 | 2012-11-08 | Isis Pharmaceuticals, Inc. | Antisense compounds targeting genes associated with usher syndrome |
| RU2620980C2 (en) | 2011-06-09 | 2017-05-30 | Курна, Инк. | Treatment of diseases associated with frataxin (fxn), by inhibiting natural antisense fxn transcript |
| WO2012170347A1 (en) | 2011-06-09 | 2012-12-13 | Isis Pharmaceuticals, Inc. | Bicyclic nucleosides and oligomeric compounds prepared therefrom |
| WO2012170947A2 (en) | 2011-06-10 | 2012-12-13 | Isis Pharmaceuticals, Inc. | Methods for modulating factor 12 expression |
| WO2012170945A2 (en) | 2011-06-10 | 2012-12-13 | Isis Pharmaceuticals, Inc. | Methods for modulating kallikrein (klkb1) expression |
| EP2721156B1 (en) | 2011-06-16 | 2016-12-21 | Ionis Pharmaceuticals, Inc. | Antisense modulation of fibroblast growth factor receptor 4 expression |
| KR102395085B1 (en) | 2011-06-21 | 2022-05-09 | 알닐람 파마슈티칼스 인코포레이티드 | Angiopoietin-like 3(angptl3) irna compostions and methods of use thereof |
| US20140127159A1 (en) | 2011-06-23 | 2014-05-08 | Stella Aps | HCV Combination Therapy |
| WO2012178033A2 (en) | 2011-06-23 | 2012-12-27 | Alnylam Pharmaceuticals, Inc. | Serpina1 sirnas: compositions of matter and methods of treatment |
| AU2012275096B2 (en) | 2011-06-29 | 2016-02-04 | Ionis Pharmaceuticals, Inc. | Methods for modulating kallikrein (KLKB1) expression |
| EP2726611A1 (en) | 2011-06-30 | 2014-05-07 | Stella ApS | Hcv combination therapy |
| WO2013000855A1 (en) | 2011-06-30 | 2013-01-03 | Santaris Pharma A/S | Hcv combination therapy |
| EP2739735A2 (en) | 2011-08-01 | 2014-06-11 | Alnylam Pharmaceuticals, Inc. | Method for improving the success rate of hematopoietic stem cell transplants |
| EP2751269B1 (en) | 2011-08-29 | 2016-03-23 | Ionis Pharmaceuticals, Inc. | Methods and compounds useful in conditions related to repeat expansion |
| DK2751270T3 (en) | 2011-08-29 | 2018-10-29 | Ionis Pharmaceuticals Inc | OLIGOMER-CONJUGATE COMPLEXES AND THEIR USE |
| US10583128B2 (en) | 2011-09-06 | 2020-03-10 | Curna, Inc. | Treatment of diseases related to alpha subunits of sodium channels, voltage-gated (SCNxA) with small molecules |
| EP2756080B1 (en) | 2011-09-14 | 2019-02-20 | Translate Bio MA, Inc. | Multimeric oligonucleotide compounds |
| EP3401401B1 (en) | 2011-09-20 | 2020-04-15 | Ionis Pharmaceuticals, Inc. | Antisense modulation of gcgr expression |
| CN103906838A (en) | 2011-10-25 | 2014-07-02 | Isis制药公司 | Antisense modulation of GCCR expression |
| EP2773777B1 (en) | 2011-10-31 | 2020-05-13 | University of Utah Research Foundation | Genetic alterations in glioblastoma |
| WO2013068347A1 (en) | 2011-11-07 | 2013-05-16 | Santaris Pharma A/S | Prognostic method for checking efficacy of micro rna-122 inhibitors in hcv+ patients |
| ES2761343T3 (en) | 2011-11-07 | 2020-05-19 | Ionis Pharmaceuticals Inc | Modulation of TMPRSS6 expression |
| KR20140091587A (en) | 2011-11-11 | 2014-07-21 | 산타리스 팔마 에이/에스 | Compounds for the modulation of smn2 splicing |
| US9243291B1 (en) | 2011-12-01 | 2016-01-26 | Isis Pharmaceuticals, Inc. | Methods of predicting toxicity |
| EP2790736B1 (en) | 2011-12-12 | 2018-01-31 | Oncoimmunin, Inc. | In vivo delivery of oligonucleotides |
| EP4144378A1 (en) | 2011-12-16 | 2023-03-08 | ModernaTX, Inc. | Modified nucleoside, nucleotide, and nucleic acid compositions |
| WO2013096837A1 (en) | 2011-12-22 | 2013-06-27 | Isis Pharmaceuticals, Inc. | Methods for modulating metastasis-associated-in-lung-adenocarcinoma-transcript-1(malat-1) expression |
| WO2013106770A1 (en) | 2012-01-11 | 2013-07-18 | Isis Pharmaceuticals, Inc. | Compositions and methods for modulation of ikbkap splicing |
| US9737480B2 (en) | 2012-02-06 | 2017-08-22 | President And Fellows Of Harvard College | ARRDC1-mediated microvesicles (ARMMs) and uses thereof |
| WO2013120003A1 (en) | 2012-02-08 | 2013-08-15 | Isis Pharmaceuticals, Inc. | Modulation of rna by repeat targeting |
| JP2015511494A (en) | 2012-03-15 | 2015-04-20 | キュアナ,インク. | Treatment of BDNF-related diseases by inhibition of natural antisense transcripts against brain-derived neurotrophic factor (BDNF) |
| WO2013142514A1 (en) | 2012-03-19 | 2013-09-26 | Isis Pharmaceuticals, Inc. | Methods and compositions for modulating alpha-1-antitrypsin expression |
| AU2013202595B2 (en) | 2012-03-30 | 2016-04-21 | Biogen Ma Inc. | Methods for modulating Tau expression for reducing seizure and modifying a neurodegenerative syndrome |
| CA2868391A1 (en) | 2012-04-02 | 2013-10-10 | Stephane Bancel | Polynucleotides comprising n1-methyl-pseudouridine and methods for preparing the same |
| JP2015513914A (en) | 2012-04-02 | 2015-05-18 | モデルナ セラピューティクス インコーポレイテッドModerna Therapeutics,Inc. | Modified polynucleotides for the production of secreted proteins |
| WO2013154799A1 (en) | 2012-04-09 | 2013-10-17 | Isis Pharmaceuticals, Inc. | Tricyclic nucleosides and oligomeric compounds prepared therefrom |
| EP2850092B1 (en) | 2012-04-09 | 2017-03-01 | Ionis Pharmaceuticals, Inc. | Tricyclic nucleic acid analogs |
| US9133461B2 (en) | 2012-04-10 | 2015-09-15 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for inhibiting expression of the ALAS1 gene |
| KR20220028183A (en) | 2012-04-25 | 2022-03-08 | 사노피 | Microrna compounds and methods for modulating mir-21 activity |
| US9127274B2 (en) | 2012-04-26 | 2015-09-08 | Alnylam Pharmaceuticals, Inc. | Serpinc1 iRNA compositions and methods of use thereof |
| AU2013262663A1 (en) | 2012-05-16 | 2015-01-22 | The General Hospital Corporation D/B/A Massachusetts General Hospital | Compositions and methods for modulating gene expression |
| EA201492123A1 (en) | 2012-05-16 | 2015-10-30 | Рана Терапьютикс, Инк. | COMPOSITIONS AND METHODS FOR MODULATING THE EXPRESSION OF THE SMN GENES FAMILY |
| US20160002624A1 (en) | 2012-05-17 | 2016-01-07 | Isis Pharmaceuticals, Inc. | Antisense oligonucleotide compositions |
| US9574193B2 (en) | 2012-05-17 | 2017-02-21 | Ionis Pharmaceuticals, Inc. | Methods and compositions for modulating apolipoprotein (a) expression |
| EP2852606B1 (en) | 2012-05-22 | 2019-08-07 | Ionis Pharmaceuticals, Inc. | Modulation of enhancer rna mediated gene expression |
| RU2624028C2 (en) | 2012-05-24 | 2017-06-30 | Ионис Фармасьютикалз, Инк. | Methods and compositions for modeling expression of apolipoprotein (a) |
| US9828602B2 (en) | 2012-06-01 | 2017-11-28 | Ionis Pharmaceuticals, Inc. | Antisense compounds targeting genes associated with fibronectin |
| US9487780B2 (en) | 2012-06-01 | 2016-11-08 | Ionis Pharmaceuticals, Inc. | Antisense compounds targeting genes associated with fibronectin |
| WO2014004572A2 (en) | 2012-06-25 | 2014-01-03 | Isis Pharmaceuticals, Inc. | Modulation of ube3a-ats expression |
| SG11201500243WA (en) | 2012-07-13 | 2015-04-29 | Shin Nippon Biomedical Lab Ltd | Chiral nucleic acid adjuvant |
| US20150297629A1 (en) | 2012-07-27 | 2015-10-22 | Isis Pharmaceuticals, Inc. | Modulation of renin-angiotensin system (ras) related diseases by angiotensinogen |
| CN104736551B (en) | 2012-08-15 | 2017-07-28 | Ionis制药公司 | The method for preparing oligomeric compounds using improved end-blocking scheme |
| AR092982A1 (en) | 2012-10-11 | 2015-05-13 | Isis Pharmaceuticals Inc | MODULATION OF THE EXPRESSION OF ANDROGEN RECEIVERS |
| WO2014059364A1 (en) | 2012-10-11 | 2014-04-17 | Isis Pharmaceuticals, Inc. | Methods of treating kennedy's disease |
| EP4144845A1 (en) | 2012-10-12 | 2023-03-08 | Ionis Pharmaceuticals, Inc. | Antisense compounds and uses thereof |
| DK2906256T3 (en) | 2012-10-12 | 2018-11-19 | Ionis Pharmaceuticals Inc | SELECTIVE ANTISENSE COMPOUNDS AND APPLICATIONS THEREOF |
| WO2014062691A2 (en) | 2012-10-15 | 2014-04-24 | Isis Pharmaceuticals, Inc. | Compositions for modulating c9orf72 expression |
| ES2762326T5 (en) | 2012-10-15 | 2023-04-27 | Ionis Pharmaceuticals Inc | Methods to modulate the expression of C9ORF72 |
| EP2906697A4 (en) | 2012-10-15 | 2016-06-22 | Ionis Pharmaceuticals Inc | Methods for monitoring c9orf72 expression |
| US9029335B2 (en) | 2012-10-16 | 2015-05-12 | Isis Pharmaceuticals, Inc. | Substituted 2′-thio-bicyclic nucleosides and oligomeric compounds prepared therefrom |
| US10723754B2 (en) | 2012-10-22 | 2020-07-28 | Idenix Pharmaceuticals Llc | 2′,4′-bridged nucleosides for HCV infection |
| NZ706540A (en) | 2012-10-31 | 2018-11-30 | Ionis Pharmaceuticals Inc | Dosage regimens for treatment of cancer |
| EP2914621B1 (en) | 2012-11-05 | 2023-06-07 | Foundation Medicine, Inc. | Novel ntrk1 fusion molecules and uses thereof |
| WO2014080004A1 (en) | 2012-11-26 | 2014-05-30 | Santaris Pharma A/S | Compositions and methods for modulation of fgfr3 expression |
| SI2922554T1 (en) | 2012-11-26 | 2022-06-30 | Modernatx, Inc. | Terminally modified rna |
| US9695475B2 (en) | 2012-12-11 | 2017-07-04 | Ionis Pharmaceuticals, Inc. | Competitive modulation of microRNAs |
| US20150337002A1 (en) * | 2013-01-15 | 2015-11-26 | Shionogi & Co., Ltd. | Nucleoside and nucleotide having sulfonamide structure |
| EP3434774A1 (en) | 2013-01-17 | 2019-01-30 | ModernaTX, Inc. | Signal-sensor polynucleotides for the alteration of cellular phenotypes |
| CA3150658A1 (en) | 2013-01-18 | 2014-07-24 | Foundation Medicine, Inc. | Methods of treating cholangiocarcinoma |
| WO2014118272A1 (en) | 2013-01-30 | 2014-08-07 | Santaris Pharma A/S | Antimir-122 oligonucleotide carbohydrate conjugates |
| US9701708B2 (en) | 2013-01-31 | 2017-07-11 | Ionis Pharmaceuticals, Inc. | Method of preparing oligomeric compounds using modified coupling protocols |
| WO2014121287A2 (en) | 2013-02-04 | 2014-08-07 | Isis Pharmaceuticals, Inc. | Selective antisense compounds and uses thereof |
| WO2014127268A2 (en) | 2013-02-14 | 2014-08-21 | Isis Pharmaceuticals, Inc. | Modulation of apolipoprotein c-iii (apociii) expression in lipoprotein lipase deficient (lpld) populations |
| WO2014130922A1 (en) | 2013-02-25 | 2014-08-28 | Trustees Of Boston University | Compositions and methods for treating fungal infections |
| WO2014143158A1 (en) | 2013-03-13 | 2014-09-18 | The Broad Institute, Inc. | Compositions and methods for labeling of agents |
| WO2014159813A1 (en) | 2013-03-13 | 2014-10-02 | Moderna Therapeutics, Inc. | Long-lived polynucleotide molecules |
| KR20150130430A (en) | 2013-03-14 | 2015-11-23 | 아이시스 파마수티컬즈 인코포레이티드 | Compositions and methods for modulating tau expression |
| EP2971010B1 (en) | 2013-03-14 | 2020-06-10 | ModernaTX, Inc. | Formulation and delivery of modified nucleoside, nucleotide, and nucleic acid compositions |
| BR112015022621A2 (en) | 2013-03-14 | 2017-10-31 | Andes Biotechnologies S A | methods for detection and treatment of multiple myeloma |
| KR102605775B1 (en) | 2013-03-14 | 2023-11-29 | 알닐람 파마슈티칼스 인코포레이티드 | Complement component c5 irna compositions and methods of use thereof |
| ES2708650T3 (en) | 2013-03-14 | 2019-04-10 | Andes Biotechnologies Global Inc | Antisense oligonucleotides for the treatment of tumor stem cells |
| US9273349B2 (en) | 2013-03-14 | 2016-03-01 | Affymetrix, Inc. | Detection of nucleic acids |
| US8980864B2 (en) | 2013-03-15 | 2015-03-17 | Moderna Therapeutics, Inc. | Compositions and methods of altering cholesterol levels |
| WO2014172698A1 (en) | 2013-04-19 | 2014-10-23 | Isis Pharmaceuticals, Inc. | Compositions and methods for modulation nucleic acids through nonsense mediated decay |
| TWI680767B (en) | 2013-05-01 | 2020-01-01 | 美商雷格勒斯治療公司 | Compounds and methods for enhanced cellular uptake |
| US9309513B2 (en) | 2013-05-01 | 2016-04-12 | Regulus Therapeutics Inc. | MicroRNA compounds and methods for modulating miR-122 |
| RS57418B1 (en) | 2013-05-22 | 2018-09-28 | Alnylam Pharmaceuticals Inc | Serpina1 irna compositions and methods of use thereof |
| TWI727917B (en) | 2013-05-22 | 2021-05-21 | 美商阿尼拉製藥公司 | TMPRSS6 iRNA COMPOSITIONS AND METHODS OF USE THEREOF |
| DK3004352T3 (en) | 2013-05-24 | 2017-11-27 | Roche Innovation Ct Copenhagen As | B-cell Cl1 / Lymphoma 11a (bcl11a) oligonucleotide modulators and uses thereof |
| WO2014197835A2 (en) | 2013-06-06 | 2014-12-11 | The General Hospital Corporation | Methods and compositions for the treatment of cancer |
| EP3656386A1 (en) | 2013-06-21 | 2020-05-27 | Ionis Pharmaceuticals, Inc. | Compounds and methods for modulating apolipoprotein c-iii expression for improving a diabetic profile |
| KR102236784B1 (en) | 2013-07-02 | 2021-04-05 | 아이오니스 파마수티컬즈, 인코포레이티드 | Modulators of growth hormone receptor |
| JP7019233B2 (en) | 2013-07-11 | 2022-02-15 | モデルナティエックス インコーポレイテッド | Compositions and Methods of Use Containing Synthetic polynucleotides and Synthetic sgRNAs Encoding CRISPR-Related Proteins |
| EP3022176B8 (en) | 2013-07-15 | 2019-12-25 | The Regents of the University of California | Azacyclic constrained analogs of fty720 |
| TWI657819B (en) | 2013-07-19 | 2019-05-01 | 美商Ionis製藥公司 | Compositions for modulating tau expression |
| US10435430B2 (en) | 2013-07-31 | 2019-10-08 | Ionis Pharmaceuticals, Inc. | Methods and compounds useful in conditions related to repeat expansion |
| ES2773547T3 (en) | 2013-08-08 | 2020-07-13 | Scripps Research Inst | An in vitro nucleic acid site specific enzymatic labeling procedure by incorporating unnatural nucleotides |
| TW201536329A (en) | 2013-08-09 | 2015-10-01 | Isis Pharmaceuticals Inc | Compounds and methods for modulation of dystrophia myotonica-protein kinase (DMPK) expression |
| KR102365486B1 (en) | 2013-08-28 | 2022-02-18 | 아이오니스 파마수티컬즈, 인코포레이티드 | Modulation of prekallikrein (pkk) expression |
| US20160194625A1 (en) | 2013-09-03 | 2016-07-07 | Moderna Therapeutics, Inc. | Chimeric polynucleotides |
| US20160194368A1 (en) | 2013-09-03 | 2016-07-07 | Moderna Therapeutics, Inc. | Circular polynucleotides |
| PE20190354A1 (en) | 2013-09-13 | 2019-03-07 | Ionis Pharmaceuticals Inc | COMPLEMENT B FACTOR MODULATORS |
| CN105793423A (en) | 2013-10-02 | 2016-07-20 | 阿尔尼拉姆医药品有限公司 | Compositions and methods for inhibiting expression of the LECT2 gene |
| EP3052521A1 (en) | 2013-10-03 | 2016-08-10 | Moderna Therapeutics, Inc. | Polynucleotides encoding low density lipoprotein receptor |
| JP6613227B2 (en) | 2013-10-04 | 2019-11-27 | アルナイラム ファーマシューティカルズ, インコーポレイテッド | Composition and method for inhibiting the expression of ALAS1 gene |
| CN105637090B (en) | 2013-10-11 | 2020-10-16 | Ionis制药公司 | Compositions for modulating expression of C9ORF72 |
| US11162096B2 (en) | 2013-10-14 | 2021-11-02 | Ionis Pharmaceuticals, Inc | Methods for modulating expression of C9ORF72 antisense transcript |
| CN105899679A (en) | 2013-10-21 | 2016-08-24 | 通用医疗公司 | Methods relating to circulating tumor cell clusters and the treatment of cancer |
| WO2015061246A1 (en) | 2013-10-21 | 2015-04-30 | Isis Pharmaceuticals, Inc. | Method for solution phase detritylation of oligomeric compounds |
| WO2015061536A1 (en) | 2013-10-25 | 2015-04-30 | Regulus Therapeutics Inc. | Microrna compounds and methods for modulating mir-21 activity |
| US10752940B2 (en) | 2013-11-08 | 2020-08-25 | Ionis Pharmaceuticals, Inc. | Compounds and methods for detecting oligonucleotides |
| ES2797679T3 (en) | 2013-12-02 | 2020-12-03 | Ionis Pharmaceuticals Inc | Antisense compounds and their uses |
| CA2844640A1 (en) | 2013-12-06 | 2015-06-06 | The University Of British Columbia | Method for treatment of castration-resistant prostate cancer |
| IL282401B (en) | 2013-12-12 | 2022-08-01 | Alnylam Pharmaceuticals Inc | Complement component irna compositions and methods of use thereof |
| CN111729090A (en) | 2013-12-20 | 2020-10-02 | 通用医疗公司 | Methods and assays relating to circulating tumor cells |
| NZ720286A (en) | 2013-12-24 | 2022-12-23 | Ionis Pharmaceuticals Inc | Modulation of angiopoietin-like 3 expression |
| JPWO2015108046A1 (en) | 2014-01-15 | 2017-03-23 | 株式会社新日本科学 | Chiral nucleic acid adjuvant and antiallergic agent having antiallergic action |
| WO2015123264A1 (en) | 2014-02-11 | 2015-08-20 | Alnylam Pharmaceuticals, Inc. | Ketohexokinase (khk) irna compositions and methods of use thereof |
| EP3119789B1 (en) | 2014-03-17 | 2020-04-22 | Ionis Pharmaceuticals, Inc. | Bicyclic carbocyclic nucleosides and oligomeric compounds prepared therefrom |
| RU2019130898A (en) | 2014-03-19 | 2019-11-11 | Ионис Фармасьютикалз, Инк. | COMPOSITIONS FOR MODULATION OF ATAXIN 2 EXPRESSION |
| US10006027B2 (en) | 2014-03-19 | 2018-06-26 | Ionis Pharmaceuticals, Inc. | Methods for modulating Ataxin 2 expression |
| RU2704619C2 (en) | 2014-04-01 | 2019-10-30 | Биоген Ма Инк. | Compositions for modulating expression of sod-1 |
| EP4162940A1 (en) | 2014-04-17 | 2023-04-12 | Biogen MA Inc. | Compositions and methods for modulation of smn2 splicing in a subject |
| DK3137476T3 (en) | 2014-04-28 | 2019-11-18 | Ionis Pharmaceuticals Inc | LINKER-MODIFIED OLIGOMER COMPOUNDS |
| EP3811977A1 (en) | 2014-05-01 | 2021-04-28 | Ionis Pharmaceuticals, Inc. | Method for synthesis of reactive conjugate clusters |
| WO2015175510A1 (en) | 2014-05-12 | 2015-11-19 | Alnylam Pharmaceuticals, Inc. | Methods and compositions for treating a serpinc1-associated disorder |
| GB201408623D0 (en) | 2014-05-15 | 2014-07-02 | Santaris Pharma As | Oligomers and oligomer conjugates |
| US10570169B2 (en) | 2014-05-22 | 2020-02-25 | Ionis Pharmaceuticals, Inc. | Conjugated antisense compounds and their use |
| EA201692370A1 (en) | 2014-05-22 | 2017-03-31 | Элнилэм Фармасьютикалз, Инк. | COMPOSITIONS of mRNA ANGIOTENZINOGENA (AGT) AND METHODS OF THEIR USE |
| BR112016028211A2 (en) | 2014-06-02 | 2017-10-24 | Childrens Medical Center | methods and compositions for immunomodulation |
| USD766120S1 (en) | 2014-06-09 | 2016-09-13 | Gambro Lundia Ab | Status light bar |
| GB201410693D0 (en) | 2014-06-16 | 2014-07-30 | Univ Southampton | Splicing modulation |
| CA2955250A1 (en) | 2014-07-16 | 2016-01-21 | Moderna Therapeutics, Inc. | Chimeric polynucleotides |
| WO2016014846A1 (en) | 2014-07-23 | 2016-01-28 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of intrabodies |
| CN106559995A (en) | 2014-08-07 | 2017-04-05 | 莱古路斯治疗法股份有限公司 | The Microrna of targeting metabolic disorder |
| WO2016024205A1 (en) | 2014-08-15 | 2016-02-18 | Pfizer Inc. | Oligomers targeting hexanucleotide repeat expansion in human c9orf72 gene |
| CN107106704A (en) | 2014-08-29 | 2017-08-29 | 儿童医疗中心有限公司 | Method and composition for treating cancer |
| EP3191591A1 (en) | 2014-09-12 | 2017-07-19 | Alnylam Pharmaceuticals, Inc. | Polynucleotide agents targeting complement component c5 and methods of use thereof |
| KR102620328B1 (en) | 2014-10-03 | 2024-01-02 | 콜드스프링하버러보러토리 | Targeted augmentation of nuclear gene output |
| JOP20200115A1 (en) | 2014-10-10 | 2017-06-16 | Alnylam Pharmaceuticals Inc | Compositions And Methods For Inhibition Of HAO1 (Hydroxyacid Oxidase 1 (Glycolate Oxidase)) Gene Expression |
| CN106795200B (en) | 2014-10-10 | 2020-06-19 | 豪夫迈·罗氏有限公司 | Galnac phosphoramidites, nucleic acid conjugates thereof, and uses thereof |
| EP3207138B1 (en) | 2014-10-17 | 2020-07-15 | Alnylam Pharmaceuticals, Inc. | Polynucleotide agents targeting aminolevulinic acid synthase-1 (alas1) and uses thereof |
| EP3212794B1 (en) | 2014-10-30 | 2021-04-07 | Genzyme Corporation | Polynucleotide agents targeting serpinc1 (at3) and methods of use thereof |
| US9816080B2 (en) | 2014-10-31 | 2017-11-14 | President And Fellows Of Harvard College | Delivery of CAS9 via ARRDC1-mediated microvesicles (ARMMs) |
| JOP20200092A1 (en) | 2014-11-10 | 2017-06-16 | Alnylam Pharmaceuticals Inc | HEPATITIS B VIRUS (HBV) iRNA COMPOSITIONS AND METHODS OF USE THEREOF |
| WO2016077540A1 (en) | 2014-11-12 | 2016-05-19 | Ionis Pharmaceuticals, Inc. | Compounds and methods for the modulation of comp |
| US10364433B2 (en) | 2014-11-14 | 2019-07-30 | The Regents Of The University Of California | Modulation of AGPAT5 expression |
| EP3020813A1 (en) | 2014-11-16 | 2016-05-18 | Neurovision Pharma GmbH | Antisense-oligonucleotides as inhibitors of TGF-R signaling |
| CN107250362B (en) | 2014-11-17 | 2021-10-22 | 阿尔尼拉姆医药品有限公司 | Apolipoprotein C3(APOC3) iRNA compositions and methods of use thereof |
| WO2016086104A1 (en) | 2014-11-25 | 2016-06-02 | Ionis Pharmaceuticals, Inc. | Modulation of ube3a-ats expression |
| EP3229842B1 (en) | 2014-12-08 | 2022-07-06 | The Board of Regents of The University of Texas System | Lipocationic polymers and uses thereof |
| WO2016096938A1 (en) | 2014-12-16 | 2016-06-23 | Roche Innovation Center Copenhagen A/S | Chiral toxicity screening method |
| US9688707B2 (en) | 2014-12-30 | 2017-06-27 | Ionis Pharmaceuticals, Inc. | Bicyclic morpholino compounds and oligomeric compounds prepared therefrom |
| WO2016112132A1 (en) | 2015-01-06 | 2016-07-14 | Ionis Pharmaceuticals, Inc. | Compositions for modulating expression of c9orf72 antisense transcript |
| WO2016115490A1 (en) | 2015-01-16 | 2016-07-21 | Ionis Pharmaceuticals, Inc. | Compounds and methods for modulation of dux4 |
| US11761951B2 (en) | 2015-02-04 | 2023-09-19 | Bristol-Myers Squibb Company | Methods of selecting therapeutic molecules |
| DK3253875T3 (en) | 2015-02-04 | 2020-04-14 | H Hoffmann La Roche Ag | Tau antisense oligomers and uses thereof |
| EP3256591A4 (en) | 2015-02-13 | 2018-08-08 | Translate Bio Ma, Inc. | Hybrid oligonucleotides and uses thereof |
| CA2976445A1 (en) | 2015-02-13 | 2016-08-18 | Alnylam Pharmaceuticals, Inc. | Patatin-like phospholipase domain containing 3 (pnpla3) irna compositions and methods of use thereof |
| WO2016138353A1 (en) | 2015-02-26 | 2016-09-01 | Ionis Pharmaceuticals, Inc. | Allele specific modulators of p23h rhodopsin |
| WO2016141236A1 (en) | 2015-03-03 | 2016-09-09 | Ionis Pharmaceuticals, Inc. | Compositions for modulating mecp2 expression |
| US11129844B2 (en) | 2015-03-03 | 2021-09-28 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating MECP2 expression |
| WO2016141145A1 (en) | 2015-03-03 | 2016-09-09 | Ionis Pharmaceuticals, Inc. | Methods for modulating mecp2 expression |
| EP3273974A4 (en) | 2015-03-26 | 2018-11-07 | Women and Infants Hospital of Rhode Island Inc. | Therapy for malignant disease |
| MX2017012426A (en) | 2015-04-03 | 2018-01-26 | Ionis Pharmaceuticals Inc | Compounds and methods for modulating tmprss6 expression. |
| WO2016164746A1 (en) | 2015-04-08 | 2016-10-13 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for inhibiting expression of the lect2 gene |
| WO2016167780A1 (en) | 2015-04-16 | 2016-10-20 | Ionis Pharmaceuticals, Inc. | Compositions for modulating expression of c9orf72 antisense transcript |
| RS60230B1 (en) | 2015-04-16 | 2020-06-30 | Ionis Pharmaceuticals Inc | Compositions for modulating c9orf72 expression |
| EP3307316A1 (en) | 2015-06-12 | 2018-04-18 | Alnylam Pharmaceuticals, Inc. | Complement component c5 irna compositions and methods of use thereof |
| WO2016205323A1 (en) | 2015-06-18 | 2016-12-22 | Alnylam Pharmaceuticals, Inc. | Polynucleotde agents targeting hydroxyacid oxidase (glycolate oxidase, hao1) and methods of use thereof |
| WO2016205681A1 (en) | 2015-06-19 | 2016-12-22 | University Of Rochester | Septin proteins as novel biomarkers for detection and treatment of müllerian cancers |
| WO2016209862A1 (en) | 2015-06-23 | 2016-12-29 | Alnylam Pharmaceuticals, Inc. | Glucokinase (gck) irna compositions and methods of use thereof |
| EP3313989A4 (en) | 2015-06-29 | 2018-12-05 | Ionis Pharmaceuticals, Inc. | Modified crispr rna and modified single crispr rna and uses thereof |
| MY192997A (en) | 2015-07-10 | 2022-09-20 | Ionis Pharmaceuticals Inc | Modulators of diacyglycerol acyltransferase 2 (dgat2) |
| US10494632B2 (en) | 2015-07-10 | 2019-12-03 | Alnylam Pharmaceuticals, Inc. | Insulin-like growth factor binding protein, acid labile subunit (IGFALS) compositions and methods of use thereof |
| WO2017024111A1 (en) | 2015-08-04 | 2017-02-09 | The University Of Chicago | Inhibitors of cacna1a/alpha1a subunit internal ribosomal entry site (ires) and methods of treating spinocerebellar ataxia type 6 |
| CN114525280A (en) | 2015-09-02 | 2022-05-24 | 阿尔尼拉姆医药品有限公司 | iRNA compositions of programmed cell death 1 ligand 1(PD-L1) and methods of use thereof |
| CN108271360B (en) | 2015-09-14 | 2023-01-24 | 得克萨斯州大学系统董事会 | Lipophilic cationic dendritic polymer and use thereof |
| CA2999177A1 (en) | 2015-09-24 | 2017-03-30 | The Regents Of The University Of California | Synthetic sphingolipid-like molecules, drugs, methods of their synthesis and methods of treatment |
| RU2018113709A (en) | 2015-09-24 | 2019-10-30 | Айонис Фармасьютикалз, Инк. | KRAS EXPRESSION MODULATORS |
| WO2017053781A1 (en) | 2015-09-25 | 2017-03-30 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating ataxin 3 expression |
| CA2998898A1 (en) | 2015-10-08 | 2017-04-13 | Ionis Pharmaceuticals, Inc. | Compounds and methods for modulating angiotensinogen expression |
| KR102422625B1 (en) | 2015-10-09 | 2022-07-20 | 유니버시티 오브 사우스앰톤 | Regulation of gene expression and screening of deregulated protein expression |
| US11078528B2 (en) | 2015-10-12 | 2021-08-03 | Advanced Cell Diagnostics, Inc. | In situ detection of nucleotide variants in high noise samples, and compositions and methods related thereto |
| US10955407B2 (en) | 2015-10-22 | 2021-03-23 | Roche Innovation Center Copenhagen A/S | In vitro toxicity screening assay |
| US11260073B2 (en) | 2015-11-02 | 2022-03-01 | Ionis Pharmaceuticals, Inc. | Compounds and methods for modulating C90RF72 |
| US20190046555A1 (en) | 2015-11-06 | 2019-02-14 | Ionis Pharmaceuticals, Inc. | Conjugated antisense compounds for use in therapy |
| WO2017096395A1 (en) | 2015-12-04 | 2017-06-08 | Ionis Pharmaceuticals, Inc. | Methods of treating breast cancer |
| SG11201804443UA (en) | 2015-12-14 | 2018-06-28 | Cold Spring Harbor Laboratory | Antisense oligomers for treatment of autosomal dominant mental retardation-5 and dravet syndrome |
| US11096956B2 (en) | 2015-12-14 | 2021-08-24 | Stoke Therapeutics, Inc. | Antisense oligomers and uses thereof |
| WO2017111137A1 (en) * | 2015-12-22 | 2017-06-29 | 味の素株式会社 | Oligonucleotide manufacturing method |
| AU2017205462A1 (en) | 2016-01-05 | 2018-06-07 | Ionis Pharmaceuticals, Inc. | Methods for reducing LRRK2 expression |
| CN108779488B (en) | 2016-02-26 | 2022-01-21 | 小利兰·斯坦福大学托管委员会 | Multiplex single-molecule RNA visualization using a dual-probe proximity ligation system |
| CA3016592A1 (en) | 2016-03-04 | 2017-09-08 | Rhode Island Hospital | Targeting microrna for cancer treatment |
| US11136577B2 (en) | 2016-03-09 | 2021-10-05 | Ionis Pharmaceuticals, Inc. | Methods and compositions for inhibiting PMP22 expression |
| AU2017234678A1 (en) | 2016-03-16 | 2018-08-16 | Ionis Pharmaceuticals, Inc. | Methods of modulating KEAP1 |
| WO2017161168A1 (en) | 2016-03-16 | 2017-09-21 | Ionis Pharmaceuticals, Inc. | Modulation of dyrk1b expression |
| ES2933435T3 (en) | 2016-04-13 | 2023-02-08 | Ionis Pharmaceuticals Inc | Methods to reduce the expression of C9ORF72 |
| MA45295A (en) | 2016-04-19 | 2019-02-27 | Alnylam Pharmaceuticals Inc | HIGH DENSITY LIPOPROTEIN BINDING PROTEIN (HDLBP / VIGILINE) RNA COMPOSITION AND METHODS FOR USING THEM |
| CN109414408B (en) | 2016-05-16 | 2022-03-29 | 得克萨斯州大学系统董事会 | Cationic sulfonamide amino lipids and amphiphilic zwitterionic amino lipids |
| JP6876383B2 (en) * | 2016-06-07 | 2021-05-26 | 富士通オプティカルコンポーネンツ株式会社 | Tunable light source |
| US20190256845A1 (en) | 2016-06-10 | 2019-08-22 | Alnylam Pharmaceuticals, Inc. | COMPLEMENT COMPONENT C5 iRNA COMPOSITIONS AND METHODS OF USE THEREOF FOR TREATING PAROXYSMAL NOCTURNAL HEMOGLOBINURIA (PNH) |
| BR112018075667B1 (en) | 2016-06-14 | 2024-02-20 | Biogen Ma, Inc | Methods for separating a target oligonucleotide from a mixture containing the target oligonucleotide and a product-related impurity |
| EP3471781A4 (en) | 2016-06-17 | 2020-05-06 | Ionis Pharmaceuticals, Inc. | Modulation of gys1 expression |
| EP3472348B1 (en) | 2016-06-17 | 2022-06-29 | F. Hoffmann-La Roche AG | In vitro nephrotoxicity screening assay |
| CN109312403B (en) | 2016-06-17 | 2023-06-27 | 豪夫迈·罗氏有限公司 | In vitro nephrotoxicity screening assay |
| EP3507367A4 (en) | 2016-07-05 | 2020-03-25 | Aduro BioTech, Inc. | Locked nucleic acid cyclic dinucleotide compounds and uses thereof |
| US11253601B2 (en) | 2016-07-11 | 2022-02-22 | Translate Bio Ma, Inc. | Nucleic acid conjugates and uses thereof |
| RS63928B1 (en) | 2016-07-15 | 2023-02-28 | Ionis Pharmaceuticals Inc | Compounds and methods for modulation of smn2 |
| JOP20190065A1 (en) | 2016-09-29 | 2019-03-28 | Ionis Pharmaceuticals Inc | Compounds and methods for reducing tau expression |
| WO2018067546A1 (en) | 2016-10-03 | 2018-04-12 | President And Fellows Of Harvard College | Delivery of therapeutic rnas via arrdc1-mediated microvesicles |
| JOP20190104A1 (en) | 2016-11-10 | 2019-05-07 | Ionis Pharmaceuticals Inc | Compounds and methods for reducing atxn3 expression |
| TWI788312B (en) | 2016-11-23 | 2023-01-01 | 美商阿尼拉製藥公司 | SERPINA1 iRNA COMPOSITIONS AND METHODS OF USE THEREOF |
| EP3548005A4 (en) | 2016-11-29 | 2020-06-17 | Puretech Health LLC | Exosomes for delivery of therapeutic agents |
| US11033570B2 (en) | 2016-12-02 | 2021-06-15 | Cold Spring Harbor Laboratory | Modulation of Lnc05 expression |
| KR20230166146A (en) | 2016-12-16 | 2023-12-06 | 알닐람 파마슈티칼스 인코포레이티드 | Methods for treating or preventing ttr-associated diseases using transthyretin(ttr) irna compositions |
| US11180756B2 (en) | 2017-03-09 | 2021-11-23 | Ionis Pharmaceuticals | Morpholino modified oligomeric compounds |
| JOP20190215A1 (en) | 2017-03-24 | 2019-09-19 | Ionis Pharmaceuticals Inc | Modulators of pcsk9 expression |
| AU2018254437A1 (en) | 2017-04-18 | 2019-11-28 | Alnylam Pharmaceuticals, Inc. | Methods for the treatment of subjects having a hepatitis B virus (HBV) infection |
| WO2018226788A1 (en) | 2017-06-07 | 2018-12-13 | University Of Massachusetts | Anti-adam33 oligonucleotides and related methods |
| WO2019014530A1 (en) | 2017-07-13 | 2019-01-17 | Alnylam Pharmaceuticals Inc. | Lactate dehydrogenase a (ldha) irna compositions and methods of use thereof |
| WO2019036613A1 (en) | 2017-08-18 | 2019-02-21 | Ionis Pharmaceuticals, Inc. | Modulation of the notch signaling pathway for treatment of respiratory disorders |
| PL3673080T3 (en) | 2017-08-25 | 2024-03-11 | Stoke Therapeutics, Inc. | Antisense oligomers for treatment of conditions and diseases |
| US10517889B2 (en) | 2017-09-08 | 2019-12-31 | Ionis Pharmaceuticals, Inc. | Modulators of SMAD7 expression |
| WO2019089922A1 (en) | 2017-11-01 | 2019-05-09 | Alnylam Pharmaceuticals, Inc. | Complement component c3 irna compositions and methods of use thereof |
| TWI809004B (en) | 2017-11-09 | 2023-07-21 | 美商Ionis製藥公司 | Compounds and methods for reducing snca expression |
| US20200385719A1 (en) | 2017-11-16 | 2020-12-10 | Alnylam Pharmaceuticals, Inc. | Kisspeptin 1 (kiss1) irna compositions and methods of use thereof |
| EP3714054A1 (en) | 2017-11-20 | 2020-09-30 | Alnylam Pharmaceuticals, Inc. | Serum amyloid p component (apcs) irna compositions and methods of use thereof |
| WO2019118916A1 (en) | 2017-12-14 | 2019-06-20 | Ionis Pharmaceuticals, Inc. | Conjugated antisense compounds and their use |
| AU2018388484A1 (en) | 2017-12-18 | 2020-07-30 | Alnylam Pharmaceuticals, Inc. | High mobility group box-1 (HMGB1) iRNA compositions and methods of use thereof |
| WO2019126641A2 (en) | 2017-12-21 | 2019-06-27 | Ionis Pharmaceuticals, Inc. | Modulation of frataxin expression |
| MX2020007369A (en) | 2018-01-15 | 2020-10-28 | Ionis Pharmaceuticals Inc | Modulators of dnm2 expression. |
| JP7317029B2 (en) | 2018-02-12 | 2023-07-28 | アイオーニス ファーマシューティカルズ, インコーポレーテッド | Modified compounds and uses thereof |
| TW202000199A (en) | 2018-03-02 | 2020-01-01 | 美商Ionis製藥公司 | Modulators of IRF4 expression |
| US11732260B2 (en) | 2018-03-02 | 2023-08-22 | Ionis Pharmaceuticals, Inc. | Compounds and methods for the modulation of amyloid-β precursor protein |
| US11661601B2 (en) | 2018-03-22 | 2023-05-30 | Ionis Pharmaceuticals, Inc. | Methods for modulating FMR1 expression |
| CN112292457A (en) | 2018-04-09 | 2021-01-29 | 领先细胞医疗诊断有限公司 | Method for further enhancing signal amplification for in situ detection of nucleic acids |
| SG11202008660TA (en) | 2018-04-11 | 2020-10-29 | Ionis Pharmaceuticals Inc | Modulators of ezh2 expression |
| CR20200604A (en) | 2018-05-09 | 2021-02-09 | Ionis Pharmaceuticals Inc | Compounds and methods for reducing atxn3 expression |
| BR112020020957B1 (en) | 2018-05-09 | 2022-05-10 | Ionis Pharmaceuticals, Inc | Oligomeric compounds, population and pharmaceutical composition thereof and their uses |
| TW202016304A (en) | 2018-05-14 | 2020-05-01 | 美商阿尼拉製藥公司 | Angiotensinogen (agt) irna compositions and methods of use thereof |
| US11833168B2 (en) | 2018-06-14 | 2023-12-05 | Ionis Pharmaceuticals, Inc. | Compounds and methods for increasing STMN2 expression |
| US11332746B1 (en) | 2018-06-27 | 2022-05-17 | Ionis Pharmaceuticals, Inc. | Compounds and methods for reducing LRRK2 expression |
| MX2021000922A (en) | 2018-07-25 | 2021-03-31 | Ionis Pharmaceuticals Inc | Compounds and methods for reducing atxn2 expression. |
| BR112021001613A2 (en) | 2018-08-13 | 2021-05-04 | Alnylam Pharmaceuticals, Inc. | double-stranded ribonucleic acid agents, cell, pharmaceutical compositions, methods of inhibiting gene expression, inhibiting replication and treating a subject, methods for reducing the level of an antigen and for reducing viral load, and use of an agent dsrna |
| KR20210043634A (en) | 2018-08-15 | 2021-04-21 | 일루미나, 인코포레이티드 | Compositions and methods for improving library enrichment |
| US20210348162A1 (en) | 2018-08-16 | 2021-11-11 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for inhibiting expression of the lect2 gene |
| AU2019325255A1 (en) | 2018-08-20 | 2021-04-15 | Rogcon, Inc. | Antisense oligonucleotides targeting SCN2A for the treatment of SCN1A encephalopathies |
| EP3620520A1 (en) | 2018-09-10 | 2020-03-11 | Universidad del Pais Vasco | Novel target to treat a metabolic disease in an individual |
| US20210332367A1 (en) | 2018-09-18 | 2021-10-28 | Alnylam Pharmaceuticals, Inc. | KETOHEXOKINASE (KHK) iRNA COMPOSITIONS AND METHODS OF USE THEREOF |
| TW202023573A (en) | 2018-09-19 | 2020-07-01 | 美商Ionis製藥公司 | Modulators of pnpla3 expression |
| US10913951B2 (en) | 2018-10-31 | 2021-02-09 | University of Pittsburgh—of the Commonwealth System of Higher Education | Silencing of HNF4A-P2 isoforms with siRNA to improve hepatocyte function in liver failure |
| MX2021005357A (en) * | 2018-11-08 | 2021-06-30 | Aligos Therapeutics Inc | S-antigen transport inhibiting oligonucleotide polymers and methods. |
| TW202028222A (en) | 2018-11-14 | 2020-08-01 | 美商Ionis製藥公司 | Modulators of foxp3 expression |
| BR112021008967A2 (en) | 2018-11-15 | 2021-08-17 | Ionis Pharmaceuticals, Inc. | irf5 expression modulators |
| TW202039841A (en) | 2018-11-21 | 2020-11-01 | 美商Ionis製藥公司 | Compounds and methods for reducing prion expression |
| WO2020132521A1 (en) | 2018-12-20 | 2020-06-25 | Praxis Precision Medicines, Inc. | Compositions and methods for the treatment of kcnt1 related disorders |
| EP4285929A3 (en) | 2018-12-20 | 2024-03-06 | Humabs Biomed SA | Combination hbv therapy |
| SG11202107669WA (en) | 2019-01-16 | 2021-08-30 | Genzyme Corp | Serpinc1 irna compositions and methods of use thereof |
| JP2022519532A (en) | 2019-01-31 | 2022-03-24 | アイオーニス ファーマシューティカルズ, インコーポレーテッド | Modulator of YAP1 expression |
| CN114555825A (en) | 2019-02-15 | 2022-05-27 | 领先细胞医疗诊断有限公司 | Method for multiplexed detection of nucleic acids by in situ hybridization |
| AU2020227824A1 (en) | 2019-02-27 | 2021-08-26 | Ionis Pharmaceuticals, Inc. | Modulators of MALAT1 expression |
| WO2020205463A1 (en) | 2019-03-29 | 2020-10-08 | Ionis Pharmaceuticals, Inc. | Compounds and methods for modulating ube3a-ats |
| SG11202110674XA (en) | 2019-03-29 | 2021-10-28 | Mitsubishi Tanabe Pharma Corp | Compound, method and pharmaceutical composition for modulating expression of dux4 |
| KR20220004675A (en) | 2019-05-03 | 2022-01-11 | 다이서나 파마수이티컬, 인크. | Double-Stranded Nucleic Acid Inhibitor Molecules with a Single Sense Strand |
| WO2020227395A2 (en) | 2019-05-06 | 2020-11-12 | University Of Massachusetts | Anti-c9orf72 oligonucleotides and related methods |
| BR112021022806A2 (en) | 2019-05-13 | 2022-01-25 | Vir Biotechnology Inc | Method for treating chronic hbv infection, sirna, uses of a sirna, and of a sirna and peg-ifna and of a sirna, peg-ifna and an nrti, method, composition for use or use and kit |
| EP3956450A4 (en) | 2019-07-26 | 2022-11-16 | Ionis Pharmaceuticals, Inc. | Compounds and methods for modulating gfap |
| WO2021022109A1 (en) | 2019-08-01 | 2021-02-04 | Alnylam Pharmaceuticals, Inc. | SERPIN FAMILY F MEMBER 2 (SERPINF2) iRNA COMPOSITIONS AND METHODS OF USE THEREOF |
| WO2021022108A2 (en) | 2019-08-01 | 2021-02-04 | Alnylam Pharmaceuticals, Inc. | CARBOXYPEPTIDASE B2 (CPB2) iRNA COMPOSITIONS AND METHODS OF USE THEREOF |
| WO2021030522A1 (en) | 2019-08-13 | 2021-02-18 | Alnylam Pharmaceuticals, Inc. | SMALL RIBOSOMAL PROTEIN SUBUNIT 25 (RPS25) iRNA AGENT COMPOSITIONS AND METHODS OF USE THEREOF |
| EP4013767A4 (en) | 2019-08-15 | 2023-10-25 | Ionis Pharmaceuticals, Inc. | Linkage modified oligomeric compounds and uses thereof |
| EP4025694A1 (en) | 2019-09-03 | 2022-07-13 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for inhibiting expression of the lect2 gene |
| US20220389429A1 (en) | 2019-10-04 | 2022-12-08 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for silencing ugt1a1 gene expression |
| JP2022552249A (en) | 2019-10-14 | 2022-12-15 | アストラゼネカ・アクチエボラーグ | Modulators of PNPLA3 expression |
| WO2021076828A1 (en) | 2019-10-18 | 2021-04-22 | Alnylam Pharmaceuticals, Inc. | Solute carrier family member irna compositions and methods of use thereof |
| AU2020369515A1 (en) | 2019-10-22 | 2022-04-21 | Alnylam Pharmaceuticals, Inc. | Complement component C3 iRNA compositions and methods of use thereof |
| TW202132567A (en) | 2019-11-01 | 2021-09-01 | 美商阿尼拉製藥公司 | Huntingtin (htt) irna agent compositions and methods of use thereof |
| EP4051796A1 (en) | 2019-11-01 | 2022-09-07 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for silencing dnajb1-prkaca fusion gene expression |
| WO2021096763A1 (en) | 2019-11-13 | 2021-05-20 | Alnylam Pharmaceuticals, Inc. | Methods and compositions for treating an angiotensinogen- (agt-) associated disorder |
| US20230056569A1 (en) | 2019-11-22 | 2023-02-23 | Alnylam Pharmaceuticals, Inc. | Ataxin3 (atxn3) rnai agent compositions and methods of use thereof |
| MX2022006433A (en) | 2019-12-13 | 2022-06-23 | Alnylam Pharmaceuticals Inc | Human chromosome 9 open reading frame 72 (c9orf72) irna agent compositions and methods of use thereof. |
| WO2021126734A1 (en) | 2019-12-16 | 2021-06-24 | Alnylam Pharmaceuticals, Inc. | Patatin-like phospholipase domain containing 3 (pnpla3) irna compositions and methods of use thereof |
| WO2021154941A1 (en) | 2020-01-31 | 2021-08-05 | Alnylam Pharmaceuticals, Inc. | Complement component c5 irna compositions for use in the treatment of amyotrophic lateral sclerosis (als) |
| CN115427571A (en) | 2020-02-10 | 2022-12-02 | 阿尔尼拉姆医药品有限公司 | Compositions and methods for silencing VEGF-A expression |
| MX2022010052A (en) | 2020-02-18 | 2022-09-05 | Alnylam Pharmaceuticals Inc | Apolipoprotein c3 (apoc3) irna compositions and methods of use thereof. |
| TW202140787A (en) | 2020-02-28 | 2021-11-01 | 美商Ionis製藥公司 | Compounds and methods for modulating smn2 |
| JPWO2021177267A1 (en) | 2020-03-02 | 2021-09-10 | ||
| EP4114947A1 (en) | 2020-03-05 | 2023-01-11 | Alnylam Pharmaceuticals, Inc. | Complement component c3 irna compositions and methods of use thereof for treating or preventing complement component c3-associated diseases |
| CA3174725A1 (en) | 2020-03-06 | 2021-09-10 | Alnylam Pharmaceuticals, Inc. | Ketohexokinase (khk) irna compositions and methods of use thereof |
| KR20220152270A (en) | 2020-03-06 | 2022-11-15 | 알닐람 파마슈티칼스 인코포레이티드 | Compositions and methods for inhibiting the expression of transthyretin (TTR) |
| EP4121534A1 (en) | 2020-03-18 | 2023-01-25 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for treating subjects having a heterozygous alanine-glyoxylate aminotransferase gene (agxt) variant |
| WO2021195307A1 (en) | 2020-03-26 | 2021-09-30 | Alnylam Pharmaceuticals, Inc. | Coronavirus irna compositions and methods of use thereof |
| US20230190785A1 (en) | 2020-03-30 | 2023-06-22 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for silencing dnajc15 gene expression |
| US20230295622A1 (en) | 2020-04-06 | 2023-09-21 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for silencing myoc expression |
| EP4133077A1 (en) | 2020-04-07 | 2023-02-15 | Alnylam Pharmaceuticals, Inc. | Transmembrane serine protease 2 (tmprss2) irna compositions and methods of use thereof |
| CN116134135A (en) | 2020-04-07 | 2023-05-16 | 阿尔尼拉姆医药品有限公司 | Compositions and methods for silencing SCN9A expression |
| WO2021206917A1 (en) | 2020-04-07 | 2021-10-14 | Alnylam Pharmaceuticals, Inc. | ANGIOTENSIN-CONVERTING ENZYME 2 (ACE2) iRNA COMPOSITIONS AND METHODS OF USE THEREOF |
| BR112022021813A2 (en) | 2020-04-27 | 2023-01-17 | Alnylam Pharmaceuticals Inc | APOLIPOPROTEIN AND (APOE) IRNA AGENT COMPOSITIONS AND METHODS OF USE THEREOF |
| IL297680A (en) | 2020-04-30 | 2022-12-01 | Alnylam Pharmaceuticals Inc | Complement factor b (cfb) irna compositions and methods of use thereof |
| EP4143321A2 (en) | 2020-05-01 | 2023-03-08 | Ionis Pharmaceuticals, Inc. | Compounds and methods for modulating atxn1 |
| WO2021231107A1 (en) | 2020-05-11 | 2021-11-18 | Stoke Therapeutics, Inc. | Opa1 antisense oligomers for treatment of conditions and diseases |
| CN115667513A (en) | 2020-05-12 | 2023-01-31 | 田边三菱制药株式会社 | Compounds, methods and pharmaceutical compositions for modulating Ataxin 3 expression |
| EP4150088A1 (en) | 2020-05-15 | 2023-03-22 | Korro Bio, Inc. | Methods and compositions for the adar-mediated editing of argininosuccinate synthetase (ass1) |
| EP4150078A1 (en) | 2020-05-15 | 2023-03-22 | Korro Bio, Inc. | Methods and compositions for the adar-mediated editing of argininosuccinate lyase (asl) |
| WO2021231691A1 (en) | 2020-05-15 | 2021-11-18 | Korro Bio, Inc. | Methods and compositions for the adar-mediated editing of retinoschisin 1 (rsi) |
| EP4150087A1 (en) | 2020-05-15 | 2023-03-22 | Korro Bio, Inc. | Methods and compositions for the adar-mediated editing of gap junction protein beta 2 (gjb2) |
| EP4150076A1 (en) | 2020-05-15 | 2023-03-22 | Korro Bio, Inc. | Methods and compositions for the adar-mediated editing of methyl-cpg binding protein 2 (mecp2) |
| EP4150090A1 (en) | 2020-05-15 | 2023-03-22 | Korro Bio, Inc. | Methods and compositions for the adar-mediated editing of otoferlin (otof) |
| WO2021231673A1 (en) | 2020-05-15 | 2021-11-18 | Korro Bio, Inc. | Methods and compositions for the adar-mediated editing of leucine rich repeat kinase 2 (lrrk2) |
| WO2021231685A1 (en) | 2020-05-15 | 2021-11-18 | Korro Bio, Inc. | Methods and compositions for the adar-mediated editing of transmembrane channel-like protein 1 (tmc1) |
| EP4153746A1 (en) | 2020-05-21 | 2023-03-29 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for inhibiting marc1 gene expression |
| AR122534A1 (en) | 2020-06-03 | 2022-09-21 | Triplet Therapeutics Inc | METHODS FOR THE TREATMENT OF NUCLEOTIDE REPEAT EXPANSION DISORDERS ASSOCIATED WITH MSH3 ACTIVITY |
| EP4162050A1 (en) | 2020-06-09 | 2023-04-12 | Alnylam Pharmaceuticals, Inc. | Rnai compositions and methods of use thereof for delivery by inhalation |
| BR112022024420A2 (en) | 2020-06-18 | 2023-01-17 | Alnylam Pharmaceuticals Inc | XANTHINE DEHYDROGENASE (XDH) IRNA COMPOSITIONS AND METHODS OF USE THEREOF |
| CA3182458A1 (en) | 2020-06-24 | 2021-12-30 | Laura ROSEN | Engineered hepatitis b virus neutralizing antibodies and uses thereof |
| JP2023532518A (en) | 2020-06-29 | 2023-07-28 | アイオーニス ファーマシューティカルズ, インコーポレーテッド | Compounds and methods for modulating PLP1 |
| EP4183416A1 (en) | 2020-07-17 | 2023-05-24 | Mitsubishi Tanabe Pharma Corporation | Agent for preventing or treating muscular disease |
| TW202227102A (en) | 2020-09-22 | 2022-07-16 | 瑞典商阿斯特捷利康公司 | Method of treating fatty liver disease |
| EP4217489A1 (en) | 2020-09-24 | 2023-08-02 | Alnylam Pharmaceuticals, Inc. | Dipeptidyl peptidase 4 (dpp4) irna compositions and methods of use thereof |
| TW202229552A (en) | 2020-10-05 | 2022-08-01 | 美商艾拉倫製藥股份有限公司 | G protein-coupled receptor 75 (gpr75) irna compositions and methods of use thereof |
| EP4232581A1 (en) | 2020-10-21 | 2023-08-30 | Alnylam Pharmaceuticals, Inc. | Methods and compositions for treating primary hyperoxaluria |
| EP4232582A1 (en) | 2020-10-23 | 2023-08-30 | Alnylam Pharmaceuticals, Inc. | Mucin 5b (muc5b) irna compositions and methods of use thereof |
| JP2023549500A (en) | 2020-11-13 | 2023-11-27 | アルナイラム ファーマシューティカルズ, インコーポレイテッド | Coagulation factor V (F5) iRNA composition and method of use thereof |
| EP4247950A1 (en) | 2020-11-18 | 2023-09-27 | Lemba BV | Umlilo antisense transcription inhibitors |
| US11447521B2 (en) | 2020-11-18 | 2022-09-20 | Ionis Pharmaceuticals, Inc. | Compounds and methods for modulating angiotensinogen expression |
| CA3202708A1 (en) | 2020-11-23 | 2022-05-27 | Alpha Anomeric Sas | Nucleic acid duplexes |
| CA3201452A1 (en) | 2020-12-01 | 2022-06-09 | Alnylam Pharmaceuticals, Inc. | Methods and compositions for inhibition of hao1 (hydroxyacid oxidase 1 (glycolate oxidase)) gene expression |
| EP4259795A1 (en) | 2020-12-08 | 2023-10-18 | Alnylam Pharmaceuticals, Inc. | Coagulation factor x (f10) irna compositions and methods of use thereof |
| GB2603454A (en) | 2020-12-09 | 2022-08-10 | Ucl Business Ltd | Novel therapeutics for the treatment of neurodegenerative disorders |
| WO2022150260A1 (en) | 2021-01-05 | 2022-07-14 | Alnylam Pharmaceuticals, Inc. | COMPLEMENT COMPONENT 9 (C9) iRNA COMPOSITIONS AND METHODS OF USE THEREOF |
| TW202305131A (en) | 2021-02-12 | 2023-02-01 | 美商艾拉倫製藥股份有限公司 | SUPEROXIDE DISMUTASE 1 (SOD1) iRNA COMPOSITIONS AND METHODS OF USE THEREOF FOR TREATING OR PREVENTING SUPEROXIDE DISMUTASE 1- (SOD1-) ASSOCIATED NEURODEGENERATIVE DISEASES |
| CN117222739A (en) | 2021-02-25 | 2023-12-12 | 阿尔尼拉姆医药品有限公司 | Prion protein (PRNP) IRNA compositions and methods of use thereof |
| WO2022182574A1 (en) | 2021-02-26 | 2022-09-01 | Alnylam Pharmaceuticals, Inc. | KETOHEXOKINASE (KHK) iRNA COMPOSITIONS AND METHODS OF USE THEREOF |
| AU2022231003A1 (en) | 2021-03-04 | 2023-09-14 | Alnylam Pharmaceuticals, Inc. | Angiopoietin-like 3 (angptl3) irna compositions and methods of use thereof |
| WO2022192519A1 (en) | 2021-03-12 | 2022-09-15 | Alnylam Pharmaceuticals, Inc. | Glycogen synthase kinase 3 alpha (gsk3a) irna compositions and methods of use thereof |
| JP2024512635A (en) | 2021-03-29 | 2024-03-19 | アルナイラム ファーマシューティカルズ, インコーポレイテッド | Huntingtin (HTT) iRNA agent composition and method of use thereof |
| WO2022212153A1 (en) | 2021-04-01 | 2022-10-06 | Alnylam Pharmaceuticals, Inc. | Proline dehydrogenase 2 (prodh2) irna compositions and methods of use thereof |
| TW202309291A (en) | 2021-04-07 | 2023-03-01 | 法商新植物Sas公司 | Compositions and methods for indoor air remediation |
| KR20240001207A (en) | 2021-04-26 | 2024-01-03 | 알닐람 파마슈티칼스 인코포레이티드 | Transmembrane protease, serine 6 (TMPRSS6) iRNA compositions and methods of using the same |
| WO2022232343A1 (en) | 2021-04-29 | 2022-11-03 | Alnylam Pharmaceuticals, Inc. | Signal transducer and activator of transcription factor 6 (stat6) irna compositions and methods of use thereof |
| EP4331606A1 (en) | 2021-04-30 | 2024-03-06 | Kyoto University | Prevention or treatment of myopathy using mir-33b inhibitor |
| WO2022245583A1 (en) | 2021-05-18 | 2022-11-24 | Alnylam Pharmaceuticals, Inc. | Sodium-glucose cotransporter-2 (sglt2) irna compositions and methods of use thereof |
| EP4341405A1 (en) | 2021-05-20 | 2024-03-27 | Korro Bio, Inc. | Methods and compositions for adar-mediated editing |
| CA3221935A1 (en) | 2021-05-29 | 2022-12-08 | 1Globe Health Institute Llc | Asymmetric short duplex dna as a novel gene silencing technology and use thereof |
| WO2022256354A1 (en) | 2021-05-29 | 2022-12-08 | 1Globe Health Institute Llc | Short duplex dna as a novel gene silencing technology and use thereof |
| WO2022256283A2 (en) | 2021-06-01 | 2022-12-08 | Korro Bio, Inc. | Methods for restoring protein function using adar |
| WO2022256395A1 (en) | 2021-06-02 | 2022-12-08 | Alnylam Pharmaceuticals, Inc. | Patatin-like phospholipase domain containing 3 (pnpla3) irna compositions and methods of use thereof |
| WO2022256290A2 (en) | 2021-06-04 | 2022-12-08 | Alnylam Pharmaceuticals, Inc. | HUMAN CHROMOSOME 9 OPEN READING FRAME 72 (C9ORF72) iRNA AGENT COMPOSITIONS AND METHODS OF USE THEREOF |
| AR126070A1 (en) | 2021-06-08 | 2023-09-06 | Alnylam Pharmaceuticals Inc | COMPOSITIONS AND METHODS FOR TREATING OR PREVENTING STARGARDT DISEASE AND/OR DISORDERS ASSOCIATED WITH RETINOL BORDER PROTEIN 4 (RBP4) |
| EP4105328A1 (en) | 2021-06-15 | 2022-12-21 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Antisense-oligonucleotides for prevention of kidney dysfunction promoted by endothelial dysfunction by ephrin-b2 suppression |
| CN117500815A (en) | 2021-06-18 | 2024-02-02 | Ionis制药公司 | Compounds and methods for reducing IFNAR1 expression |
| WO2023278410A1 (en) | 2021-06-29 | 2023-01-05 | Korro Bio, Inc. | Methods and compositions for adar-mediated editing |
| US20230194709A9 (en) | 2021-06-29 | 2023-06-22 | Seagate Technology Llc | Range information detection using coherent pulse sets with selected waveform characteristics |
| CA3225469A1 (en) | 2021-06-30 | 2023-01-05 | Alnylam Pharmaceuticals, Inc. | Methods and compositions for treating an angiotensinogen- (agt-) associated disorder |
| TW202333748A (en) | 2021-07-19 | 2023-09-01 | 美商艾拉倫製藥股份有限公司 | Methods and compositions for treating subjects having or at risk of developing a non-primary hyperoxaluria disease or disorder |
| IL309905A (en) | 2021-07-23 | 2024-03-01 | Alnylam Pharmaceuticals Inc | Beta-catenin (ctnnb1) irna compositions and methods of use thereof |
| WO2023009687A1 (en) | 2021-07-29 | 2023-02-02 | Alnylam Pharmaceuticals, Inc. | 3-hydroxy-3-methylglutaryl-coa reductase (hmgcr) irna compositions and methods of use thereof |
| IL310244A (en) | 2021-08-03 | 2024-03-01 | Alnylam Pharmaceuticals Inc | Transthyretin (ttr) irna compositions and methods of use thereof |
| CA3228255A1 (en) | 2021-08-04 | 2023-02-09 | Alnylam Pharmaceuticals, Inc. | Irna compositions and methods for silencing angiotensinogen (agt) |
| AU2022328347A1 (en) | 2021-08-13 | 2024-02-08 | Alnylam Pharmaceuticals, Inc. | Factor xii (f12) irna compositions and methods of use thereof |
| WO2023034870A2 (en) | 2021-09-01 | 2023-03-09 | Ionis Pharmaceuticals, Inc. | Compounds and methods for reducing dmpk expression |
| WO2023044370A2 (en) | 2021-09-17 | 2023-03-23 | Alnylam Pharmaceuticals, Inc. | Irna compositions and methods for silencing complement component 3 (c3) |
| CA3232420A1 (en) | 2021-09-20 | 2023-03-23 | Alnylam Pharmaceuticals, Inc. | Inhibin subunit beta e (inhbe) modulator compositions and methods of use thereof |
| WO2023056329A1 (en) | 2021-09-30 | 2023-04-06 | Akouos, Inc. | Compositions and methods for treating kcnq4-associated hearing loss |
| WO2023069603A1 (en) | 2021-10-22 | 2023-04-27 | Korro Bio, Inc. | Methods and compositions for disrupting nrf2-keap1 protein interaction by adar mediated rna editing |
| WO2023076451A1 (en) | 2021-10-29 | 2023-05-04 | Alnylam Pharmaceuticals, Inc. | Complement factor b (cfb) irna compositions and methods of use thereof |
| TW202334418A (en) | 2021-10-29 | 2023-09-01 | 美商艾拉倫製藥股份有限公司 | Huntingtin (htt) irna agent compositions and methods of use thereof |
| WO2023086295A2 (en) | 2021-11-10 | 2023-05-19 | University Of Rochester | Antisense oligonucleotides for modifying protein expression |
| WO2023086292A2 (en) | 2021-11-10 | 2023-05-19 | University Of Rochester | Gata4-targeted therapeutics for treatment of cardiac hypertrophy |
| GB202117758D0 (en) | 2021-12-09 | 2022-01-26 | Ucl Business Ltd | Therapeutics for the treatment of neurodegenerative disorders |
| WO2023127857A1 (en) * | 2021-12-27 | 2023-07-06 | 株式会社理研ジェネシス | Novel artificial nucleic acid, method for producing same, and use of same |
| WO2023141314A2 (en) | 2022-01-24 | 2023-07-27 | Alnylam Pharmaceuticals, Inc. | Heparin sulfate biosynthesis pathway enzyme irna agent compositions and methods of use thereof |
| US11879125B2 (en) | 2022-03-16 | 2024-01-23 | Empirico Inc. | GalNAc compositions for improving siRNA bioavailability |
| WO2024026474A1 (en) | 2022-07-29 | 2024-02-01 | Regeneron Pharmaceuticals, Inc. | Compositions and methods for transferrin receptor (tfr)-mediated delivery to the brain and muscle |
| WO2024040041A1 (en) | 2022-08-15 | 2024-02-22 | Dicerna Pharmaceuticals, Inc. | Regulation of activity of rnai molecules |
| WO2024039776A2 (en) | 2022-08-18 | 2024-02-22 | Alnylam Pharmaceuticals, Inc. | Universal non-targeting sirna compositions and methods of use thereof |
| WO2024050261A1 (en) | 2022-08-29 | 2024-03-07 | University Of Rochester | Antisense oligonucleotide-based anti-fibrotic therapeutics |
| WO2024059165A1 (en) | 2022-09-15 | 2024-03-21 | Alnylam Pharmaceuticals, Inc. | 17b-hydroxysteroid dehydrogenase type 13 (hsd17b13) irna compositions and methods of use thereof |
Family Cites Families (138)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3687808A (en) | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| US4500707A (en) | 1980-02-29 | 1985-02-19 | University Patents, Inc. | Nucleosides useful in the preparation of polynucleotides |
| US5132418A (en) | 1980-02-29 | 1992-07-21 | University Patents, Inc. | Process for preparing polynucleotides |
| US4458066A (en) | 1980-02-29 | 1984-07-03 | University Patents, Inc. | Process for preparing polynucleotides |
| US4469863A (en) | 1980-11-12 | 1984-09-04 | Ts O Paul O P | Nonionic nucleic acid alkyl and aryl phosphonates and processes for manufacture and use thereof |
| US4668777A (en) | 1981-03-27 | 1987-05-26 | University Patents, Inc. | Phosphoramidite nucleoside compounds |
| US4415732A (en) | 1981-03-27 | 1983-11-15 | University Patents, Inc. | Phosphoramidite compounds and processes |
| US4973679A (en) | 1981-03-27 | 1990-11-27 | University Patents, Inc. | Process for oligonucleo tide synthesis using phosphormidite intermediates |
| US5023243A (en) | 1981-10-23 | 1991-06-11 | Molecular Biosystems, Inc. | Oligonucleotide therapeutic agent and method of making same |
| US4476301A (en) | 1982-04-29 | 1984-10-09 | Centre National De La Recherche Scientifique | Oligonucleotides, a process for preparing the same and their application as mediators of the action of interferon |
| DE3329892A1 (en) | 1983-08-18 | 1985-03-07 | Köster, Hubert, Prof. Dr., 2000 Hamburg | METHOD FOR PRODUCING OLIGONUCLEOTIDES |
| USRE34069E (en) | 1983-08-18 | 1992-09-15 | Biosyntech Gmbh | Process for the preparation of oligonucleotides |
| US5118800A (en) | 1983-12-20 | 1992-06-02 | California Institute Of Technology | Oligonucleotides possessing a primary amino group in the terminal nucleotide |
| US5550111A (en) | 1984-07-11 | 1996-08-27 | Temple University-Of The Commonwealth System Of Higher Education | Dual action 2',5'-oligoadenylate antiviral derivatives and uses thereof |
| FR2567892B1 (en) | 1984-07-19 | 1989-02-17 | Centre Nat Rech Scient | NOVEL OLIGONUCLEOTIDES, THEIR PREPARATION PROCESS AND THEIR APPLICATIONS AS MEDIATORS IN DEVELOPING THE EFFECTS OF INTERFERONS |
| US5367066A (en) | 1984-10-16 | 1994-11-22 | Chiron Corporation | Oligonucleotides with selectably cleavable and/or abasic sites |
| FR2575751B1 (en) | 1985-01-08 | 1987-04-03 | Pasteur Institut | NOVEL ADENOSINE DERIVATIVE NUCLEOSIDES, THEIR PREPARATION AND THEIR BIOLOGICAL APPLICATIONS |
| US5185444A (en) | 1985-03-15 | 1993-02-09 | Anti-Gene Deveopment Group | Uncharged morpolino-based polymers having phosphorous containing chiral intersubunit linkages |
| US5166315A (en) | 1989-12-20 | 1992-11-24 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5235033A (en) | 1985-03-15 | 1993-08-10 | Anti-Gene Development Group | Alpha-morpholino ribonucleoside derivatives and polymers thereof |
| US5405938A (en) | 1989-12-20 | 1995-04-11 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5034506A (en) | 1985-03-15 | 1991-07-23 | Anti-Gene Development Group | Uncharged morpholino-based polymers having achiral intersubunit linkages |
| DE3788914T2 (en) | 1986-09-08 | 1994-08-25 | Ajinomoto Kk | Compounds for cleaving RNA at a specific position, oligomers used in the preparation of these compounds and starting materials for the synthesis of these oligomers. |
| US5264423A (en) | 1987-03-25 | 1993-11-23 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5276019A (en) | 1987-03-25 | 1994-01-04 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| DE3851889T2 (en) | 1987-06-24 | 1995-04-13 | Florey Howard Inst | NUCLEOSIDE DERIVATIVES. |
| US5188897A (en) | 1987-10-22 | 1993-02-23 | Temple University Of The Commonwealth System Of Higher Education | Encapsulated 2',5'-phosphorothioate oligoadenylates |
| US4924624A (en) | 1987-10-22 | 1990-05-15 | Temple University-Of The Commonwealth System Of Higher Education | 2,',5'-phosphorothioate oligoadenylates and plant antiviral uses thereof |
| DE3855864T2 (en) | 1987-11-30 | 1997-09-25 | Univ Iowa Res Found | DNA MOLECULES STABILIZED BY MODIFICATIONS ON THE 3'-TERMINAL PHOSPHODIESTERBINDING, THEIR USE AS NUCLEIC ACID PROBE AND AS A THERAPEUTIC AGENT FOR INHIBITING THE EXPRESSION OF SPECIFIC TARGET GENES |
| US5403711A (en) | 1987-11-30 | 1995-04-04 | University Of Iowa Research Foundation | Nucleic acid hybridization and amplification method for detection of specific sequences in which a complementary labeled nucleic acid probe is cleaved |
| JPH03503894A (en) | 1988-03-25 | 1991-08-29 | ユニバーシィティ オブ バージニア アランミ パテンツ ファウンデイション | Oligonucleotide N-alkylphosphoramidate |
| US5278302A (en) | 1988-05-26 | 1994-01-11 | University Patents, Inc. | Polynucleotide phosphorodithioates |
| US5216141A (en) | 1988-06-06 | 1993-06-01 | Benner Steven A | Oligonucleotide analogs containing sulfur linkages |
| US5175273A (en) | 1988-07-01 | 1992-12-29 | Genentech, Inc. | Nucleic acid intercalating agents |
| US5194599A (en) | 1988-09-23 | 1993-03-16 | Gilead Sciences, Inc. | Hydrogen phosphonodithioate compositions |
| JP2863922B2 (en) | 1988-11-28 | 1999-03-03 | マツダ株式会社 | Rear wheel suspension system for rear wheel steering vehicles |
| US5256775A (en) | 1989-06-05 | 1993-10-26 | Gilead Sciences, Inc. | Exonuclease-resistant oligonucleotides |
| US5134066A (en) | 1989-08-29 | 1992-07-28 | Monsanto Company | Improved probes using nucleosides containing 3-dezauracil analogs |
| US5591722A (en) | 1989-09-15 | 1997-01-07 | Southern Research Institute | 2'-deoxy-4'-thioribonucleosides and their antiviral activity |
| US5721218A (en) | 1989-10-23 | 1998-02-24 | Gilead Sciences, Inc. | Oligonucleotides with inverted polarity |
| US5399676A (en) | 1989-10-23 | 1995-03-21 | Gilead Sciences | Oligonucleotides with inverted polarity |
| EP0497875B1 (en) | 1989-10-24 | 2000-03-22 | Isis Pharmaceuticals, Inc. | 2' modified oligonucleotides |
| US5264562A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences, Inc. | Oligonucleotide analogs with novel linkages |
| US5264564A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences | Oligonucleotide analogs with novel linkages |
| US5177198A (en) | 1989-11-30 | 1993-01-05 | University Of N.C. At Chapel Hill | Process for preparing oligoribonucleoside and oligodeoxyribonucleoside boranophosphates |
| US5130302A (en) | 1989-12-20 | 1992-07-14 | Boron Bilogicals, Inc. | Boronated nucleoside, nucleotide and oligonucleotide compounds, compositions and methods for using same |
| US5459255A (en) | 1990-01-11 | 1995-10-17 | Isis Pharmaceuticals, Inc. | N-2 substituted purines |
| US5681941A (en) | 1990-01-11 | 1997-10-28 | Isis Pharmaceuticals, Inc. | Substituted purines and oligonucleotide cross-linking |
| US5587470A (en) * | 1990-01-11 | 1996-12-24 | Isis Pharmaceuticals, Inc. | 3-deazapurines |
| US5623065A (en) | 1990-08-13 | 1997-04-22 | Isis Pharmaceuticals, Inc. | Gapped 2' modified oligonucleotides |
| US5587361A (en) | 1991-10-15 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides having phosphorothioate linkages of high chiral purity |
| US5646265A (en) | 1990-01-11 | 1997-07-08 | Isis Pharmceuticals, Inc. | Process for the preparation of 2'-O-alkyl purine phosphoramidites |
| US5670633A (en) | 1990-01-11 | 1997-09-23 | Isis Pharmaceuticals, Inc. | Sugar modified oligonucleotides that detect and modulate gene expression |
| US5149797A (en) | 1990-02-15 | 1992-09-22 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of rna and production of encoded polypeptides |
| US5220007A (en) | 1990-02-15 | 1993-06-15 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of RNA and production of encoded polypeptides |
| US5321131A (en) | 1990-03-08 | 1994-06-14 | Hybridon, Inc. | Site-specific functionalization of oligodeoxynucleotides for non-radioactive labelling |
| US5470967A (en) | 1990-04-10 | 1995-11-28 | The Dupont Merck Pharmaceutical Company | Oligonucleotide analogs with sulfamate linkages |
| GB9009980D0 (en) | 1990-05-03 | 1990-06-27 | Amersham Int Plc | Phosphoramidite derivatives,their preparation and the use thereof in the incorporation of reporter groups on synthetic oligonucleotides |
| ES2116977T3 (en) | 1990-05-11 | 1998-08-01 | Microprobe Corp | SOLID SUPPORTS FOR NUCLEIC ACID HYBRIDIZATION TESTS AND METHODS TO IMMOBILIZE OLIGONUCLEOTIDES IN A COVALENT WAY. |
| US5610289A (en) | 1990-07-27 | 1997-03-11 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogues |
| DE69126530T2 (en) | 1990-07-27 | 1998-02-05 | Isis Pharmaceuticals Inc | NUCLEASE RESISTANT, PYRIMIDINE MODIFIED OLIGONUCLEOTIDES THAT DETECT AND MODULE GENE EXPRESSION |
| US5608046A (en) | 1990-07-27 | 1997-03-04 | Isis Pharmaceuticals, Inc. | Conjugated 4'-desmethyl nucleoside analog compounds |
| US5677437A (en) | 1990-07-27 | 1997-10-14 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5623070A (en) | 1990-07-27 | 1997-04-22 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5602240A (en) | 1990-07-27 | 1997-02-11 | Ciba Geigy Ag. | Backbone modified oligonucleotide analogs |
| US5223618A (en) | 1990-08-13 | 1993-06-29 | Isis Pharmaceuticals, Inc. | 4'-desmethyl nucleoside analog compounds |
| US5541307A (en) | 1990-07-27 | 1996-07-30 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs and solid phase synthesis thereof |
| US5386023A (en) | 1990-07-27 | 1995-01-31 | Isis Pharmaceuticals | Backbone modified oligonucleotide analogs and preparation thereof through reductive coupling |
| US5378825A (en) | 1990-07-27 | 1995-01-03 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs |
| US5618704A (en) | 1990-07-27 | 1997-04-08 | Isis Pharmacueticals, Inc. | Backbone-modified oligonucleotide analogs and preparation thereof through radical coupling |
| US5489677A (en) | 1990-07-27 | 1996-02-06 | Isis Pharmaceuticals, Inc. | Oligonucleoside linkages containing adjacent oxygen and nitrogen atoms |
| EP0541722B1 (en) | 1990-08-03 | 1995-12-20 | Sterling Winthrop Inc. | Compounds and methods for inhibiting gene expression |
| US5177196A (en) | 1990-08-16 | 1993-01-05 | Microprobe Corporation | Oligo (α-arabinofuranosyl nucleotides) and α-arabinofuranosyl precursors thereof |
| US5214134A (en) | 1990-09-12 | 1993-05-25 | Sterling Winthrop Inc. | Process of linking nucleosides with a siloxane bridge |
| US5561225A (en) | 1990-09-19 | 1996-10-01 | Southern Research Institute | Polynucleotide analogs containing sulfonate and sulfonamide internucleoside linkages |
| US5596086A (en) | 1990-09-20 | 1997-01-21 | Gilead Sciences, Inc. | Modified internucleoside linkages having one nitrogen and two carbon atoms |
| US5432272A (en) | 1990-10-09 | 1995-07-11 | Benner; Steven A. | Method for incorporating into a DNA or RNA oligonucleotide using nucleotides bearing heterocyclic bases |
| US5672697A (en) * | 1991-02-08 | 1997-09-30 | Gilead Sciences, Inc. | Nucleoside 5'-methylene phosphonates |
| US5571799A (en) | 1991-08-12 | 1996-11-05 | Basco, Ltd. | (2'-5') oligoadenylate analogues useful as inhibitors of host-v5.-graft response |
| DE59208572D1 (en) | 1991-10-17 | 1997-07-10 | Ciba Geigy Ag | Bicyclic nucleosides, oligonucleotides, processes for their preparation and intermediates |
| US5594121A (en) | 1991-11-07 | 1997-01-14 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified purines |
| US5484908A (en) | 1991-11-26 | 1996-01-16 | Gilead Sciences, Inc. | Oligonucleotides containing 5-propynyl pyrimidines |
| TW393513B (en) | 1991-11-26 | 2000-06-11 | Isis Pharmaceuticals Inc | Enhanced triple-helix and double-helix formation with oligomers containing modified pyrimidines |
| ATE226093T1 (en) | 1991-11-26 | 2002-11-15 | Isis Pharmaceuticals Inc | INCREASED FORMATION OF TRIPLE AND DOUBLE HELICES FROM OLIGOMERS WITH MODIFIED PYRIMIDINES |
| US5792608A (en) * | 1991-12-12 | 1998-08-11 | Gilead Sciences, Inc. | Nuclease stable and binding competent oligomers and methods for their use |
| US5359044A (en) | 1991-12-13 | 1994-10-25 | Isis Pharmaceuticals | Cyclobutyl oligonucleotide surrogates |
| US5700922A (en) | 1991-12-24 | 1997-12-23 | Isis Pharmaceuticals, Inc. | PNA-DNA-PNA chimeric macromolecules |
| FR2687679B1 (en) | 1992-02-05 | 1994-10-28 | Centre Nat Rech Scient | OLIGOTHIONUCLEOTIDES. |
| WO1993017692A1 (en) * | 1992-03-12 | 1993-09-16 | Temple University - Of The Commonwealth System Of Higher Education | Dual action 2',5'-oligoadenylate antiviral derivatives and uses thereof |
| US5633360A (en) | 1992-04-14 | 1997-05-27 | Gilead Sciences, Inc. | Oligonucleotide analogs capable of passive cell membrane permeation |
| US5434257A (en) | 1992-06-01 | 1995-07-18 | Gilead Sciences, Inc. | Binding compentent oligomers containing unsaturated 3',5' and 2',5' linkages |
| NL9300058A (en) | 1992-06-18 | 1994-01-17 | Stichting Rega V Z W | 1,5-ANHYDROHEXITOL NUCLEOSIDE ANALOGA AND PHARMACEUTICAL USE THEREOF. |
| EP0577558A2 (en) | 1992-07-01 | 1994-01-05 | Ciba-Geigy Ag | Carbocyclic nucleosides having bicyclic rings, oligonucleotides therefrom, process for their preparation, their use and intermediates |
| US5652355A (en) | 1992-07-23 | 1997-07-29 | Worcester Foundation For Experimental Biology | Hybrid oligonucleotide phosphorothioates |
| DE69316369D1 (en) | 1992-07-27 | 1998-02-19 | Hybridon Inc | Oligonukleotid alkylphosphonothiate |
| DE69400208T2 (en) | 1993-01-25 | 1996-11-28 | Hybridon Inc | OLIONUCLEOTIDALKYLPHOSPHONATES AND PHOSPHONOTHIOATES |
| US5476925A (en) | 1993-02-01 | 1995-12-19 | Northwestern University | Oligodeoxyribonucleotides including 3'-aminonucleoside-phosphoramidate linkages and terminal 3'-amino groups |
| GB9304618D0 (en) | 1993-03-06 | 1993-04-21 | Ciba Geigy Ag | Chemical compounds |
| GB9304620D0 (en) | 1993-03-06 | 1993-04-21 | Ciba Geigy Ag | Compounds |
| ATE155467T1 (en) | 1993-03-30 | 1997-08-15 | Sanofi Sa | ACYCLIC NUCLEOSIDE ANALOGUES AND OLIGONUCLEOTIDE SEQUENCES CONTAINING THEM |
| JPH08508491A (en) | 1993-03-31 | 1996-09-10 | スターリング ウインスロップ インコーポレイティド | Oligonucleotides with phosphodiester bonds replaced by amide bonds |
| DE4311944A1 (en) * | 1993-04-10 | 1994-10-13 | Degussa | Coated sodium percarbonate particles, process for their preparation and detergent, cleaning and bleaching compositions containing them |
| US5502177A (en) | 1993-09-17 | 1996-03-26 | Gilead Sciences, Inc. | Pyrimidine derivatives for labeled binding partners |
| US5457187A (en) | 1993-12-08 | 1995-10-10 | Board Of Regents University Of Nebraska | Oligonucleotides containing 5-fluorouracil |
| US5446137B1 (en) | 1993-12-09 | 1998-10-06 | Behringwerke Ag | Oligonucleotides containing 4'-substituted nucleotides |
| JP3585238B2 (en) | 1993-12-09 | 2004-11-04 | トーマス ジェファーソン ユニバーシティー | Compounds and methods for site-directed mutagenesis in eukaryotic cells |
| US5519134A (en) | 1994-01-11 | 1996-05-21 | Isis Pharmaceuticals, Inc. | Pyrrolidine-containing monomers and oligomers |
| US5596091A (en) | 1994-03-18 | 1997-01-21 | The Regents Of The University Of California | Antisense oligonucleotides comprising 5-aminoalkyl pyrimidine nucleotides |
| US5627053A (en) | 1994-03-29 | 1997-05-06 | Ribozyme Pharmaceuticals, Inc. | 2'deoxy-2'-alkylnucleotide containing nucleic acid |
| US5625050A (en) | 1994-03-31 | 1997-04-29 | Amgen Inc. | Modified oligonucleotides and intermediates useful in nucleic acid therapeutics |
| US5646269A (en) | 1994-04-28 | 1997-07-08 | Gilead Sciences, Inc. | Method for oligonucleotide analog synthesis |
| US5525711A (en) | 1994-05-18 | 1996-06-11 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Pteridine nucleotide analogs as fluorescent DNA probes |
| US5597909A (en) | 1994-08-25 | 1997-01-28 | Chiron Corporation | Polynucleotide reagents containing modified deoxyribose moieties, and associated methods of synthesis and use |
| US5792747A (en) * | 1995-01-24 | 1998-08-11 | The Administrators Of The Tulane Educational Fund | Highly potent agonists of growth hormone releasing hormone |
| US5652356A (en) | 1995-08-17 | 1997-07-29 | Hybridon, Inc. | Inverted chimeric and hybrid oligonucleotides |
| WO2005121371A2 (en) | 2004-06-03 | 2005-12-22 | Isis Pharmaceuticals, Inc. | Double strand compositions comprising differentially modified strands for use in gene modulation |
| AU4966997A (en) | 1996-11-18 | 1998-06-10 | Takeshi Imanishi | Novel nucleotide analogues |
| US6770748B2 (en) * | 1997-03-07 | 2004-08-03 | Takeshi Imanishi | Bicyclonucleoside and oligonucleotide analogue |
| JP3756313B2 (en) * | 1997-03-07 | 2006-03-15 | 武 今西 | Novel bicyclonucleosides and oligonucleotide analogues |
| EP2341057A3 (en) * | 1997-09-12 | 2011-11-23 | Exiqon A/S | Oligonucleotide Analogues |
| US6794499B2 (en) | 1997-09-12 | 2004-09-21 | Exiqon A/S | Oligonucleotide analogues |
| US6043352A (en) | 1998-08-07 | 2000-03-28 | Isis Pharmaceuticals, Inc. | 2'-O-Dimethylaminoethyloxyethyl-modified oligonucleotides |
| AU758956B2 (en) | 1999-02-12 | 2003-04-03 | Daiichi Sankyo Company, Limited | Novel nucleosides and oligonucleotide analogues |
| US6436640B1 (en) | 1999-03-18 | 2002-08-20 | Exiqon A/S | Use of LNA in mass spectrometry |
| US7084125B2 (en) | 1999-03-18 | 2006-08-01 | Exiqon A/S | Xylo-LNA analogues |
| ATE356824T1 (en) * | 1999-05-04 | 2007-04-15 | Santaris Pharma As | L-RIBO-LNA ANALOGUE |
| JP4151751B2 (en) | 1999-07-22 | 2008-09-17 | 第一三共株式会社 | New bicyclonucleoside analogues |
| US7053199B2 (en) | 2000-08-29 | 2006-05-30 | Takeshi Imanishi | Nucleoside analogs and oligonucleotide derivatives containing these analogs |
| US6426220B1 (en) | 2000-10-30 | 2002-07-30 | Isis Pharmaceuticals, Inc. | Antisense modulation of calreticulin expression |
| WO2003020739A2 (en) | 2001-09-04 | 2003-03-13 | Exiqon A/S | Novel lna compositions and uses thereof |
| JP3712374B2 (en) * | 2001-12-06 | 2005-11-02 | 富士写真フイルム株式会社 | Small magnetic disk cartridge |
| US7655456B2 (en) | 2002-01-18 | 2010-02-02 | Arkray, Inc. | Analytical device having temperature detection unit |
| US7569575B2 (en) | 2002-05-08 | 2009-08-04 | Santaris Pharma A/S | Synthesis of locked nucleic acid derivatives |
| US20040219565A1 (en) | 2002-10-21 | 2004-11-04 | Sakari Kauppinen | Oligonucleotides useful for detecting and analyzing nucleic acids of interest |
| US7456393B2 (en) | 2003-04-10 | 2008-11-25 | Ge Homeland Protection, Inc. | Device for testing surfaces of articles for traces of explosives and/or drugs |
| JP5342881B2 (en) * | 2006-01-27 | 2013-11-13 | アイシス ファーマシューティカルズ, インコーポレーテッド | 6-modified bicyclic nucleic acid analogues |
| US8935416B2 (en) | 2006-04-21 | 2015-01-13 | Fortinet, Inc. | Method, apparatus, signals and medium for enforcing compliance with a policy on a client computer |
| US9400902B2 (en) | 2012-05-22 | 2016-07-26 | Trimble Navigation Limited | Multi-modal entity tracking and display |
-
2007
- 2007-01-27 JP JP2008552604A patent/JP5342881B2/en active Active
- 2007-01-27 US US11/627,964 patent/US7399845B2/en active Active
- 2007-01-27 ES ES10176867.9T patent/ES2516815T3/en active Active
- 2007-01-27 KR KR1020137007796A patent/KR20130042043A/en not_active Application Discontinuation
- 2007-01-27 AT AT07762892T patent/ATE482965T1/en active
- 2007-01-27 AU AU2007211080A patent/AU2007211080B9/en active Active
- 2007-01-27 EP EP10179781.9A patent/EP2332951A3/en not_active Withdrawn
- 2007-01-27 CA CA2640171A patent/CA2640171C/en active Active
- 2007-01-27 PL PL10176867T patent/PL2314594T3/en unknown
- 2007-01-27 EP EP07762892A patent/EP1984381B1/en active Active
- 2007-01-27 CN CN201210112347.6A patent/CN102908630B/en active Active
- 2007-01-27 DK DK10176867.9T patent/DK2314594T3/en active
- 2007-01-27 KR KR1020087020907A patent/KR101304071B1/en active IP Right Grant
- 2007-01-27 DE DE602007009487T patent/DE602007009487D1/en active Active
- 2007-01-27 DK DK07762892.3T patent/DK1984381T3/en active
- 2007-01-27 EP EP10176867.9A patent/EP2314594B1/en active Active
- 2007-01-27 CN CN201210111246.7A patent/CN102766630B/en active Active
- 2007-01-27 WO PCT/US2007/061183 patent/WO2007090071A2/en active Application Filing
- 2007-01-27 PL PL07762892T patent/PL1984381T3/en unknown
-
2008
- 2008-05-29 US US12/129,154 patent/US7741457B2/en active Active
-
2010
- 2010-05-05 US US12/774,527 patent/US8022193B2/en active Active
-
2013
- 2013-01-31 JP JP2013017584A patent/JP2013147498A/en active Pending
Non-Patent Citations (1)
| Title |
|---|
| None |
Cited By (302)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| USRE44760E1 (en) | 2002-11-13 | 2014-02-11 | Genzyme Corporation | Antisense modulation of apolipoprotein B-expression |
| US8916694B2 (en) | 2004-05-05 | 2014-12-23 | Genzyme Corporation | SNPs of apolipoprotein B and modulation of their expression |
| US9045754B2 (en) | 2006-05-05 | 2015-06-02 | Isis Pharmaceuticals, Inc. | Short antisense compounds with gapmer configuration |
| EP2363481B1 (en) | 2006-05-05 | 2017-04-12 | Ionis Pharmaceuticals, Inc. | Compounds and methods for modulating gene expression |
| US8143230B2 (en) | 2006-05-05 | 2012-03-27 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression of PCSK9 |
| EP2527442A2 (en) * | 2006-05-05 | 2012-11-28 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating gene expression |
| EP3916095A1 (en) | 2006-10-18 | 2021-12-01 | Ionis Pharmaceuticals, Inc. | Antisense compounds |
| US9550988B2 (en) | 2006-10-18 | 2017-01-24 | Ionis Pharmaceuticals, Inc. | Antisense compounds |
| EP3202905A1 (en) | 2006-10-18 | 2017-08-09 | Ionis Pharmaceuticals, Inc. | Antisense compounds |
| EP2410054A1 (en) | 2006-10-18 | 2012-01-25 | Isis Pharmaceuticals, Inc. | Antisense compounds |
| EP2410053A1 (en) | 2006-10-18 | 2012-01-25 | Isis Pharmaceuticals, Inc. | Antisense compounds |
| US8093222B2 (en) | 2006-11-27 | 2012-01-10 | Isis Pharmaceuticals, Inc. | Methods for treating hypercholesterolemia |
| US11530410B2 (en) | 2006-11-27 | 2022-12-20 | Ionis Pharmaceuticals, Inc. | Methods for treating hypercholesterolemia |
| US8912160B2 (en) | 2006-11-27 | 2014-12-16 | Isis Pharmaceuticals, Inc. | Methods for treating hypercholesterolemia |
| US9650636B2 (en) | 2006-11-27 | 2017-05-16 | Ionis Pharmaceuticals, Inc. | Methods for treating hypercholesterolemia |
| US8084437B2 (en) | 2006-11-27 | 2011-12-27 | Isis Pharmaceuticals, Inc. | Methods for treating hypercholesterolemia |
| US8664190B2 (en) | 2006-11-27 | 2014-03-04 | Isis Pharmaceuticals, Inc. | Methods for treating hypercholesterolemia |
| US9347061B2 (en) | 2007-03-24 | 2016-05-24 | Genzyme Corporation | Administering antisense oligonucleotides complementary to human apolipoprotein B |
| EP2176280B2 (en) † | 2007-07-05 | 2015-06-24 | Isis Pharmaceuticals, Inc. | 6-disubstituted bicyclic nucleic acid analogs |
| WO2009124295A2 (en) * | 2008-04-04 | 2009-10-08 | Isis Pharmaceuticals, Inc. | Oligomeric compounds comprising bicyclic nucleosides and having reduced toxicity |
| WO2009124295A3 (en) * | 2008-04-04 | 2009-12-30 | Isis Pharmaceuticals, Inc. | Oligomeric compounds comprising bicyclic nucleosides and having reduced toxicity |
| WO2010036698A1 (en) * | 2008-09-24 | 2010-04-01 | Isis Pharmaceuticals, Inc. | Substituted alpha-l-bicyclic nucleosides |
| US10329318B2 (en) | 2008-12-02 | 2019-06-25 | Wave Life Sciences Ltd. | Method for the synthesis of phosphorus atom modified nucleic acids |
| WO2010077578A1 (en) * | 2008-12-09 | 2010-07-08 | Isis Pharmaceuticals, Inc. | Bis-modified bicyclic nucleic acid analogs |
| US9107933B2 (en) | 2009-03-16 | 2015-08-18 | Isis Pharmaceuticals, Inc. | Compositions and methods of targeting apolipoprotein B for the reduction of apolipoprotein C-III |
| WO2010150789A1 (en) | 2009-06-23 | 2010-12-29 | 武田薬品工業株式会社 | Method for synthesizing nucleic acid |
| US10307434B2 (en) | 2009-07-06 | 2019-06-04 | Wave Life Sciences Ltd. | Nucleic acid prodrugs and methods of use thereof |
| US8563528B2 (en) | 2009-07-21 | 2013-10-22 | Santaris Pharma A/S | Antisense oligomers targeting PCSK9 |
| EP2580228B1 (en) * | 2010-06-08 | 2016-03-23 | Ionis Pharmaceuticals, Inc. | Substituted 2'-amino and 2'-thio-bicyclic nucleosides and oligomeric compounds prepared therefrom |
| US10428019B2 (en) | 2010-09-24 | 2019-10-01 | Wave Life Sciences Ltd. | Chiral auxiliaries |
| US10017764B2 (en) | 2011-02-08 | 2018-07-10 | Ionis Pharmaceuticals, Inc. | Oligomeric compounds comprising bicyclic nucleotides and uses thereof |
| US10280192B2 (en) | 2011-07-19 | 2019-05-07 | Wave Life Sciences Ltd. | Methods for the synthesis of functionalized nucleic acids |
| WO2013022967A1 (en) | 2011-08-11 | 2013-02-14 | Isis Pharmaceuticals, Inc. | Gapped oligomeric compounds comprising 5'-modified deoxyribonucleosides in the gap and uses thereof |
| US11732261B2 (en) | 2011-08-11 | 2023-08-22 | Ionis Pharmaceuticals, Inc. | Selective antisense compounds and uses thereof |
| US9752142B2 (en) | 2011-08-11 | 2017-09-05 | Ionis Pharmaceuticals, Inc. | Gapped oligomeric compounds comprising 5′-modified deoxyribonucleosides in the gap and uses thereof |
| WO2013022966A1 (en) | 2011-08-11 | 2013-02-14 | Isis Pharmaceuticals, Inc. | Linkage modified gapped oligomeric compounds and uses thereof |
| WO2013068348A1 (en) | 2011-11-07 | 2013-05-16 | Santaris Pharma A/S | Lna oligomers for improvement in hepatic function |
| US9914922B2 (en) | 2012-04-20 | 2018-03-13 | Ionis Pharmaceuticals, Inc. | Oligomeric compounds comprising bicyclic nucleotides and uses thereof |
| US11566245B2 (en) | 2012-04-20 | 2023-01-31 | Ionis Pharmaceuticals, Inc. | Oligomeric compounds comprising bicyclic nucleotides and uses thereof |
| US10590413B2 (en) | 2012-07-13 | 2020-03-17 | Wave Life Sciences Ltd. | Chiral control |
| US9982257B2 (en) | 2012-07-13 | 2018-05-29 | Wave Life Sciences Ltd. | Chiral control |
| US10167309B2 (en) | 2012-07-13 | 2019-01-01 | Wave Life Sciences Ltd. | Asymmetric auxiliary group |
| US9695418B2 (en) | 2012-10-11 | 2017-07-04 | Ionis Pharmaceuticals, Inc. | Oligomeric compounds comprising bicyclic nucleosides and uses thereof |
| WO2014076195A1 (en) | 2012-11-15 | 2014-05-22 | Santaris Pharma A/S | Oligonucleotide conjugates |
| EP3406718A1 (en) | 2012-11-15 | 2018-11-28 | Roche Innovation Center Copenhagen A/S | Oligonucleotide conjugates |
| US11155816B2 (en) | 2012-11-15 | 2021-10-26 | Roche Innovation Center Copenhagen A/S | Oligonucleotide conjugates |
| US10077443B2 (en) | 2012-11-15 | 2018-09-18 | Roche Innovation Center Copenhagen A/S | Oligonucleotide conjugates |
| WO2014118267A1 (en) | 2013-01-30 | 2014-08-07 | Santaris Pharma A/S | Lna oligonucleotide carbohydrate conjugates |
| US10883104B2 (en) | 2013-05-01 | 2021-01-05 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating apolipoprotein (a) expression |
| EP3690049A1 (en) | 2013-05-01 | 2020-08-05 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating apolipoprotein c-iii expression |
| US9714421B2 (en) | 2013-05-01 | 2017-07-25 | Ionis Pharmaceuticals, Inc. | Compositions and methods |
| US9932580B2 (en) | 2013-05-01 | 2018-04-03 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating HBV expression |
| US9932581B2 (en) | 2013-05-01 | 2018-04-03 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating apolipoprotein C-III expression |
| WO2014179626A2 (en) | 2013-05-01 | 2014-11-06 | Isis Pharmaceuticals, Inc. | Compositions and methods for modulating apolipoprotein c-iii expression |
| US9957504B2 (en) | 2013-05-01 | 2018-05-01 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating apolipoprotein (a) expression |
| EP3828275A1 (en) | 2013-05-01 | 2021-06-02 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating ttr expression |
| EP3633039A1 (en) | 2013-05-01 | 2020-04-08 | Ionis Pharmaceuticals, Inc. | Compositions and methods |
| US11299736B1 (en) | 2013-05-01 | 2022-04-12 | Ionis Pharmaceuticals, Inc. | Conjugated antisense compounds and their use |
| US10683499B2 (en) | 2013-05-01 | 2020-06-16 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating TTR expression |
| WO2014179629A2 (en) | 2013-05-01 | 2014-11-06 | Isis Pharmaceuticals, Inc. | Compositions and methods |
| WO2014179627A2 (en) | 2013-05-01 | 2014-11-06 | Isis Pharmaceuticals, Inc. | Compositions and methods for modulating hbv and ttr expression |
| WO2014179625A1 (en) | 2013-05-01 | 2014-11-06 | Isis Pharmaceuticals, Inc. | COMPOSITIONS AND METHODS FOR MODULATING APOLIPOPROTEIN (a) EXPRESSION |
| EP3524680A1 (en) | 2013-05-01 | 2019-08-14 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating ttr expression |
| EP3730619A1 (en) | 2013-06-21 | 2020-10-28 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulation of target nucleic acids |
| US10385342B2 (en) | 2013-06-27 | 2019-08-20 | Roche Innovation Center Copenhagen A/S | Methods of treatment using antisense oligomers and conjugates targeting PCSK9 |
| US11739332B2 (en) | 2013-06-27 | 2023-08-29 | Roche Innovation Center Copenhagen A/S | Antisense oligomers targeting PCSK9 |
| US10443058B2 (en) | 2013-06-27 | 2019-10-15 | Roche Innovation Center Copenhagen A/S | Antisense oligomers targeting PCSK9 |
| US10370668B2 (en) | 2013-06-27 | 2019-08-06 | Roche Innovation Center Copenhagen A/S | Manufacture of antisense oligomers and conjugates targeting PCSK9 |
| US9879265B2 (en) | 2013-06-27 | 2018-01-30 | Roche Innovation Center Copenhagen A/S | Oligonucleotide conjugates |
| US9943604B2 (en) | 2013-09-20 | 2018-04-17 | Ionis Pharmaceuticals, Inc. | Targeted therapeutic nucleosides and their use |
| US10149905B2 (en) | 2014-01-15 | 2018-12-11 | Shin Nippon Biomedical Laboratories, Ltd. | Chiral nucleic acid adjuvant having antitumor effect and antitumor agent |
| US10144933B2 (en) | 2014-01-15 | 2018-12-04 | Shin Nippon Biomedical Laboratories, Ltd. | Chiral nucleic acid adjuvant having immunity induction activity, and immunity induction activator |
| US10160969B2 (en) | 2014-01-16 | 2018-12-25 | Wave Life Sciences Ltd. | Chiral design |
| EP3943607A1 (en) | 2014-04-09 | 2022-01-26 | The Scripps Research Institute | Import of unnatural or modified nucleoside triphosphates into cells via nucleic acid triphosphate transporters |
| US11466279B2 (en) | 2014-04-09 | 2022-10-11 | The Scripps Research Institute | Import of unnatural or modified nucleoside triphosphates into cells via nucleic acid triphosphate transporters |
| US10513706B2 (en) | 2014-04-09 | 2019-12-24 | The Scripps Research Institute | Import of unnatural or modified nucleoside triphosphates into cells via nucleic acid triphosphate transporters |
| WO2015164693A1 (en) | 2014-04-24 | 2015-10-29 | Isis Pharmaceuticals, Inc. | Oligomeric compounds comprising alpha-beta-constrained nucleic acid |
| US10221416B2 (en) | 2014-04-24 | 2019-03-05 | Ionis Pharmaceuticals, Inc. | Oligomeric compounds comprising alpha-beta-constrained nucleic acid |
| EP3845547A1 (en) | 2014-05-01 | 2021-07-07 | Ionis Pharmaceuticals, Inc. | Galnac3 conjugated modified oligonucleotide for modulating angiopoietin-like 3 expression |
| US10280423B2 (en) | 2014-05-01 | 2019-05-07 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating complement factor B expression |
| US10875884B2 (en) | 2014-05-01 | 2020-12-29 | Isis Pharmaceuticals, Inc. | Compositions and methods for modulating angiopoietin-like 3 expression |
| US11732265B2 (en) | 2014-05-01 | 2023-08-22 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating complement factor B expression |
| EP3862362A2 (en) | 2014-05-01 | 2021-08-11 | Ionis Pharmaceuticals, Inc. | Conjugates of modified antisense oligonucleotides and their use for modulating pkk expression |
| US10793862B2 (en) | 2014-05-01 | 2020-10-06 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating growth hormone receptor expression |
| US11312964B2 (en) | 2014-05-01 | 2022-04-26 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating growth hormone receptor expression |
| EP3757215A2 (en) | 2014-05-01 | 2020-12-30 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating growth hormone receptor expression |
| WO2015168618A2 (en) | 2014-05-01 | 2015-11-05 | Isis Pharmaceuticals, Inc. | Compositions and methods for modulating growth hormone receptor expression |
| EP4219718A2 (en) | 2014-05-01 | 2023-08-02 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating complement factor b expression |
| WO2015168589A2 (en) | 2014-05-01 | 2015-11-05 | Isis Pharmaceuticals, Inc. | Compositions and methods for modulating angiopoietin-like 3 expression |
| US9994855B2 (en) | 2014-05-01 | 2018-06-12 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating growth hormone receptor expression |
| EP3974534A1 (en) | 2014-05-01 | 2022-03-30 | Ionis Pharmaceuticals, Inc. | Compositions and methods for modulating growth hormone receptor expression |
| WO2017015555A1 (en) | 2015-07-22 | 2017-01-26 | Wave Life Sciences Ltd. | Oligonucleotide compositions and methods thereof |
| US11591594B2 (en) | 2015-08-24 | 2023-02-28 | Roche Innovation Center Copenhagen A/S | LNA-G process |
| WO2017032726A1 (en) | 2015-08-24 | 2017-03-02 | Roche Innovation Center Copenhagen A/S | Lna-g process |
| WO2017068087A1 (en) | 2015-10-22 | 2017-04-27 | Roche Innovation Center Copenhagen A/S | Oligonucleotide detection method |
| US11319536B2 (en) | 2015-11-06 | 2022-05-03 | Ionis Pharmacueticals, Inc. | Modulating apolipoprotein (a) expression |
| US10557137B2 (en) | 2015-11-06 | 2020-02-11 | Ionis Pharmaceuticals, Inc. | Modulating apolipoprotein (a) expression |
| US10718753B2 (en) | 2015-11-12 | 2020-07-21 | Hoffman-La Roche Inc. | Oligonucleotides for inducing paternal UBE3A expression |
| US10739332B2 (en) | 2015-11-12 | 2020-08-11 | Hoffmann-La Roche Inc. | Oligonucleotides for inducing paternal UBE3A expression |
| EP4220360A2 (en) | 2015-11-12 | 2023-08-02 | F. Hoffmann-La Roche AG | Oligonucleotides for inducing paternal ube3a expression |
| EP3798307A1 (en) | 2015-11-12 | 2021-03-31 | F. Hoffmann-La Roche AG | Oligonucleotides for inducing paternal ube3a expression |
| US10494633B2 (en) | 2015-11-12 | 2019-12-03 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for inducing paternal UBE3A expression |
| US11320421B2 (en) | 2015-11-12 | 2022-05-03 | Hoffmann-La Roche Inc. | Oligonucleotides for inducing paternal UBE3A expression |
| US11761007B2 (en) | 2015-12-18 | 2023-09-19 | The Scripps Research Institute | Production of unnatural nucleotides using a CRISPR/Cas9 system |
| US10829555B2 (en) | 2016-03-14 | 2020-11-10 | Hoffman-La Roche Inc. | Oligonucleotides for reduction of PD-L1 expression |
| US10745480B2 (en) | 2016-03-14 | 2020-08-18 | Hoffmann-La Roche, Inc. | Oligonucleotides for reduction of PD-L1 expression |
| WO2017157899A1 (en) | 2016-03-14 | 2017-09-21 | F. Hoffmann-La Roche Ag | Oligonucleotides for reduction of pd-l1 expression |
| EP3786297A1 (en) | 2016-03-14 | 2021-03-03 | F. Hoffmann-La Roche AG | Oligonucleotides for reduction of pd-l1 expression |
| US11466081B2 (en) | 2016-03-14 | 2022-10-11 | Hoffmann-La Roche Inc. | Oligonucleotides for reduction of PD-L1 expression |
| WO2017157672A1 (en) | 2016-03-18 | 2017-09-21 | Roche Innovation Center Copenhagen A/S | Acyl-protected l-lna-guanosine monomers |
| US11267843B2 (en) | 2016-03-18 | 2022-03-08 | Roche Innovation Center Copenhagen A/S | Stereodefining L-monomers |
| WO2017178656A1 (en) | 2016-04-14 | 2017-10-19 | Roche Innovation Center Copenhagen A/S | TRITYL-MONO-GalNAc COMPOUNDS AND THEIR USE |
| US11248019B2 (en) | 2016-04-14 | 2022-02-15 | Hoffmann-La Roche Inc. | Trityl-mono-GalNAc compounds and their use |
| US11261209B2 (en) | 2016-05-12 | 2022-03-01 | Roche Innovation Center Copenhagen A/S | Enhanced coupling of stereodefined oxazaphospholidine phosphoramidite monomers to nucleoside or oligonucleotide |
| US11534452B2 (en) | 2016-06-17 | 2022-12-27 | Hoffmann-La Roche Inc. | Nucleic acid molecules for reduction of PAPD5 or PAPD7 mRNA for treating hepatitis B infection |
| US11191775B2 (en) | 2016-06-17 | 2021-12-07 | Hoffmann-La Roche Inc. | PAPD5 and PAPD7 inhibitors for treating a hepatitis B infection |
| WO2017216390A1 (en) | 2016-06-17 | 2017-12-21 | F. Hoffmann-La Roche Ag | Nucleic acid molecules for reduction of papd5 or papd7 mrna for treating hepatitis b infection |
| US10696720B2 (en) | 2016-06-24 | 2020-06-30 | The Scripps Research Institute | Nucleoside triphosphate transporter and uses thereof |
| EP4163293A1 (en) | 2016-06-24 | 2023-04-12 | The Scripps Research Institute | Novel nucleoside triphosphate transporter and uses thereof |
| US10696719B2 (en) | 2016-06-24 | 2020-06-30 | The Scripps Research Institute | Nucleoside triphosphate transporter and uses thereof |
| US11834479B2 (en) | 2016-06-24 | 2023-12-05 | The Scripps Research Institute | Nucleoside triphosphate transporter and uses thereof |
| WO2017223528A1 (en) | 2016-06-24 | 2017-12-28 | The Scripps Research Institute | Novel nucleoside triphosphate transporter and uses thereof |
| WO2018002105A1 (en) | 2016-07-01 | 2018-01-04 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides for modulating htra1 expression |
| US10519450B2 (en) | 2016-07-01 | 2019-12-31 | Hoffmann-La Roche Inc. | Antisense oligonucleotides for modulating HTRA1 expression |
| US11400161B2 (en) | 2016-10-06 | 2022-08-02 | Ionis Pharmaceuticals, Inc. | Method of conjugating oligomeric compounds |
| WO2018087200A1 (en) | 2016-11-11 | 2018-05-17 | Roche Innovation Center Copenhagen A/S | Therapeutic oligonucleotides capture and detection |
| WO2018130581A1 (en) | 2017-01-13 | 2018-07-19 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides for modulating rela expression |
| WO2018130583A1 (en) | 2017-01-13 | 2018-07-19 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides for modulating nfkb1 expression |
| WO2018130584A1 (en) | 2017-01-13 | 2018-07-19 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides for modulating nfkb2 expression |
| WO2018130582A1 (en) | 2017-01-13 | 2018-07-19 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides for modulating rel expression |
| WO2018130585A1 (en) | 2017-01-13 | 2018-07-19 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides for modulating relb expression |
| US11591362B2 (en) | 2017-03-29 | 2023-02-28 | Roche Innovation Center Copenhagen A/S | Orthogonal protecting groups for the preparation of stereodefined phosphorothioate oligonucleotides |
| WO2018177825A1 (en) | 2017-03-29 | 2018-10-04 | Roche Innovation Center Copenhagen A/S | Orthogonal protecting groups for the preparation of stereodefined phosphorothioate oligonucleotides |
| WO2018220034A1 (en) | 2017-06-01 | 2018-12-06 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides for modulating htra1 expression |
| US11814407B2 (en) | 2017-06-28 | 2023-11-14 | Roche Innovation Center Copenhagen A/S | Multiple coupling and oxidation method |
| WO2019002237A1 (en) | 2017-06-28 | 2019-01-03 | Roche Innovation Center Copenhagen A/S | Multiple coupling & oxidation method |
| US11834689B2 (en) | 2017-07-11 | 2023-12-05 | The Scripps Research Institute | Incorporation of unnatural nucleotides and methods thereof |
| US11701407B2 (en) | 2017-08-03 | 2023-07-18 | Synthorx, Inc. | Cytokine conjugates for the treatment of proliferative and infectious diseases |
| US10610571B2 (en) | 2017-08-03 | 2020-04-07 | Synthorx, Inc. | Cytokine conjugates for the treatment of proliferative and infectious diseases |
| US11622993B2 (en) | 2017-08-03 | 2023-04-11 | Synthorx, Inc. | Cytokine conjugates for the treatment of autoimmune diseases |
| WO2019030313A2 (en) | 2017-08-11 | 2019-02-14 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for modulating ube3c expression |
| WO2019038228A1 (en) | 2017-08-22 | 2019-02-28 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for modulating tom1 expression |
| WO2019073018A1 (en) | 2017-10-13 | 2019-04-18 | Roche Innovation Center Copenhagen A/S | Methods for identifying improved stereodefined phosphorothioate oligonucleotide variants of antisense oligonucleotides utilising sub-libraries of partially stereodefined oligonucleotides |
| US11484546B2 (en) | 2017-10-16 | 2022-11-01 | Hoffman-La Roche Inc. | Nucleic acid molecule for reduction of PAPD5 and PAPD7 mRNA for treating hepatitis B infection |
| US10953034B2 (en) | 2017-10-16 | 2021-03-23 | Hoffmann-La Roche Inc. | Nucleic acid molecule for reduction of PAPD5 and PAPD7 mRNA for treating hepatitis B infection |
| WO2019076842A1 (en) | 2017-10-16 | 2019-04-25 | F. Hoffmann-La Roche Ag | NUCLEIC ACID MOLECULE FOR REDUCTION OF PAPD5 AND PAPD7 mRNA FOR TREATING HEPATITIS B INFECTION |
| WO2019115416A2 (en) | 2017-12-11 | 2019-06-20 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for modulating fndc3b expression |
| WO2019115417A2 (en) | 2017-12-12 | 2019-06-20 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for modulating rb1 expression |
| WO2019121838A1 (en) | 2017-12-21 | 2019-06-27 | F. Hoffmann-La Roche Ag | Companion diagnostic for htra1 rna antagonists |
| WO2019122277A1 (en) | 2017-12-22 | 2019-06-27 | Roche Innovation Center Copenhagen A/S | Novel thiophosphoramidites |
| WO2019122279A1 (en) | 2017-12-22 | 2019-06-27 | Roche Innovation Center Copenhagen A/S | Oligonucleotides comprising a phosphorodithioate internucleoside linkage |
| WO2019122282A1 (en) | 2017-12-22 | 2019-06-27 | Roche Innovation Center Copenhagen A/S | Gapmer oligonucleotides comprising a phosphorodithioate internucleoside linkage |
| EP4092117A1 (en) | 2017-12-22 | 2022-11-23 | Roche Innovation Center Copenhagen A/S | Gapmer oligonucleotides comprising a phosphorodithioate internucleoside linkage |
| US11597926B2 (en) | 2017-12-22 | 2023-03-07 | Roche Innovation Center Copenhagen A/S | Thiophosphoramidites |
| WO2019137883A1 (en) | 2018-01-10 | 2019-07-18 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for modulating pias4 expression |
| WO2019138057A1 (en) | 2018-01-12 | 2019-07-18 | Roche Innovation Center Copenhagen A/S | Alpha-synuclein antisense oligonucleotides and uses thereof |
| WO2019137974A1 (en) | 2018-01-12 | 2019-07-18 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for modulating gsk3b expression |
| WO2019140231A1 (en) | 2018-01-12 | 2019-07-18 | Bristol-Myers Squibb Company | Antisense oligonucleotides targeting alpha-synuclein and uses thereof |
| WO2019140236A1 (en) | 2018-01-12 | 2019-07-18 | Bristol-Myers Squibb Company | Antisense oligonucleotides targeting alpha-synuclein and uses thereof |
| US11447775B2 (en) | 2018-01-12 | 2022-09-20 | Bristol-Myers Squibb Company | Antisense oligonucleotides targeting alpha-synuclein and uses thereof |
| US11359197B2 (en) | 2018-01-12 | 2022-06-14 | Bristol-Myers Squibb Company | Antisense oligonucleotides targeting alpha-synuclein and uses thereof |
| WO2019141656A1 (en) | 2018-01-17 | 2019-07-25 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for modulating erc1 expression |
| WO2019141723A1 (en) | 2018-01-18 | 2019-07-25 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting srebp1 |
| WO2019145386A1 (en) | 2018-01-26 | 2019-08-01 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for modulating csnk1d expression |
| WO2019154979A1 (en) | 2018-02-09 | 2019-08-15 | Genentech, Inc. | Oligonucleotides for modulating tmem106b expression |
| WO2019165067A1 (en) | 2018-02-21 | 2019-08-29 | Bristol-Myers Squibb Company | Camk2d antisense oligonucleotides and uses thereof |
| US11058767B2 (en) | 2018-02-21 | 2021-07-13 | Bristol-Myers Squibb Company | CAMK2D antisense oligonucleotides and uses thereof |
| WO2019165453A1 (en) | 2018-02-26 | 2019-08-29 | Synthorx, Inc. | Il-15 conjugates and uses thereof |
| US11919934B2 (en) | 2018-02-26 | 2024-03-05 | Synthorx, Inc. | IL-15 conjugates and uses thereof |
| WO2019182037A1 (en) | 2018-03-20 | 2019-09-26 | 国立大学法人東京工業大学 | Antisense oligonucleotide having reduced toxicity |
| US11732262B2 (en) | 2018-04-05 | 2023-08-22 | Hoffmann—La Roche, Inc. | Use of FUBP1 inhibitors for treating hepatitis B virus infection |
| WO2019193165A1 (en) | 2018-04-05 | 2019-10-10 | F. Hoffmann-La Roche Ag | Use of fubp1 inhibitors for treating hepatitis b virus infection |
| WO2019215067A1 (en) | 2018-05-07 | 2019-11-14 | Roche Innovation Center Copenhagen A/S | Massively parallel discovery methods for oligonucleotide therapeutics |
| WO2019215066A1 (en) | 2018-05-07 | 2019-11-14 | Roche Innovation Center Copenhagen A/S | Quality control of lna oligonucleotide therapeutics using massively parallel sequencing |
| WO2019215065A1 (en) | 2018-05-07 | 2019-11-14 | Roche Innovation Center Copenhagen A/S | Massively parallel discovery methods for oligonucleotide therapeutics |
| WO2019215175A1 (en) | 2018-05-08 | 2019-11-14 | Roche Innovation Center Copenhagen A/S | Oligonucleotides for modulating myh7 expression |
| WO2019219723A1 (en) | 2018-05-18 | 2019-11-21 | F. Hoffmann-La Roche Ag | Pharmaceutical compositions for treatment of microrna related diseases |
| WO2019224172A1 (en) | 2018-05-25 | 2019-11-28 | Roche Innovation Center Copenhagen A/S | Novel process for making allofuranose from glucofuranose |
| WO2019233922A1 (en) | 2018-06-05 | 2019-12-12 | F. Hoffmann-La Roche Ag | Oligonucleotides for modulating atxn2 expression |
| WO2019233921A1 (en) | 2018-06-05 | 2019-12-12 | F. Hoffmann-La Roche Ag | Oligonucleotides for modulating atxn2 expression |
| US11066669B2 (en) | 2018-06-05 | 2021-07-20 | Hoffmann-La Roche Inc. | Oligonucleotides for modulating ATXN2 expression |
| WO2020007700A1 (en) | 2018-07-02 | 2020-01-09 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting spi1 |
| WO2020007772A1 (en) | 2018-07-02 | 2020-01-09 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting gbp-1 |
| WO2020007702A1 (en) | 2018-07-02 | 2020-01-09 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting bcl2l11 |
| US11279929B2 (en) | 2018-07-03 | 2022-03-22 | Hoffmann-La Roche, Inc. | Oligonucleotides for modulating Tau expression |
| US11753640B2 (en) | 2018-07-03 | 2023-09-12 | Hoffmann-La Roche Inc. | Oligonucleotides for modulating Tau expression |
| WO2020007892A1 (en) | 2018-07-03 | 2020-01-09 | F. Hoffmann-La Roche Ag | Oligonucleotides for modulating tau expression |
| WO2020007826A1 (en) | 2018-07-05 | 2020-01-09 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting mbtps1 |
| WO2020007889A1 (en) | 2018-07-05 | 2020-01-09 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting stat1 |
| WO2020011743A1 (en) | 2018-07-09 | 2020-01-16 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting mafb |
| WO2020011653A1 (en) | 2018-07-09 | 2020-01-16 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting kynu |
| WO2020011869A2 (en) | 2018-07-11 | 2020-01-16 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting tlr2 |
| WO2020011744A2 (en) | 2018-07-11 | 2020-01-16 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting cers5 |
| WO2020011745A2 (en) | 2018-07-11 | 2020-01-16 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting cers6 |
| WO2020011902A1 (en) | 2018-07-13 | 2020-01-16 | F. Hoffmann-La Roche Ag | Oligonucleotides for modulating rtel1 expression |
| WO2020025527A1 (en) | 2018-07-31 | 2020-02-06 | Roche Innovation Center Copenhagen A/S | Oligonucleotides comprising a phosphorotrithioate internucleoside linkage |
| WO2020025563A1 (en) | 2018-07-31 | 2020-02-06 | Roche Innovation Center Copenhagen A/S | Oligonucleotides comprising a phosphorotrithioate internucleoside linkage |
| WO2020038968A1 (en) | 2018-08-23 | 2020-02-27 | Roche Innovation Center Copenhagen A/S | Microrna-134 biomarker |
| WO2020038976A1 (en) | 2018-08-23 | 2020-02-27 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting usp8 |
| WO2020038971A1 (en) | 2018-08-23 | 2020-02-27 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting vcan |
| WO2020038973A1 (en) | 2018-08-23 | 2020-02-27 | Roche Innovation Center Copenhagen A/S | Antisense oligonucleotides targeting sptlc1 |
| WO2020043750A1 (en) | 2018-08-28 | 2020-03-05 | Roche Innovation Center Copenhagen A/S | Neoantigen engineering using splice modulating compounds |
| EP3620519A1 (en) | 2018-09-04 | 2020-03-11 | F. Hoffmann-La Roche AG | Use of isolated milk extracellular vesicles for delivering oligonucleotides orally |
| WO2020089260A1 (en) | 2018-11-01 | 2020-05-07 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides targeting tia1 |
| WO2020104492A1 (en) | 2018-11-22 | 2020-05-28 | Roche Innovation Center Copenhagen A/S | Pyridinium salts as activators in the synthesis of stereodefined oligonucleotides |
| WO2020109344A1 (en) | 2018-11-29 | 2020-06-04 | F. Hoffmann-La Roche Ag | Occular administration device for antisense oligonucleotides |
| WO2020109343A1 (en) | 2018-11-29 | 2020-06-04 | F. Hoffmann-La Roche Ag | Combination therapy for treatment of macular degeneration |
| WO2020136125A2 (en) | 2018-12-21 | 2020-07-02 | Boehringer Ingelheim International Gmbh | Antisense oligonucleotides targeting card9 |
| WO2020152303A1 (en) | 2019-01-25 | 2020-07-30 | F. Hoffmann-La Roche Ag | Lipid vesicle for oral drug delivery |
| US11077195B2 (en) | 2019-02-06 | 2021-08-03 | Synthorx, Inc. | IL-2 conjugates and methods of use thereof |
| WO2020169696A1 (en) | 2019-02-20 | 2020-08-27 | Roche Innovation Center Copenhagen A/S | Novel phosphoramidites |
| WO2020169695A1 (en) | 2019-02-20 | 2020-08-27 | Roche Innovation Center Copenhagen A/S | Phosphonoacetate gapmer oligonucleotides |
| WO2020173845A1 (en) | 2019-02-26 | 2020-09-03 | Roche Innovation Center Copenhagen A/S | Oligonucleotide formulation method |
| WO2020178258A1 (en) | 2019-03-05 | 2020-09-10 | F. Hoffmann-La Roche Ag | Intracellular targeting of molecules |
| WO2020191377A1 (en) | 2019-03-21 | 2020-09-24 | Codiak Biosciences, Inc. | Extracellular vesicle conjugates and uses thereof |
| WO2020206115A2 (en) | 2019-04-03 | 2020-10-08 | Bristol-Myers Squibb Company | Angptl2 antisense oligonucleotides and uses thereof |
| WO2020201339A1 (en) | 2019-04-04 | 2020-10-08 | F. Hoffmann-La Roche Ag | Oligonucleotides for modulating atxn2 expression |
| US11286485B2 (en) | 2019-04-04 | 2022-03-29 | Hoffmann-La Roche Inc. | Oligonucleotides for modulating ATXN2 expression |
| WO2020212301A1 (en) | 2019-04-16 | 2020-10-22 | Roche Innovation Center Copenhagen A/S | Novel process for preparing nucleotide p(v) monomers |
| WO2020221705A1 (en) | 2019-04-30 | 2020-11-05 | Roche Innovation Center Copenhagen A/S | Novel process for preparing rhenium chelated mag3 oligonucleotides |
| WO2020245233A1 (en) | 2019-06-06 | 2020-12-10 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides targeting atxn3 |
| US11542501B2 (en) | 2019-06-06 | 2023-01-03 | Hoffmann-La Roche Inc. | Antisense oligonucleotides targeting ATXN3 |
| US11879145B2 (en) | 2019-06-14 | 2024-01-23 | The Scripps Research Institute | Reagents and methods for replication, transcription, and translation in semi-synthetic organisms |
| WO2021030769A1 (en) | 2019-08-14 | 2021-02-18 | Codiak Biosciences, Inc. | Extracellular vesicles with nras antisense oligonucleotides |
| WO2021030780A1 (en) | 2019-08-14 | 2021-02-18 | Codiak Biosciences, Inc. | Extracellular vesicle-aso constructs targeting cebp/beta |
| WO2021030773A1 (en) | 2019-08-14 | 2021-02-18 | Codiak Biosciences, Inc. | Extracellular vesicle-nlrp3 antagonist |
| WO2021030781A1 (en) | 2019-08-14 | 2021-02-18 | Codiak Biosciences, Inc. | Extracellular vesicles with antisense oligonucleotides targeting kras |
| WO2021030768A1 (en) | 2019-08-14 | 2021-02-18 | Codiak Biosciences, Inc. | Extracellular vesicles with stat3-antisense oligonucleotides |
| WO2021030776A1 (en) | 2019-08-14 | 2021-02-18 | Codiak Biosciences, Inc. | Extracellular vesicle-aso constructs targeting stat6 |
| WO2021030777A1 (en) | 2019-08-14 | 2021-02-18 | Codiak Biosciences, Inc. | Extracellular vesicle linked to molecules and uses thereof |
| WO2021030706A1 (en) | 2019-08-15 | 2021-02-18 | Synthorx, Inc. | Immuno oncology combination therapies with il-2 conjugates |
| WO2021041206A1 (en) | 2019-08-23 | 2021-03-04 | Synthorx, Inc. | Il-15 conjugates and uses thereof |
| WO2021050554A1 (en) | 2019-09-10 | 2021-03-18 | Synthorx, Inc. | Il-2 conjugates and methods of use to treat autoimmune diseases |
| WO2021062058A1 (en) | 2019-09-25 | 2021-04-01 | Codiak Biosciences, Inc. | Sting agonist comprising exosomes for treating neuroimmunological disorders |
| WO2021091986A1 (en) | 2019-11-04 | 2021-05-14 | Synthorx, Inc. | Interleukin 10 conjugates and uses thereof |
| WO2021122735A1 (en) | 2019-12-19 | 2021-06-24 | F. Hoffmann-La Roche Ag | Use of sept9 inhibitors for treating hepatitis b virus infection |
| WO2021122910A1 (en) | 2019-12-19 | 2021-06-24 | F. Hoffmann-La Roche Ag | Use of sbds inhibitors for treating hepatitis b virus infection |
| WO2021122921A1 (en) | 2019-12-19 | 2021-06-24 | F. Hoffmann-La Roche Ag | Use of cops3 inhibitors for treating hepatitis b virus infection |
| WO2021122869A1 (en) | 2019-12-19 | 2021-06-24 | F. Hoffmann-La Roche Ag | Use of scamp3 inhibitors for treating hepatitis b virus infection |
| WO2021122993A1 (en) | 2019-12-19 | 2021-06-24 | F. Hoffmann-La Roche Ag | Use of saraf inhibitors for treating hepatitis b virus infection |
| WO2021123086A1 (en) | 2019-12-20 | 2021-06-24 | F. Hoffmann-La Roche Ag | Enhanced oligonucleotides for inhibiting scn9a expression |
| WO2021130270A1 (en) | 2019-12-24 | 2021-07-01 | F. Hoffmann-La Roche Ag | Pharmaceutical combination of antiviral agents targeting hbv and/or an immune modulator for treatment of hbv |
| WO2021130266A1 (en) | 2019-12-24 | 2021-07-01 | F. Hoffmann-La Roche Ag | Pharmaceutical combination of a therapeutic oligonucleotide targeting hbv and a tlr7 agonist for treatment of hbv |
| WO2021158810A1 (en) | 2020-02-05 | 2021-08-12 | Bristol-Myers Squibb Company | Oligonucleotides for splice modulation of camk2d |
| WO2021170697A1 (en) | 2020-02-28 | 2021-09-02 | F. Hoffmann-La Roche Ag | Oligonucleotides for modulating cd73 exon 7 splicing |
| WO2021184020A1 (en) | 2020-03-13 | 2021-09-16 | Codiak Biosciences, Inc. | Methods of treating neuroinflammation |
| WO2021184021A1 (en) | 2020-03-13 | 2021-09-16 | Codiak Biosciences, Inc. | Extracellular vesicle-aso constructs targeting pmp22 |
| WO2021231211A1 (en) | 2020-05-11 | 2021-11-18 | Genentech, Inc. | Complement component c1s inhibitors for treating a neurological disease, and related compositions, systems and methods of using same |
| WO2021231210A1 (en) | 2020-05-11 | 2021-11-18 | Genentech, Inc. | Complement component c1r inhibitors for treating a neurological disease, and related compositions, systems and methods of using same |
| WO2021231204A1 (en) | 2020-05-11 | 2021-11-18 | Genentech, Inc. | Complement component 4 inhibitors for treating neurological diseases, and related compositons, systems and methods of using same |
| WO2021229036A1 (en) | 2020-05-13 | 2021-11-18 | F. Hoffmann-La Roche Ag | Oligonucleotide agonists targeting progranulin |
| WO2021233551A1 (en) | 2020-05-22 | 2021-11-25 | F.Hoffmann-La Roche Ag | Oligonucleotides for splice modulation of card9 |
| WO2021249993A1 (en) | 2020-06-09 | 2021-12-16 | Roche Innovation Center Copenhagen A/S | Guanosine analogues for use in therapeutic polynucleotides |
| WO2021263026A1 (en) | 2020-06-25 | 2021-12-30 | Synthorx, Inc. | Immuno oncology combination therapy with il-2 conjugates and anti-egfr antibodies |
| WO2021260197A1 (en) | 2020-06-26 | 2021-12-30 | F. Hoffmann-La Roche Ag | Enhanced oligonucleotides for modulating fubp1 expression |
| WO2022018155A1 (en) | 2020-07-23 | 2022-01-27 | F. Hoffmann-La Roche Ag | Lna oligonucleotides for splice modulation of stmn2 |
| WO2022018187A1 (en) | 2020-07-23 | 2022-01-27 | F. Hoffmann-La Roche Ag | Oligonucleotides targeting rna binding protein sites |
| WO2022038211A2 (en) | 2020-08-21 | 2022-02-24 | F. Hoffmann-La Roche Ag | Use of a1cf inhibitors for treating hepatitis b virus infection |
| WO2022076596A1 (en) | 2020-10-06 | 2022-04-14 | Codiak Biosciences, Inc. | Extracellular vesicle-aso constructs targeting stat6 |
| WO2022076853A1 (en) | 2020-10-09 | 2022-04-14 | Synthorx, Inc. | Immuno oncology combination therapy with il-2 conjugates and pembrolizumab |
| WO2022076859A1 (en) | 2020-10-09 | 2022-04-14 | Synthorx, Inc. | Immuno oncology therapies with il-2 conjugates |
| WO2022117747A2 (en) | 2020-12-03 | 2022-06-09 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides targeting atxn3 |
| WO2022117745A1 (en) | 2020-12-03 | 2022-06-09 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides targeting atxn3 |
| WO2022122613A1 (en) | 2020-12-08 | 2022-06-16 | F. Hoffmann-La Roche Ag | Novel synthesis of phosphorodithioate oligonucleotides |
| WO2022129320A1 (en) | 2020-12-18 | 2022-06-23 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides for targeting progranulin |
| WO2022136140A1 (en) | 2020-12-22 | 2022-06-30 | F. Hoffmann-La Roche Ag | Oligonucleotides targeting xbp1 |
| US11898145B2 (en) | 2021-02-02 | 2024-02-13 | Hoffmann-La Roche Inc. | Enhanced oligonucleotides for inhibiting RTEL1 expression |
| WO2022167456A1 (en) | 2021-02-02 | 2022-08-11 | F. Hoffmann-La Roche Ag | Enhanced oligonucleotides for inhibiting rtel1 expression |
| WO2022174102A1 (en) | 2021-02-12 | 2022-08-18 | Synthorx, Inc. | Lung cancer combination therapy with il-2 conjugates and an anti-pd-1 antibody or antigen-binding fragment thereof |
| WO2022174101A1 (en) | 2021-02-12 | 2022-08-18 | Synthorx, Inc. | Skin cancer combination therapy with il-2 conjugates and cemiplimab |
| WO2022178149A2 (en) | 2021-02-17 | 2022-08-25 | Codiak Biosciences, Inc. | Extracellular vesicle-nlrp3 antagonist |
| WO2022178180A1 (en) | 2021-02-17 | 2022-08-25 | Codiak Biosciences, Inc. | Extracellular vesicle linked to a biologically active molecule via an optimized linker and an anchoring moiety |
| WO2022213118A1 (en) | 2021-03-31 | 2022-10-06 | Entrada Therapeutics, Inc. | Cyclic cell penetrating peptides |
| WO2022212884A1 (en) | 2021-04-01 | 2022-10-06 | Codiak Biosciences, Inc. | Extracellular vesicle compositions |
| WO2022241408A1 (en) | 2021-05-10 | 2022-11-17 | Entrada Therapeutics, Inc. | Compositions and methods for modulating tissue distribution of intracellular therapeutics |
| WO2022240760A2 (en) | 2021-05-10 | 2022-11-17 | Entrada Therapeutics, Inc. | COMPOSITIONS AND METHODS FOR MODULATING mRNA SPLICING |
| WO2022240721A1 (en) | 2021-05-10 | 2022-11-17 | Entrada Therapeutics, Inc. | Compositions and methods for modulating interferon regulatory factor-5 (irf-5) activity |
| WO2022256538A1 (en) | 2021-06-03 | 2022-12-08 | Synthorx, Inc. | Head and neck cancer combination therapy comprising an il-2 conjugate and cetuximab |
| WO2022256534A1 (en) | 2021-06-03 | 2022-12-08 | Synthorx, Inc. | Head and neck cancer combination therapy comprising an il-2 conjugate and pembrolizumab |
| WO2022258555A1 (en) | 2021-06-08 | 2022-12-15 | F. Hoffmann-La Roche Ag | Oligonucleotide progranulin agonists |
| WO2022271818A1 (en) | 2021-06-23 | 2022-12-29 | Entrada Therapeutics, Inc. | Antisense compounds and methods for targeting cug repeats |
| WO2023021046A1 (en) | 2021-08-16 | 2023-02-23 | Vib Vzw | Oligonucleotides for modulating synaptogyrin-3 expression |
| WO2023034817A1 (en) | 2021-09-01 | 2023-03-09 | Entrada Therapeutics, Inc. | Compounds and methods for skipping exon 44 in duchenne muscular dystrophy |
| WO2023052317A1 (en) | 2021-09-29 | 2023-04-06 | F. Hoffmann-La Roche Ag | Rna editing |
| WO2023078883A1 (en) | 2021-11-03 | 2023-05-11 | F. Hoffmann-La Roche Ag | Oligonucleotides for modulating apolipoprotein e4 expression |
| WO2023083906A2 (en) | 2021-11-11 | 2023-05-19 | F. Hoffmann-La Roche Ag | Pharmaceutical combinations for treatment of hbv |
| WO2023104693A1 (en) | 2021-12-07 | 2023-06-15 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides targeting actl6b |
| WO2023111336A1 (en) | 2021-12-17 | 2023-06-22 | F. Hoffmann-La Roche Ag | Oligonucleotide gba agonists |
| WO2023111210A1 (en) | 2021-12-17 | 2023-06-22 | F. Hoffmann-La Roche Ag | Combination of oligonucleotides for modulating rtel1 and fubp1 |
| WO2023117738A1 (en) | 2021-12-20 | 2023-06-29 | F. Hoffmann-La Roche Ag | Threose nucleic acid antisense oligonucleotides and methods thereof |
| WO2023122573A1 (en) | 2021-12-20 | 2023-06-29 | Synthorx, Inc. | Head and neck cancer combination therapy comprising an il-2 conjugate and pembrolizumab |
| WO2023122762A1 (en) | 2021-12-22 | 2023-06-29 | Camp4 Therapeutics Corporation | Modulation of gene transcription using antisense oligonucleotides targeting regulatory rnas |
| WO2023122750A1 (en) | 2021-12-23 | 2023-06-29 | Synthorx, Inc. | Cancer combination therapy with il-2 conjugates and cetuximab |
| WO2023141507A1 (en) | 2022-01-20 | 2023-07-27 | Genentech, Inc. | Antisense oligonucleotides for modulating tmem106b expression |
| WO2023156652A1 (en) | 2022-02-21 | 2023-08-24 | F. Hoffmann-La Roche Ag | Antisense oligonucleotide |
| WO2023217890A1 (en) | 2022-05-10 | 2023-11-16 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides targeting cfp-elk1 intergene region |
| WO2023222858A1 (en) | 2022-05-18 | 2023-11-23 | F. Hoffmann-La Roche Ag | Improved oligonucleotides targeting rna binding protein sites |
| WO2023240277A2 (en) | 2022-06-10 | 2023-12-14 | Camp4 Therapeutics Corporation | Methods of modulating progranulin expression using antisense oligonucleotides targeting regulatory rnas |
| WO2023242324A1 (en) | 2022-06-17 | 2023-12-21 | F. Hoffmann-La Roche Ag | Antisense oligonucleotides for targeting progranulin |
| EP4332221A1 (en) | 2022-08-29 | 2024-03-06 | Roche Innovation Center Copenhagen A/S | Threose nucleic acid antisense oligonucleotides and methods thereof |
| WO2024046937A1 (en) | 2022-08-29 | 2024-03-07 | F. Hoffmann-La Roche Ag | Threose nucleic acid antisense oligonucleotides and methods thereof |
| WO2024052403A1 (en) | 2022-09-06 | 2024-03-14 | F. Hoffmann-La Roche Ag | Double-stranded rna molecule for administration to the eye |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2314594B1 (en) | 6-modified bicyclic nucleic acid analogs | |
| US8268980B2 (en) | 5′-modified bicyclic nucleic acid analogs | |
| AU2008272918B2 (en) | 6-disubstituted bicyclic nucleic acid analogs | |
| EP2173760B1 (en) | Carbocyclic bicyclic nucleic acid analogs | |
| EP2361256B1 (en) | Cyclohexenyl nucleic acid analogs | |
| EP2285819B1 (en) | Oligomeric compounds comprising neutrally linked terminal bicyclic nucleosides | |
| EP2376516A1 (en) | Bis-modified bicyclic nucleic acid analogs | |
| WO2011115818A1 (en) | 5'-substituted bicyclic nucleosides and oligomeric compounds prepared therefrom | |
| AU2012201664A1 (en) | 6-modified bicyclic nucleic acid analogs |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 11745429 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2640171 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008552604 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007211080 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007762892 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4428/CHENP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087020907 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780010566.0 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020137007796 Country of ref document: KR |