WO2010126551A1 - Potent conjugates and hydrophilic linkers - Google Patents
Potent conjugates and hydrophilic linkers Download PDFInfo
- Publication number
- WO2010126551A1 WO2010126551A1 PCT/US2009/059620 US2009059620W WO2010126551A1 WO 2010126551 A1 WO2010126551 A1 WO 2010126551A1 US 2009059620 W US2009059620 W US 2009059620W WO 2010126551 A1 WO2010126551 A1 WO 2010126551A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cell
- bond
- antibody
- maytansinoid
- binding agent
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/18—Bridged systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
Definitions
- the present invention relates to new linkers to link drugs (e.g. cytotoxic agents) to cell-binding agents (e.g., antibodies) in such a way that the linker contributes in increasing the activity of the drug.
- the present invention relates to the use of novel hydrophilic linkers, wherein such linkers enhance the potency or the efficacy of the cell-binding agent-drug conjugates by several fold in a variety of cancer cell types, including those expressing a low number of antigens on the cell surface or cancers that are resistant to treatment.
- the present invention also relates to a method for preparing maytansinoids bearing a thioether moiety and a reactive group which allows the maytansinoid to be linked to a cell-binding agent.
- Antibody conjugates of cytotoxic drugs are being developed as target-specific therapeutic agents.
- Antibodies against various cancer cell-surface antigens have been conjugated with different cytotoxic agents that inhibit various essential cellular targets such as microtubules (maytansinoids, auristatins, taxanes: U.S. Patent Nos. 5,208,020; 5,416,064; 6.333,410; 6,441,163; 6,340,701 ; 6,372,738; 6,436,931 ; 6,596,757; 7,276.497), DNA (calicheamicin, doxorubicin, CC-1065 analogs; U.S. Patent Nos.
- the antibody-cytotoxic agent conjugates typically are prepared by the initial modification of reactive moieties on antibodies, such as lysine amino groups, or cysteine groups (generated by reduction of native disulfide bonds or by engineering of additional non-native cysteine residues on to antibodies using molecular biology methods).
- antibodies are first modified with a heterobifunctional linker reagent, such as those previously described, exemplified by SPDB, SMCC and SIAB (U.S. Patent No. 6,913,758 and U.S. Patent Publication No. 20050169933) to incorporate a linker with a reactive group such as mixed pyridyldisulfide, maleimide or haloacetamide.
- the incorporated reactive linker group in the antibody is subsequently conjugated with a cytotoxic agent containing a reactive moiety such as a thiol group.
- a cytotoxic agent containing a reactive moiety such as a thiol group.
- Another conjugation route is by reaction of a cytotoxic agent derivative containing a thiol-reactive group (such as haloacetamide, or maleimide) with thiol groups on the cell-binding agent.
- Thiol groups are incorporated on cell-binding agents such as an antibody by reduction of native disulfide residues (R. Singh et al., Anal.
- antibody-cytotoxic agent conjugates with disulfide or thioether linkages are cleaved intracellularly, presumably in lysosomes, to deliver the active cytotoxic agent inside the cancer cell (H. K. Erickson et al., 2006, Cancer Research, 66, 4626-4433).
- antibody-cytotoxic agent conjugates with reducible disulfide linkage also kill proximate antigen-negative cells in mixed populations of antigen-negative and antigen-positive cells in vitro and in vivo in xenograft models, suggesting the role of target-cell released cytotoxic agent in improving potency against neighboring non-antigen-expressing cells in tumors with heterogeneous antigen expression (Y. V.
- the method described in the art for preparing a cytotoxic conjugate of a cell binding agent and a drug via non-cleavable linker requires two reaction steps (US Patent 5,208,020 & US Publication No. 2005/0169933).
- the cell binding agent such as an antibody
- a bifunctional crosslinker that undergoes reaction with the reactive groups of the cell binding agent, such as the amine group on lysine residues or the sulfhydryl group on cysteine residues, to form covalent chemical bonds.
- the product is purified to separate the desired modified cell binding agent from unreacted crosslinker.
- a reactive drug derivative such as a thiol-containing maytansinoid
- a reactive drug derivative such as a thiol-containing maytansinoid
- an additional purification is required to remove any unreacted drug species and other byproducts from the final conjugate.
- the present invention addresses the problem of resistance by designing new linkers to link drugs to cell-binding agents in such a way that the linker contributes in increasing the activity of the drug.
- the present invention improves the manner in which drugs are linked to a cell-binding agent such that the linker design provides conjugates that are active across a broad spectrum of tumors, particularly in low antigen expressing or drug resistant tumors.
- the present invention is based on the novel finding that when traditional linkers (e.g. SMCC, SIAB etc, described in U.S. Patent Publication No. 20050169933) are modified to hydrophilic linkers by incorporating a polyethylene glycol [PEG n , (- CH 2 CH 2 O) n )] spacer, the potency or the efficacy of the cell-binding agent-drug conjugates is surprisingly enhanced several fold in a variety of cancer cell types, including those expressing a low number of antigens on the cell surface. [09] Also, these PEG-containing conjugates unexpectedly are more potent than the previously described conjugates toward cell lines that are resistant to treatment.
- traditional linkers e.g. SMCC, SIAB etc, described in U.S. Patent Publication No. 20050169933
- PEG n polyethylene glycol
- the present invention discloses processes for the synthesis of novel maytansinoid derivatives bearing a thioether-moiety and a reactive group . These novel maytansinoids are useful in the preparation of thioether-linked conjugates with cell binding agents in essentially one reaction step. Processes for the preparation of cell binding agent conjugates employing these novel reactive maytansinoids are also disclosed.
- the present invention provides a compound of formula (1) or a specific compound of formula (1'):
- Z represents a reactive functionality that can form an amide or a thioether bond with a cell-binding agent
- D represents a drug
- X represents an aliphatic, an aromatic or a heterocyclic group attached to the cell- binding agent via a thioether bond, an amide bond, a carbamate bond, or an ether bond
- Y represents an aliphatic, an aromatic or a heterocyclic group attached to the drug via a covalent bond selected from the group consisting of a thioether bond, an amide bond, a carbamate bond, an ether bond, an amine bond, a carbon-carbon bond and a hydrazone bond;
- n is an integer from 1 to 2000.
- Another aspect of the present invention is a cell-binding agent drug conjugate of formula (2) or a specific compound of formula (2'):
- CB represents a cell-binding agent
- D represents a drug
- X represents an aliphatic, an aromatic or a heterocyclic group attached to the cell- binding agent via a thioether bond, an amide bond, a carbamate bond, or an ether bond
- Y represents an aliphatic, an aromatic, or a heterocyclic group attached to the drug via a covalent bond selected from the group consisting of a thioether bond, an amide bond, a carbamate bond, an ether bond, an amine bond, a carbon-carbon bond and a hydrazone bond
- 1 is 0 or 1
- p is 0 or 1
- m is an integer from 2 to 15
- n is an integer from 1 to 2000.
- Another aspect of the present invention is a compound of formula (3) or a specific compound of formula (3'): Z-X,-(-CH 2 -CH 2 O-) n -Y-D (3)
- Z represents a reactive functionality that can form an amide or a thioether bond with a cell-binding agent
- D represents a drug
- X represents an aliphatic, an aromatic or a heterocyclic group attached to the cell- binding agent via a thioether bond, an amide bond, a carbamate bond, or an ether bond;
- Y represents an aliphatic, non-aromatic heterocyclic or aromatic heterocyclic group attached to the drug via a disulfide bond
- n is an integer from 1 to 14.
- Another aspect of the present invention is a cell-binding agent drug conjugate of formula (4) or a specific compound of formula (4'):
- CB represents a cell -binding agent
- D represents a drug
- X represents an aliphatic, an aromatic or a heterocyclic group attached to the cell- binding agent via a thioether bond, an amide bond, a carbamate bond, or an ether bond;
- Y represents an aliphatic, an aromatic or a heterocyclic group attached to the drug via a disulfide bond
- 1 is 0 or 1 ; and m is an integer from 3 to 8; and n is an integer from 1 to 14.
- An even further aspect of the present invention is a method for treating cancer sensitive to treatment with said method, said method comprising parenterally administering to a patient in need thereof an effective dose of a composition comprising the conjugate of formula (2) or (4).
- novel maytansinoids having a thioether moiety that bears a reactive group and that are represented by the formula (5):
- D' represents a sulfhydryl-bearing maytansinoid, such as N 2 -deacetyl-iV 2 -(3-mercapto- l-oxopropyl)-maytansine (DMl) or TV 2 -deacetyl-N 2 -(4-mercapto-4-methyl-l- oxopentyl)maytansine (DM4);
- Y' represents a thioether bond
- V is an optional linear, branched or cyclic alkyl, alkenyl or alkynyl group having from 1 to 10 carbon atoms;
- Q represents an optional aromatic or a heterocyclic moiety
- W is an optional linear, branched or cyclic alkyl, alkenyl or alkynyl group having from 1 to 10 carbon atoms;
- Z' represents an amine or sulfhydryl reactive group.
- the reactive maytansinoid derivative bearing a thioether moiety is prepared from a sulfhydryl-bearing maytansinoid (such as DMl and DM4) and a heterobifunctional crosslinker. The reaction is represented by the following chemical equation:
- D' represents a sulfhydryl-bearing maytansinoid such as N 2 -deacetyl-TV 2 -(3-mercapto-l- oxopropyl)-maytansine (DMl) or N 2 -deacetyl-N 2 -(4-mercapto-4-methyl-l- oxopentyl)maytansine (DM4);
- V is an optional linear, branched or cyclic alkyl, alkenyl or alkynyl group having from 1 to 10 carbon atoms;
- Q represents an optional aromatic or a heterocyclic moiety
- W is an optional linear, branched or cyclic alkyl, alkenyl or alkynyl group having from 1 to 10 carbon atoms;
- Z' is an amine or sulfhydryl reactive group
- Y represents a sulfhydryl -reactive moiety
- Y' represents a thioether bond between the sulfhydryl-bearing maytansinoid and the crosslinker.
- the present invention also discloses a process for the preparation of cytotoxic conjugates of maytansinoids and cell binding agents linked via a non-cleavable bond
- Z' represents an amine or sulfhydryl reactive group
- W is an optional linear, branched or cyclic alkyl, alkenyl or alkynyl group having from 1 to 10 carbon atoms;
- Q represents an optional aromatic or a heterocyclic moiety
- V is an optional linear, branched or cyclic alkyl, alkenyl or alkynyl group having from 1 to 10 carbon atoms;
- Y' represents a thioether bond
- D' represents a sulfhydryl bearing maytansinoid, such as N 2 -deacetyl-iV 2 -(3-mercapto-
- DM1 1 -oxopropyl)-maytansine
- DM4 N 2 -deacetyl-N 2 -(4-mercapto-4-methyl-l- oxopentyl)maytansine
- CB represents a cell-binding agent
- Z" represents an amide bond; and m is an integer from 2 to 8.
- the cell-binding agent maytansinoid conjugate may be further purified.
- FIGURE 1 shows a structural representation of representative PEG-containing thiosuccinimidyl-linked conjugates of the present invention
- FIGURE 2 shows a structural representation of representative PEG-containing thioacetamidyl-linked conjugates of the present invention.
- FIGURE 3 shows a structural representation of representative PEG-containing disulfide linked compounds of the present invention.
- FIGURE 4 shows synthetic schemes for PEG-containing thiosuccinimidyl- linked conjugates of the present invention.
- FIGURE 5 shows a synthetic scheme for PEG-containing thioacetamidyl-linked conjugates of the present invention.
- FIGURE 6 shows synthetic schemes for PEG-containing disulfide linked compounds of the present invention: a.) Synthesis of the PEG-containing disulfide linked compound for 1-step conjugation to cell-binding agent; and b.) Synthesis of the heterobifunctional PEG-containing disulfide linked crosslinking compound.
- FIGURE 7 shows a conjugation procedure for PEG-containing thiosuccinimidyl- linked conjugates of the present invention (one-step conjugation).
- FIGURE 8 shows a conjugation procedure for PEG-containing thiosuccinimidyl- linked conjugate of the present invention (2-step conjugation).
- FIGURE 9 shows a conjugation procedure for PEG-containing thioether-linked
- FIGURE 10 shows a conjugation procedure for PEG-containing thioether-linked
- FIGURE 11 shows a conjugation procedure for PEG-containing disulfide linked conjugate of the present invention (1-step conjugation).
- FIGURE 12 shows a conjugation procedure for PEG-containing disulfide linked conjugate of the present invention (2-step conjugation).
- FIGURE 13 shows a synthetic scheme for PEG-containing, sulfhydryl-reactive, thiosuccinimidyl-linked compounds of the present invention.
- FIGURE 14 shows a conjugation procedure for PEG-containing thiosuccinimidyl-linked conjugate of the present invention (1-step conjugation).
- FIGURE 15 shows a conjugation procedure for PEG-containing , thiosuccinimidyl-linked conjugate of the present invention (2-step conjugation).
- FIGURE 16 shows synthetic schemes for PEG-containing, sulfhydryl-reactive, thioacetamidyl-linked compounds of the present invention; a.) Synthesis of the PEG-containing, sulfhydryl-reactive, thioacetamide linked compound for 1-step conjugation; and b.) Synthesis of the heterobifunctional PEG-containing, sulfhydryl-reactive crosslinking compound for 2-step conjugation.
- FIGURE 17 shows a conjugation procedure for PEG-containing thioacetamidyl- linked conjugates of the present invention (1-step conjugation).
- FIGURE 18 shows a conjugation procedure for PEG-containing thioacetamidyl- linked conjugates of the present invention (2-step conjugation).
- FIGURE 19 shows a synthetic scheme for the PEG-containing, sulfhydryl- reactive, thioether-linked compounds of the present invention: a.) Synthesis of the PEG- containing, sulfhydryl-reactive, thioacetamidyl-linked compound for 1-step conjugation; and b.) Synthesis of the homobifunctional PEG-containing, sulfhydryl-reactive crosslinking compound for 2-step conjugation.
- FIGURE 20 shows a conjugation procedure for PEG-containing thioacetamidyl- linked conjugate of the present invention (1-step conjugation).
- FIGURE 21 shows a conjugation procedure for PEG-containing thioacetamidyl- linked conjugate of the present invention (2-step conjugation).
- FIGURE 22 shows a mass spectrum (MS) of deglycosylated HuAb-PEG 4 MaI-DMl conjugate (10.7 DMl /Ab, average).
- FIGURE 23 shows size exclusion chromatography (SEC) of HuAb-PEG 4 MaI- DMl conjugate (10.7 DMl /Ab, average).
- FIGURE 24 shows FACS binding of HuAb-PEG4Mal-DMl conjugate (10.7 maytansinoid/antibody) is similar to that of unmodified antibody.
- FIGURE 25 shows cytotoxicity of anti-EpCAM antibody-maytansinoid conjugates on multi-drug resistant COLO205-MDR cells.
- FIGURE 26 shows cytotoxicity of anti-CanAg antibody-maytansinoid conjugates on multi-drug resistant COLO205-MDR cells.
- FIGURE 27 shows cytotoxicity of anti-CD56 antibody-maytansinoid conjugates on Molp-8 multiple myeloma cells.
- FIGURE 28 shows cytotoxicity of anti-EpCAM antibody-maytansinoid conjugates on HCTl 5 cells.
- FIGURE 29 shows cytotoxicity of anti-EpCAM antibody-maytansinoid conjugates on COLO 205 mdr cells.
- FIGURE 30 shows in vivo anti-tumor activity of anti -EpCAM antibody- maytansinoid conjugates on HCTl 5 xenografts.
- FIGURE 31 shows in vivo anti-tumor activity of anti-EpCAM antibody- maytansinoid conjugates on COLO205 mdr xenografts.
- FIGURE 32 shows in vivo anti-tumor activity of anti-EpCAM antibody- maytansinoid conjugates on COLO 205 xenografts.
- FIGURE 33 shows in vivo anti-tumor activity of anti-CanAg antibody- maytansinoid conjugates on COLO 205 mdr xenografts.
- FIGURE 34 shows the binding of anti-CanAg antibody (huC242)-PEG24-Mal-
- FIGURE 35 shows in vitro potency of Anti-CanAg antibody (huC242)-PEG24-
- FIGURE 36 shows in vitro potency of anti-CanAg antibody (huC242)-PEG24-
- FIGURE 37 shows cytotoxicity of Anti-EGFR Antibody- Maytansinoid conjugates on UO-31 Cells.
- FIGURE 38 shows plasma pharmacokinetics of Antibody-PEG4-Mal-DM 1.
- FIGURE 43 shows a synthetic scheme for amine-reactive thiosuccinimidyl- linked compounds of the present invention.
- FIGURE 44 Shows a synthetic scheme for the preparation of the amine-reactive thiosuccinimidyl-linked compounds which contain a straight chain hydrocarbon between the maleimide and NHS ester.
- FIGURE 45 Shows mass spectrum (MS) of deglycosylated mAb-SMCC-DMl conjugate prepared with compounds of the present invention (3.42 DMl/mAb, average).
- FIGURE 46 shows a synthetic scheme for the two step preparation of the amine-reactive thiosuccinimidyl-linked compounds which contain a cyclolkyl group between the maleimide and NHS ester.
- FIGURE 47 shows a synthetic scheme for the two step preparation of the amine-reactive non-cleavable thiosuccinimidyl-linked compounds containing a cycloakyl group between the maleimide and NHS ester.
- FIGURE 48 Shows a synthetic scheme for the preparation of thioacetamidyl- linked compounds of the present invention.
- FIGURE 49 Shows a synthetic scheme for the two step preparation of thioacetamidyl-linked compounds of the present invention.
- FIGURE 52 Synthetic scheme for the preparation of the sulfhydryl-reactive thiosuccinimidyl-linked compounds of the present invention.
- FIGURE 53 Shows that anti-CanAg (huC242)-Mal-(CH 2 ) 6 -Mal-DMl conjugate with 3.8 or D/A binds to antigen-positive COL205 cells.
- FIGURE 54 Shows that Anti-CanAg antibody (huC242)-Mal-(CH 2 ) 6 -Mal-DM 1 conjugate with 3.8 D/A is potent against antigen-positive COLO205 cells but less so against antigen-negative Namalwa cells.
- This invention discloses the novel findings that conjugates of cell-binding agents, such as an antibody, linked to drugs, for example, cytotoxic agents, by polyethylene glycol or polyethylene oxide linkers ((-CH 2 CH 2 O) n ) exhibit several fold greater cytotoxicity toward target cancer cells than expected based on comparison with traditional cell-binding agent drug conjugates with typical aliphatic linkers and similar drug loads.
- the conjugates described in this invention are highly potent or efficacious toward cancer cells that are multidrug resistant (mdr), which have poor sensitivity to treatment with cytotoxic drugs. Cancer therapy poses the hurdle of overcoming mechanisms of drug resistance often encountered after multiple rounds of treatment with different chemotherapeutic agents.
- multidrug resistance is caused by enhanced export of drugs by ATP- binding cassette (ABC) transporters (C. Drumond, B. I. Sikic, J Clin. Oncology, 1999, 17, 1061-1070, G, Szokacs et al., Nature Reviews, 5; 219 - 234, 2006).
- ABSC ATP- binding cassette
- Therapies that overcome these mechanisms of drug resistance, such as interfering with or overcoming this efflux of drugs by cancer cells would be highly useful.
- the cytotoxicity of the PEG- linked conjugates of cell-binding agents and cytotoxic drugs were evaluated against multidrug resistant cancer cells to test if the PEG-linkers confer any advantage against these resistant cells.
- the PEG linked conjugates of cell- binding agents and cytotoxic drugs showed unexpectedly potent cell killing of the mdr cells in comparison to the much less potent conjugates derived from conventional linkers.
- the conjugates of the present invention also display markedly higher anti-tumor activity in animal models established with multidrug resistant tumor cells.
- the use of hydrophilic polyethylene glycol or polyethylene oxide linkers also allows the incorporation of a relatively large number of drugs per cell-binding agent molecule with the high protein monomer level of greater than 90% at concentrations of greater than 1 mg/ml that are desired for therapeutic uses.
- polyethylene glycol (PEG)-linked conjugates of cell-binding agents having a range of cytotoxic drug load showed greatly enhanced cytotoxicities toward target cancer cells than expected from the stoichiometric increase in drug delivery based on increased drug load of the conjugates.
- Conjugates of cell-binding agent and drug having PEG spacers are described in this invention, which exhibited the super- stoichiometric increase in cytotoxicity toward target cancer cells by as much as a 260- 650 fold enhancement in potency (see, for example, Figure 29) as compared to traditionally prepared conjugates with similar drug loads.
- drugs with linkers having a polyethylene glycol spacer (-CH 2 CH 2 O) n and a reactive group capable of reacting with a cell-binding agent are described.
- Z represents a reactive functionality that can form an amide or a thioether bond with a cell-binding agent
- D represents a drug
- X represents an aliphatic, an aromatic or a heterocyclic group attached to the cell- binding agent via a thioether bond, an amide bond, a carbamate bond, or an ether bond
- Y represents an aliphatic, an aromatic or a heterocyclic group attached to the drug via a covalent bond selected from the group consisting of a thioether bond, an amide bond, a carbamate bond, an ether bond, an amine bond, a carbon-carbon bond and a hydrazone bond
- l is O or 1
- p is 0 or 1
- n is an integer from 1 to 2000.
- the covalent bond that attaches Y to the drug is a thioether bond or an amide bond.
- n is an integer from 1 to 100. Even more preferably, n is an integer from 1 to 14. In the most preferable aspect n is an integer from 1 to 4.
- novel conjugates of cell-binding agents and drugs with polyethylene glycol linkers (-CH 2 CH 2 O) n are described. These conjugates are more potent toward cancer cells than conjugates with traditional linkers and equivalent drug loads.
- CB represents a cell-binding agent
- D represents a drug
- X represents an aliphatic, an aromatic or a heterocyclic group attached to the cell- binding agent via a thioether bond, an amide bond, a carbamate bond, or an ether bond
- Y represents an aliphatic, an aromatic, or a heterocyclic group attached to the drug via a covalent bond selected from the group consisting of a thioether bond, an amide bond, a carbamate bond, an ether bond, an amine bond, a carbon-carbon bond and a hydrazone bond
- 1 is 0 or 1
- p is 0 or 1
- m is an integer from 2 to 15
- n is an integer from 1 to 2000.
- the covalent bond is a thioether bond or an amide bond.
- m is an integer from 3 to 8.
- n is an integer from 1 to 100. Even more preferably, n is an integer from 1 to 14. In the most preferable aspect, n is an integer from 1 to 4.
- the present invention is also based on the novel finding that in the case of antibody conjugates, wherein the antibody is linked to cytotoxic drugs via disulfide bonds, there is a critical correlation between the number of drugs linked and the length of the polyethylene glycol spacer in enhancing the potency or the efficacy of the immunoconjugate. The additional benefit of this linker design is the desired high monomer ratio and the minimal aggregation of the antibody-drug conjugate.
- the present invention is based on the critical finding that when the polyethylene glycol spacer for a disulfide-linked conjugate consists of between 2 and 8 ethyleneoxy groups and the number of drugs linked ranges from 3 to 8, it gives antibody-drug conjugates the highest biological potency or efficacy and also gives the desired high monomer content.
- cytotoxic compound of formula (3) or a specific compound of formula (3'): Z-XH-CH 2 -CH 2 O-) n -Y-D (3)
- Z represents a reactive functionality that can form an amide or a thioether bond with a cell-binding agent
- D represents a drug
- X represents an aliphatic, an aromatic or a heterocyclic group attached to the cell- binding agent via a thioether bond, an amide bond, a carbamate bond, or an ether bond;
- Y represents an aliphatic, non-aromatic heterocyclic or aromatic heterocyclic group attached to the drug via a disulfide bond
- n is an integer from 1 to 14.
- n is an integer from 2 to 8.
- CB represents a cell-binding agent
- D represents a drug
- X represents an aliphatic, an aromatic or a heterocyclic group attached to the cell- binding agent via a thioether bond, an amide bond, a carbamate bond, or an ether bond;
- Y represents an aliphatic, an aromatic or a heterocyclic group attached to the drug via a disulfide bond; 1 is 0 or 1 ; m is an integer from 3 to 8; and n is an integer from 1 to 14. [93] Preferably, m is an integer from 3 to 6. [94] Also, preferably, n is an integer from 2 to 8.
- drugs are lipophilic molecules, which when conjugated to cell- binding agents such as antibodies often result in loss of yield due to protein aggregation or precipitation. Increasing the number of drugs per cell-binding agent typically results in worse protein aggregation and precipitation, and subsequent poor monomer percentage and low yields.
- the PEG linkers result in a desirable improvement in monomer percentage (>90% monomer) and yield (>70%) of the conjugates of cell-binding agents with drugs at high concentrations of 1 mg/ml or greater that are useful for therapeutic applications. In addition, these conjugates are stable upon prolonged storage at 4 0 C.
- novel maytansinoids having a thioether moiety that bears a reactive group are disclosed such that those compounds are represented by the formula (5):
- V is an optional linear, branched or cyclic alkyl, alkenyl or alkynyl group preferably having from 1 to 10 carbon atoms; more preferably a linear alkyl having 1-5 carbon atoms, and still more preferably V is a one carbon alkyl group (CH 2 );
- W is an optional linear, branched or cyclic alkyl, alkenyl or alkynyl group having from 1 to 10 carbon atoms; more preferably having 2-8 carbon atoms, still more preferably W is a cyclohexyl group;
- D' represents a sulfhydryl-bearing maytansinoid, and more preferably it is selected from DMl, DM3 andDM4;
- Y' represents a thioether bond
- Q represents an optional aromatic or a heterocyclic moiety, and preferably Q is absent
- Z' represents an amine reactive group or a thiol reactive group selected from, but not limited to, a N-hydroxy succinimide ester, N-hydroxysulfosuccinimide ester, para or ortho-nitro phenyl ester, dinitrophenyl ester, pentafluorophenyl ester and sulfo- tetrafluorophenyl ester; a maleimide or a haloacetamide, more preferably Z is a N- hydroxysuccinimide, N-hydroxysulfosuccinimide ester or a maleimide.
- reactive maytansinoid derivatives bearing a thioether moiety are prepared from a sulfhydryl-bearing maytansinoid (such as DMl and DM4) and a heterobifunctional crosslinker.
- the reaction sequence is represented by formula (6):
- V is an optional linear, branched or cyclic alkyl, alkenyl or alkynyl group preferably having from 1 to 10 carbon atoms; more preferably a linear alkyl having 1-5 carbon atoms, and still more preferably V is a one carbon alkyl group (CH 2 );
- W is an optional linear, branched or cyclic alkyl, alkenyl or alkynyl group having from 1 to 10 carbon atoms; more preferably having 2-8 carbon atoms, still more preferably W is a cyclohexyl group;
- Y reprsents a thiol-reactive groups elected from maeimide or haloacetamide, preferably a maleimide;
- D' represents a sulfhydryl-bearing maytansinoid, and more preferably it is selected from DMl, DM3 andDM4;
- Y' represents a thioether bond
- Q represents an optional aromatic or a heterocyclic moiety, and preferably Q is absent
- Z' represents an amine reactive group or a thiol reactive group selected from, but not limited to, a N-hydroxy succinimide ester, N-hydroxysulfosuccinimide ester, para or ortho-nitro phenyl ester, dinitrophenyl ester, pentafluorophenyl ester and sulfo- tetrafluorophenyl ester; a maleimide or a haloacetamide, more preferably Z is a N- hydroxysuccinimide, N-hydroxysulfosuccinimide ester or a maleimide.
- Y' represents a thioether bond.
- the invention also provides a process for the preparation of cytotoxic conjugates of maytansinoids and cell binding agents linked via a non-cleavable bond said process comprising reacting a cell binding agent with a compound of formula Z'-W-Q-V-Y'-D' to provide a cell binding agent conjugate of formula CB-(Z"- W-Q- V- Y'— D' ) m .
- W is an optional linear, branched or cyclic alkyl, alkenyl or alkynyl group having from 1 to 10 carbon atoms; more preferably having 2-8 carbon atoms, still more preferably W is a cyclohexyl group;
- Q represents an optional aromatic or a heterocyclic moiety, and preferably Q is absent;
- V is an optional linear, branched or cyclic alkyl, alkenyl or alkynyl group preferably having from 1 to 10 carbon atoms; more preferably a linear alkyl group having 1 -5 carbon atoms, and still more preferably V is a one carbon alkyl group (CH 2 );
- Y' represents a thioether bond
- D' represents a sulfhydryl-bearing maytansinoid, and more preferably it is selected from DMl, DM3 and DM4;
- CB represents a cell binding agent selected from an antibody, a single chain antibody, an antibody fragment, a peptide, growth factor, hormone, vitamin, or ankyrin repeat proteins (DARPins), preferably the cell binding agent is an antibody or an antibody fragment or a Darpin;
- DARPins ankyrin repeat proteins
- Z' represents an amine reactive group or a thiol reactive group selected from, but not limited to, a N-hydroxy succinimide ester, N-hydroxysulfosuccinimide ester, para or ortho-nitro phenyl ester, dinitrophenyl ester, pentafluorophenyl ester and sulfo- tetrafluorophenyl ester; a maleimide or a haloacetamide, more preferably Z is a N- hydroxysuccinimide, N-hydroxysulfosuccinimide ester or a maleimide;
- Z" represents a thioether bond or an amide bond
- the process can be conducted by mixing a solution of the cell binding agent , such as an antibody, in aqueous buffer, optionally containing up to 20% organic solvent. with the compound of formula Z' -W-Q-V-Y' -D' in organic solvent or a mixture of organic solvent and aqueous buffer or water, and allowing the reaction to proceed for between 5 min to 72 hours.
- the conjugate can be further purified by chromatography, dialysis, tangential flow filtration or a combination of these
- an "aliphatic group” is defined as alkyl, alkenyl or alkynyl group.
- An alkyl group is an aliphatic hydrocarbon group which may be straight or branched, preferably having 1 to 20 carbon atoms in the chain or cyclic, preferably having 3 to 10 carbon atoms. More preferred alkyl groups have 1 to 12 carbon atoms in the chain.
- Branched means that one or more lower alkyl groups such as methyl, ethyl or propyl are attached to a linear alkyl chain.
- exemplary alkyl groups include methyl, ethyl, n- propyl, i-propyl, n-butyl, t-butyl, n-pentyl, 3-pentyl, octyl, nonyl, decyl, cyclopentyl and cyclohexyl.
- An alkenyl group is an aliphatic hydrocarbon group containing a carbon-carbon double bond and which may be straight or branched, preferably having 2 to 15 carbon atoms in the chain. More preferred alkenyl groups have 2 to 12 carbon atoms in the chain; and more preferably about 2 to 4 carbon atoms in the chain. Exemplary alkenyl groups include ethenyl, propenyl, n-butenyl, i-butenyl, 3-methylbut-2-enyl, n-pentenyl, heptenyl, octenyl, nonenyl, decenyl.
- An alkynyl group is an aliphatic hydrocarbon group containing a carbon-carbon triple bond and which may be straight or branched, preferably having 2 to 15 carbon atoms in the chain. More preferred alkynyl groups have 2 to 12 carbon atoms in the chain; and more preferably 2 to 4 carbon atoms in the chain. Exemplary alkynyl groups include ethynyl, propynyl, n-butynyl, 2-butynyl, 3-methylbutynyl, n-pentynyl, heptynyl, octynyl and decynyl.
- aromatic group means a substituted or unsubstituted aryl group consisting of an aromatic monocyclic or multicyclic hydrocarbon ring system of 6 to 14 carbon atoms, preferably of 6 to 10 carbon atoms.
- aryl groups include phenyl and naphthyl.
- Substituents include, but are not limited to, alkyl groups, halogens, nitro, amino, hydroxyl and alkoxy groups.
- Halogens include fluorine, chlorine, bromine and iodine atoms. Fluorine and chlorine atoms are preferred.
- heterocyclic group refers to a saturated, partially unsaturated or unsaturated, non-aromatic stable 3 to 14, preferably 5 to 10 membered mono, bi or multicyclic rings wherein at least one member of the ring is a hetero atom, or an aromatic, preferably 5 to 10 membered mono-, bi- or multicyclic ring having at least one hetero atom.
- hetero atoms include, but are not limited to, oxygen, nitrogen, sulfur, selenium, and phosphorus atoms.
- Preferable hetero atoms are oxygen, nitrogen and sulfur.
- Preferred heterocyclic groups include, but are not limited to, pyrrolidinyl, pyrazolidinyl, imidazolidinyl, oxiranyl, tetrahydrofuranyl, dioxolanyl, tetrahydro-pyranyl, dioxanyl, dioxolanyl, piperidyl, piperazinyl, morpholinyl, pyranyl, imidazolinyl, pyrrolinyl, pyrazolinyl, thiazolidinyl, tetrahydrothiopyranyl, dithianyl, thiomorpholinyl, dihydro- pyranyl, tetrahydropyranyl, dihydropyranyl, tetrahydro-pyridyl, dihydropyridyl, tetrahydropyrinidinyl, dihydrothiopyranyl, azepanyl, pyrrolyl, pyrid
- the aliphatic, aromatic and heterocyclic groups represented by X and Y can also possess a charged substituent.
- the charged substituent can be negatively charged selected from, but not limited to carboxylate, sulfonate and phosphates, or positively charged selected from a tertiary or quaternary amino group.
- the expression "linked to a cell-binding agent” refers to the conjugate molecule comprising at least one drug derivative bound to a cell-binding agent via a suitable linking group, or a precursor thereof.
- Preferred linking groups are thiol or disulfide bonds, or precursors thereof.
- precursor of a given group refers to any group which may lead to that group by any deprotection, chemical modification, or coupling reaction.
- a precursor could be an appropriately protected functionality exemplified by a thioester or thioether as a thiol precursor.
- the term "reactive functionality” refers to an amine-, a thiol- or a hydroxyl-reactive functionality.
- the reactive functionality can react with amine, sulfhydryl (thiol), or hydroxyl group present on cell-binding agent.
- the functionality could be a reactive carboxylic ester
- a linker is any chemical moiety that is capable of linking a drug, such as a maytansinoid, to a cell-binding agent in a stable, covalent manner.
- Linkers can be susceptible to or be substantially resistant to acid-induced cleavage, light-induced cleavage, peptidase-induced cleavage, esterase-induced cleavage, and disulfide bond cleavage, at conditions under which the drug or the cell-binding agent remains active.
- Figures 1, 2 and 3 exemplarily provide structural representations of conjugates of the present invention.
- Suitable crosslinking reagents comprising hydrophilic PEG chains that form linkers between a drug and the cell-binding agent are well known in the art, or are commercially available (for example from Quanta Biodesign, Powell, Ohio). Suitable PEG-containing crosslinkers can also be synthesized from commercially available PEGs themselves using standard synthetic chemistry techniques known to one skilled in the art. The drugs can be reacted with bifunctional PEG-containing cross linkers to give compounds of formula (1), Z -Xi-(-CH 2 -CH 2 -O-) n -Yp-D, by methods described herein.
- a thiol-containing maytansinoid drug can be reacted with a bis- maleimido crosslinking agent having a PEG spacer to give a maytansinoid drug linked via a thioether bond to the PEG spacer ( see for example Figure 13).
- This modified maytansinoid having a PEG spacer and a terminal maleimido group can then be reacted with a cell binding agent as shown for example in Figure 14, to provide a cell binding agent-drug conjugate of formula (2) of the present invention.
- the cell binding agent can be first reacted at one end of the bifunctional PEG containing cross linker bearing an amine reactive group, such as a N- hydroxysuccinimide ester, to give a modified cell binding agent covalently bonded to the linker through an amide bond (see for example Figure 15).
- the maytansinoid reacts with the maleimido substituent on the other end of the PEG spacer to give a cell-binding agent-drug conjugate of the present invention.
- Figures 16 and 17 shows by means of exemplification the synthesis of a PEG cross linking agent and its reaction with maytansinoid through a thioacetamido link.
- a maleimido substituent is then incorporated into the PEG to enable reaction with a cell binding agent via a thioether bond.
- the cell binding agent is first linked to the PEG crosslinker through a thioether bond.
- the modified cell binding agent is then reacted with a maytansinoid drug to give a conjugate.
- the synthesis of a homobifunctional PEG crosslinker, wherein both ends of the PEG spacer contain an iodoacetamido moiety that enable linkage of both the cytotoxic drug and the cell binding agent via thioether bonds to give a conjugate containing a hydrophilic PEG spacer is shown for example in Figure 19.
- 7,301,019) can be reacted with an iodoacetyl PEG (shown in Figure 5), in the presence of a base, such as pyridine or triethylamine, to provide a maytansinoid linked to the PEG via a amine link.
- a base such as pyridine or triethylamine
- the carboxy-PEG shown in Figure 5
- an amine-containing maytansinoid in the presence of a condensing agent, such as dicyclcohexylcarbodiimide, to provide an amide bonded PEG-maytansinoid.
- a condensing agent such as dicyclcohexylcarbodiimide
- the PEG is first reacted with diphosgene to provide a PEG chloroformate, which can then be reacted with an amine-containing maytansinoid, in the presence of a base such as triethylamine, to give a carbamate linked PEG-maytansinoid.
- suitable linkers include linkers having an iV-succinimidyl ester or N- sulfosuccinimidyl ester moiety for reaction with the cell-binding agent, as well as a maleimido- or haloacetyl -based moiety for reaction with the drug.
- a PEG spacer can be incorporated into any crosslinker known in the art by the methods described herein.
- Crosslinking reagents comprising a maleimido-based moiety that can be incorporated with a PEG spacer include, but is not limited to, N-succinimidyl 4-(maleimidomethyl) cyclohexanecarboxylate (SMCC), 7V-succinimidyl-4-( ⁇ '-maleimidomethyl)-cyclohexane- l-carboxy-(6-amidocaproate), which is a "long chain" analog of SMCC (LC-SMCC), K- maleimidoundecanoic acid N-succinimidyl ester (KMUA), ⁇ -maleimidobutyric acid N- succinimidyl ester (GMBS), ⁇ -maleimidocaproic acid N-hydroxysuccinimide ester (EMCS), m-maleimidobenzoyl-N-hydroxys
- Cross-linking reagents comprising a haloacetyl-based moiety include N-succinimidyl-4-(iodoacetyl)- aminobenzoate (SIAB), N-succinimidyl iodoacetate (SIA), N-succinimidyl bromoacetate (SBA), and N-succinimidyl 3-(bromoacetamido)propionate (SBAP).
- Other crosslinking reagents lacking a sulfur atom can also be used in the inventive method.
- Such linkers can be derived from dicarboxylic acid based moieties. Suitable dicarboxylic acid based moieties include, but are not limited to, ⁇ , ⁇ - dicarboxylic acids of the general formula shown below:
- A' is an optional linear or branched alkyl, alkenyl, or alkynyl group having 2 to 20 carbon atoms
- E' is an optional cycloalkyl or cycloalkenyl group having 3 to 10 carbon atoms
- G' is an optional substituted or unsubstituted aromatic group bearing 6 to 10 carbon atoms, or a substituted or unsubstituted heterocyclic group wherein the hetero atom is selected from ⁇ , O or S, and wherein p, q and r are each 0 or 1 , provided that p, q, and r are all not zero at the same time, n is an integer from 1 to 2000.
- the cell-binding agent is modified by reacting a bifunctional crosslinking reagent with the cell-binding agent, thereby resulting in the covalent attachment of a linker molecule to the cell-binding agent.
- a bifunctional crosslinking reagent is any chemical moiety that covalently links a cell- binding agent to a drug, such as the drugs described herein. In a preferred aspect of the invention, a portion of the linking moiety is provided by the drug.
- the drug comprises a linking moiety that is part of a larger linker molecule that is used to join the cell-binding agent to the drug.
- a linking moiety that is part of a larger linker molecule that is used to join the cell-binding agent to the drug.
- the side chain at the C-3 hydroxyl group of maytansine is modified to have a free sulfhydryl group (SH).
- SH free sulfhydryl group
- This thiolated form of maytansine can react with a modified cell-binding agent to form a conjugate. Therefore, the final linker is assembled from two components, one of which is provided by the crosslinking reagent, while the other is provided by the side chain from DMl .
- the drug is linked to a cell-binding agent through a disulfide bond.
- the linker molecule comprises a reactive chemical group that can react with the cell-binding agent.
- Preferred reactive chemical groups for reaction with the cell-binding agent are N-succinimidyl esters and N-sulfosuccinimidyl esters.
- the linker molecule comprises a reactive chemical group, preferably a dithiopyridyl group that can react with the drug to form a disulfide bond.
- linker molecules include, for example, N-succinimidyl 3-(2-pyridyldithio) propionate (SPDP) (see, e.g., Carlsson et al., Biochem. J, 173: 723-737 (1978)), N- succinimidyl 4-(2-pyridyldithio)butanoate (SPDB) (see, e.g., U.S. Patent No. 4,563,304), N-succinimidyl 4-(2-pyridyldithio)pentanoate (SPP) (see, e.g., CAS Registry number 341498-08-6), and other reactive cross-linkers, such as those described in U.S.
- SPDP N-succinimidyl 3-(2-pyridyldithio) propionate
- SPDB N- succinimidyl 4-(2-pyridyldithio)butanoate
- SPP N-succ
- the drug can be first modified to introduce a reactive ester suitable to react with a cell-binding agent. Reaction of these drugs containing an activated linker moiety with a cell-binding agent provides another method of producing a cell-binding agent drug conjugate.
- siRNAs can be linked to the crosslinkers of the present invention by methods commonly used for the modification of oligonucleotides (see, for example, US Patent Publications 20050107325 and 20070213292).
- siRNA in its 3' or 5'- phosphoromidite form is reacted with one end of the crosslinker bearing a hydroxyl functionality to give an ester bond between the siRNA and the crosslinker.
- reaction of the siRNA phosphoramidite with a crosslinker bearing a terminal amino group results in linkage of the crosslinker to the siRNA through an amine.
- the cell-binding agents used in this invention are proteins (e.g., immunoglobulin and non-immunoglobulin proteins) that bind specifically to target antigens on cancer cells.
- proteins e.g., immunoglobulin and non-immunoglobulin proteins
- These cell-binding agents include the following:
- -humanized or fully human antibodies are selected from, but not limited to, huMy9-6, huB4, huC242, huN901, DS6, CD38, IGF-IR, CNTO 95, B-B4, trastuzumab, bivatuzumab, sibrotuzumab, pertuzumab and rituximab (see, e.g., U.S. Patent Nos. 5,639,641 , 5,665,357, and 7,342,1 10; U.S. Provisional Patent Application No. 60/424,332, International Patent Application WO 02/16,401 , U.S. Patent Publication Number 20060045877, U.S.
- Patent Publication Number 20060127407 U.S. Patent Publication No. 200501 18183, Pedersen et al, (1994) J MoI. Biol. 235, 959-973, Roguska et al., (1994) Proceedings of the National Academy of Sciences, VoI 91 , 969-973, Colomer et al., Cancer Invest., 19: 49-56 (2001 ), Heider et al., Eur. J. Cancer, 31A: 2385-2391 (1995), Welt et al., J Clin. Oncol, 12: 1 193-1203 (1994), and Maloney et al., Blood, 90: 2188-2195 (1997).); and
- Additional cell-binding agents include other cell-binding proteins and polypeptides exemplified by, but not limited to:
- -interferons e.g. ⁇ , ⁇ , ⁇
- -lymphokines such as IL-2, IL-3, IL-4, IL-6;
- -hormones such as insulin, TRH (thyrotropin releasing hormones), MSH (melanocyte- stimulating hormone), steroid hormones, such as androgens and estrogens; and -growth factors and colony-stimulating factors such as EGF, TGF- ⁇ , IGF-I, G-CSF, M-CSF and GM-CSF (Burgess, Immunology Today 5:155-158 (1984)).
- the cell-binding agent binds to an antigen that is a polypeptide and may be a transmembrane molecule (e.g. receptor) or a ligand such as a growth factor.
- antigens include molecules such as renin; a growth hormone, including human growth hormone and bovine growth hormone; growth hormone releasing factor; parathyroid hormone; thyroid stimulating hormone; lipoproteins; alpha- 1 -antitrypsin; insulin A-chain; insulin B-chain; proinsulin; follicle stimulating hormone; calcitonin; luteinizing hormone; glucagon; clotting factors such as factor vmc, factor IX, tissue factor (TF), and von Willebrands factor; anti-clotting factors such as Protein C; atrial natriuretic factor; lung surfactant; a plasminogen activator, such as urokinase or human urine or tissue-type plasminogen activator (t-PA); bombesin;
- GM-CSF which binds to myeloid cells can be used as a cell- binding agent to diseased cells from acute myelogenous leukemia.
- IL-2 which binds to activated T-cells can be used for prevention of transplant graft rejection, for therapy and prevention of graft- versus-host disease, and for treatment of acute T-cell leukemia.
- MSH which binds to melanocytes, can be used for the treatment of melanoma.
- Folic acid can be used to target the folate receptor expressed on ovarian and other tumors.
- Epidermal growth factor can be used to target squamous cancers such as lung and head and neck.
- Somatostatin can be used to target neuroblastomas and other tumor types.
- Cancers of the breast and testes can be successfully targeted with estrogen (or estrogen analogues) or androgen (or androgen analogues) respectively as cell-binding agents.
- Preferred antigens for antibodies encompassed by the present invention include CD proteins such as CD2, CD3, CD4, CD5, CD6, CD8, CDl 1, CD 14, CD18, CD19, CD20, CD 21, CD22, CD 25, CD26, CD27, CD28, CD30, CD33, CD36, CD37, CD38, CD40, CD44, CD52, CD55, CD56, CD70, CD79, CD80, CD81, CD103, CD105, CD134, CD137, CD138, and CD152; members of the ErbB receptor family such as the EGF receptor, HER2, HER3 or HER4 receptor; cell adhesion molecules such as LFA-I, Macl, pi 50.95, VLA-4, ICAM-I, VCAM, EpCAM, alpha4/beta7 integrin, and alpha v/beta3 integrin including either alpha or beta subunits thereof (e.g.
- anti-CDl Ia, anti- CDl 8 or anti-CDl Ib antibodies growth factors such as VEGF; tissue factor (TF); TGF- ⁇ .; alpha interferon (alpha-IFN); an interleukin, such as IL-8; IgE; blood group antigens Apo2, death receptor; flk2/flt3 receptor; obesity (OB) receptor; mpl receptor; CTLA-4; protein C etc.
- growth factors such as VEGF; tissue factor (TF); TGF- ⁇ .; alpha interferon (alpha-IFN); an interleukin, such as IL-8; IgE; blood group antigens Apo2, death receptor; flk2/flt3 receptor; obesity (OB) receptor; mpl receptor; CTLA-4; protein C etc.
- the most preferred targets herein are IGF-IR, CanAg, EphA2, MUCl , MUCl 6, VEGF, TF, CDl 9, CD20, CD22, CD27, CD33, CD37, CD38, CD40, CD44, CD56, CDl 38, CA6, Her2/neu, EpCAM, CRIPTO (a protein produced at elevated levels in a majority of human breast cancer cells), darpins, alpha v /beta 3 integrin, alpha v /beta 5 integrin, alpha y/betae integrin, TGF- ⁇ , CDl Ia, CDl 8, Apo2 and C242 or an antibody which binds to one or more tumor-associated antigens or cell- surface receptors disclosed in US Publication No.
- Preferred antigens for antibodies encompassed by the present invention also include CD proteins such as CD3, CD4, CD8, CD19, CD20, CD27, CD34, CD37, CD38, CD46, CD56, CD70 and CDl 38; members of the ErbB receptor family such as the EGF receptor, HER2, HER3 or HER4 receptor; cell adhesion molecules such as LFA-I, Macl, pl50.95, VLA-4, ICAM-I, VCAM, EpCAM, alpha4/beta7 integrin, and alpha v/beta3 integrin including either alpha or beta subunits thereof (e.g.
- anti-CD 1 Ia, anti-CD 18 or anti-CD 1 Ib antibodies growth factors such as VEGF; tissue factor (TF); TGF- ⁇ .; alpha interferon (alpha-IFN); an interleukin, such as IL-8; IgE; blood group antigens Apo2, death receptor; flk2/flt3 receptor; obesity (OB) receptor; mpl receptor; CTLA-4; protein C, etc.
- growth factors such as VEGF; tissue factor (TF); TGF- ⁇ .; alpha interferon (alpha-IFN); an interleukin, such as IL-8; IgE; blood group antigens Apo2, death receptor; flk2/flt3 receptor; obesity (OB) receptor; mpl receptor; CTLA-4; protein C, etc.
- the most preferred targets herein are IGF-IR, CanAg, EGF-R, EGF-RvIII, EphA2, MUCl, MUC16, VEGF, TF, CD19, CD20, CD22, CD27, CD33, CD37, CD38, CD40, CD44, CD56, CD70, CD 138, CA6, Her2/neu, CRIPTO (a protein produced at elevated levels in a majority of human breast cancer cells), alpha v /beta 3 integrin, alpha v /beta 5 integrin, TGF- ⁇ , CDl Ia, CDl 8, Apo2, EpCAM and C242. [129] Monoclonal antibody techniques allow for the production of specific cell-binding agents in the form of monoclonal antibodies.
- the monoclonal antibody My9 is a murine IgG 2a antibody that is specific for the CD33 antigen found on Acute Myeloid Leukemia (AML) cells (Roy et al. Blood 77:2404-2412 (1991)) and can be used to treat AML patients.
- the monoclonal antibody anti-B4 is a murine IgGi that binds to the CD 19 antigen on B cells (Nadler et al, J Immunol. 131 :244-250 (1983)) and can be used if the target cells are B cells or diseased cells that express this antigen such as in non-Hodgkin's lymphoma or chronic lymphoblastic leukemia.
- the antibody N901 is a murine monoclonal IgGi antibody that binds to CD56 found on small cell lung carcinoma cells and on cells of other tumors of neuroendocrine origin (Roy et al. J Nat. Cancer Inst. 88:1136-1145 (1996)); huC242 is an antibody that binds to the CanAg antigen; Trastuzumab is an antibody that binds to HER2/neu; and anti-EGF receptor antibody binds to EGF receptor.
- the drugs used in this invention are cytotoxic drugs capable of being linked to a cell-binding agent.
- suitable drugs include maytansinoids, DNA-binding drugs such as CC-1065 and its analogs, calicheamicins, doxorubicin and its analogs, vinca alkaloids, cryptophycins, dolastatin, auristatin and analogs thereof, tubulysin, epothilones, taxoids and siRNA.
- Preferred maytansinoids are those described in U.S. Patent Nos. 5,208,020; 5,416,064; 6,333.410; 6,441,163; 6,716,821; RE39,151 and 7,276,497.
- Preferred CC- 1065 analogs are those described in U.S. Patent Nos. 5,475,092; 5,595,499; 5,846,545; 6,534,660; 6,586,618; 6,756,397 and 7,049,316.
- Preferred doxorubicins and it analogs are those described in U.S. Patent No. 6,630,579.
- Preferred taxoids are those described in U.S. Patent Nos.
- Vinca alkaloid compounds, dolastatin compounds, and cryptophycin compounds are describe in detail in WO01/24763.
- Auristatin include auristatin E, auristatin EB (AEB), auristatin EFP (AEFP), monomethyl auristatin E (MMAE) and are described in U.S. Patent No. 5,635,483, Int. J. Oncol. 15:367-72 (1999); Molecular Cancer Therapeutics, vol. 3, No. 8, pp. 921-932 (2004); U.S. Application Number 11/134826.
- Tubulysin compounds are described in U.S. Patent Publication Nos. 20050249740.
- Cryptophycin compounds are described in U.S. Patent Nos. 6,680,31 1 and 6,747,021. Epothilones are described in U.S. Patent Nos. 6,956,036 and 6,989,450.
- siRNA is described in detail in U.S. Patent Publication Numbers: 20070275465, 20070213292, 20070185050, 20070161595, 20070054279, 20060287260, 20060035254, 20060008822, 20050288244, 20050176667.
- cytotoxic agents [136]
- each of the cytotoxic agents described herein can be modified in such a manner that the resulting compound still retains the specificity and/or activity of the starting compound.
- the skilled artisan will also understand that many of these compounds can be used in place of the cytotoxic agents described herein.
- the cytotoxic agents of the present invention include analogues and derivatives of the compounds described herein.
- the cell-binding agent can be conjugated to the cytotoxic drugs by methods previously described (U.S. Patent Nos. 6,013,748; 6,441,1631, and 6,716,821 ; U.S. Patent Publication No. 20050169933; and WO2006/034488 A2).
- the cell-binding agent drug conjugates (e.g., immunoconjugates) of this invention can also be used in combination with chemotherapeutic agents. Such chemotherapeutic agents are described in U.S. Patent No. 7,303,749.
- the cell-binding agent drug conjugates (e.g., immunoconjugates) of the present invention can be administered in vitro, in vivo and/or ex vivo to treat patients and/or to modulate the growth of selected cell populations including, for example, cancer of the lung, blood, plasma, breast, colon, prostate, kidney, pancreas, brain, bones, ovary, testes, and lymphatic organs; autoimmune diseases, such as systemic lupus, rheumatoid arthritis, and multiple sclerosis; graft rejections, such as renal transplant rejection, liver transplant rejection, lung transplant rejection, cardiac transplant rejection, and bone marrow transplant rejection; graft versus host disease; viral infections, such as CMV infection, HIV infection, and AIDS; and
- the immunoconjugates and chemotherapeutic agents of the invention are administered in vitro, in vivo and/or ex vivo to treat cancer in a patient and/or to modulate the growth of cancer cells, including, for example, cancer of the blood, plasma, lung, breast, colon, prostate, kidney, pancreas, brain, bones, ovary, testes, and lymphatic organs; more preferably lung, colon prostrate, plasma, blood or colon cancer.
- the cancer is multiple myeloma.
- Modulating the growth of selected cell populations includes inhibiting the proliferation of selected cell populations (e.g., multiple myeloma cell populations, such as MOLP-8 cells, OPM2 cells, H929 cells, and the like) from dividing to produce more cells; reducing the rate of increase in cell division as compared, for example, to untreated cells; killing selected cell populations; and/or preventing selected cell populations (such as cancer cells) from metastasizing.
- the growth of selected cell populations can be modulated in vitro, in vivo or ex vivo.
- the cell-binding agent drug conjugates e.g., immunoconjugates
- the cell- binding agent drug conjugates can be used with suitable pharmaceutically acceptable carriers, diluents, and/or excipients, which are well known, and can be determined, by one of skill in the art as the clinical situation warrants.
- suitable carriers, diluents and/or excipients include: (1) Dulbecco's phosphate buffered saline, pH about 6.5, which would contain about 1 mg/ml to 25 mg/ml human serum albumin, (2) 0.9% saline (0.9% w/v NaCl), and (3) 5% (w/v) dextrose.
- compositions described herein may be administered in appropriate form, preferably parenterally, more preferably intravenously.
- the compounds or compositions can be aqueous or nonaqueous sterile solutions, suspensions or emulsions.
- Propylene glycol, vegetable oils and injectable organic esters, such as ethyl oleate, can be used as the solvent or vehicle.
- the compositions can also contain adjuvants, emulsifiers or dispersants.
- the compositions can also be in the form of sterile solid compositions that can be dissolved or dispersed in sterile water or any other injectable sterile medium.
- the "therapeutically effective amount" of the cell-binding agent drug conjugates (e.g., immunoconjugates) described herein refers to the dosage regimen for modulating the growth of selected cell populations and/or treating a patient's disease, and is selected in accordance with a variety of factors, including the age, weight, sex, diet and medical condition of the patient, the severity of the disease, the route of administration, and pharmacological considerations, such as the activity, efficacy, pharmacokinetic and toxicology profiles of the particular compound used.
- the "therapeutically effective amount” can also be determined by reference to standard medical texts, such as the Physicians Desk Reference 2004.
- the patient is preferably an animal, more preferably a mammal, most preferably a human.
- the patient can be male or female, and can be an infant, child or adult.
- cell-binding agent drug conjugates e.g., immunoconjugate
- the conjugates can be given daily for about 5 days either as an i.v., bolus each day for about 5 days, or as a continuous infusion for about 5 days.
- the conjugates can be administered once a week for six weeks or longer.
- the conjugates can be administered once every two or three weeks.
- Bolus doses are given in about 50 to about 400 ml of normal saline to which about 5 to about 10 ml of human serum albumin can be added.
- Continuous infusions are given in about 250 to about 500 ml of normal saline, to which about 25 to about 50 ml of human serum albumin can be added, per 24 hour period. Dosages will be about 10 pg to about 1000 mg/kg per person, i.v. (range of about 100 ng to about 100 mg/kg).
- the compounds and conjugates can also be used for the manufacture of a medicament useful for treating or lessening the severity of disorders, such as, characterized by abnormal growth of cells (e.g., cancer).
- the present invention also provides pharmaceutical kits comprising one or more containers filled with one or more of the ingredients of the pharmaceutical compounds and/or compositions of the present invention, including, one or more immunoconjugates and one or more chemotherapeutic agents.
- kits can also include, for example, other compounds and/or compositions, a device(s) for administering the compounds and/or compositions, and written instructions in a form prescribed by a governmental agency regulating the manufacture, use or sale of pharmaceuticals or biological products.
- Cancer therapies and their dosages, routes of administration and recommended usage are known in the art and have been described in such literature as the Physician's Desk Reference (PDR).
- the PDR discloses dosages of the agents that have been used in treatment of various cancers.
- the dosing regimen and dosages of these aforementioned chemotherapeutic drugs that are therapeutically effective will depend on the particular cancer being treated, the extent of the disease and other factors familiar to the physician of skill in the art and can be determined by the physician.
- Taxotere is an inhibitor of tubulin depolymerization
- Doxorubicin see p 786
- Doxil see p 3302
- oxaliplatin see p 29078
- Irinotecal see p. 2602
- Erbitux see p 937
- Tarceva see p 2470
- the contents of the PDR are expressly incorporated herein in their entirety by reference.
- One of skill in the art can review the PDR, using one or more of the following parameters, to determine dosing regimen and dosages of the chemotherapeutic agents and conjugates that can be used in accordance with the teachings of this invention. These parameters include:
- Novel maytansinoids having a non-cleavable thioether moiety that bears a reactive group are compounds of formula (5), D'-Y'-V-Q-W-Z'.
- Chemical groups Z' for reaction with the cell-binding agent include but are not limited to iV-succinimidyl esters, iV-sulfosuccinimidyl esters, pentafluorophenyl ester, tetrafluorosulfophenyl, and nitrophenyl ester. The method of preparation of these compounds are described herein.
- Sulfhydryl-bearing maytansinoids are covalently linked via a non-cleavable thioether bond to a heterobifuntional crosslinker that bears a reactive group and isolated prior to conjugation with a cell binding agent.
- a non-cleavable linker is any chemical moiety that is capable of linking a cytotoxic drug to a cell-binding agent in a stable, covalent manner.
- Non-cleavable linkers are substantially resistant to acid-induced cleavage, light-induced cleavage, peptidase-induced cleavage, esterase-induced cleavage, and disulfide bond cleavage.
- Examples of non-cleavable linkers include linkers having an iV-succinimidyl ester, or an TV-sulfosuccinimidyl ester moiety and maleimido- or haloacetyl -based moiety for reaction with the maytansinoid.
- Crosslinking reagents comprising a maleimido-based moiety include iV-succinimidyl 4-(maleimidomethyl)cyclohexanecarboxylate (SMCC), iV-succinimidyl-4-(N-maleimidomethyl)-cyclohexane-l-carboxy-(6-amidocaproate), which is a "long chain" analog of SMCC (LC-SMCC), ⁇ -maleimidoundecanoic acid N- succinimidyl ester (KMUA), ⁇ -maleimidobutyric acid iV-succinimidyl ester (GMBS), ⁇ - maleimidocaproic acid N-hydroxysuccinimide ester (EMCS), m-maleimidobenzoyl-iV- hydroxysuccinimide ester (MBS), N-( ⁇ -maleimidoacetoxy)-succinimide ester (AMAS), succinimidyl-6
- Cross-linking reagents comprising a haloacetyl-based moiety include iV-succinimidyl-4- (iodoacetyl)-aminobenzoate (SIAB), 7V-succinimidyl iodoacetate (SIA), iV-succinimidyl bromoacetate (SBA), and iV-succinimidyl 3-(bromoacetamido)propionate (SBAP).
- SIAB iV-succinimidyl-4- (iodoacetyl)-aminobenzoate
- SIA 7V-succinimidyl iodoacetate
- SBA iV-succinimidyl bromoacetate
- SBAP iV-succinimidyl 3-(bromoacetamido)propionate
- Maytansinoids that can be used in the present invention to produce the reactive maytansinoid derivatives bearing a thioether moiety that can be covalently linked to a cell binding agent are well known in the art and can be isolated from natural sources according to known methods or prepared synthetically according to known methods.
- DMl termed N 2 -deacetyl-N 2 -(3-mercapto-l-oxopropyl)- maytansine, represented by structural formula (l):and DM4, termed N 2 -deacetyl-N 2 -(4- mercapto-4-methyl-l-oxopentyl)maytansine represented by structural formula (2) are shown below.:
- Novel maytansinoids having a thioether moiety that bears a reactive group are prepared by the following newly described methods.
- W is an optional linear, branched or cyclic alkyl, alkenyl or alkynyl group preferably having from 1 to 10 carbon atoms; more preferably containing 1 -5 atoms.
- Q represents an optional aromatic or a heterocyclic moiety; and Z' is an amine-reactive group such as but not limited to /V-succinimidyl esters, 7V-sulfosuccinimidyl esters, pentafluorophenyl ester, tetrafluorosulfophenyl, or nitrophenyl ester.
- the reaction comprises of the sequence (6):
- the reaction may be performed in a suitable organic solvent or a mixture of aqueous buffer and organic solvent such that the thiol-bearing maytansinoid, D', and the heterobifunctional linker, V-V-Q-W-Z', are fully soluble.
- suitable organic solvents include tetrahydrofuran (THF), 1,2-dimethoxy ethane, ⁇ iV-dimethylformamide (DMF), iV,7V-dimethylacetamide (DMA), methanol and ethanol.
- Organic bases such as N,N- diisopropylethylamine (DIPEA), l,8-Diazabicyclo[5.4.0]undec-7-ene (DBU), triethylamine and 4-methylmorpholine may be used to form the desired thioether- containing maytansinoids bearing a reactive group of the formula (5) D'-Y'-V-Q-W-Z'.
- DIPEA N,N- diisopropylethylamine
- DBU l,8-Diazabicyclo[5.4.0]undec-7-ene
- triethylamine and 4-methylmorpholine may be used to form the desired thioether- containing maytansinoids bearing a reactive group of the formula (5) D'-Y'-V-Q-W-Z'.
- a compound of the formula (5) D'-Y'-V-Q-W-Z' may be isolated following completion of the reaction.
- Suitable techniques for the purification of a maytansinoid having a thioether moiety that bears an amine reactive group Z' include silica gel chromatography, normal or reverse-phase preparative high performace liquid chromatography (HPLC), preparative thin layer chromatography (TLC) and crystallization.
- HPLC normal or reverse-phase preparative high performace liquid chromatography
- TLC preparative thin layer chromatography
- the purity and identity of the isolated product of the formula (5) D'-Y'-V-Q-W-Z' may be established by analytical methods including HPLC, mass spectroscopy (MS), LC/MS, 1 H NMR and 13 C NMR.
- Novel maytansinoid derivatives containing a thioether moiety that are useful in the preparation of maytansinoids containing a non-cleavable, thioether moiety that bears a reactive group are described herein.
- V is an optional linear, branched or cyclic alkyl, alkenyl or alkynyl group preferably having from 1 to 10 carbon atoms; more preferably having 1-5 carbon atoms.
- W is an optional linear, branched or cyclic alkyl, alkenyl or alkynyl group having from 1 to 10 carbon atoms; more preferably having 1-5 carbon atoms.
- D' represents a sulfhydryl-bearing maytansinoid, DMl or DM4;
- Y' represents a thioether bond
- Q represents an optional aromatic or a heterocyclic moiety.
- V and W are as defined above
- D' is as defined above ;
- Y is as defined above;
- Y' is a thioether bond between the sulfhydryl bearing maytansinoid and the carboxylic acid, COOH;
- Q represents an optional aromatic or a heterocyclic moiety.
- Stoichiometric or excess equivalents of the carboxylic acid linker of the formula Y"- V-Q-W-COOH, over the thiol-bearing maytansinoid, D', such as DMl and DM4, may be used.
- D' such as DMl and DM4
- the reaction proceeds to completion and can be monitored by analytical methods such as HPLC and TLC.
- the reaction may also be performed with an excess of the thiol-bearing maytansinoid, D', over the carboxylic acid linker of the formula Y"-V- Q-W-COOH.
- the reaction may be performed in a suitable organic solvent or a mixture of aqueous buffer and organic solvent such that the thiol-bearing maytansinoid, D', and the carboxylic acid linker of the formula V-V-Q-W-COOH, are fully soluble.
- suitable organic solvents include tetrahydrofuran (THF), 1 ,2-dimethoxyethane, NJN- dimethylformamide (DMF), ⁇ N-dimethylacetamide (DMA), methanol or ethanol.
- a suitable pH range for this reaction is pH 3 - 10 a preferred pH is 5- 9 and a most preferred pH is pH 6-8.
- Thioether formation between the thiol-bearing maytansinoid, D', and the thiol- reactive group, Y" will also occur when using a suitable organic base and neat organic solvent, such as those described above.
- Organic bases such as N,N- diisopropylethylamine (DIPEA), l,8-Diazabicyclo[5.4.0]undec-7-ene (DBU), triethylamine and 4-methylmorpholine may be used to form the desired thioether- containing maytansinoids bearing a carboxylic acid.
- a compound of the formula (10) D' -Y' -V-Q-W-COOH may be isolated following completion of the reaction. Suitable techniques for the purification of a maytansinoid having a thioether moiety that bears a carboxylic acid include silica gel chromatography, normal or reverse-phase preparative high performance liquid chromatography (HPLC), preparative thin layer chromatography (TLC) or crystallization. f.) The purity and identity of the isolated product of the formula (10) D'-Y'V-Q-W- COOH may be established by analytical methods including HPLC, mass spectroscopy (MS), LC/MS, 1 H NMR and 13 C NMR.
- novel maytansinoid derivatives containing a thioether moiety bearing a terminal carboxylic acid of the formula (1 1) D'-Y'-V-Q- W-COOH are useful in the preparation of compounds of formula (5) D'-Y'-V-Q- W-Z'.
- Maytansinoid derivatives of the formula (1 1) D'-Y'-V-Q-W-COOH may be further derivatized to give an an amine reactive group, Z.
- the preferred amine reactive group, Z' is an active ester such as as but not limited to iV-succinimidyl esters, 7V-sulfosuccinimidyl esters, pentafluorophenyl ester, tetrafluorosulfophenyl, or nitrophenyl ester.
- active ester such as but not limited to iV-succinimidyl esters, 7V-sulfosuccinimidyl esters, pentafluorophenyl ester, tetrafluorosulfophenyl, or nitrophenyl ester.
- a maytansinoid of the formula (5) D' -Y' -V-Q-W-Z' may be prepared from the novel maytansinoid derivative of formula (11) D'-Y'-V-Q-W-COOH by known methods described herein.
- D'-Y'-V-Q-W-COOH may be reacted with a slight excess of TV-hydroxysuccinimide (X) in a dry organic solvent (such as methylene chloride, dimethylformamide, tetrahydrofuran, dioxane, diethylether) in the presence of 1 -(3-dimethylaminopropyl)-3-ethyl-carbodiimide hydrochloride (EDCHCl).
- a dry organic solvent such as methylene chloride, dimethylformamide, tetrahydrofuran, dioxane, diethylether
- EDCHCl 1 -(3-dimethylaminopropyl)-3-ethyl-carbodiimide hydrochloride
- Condensing agents other than EDCHCl can be employed for the reaction.
- Completion of reaction may be monitored using standard chemical techniques such as TLC or HPLC.
- the maytansinoid derivative that bears a reactive 7V-hydroxysuccinimidyl ester may be purified by silica gel chromatography, or by HPLC, or by crystallization.
- a process for the preparation of a non-cleavable, thioether linked maytansinoid conjugate with a cell binding agent using an isolated, reactive maytansinoid derivative having a thioether moiety that bears a reactive group of the formula (8) CB-(Z"- W-Q-V- Y'-D') m ( Figures 39 and 40) is described and comprises of the following steps: a.) Covalently linking the purified amine-reactive maytansinoid bearing a thioether moiety of the formula (5) D' -Y' -W-Q-V-Z' of this invention, to a cell binding agent, CB (antibody), via covalent amide bond formation between lysine residues of the cell binding agent and the amine-reactive group linked to the may
- the conjugation reaction may be performed at a concentration of 0.5-10 mg/mL antibody depending on the nature of the antibody.
- a stock solution of the amine-reactive non-cleavable maytansinoid bearing a thioether moiety may be prepared in neat organic solvent prior to conjugation with the antibody.
- Suitable organic solvents for stock solution preparation include methanol, ethanol, N,iV-dimethylacetamide (DMA), ⁇ iV-dimethylformamide (DMF) and dimethyl sulfoxide (DMSO) c.) Due to the hydrophobic nature of maytansinoids it may be necessary to carry out the conjugation to antibody in a mixture of aqueous buffer and organic solvent to ensure that the maytansinoid remains in solution during conjugation.
- the preferred amount of organic solvent in the aqueous buffer ranges from 0-30% (v/v) depending on the solubility of the reagent and the behavior of the antibody under these conditions.
- the conjugation may be carried out at pH 3 - 10 a preferred pH is 5- 9 and a most preferred pH is pH 6-8.
- the buffers used for the conjugation reaction are buffers with pK a values around this pH range, such as phosphate and HEPES buffer.
- a 5-50 fold excess of the amine-reactive maytansinoid bearing a thioether moiety over antibody may be used in the conjugation reaction to yield a conjugate with the desired number of maytansinoid molecules linked per molecule of antibody.
- a range of 2-8 maytansinoids linked per antibody is desired for the final conjugate.
- conjugation methods include a one-step conjugation of antibody with drugs such as maytansinoids linked via polyethylene glycol ((CH 2 CH 2 O) n ) linker by reaction at 7V-hydroxysuccinimide (NHS) reactive group.
- drugs such as maytansinoids linked via polyethylene glycol ((CH 2 CH 2 O) n ) linker by reaction at 7V-hydroxysuccinimide (NHS) reactive group.
- conjugation methods include a one-step conjugation of antibody with drugs such as maytansinoid linked with disulfide-group having polyethylene glycol ((CH 2 CH 2 O) n ) linker via reaction at a iV-hydroxysuccinimide (NHS) reactive group.
- drugs such as maytansinoid linked with disulfide-group having polyethylene glycol ((CH 2 CH 2 O) n ) linker via reaction at a iV-hydroxysuccinimide (NHS) reactive group.
- a humanized antibody was first modified with a commercially available heterobifunctional linker (SPDB) containing both an amine- reactive N-hydroxysuccinimide group (NHS group) and a thiol -reactive 2-pyridyldithio group (-SSPy group) to incorporate several molecules of the linker in the antibody molecule (as described in W. C. Widdison et al., J Med. Chem., 2006, 49, 4392-4408).
- SPDB heterobifunctional linker
- the maytansinoid DM4 or DMl with a reactive thiol group was added to the linker-modified antibody to conjugate the maytansinoid to antibody by disulfide bonds.
- a humanized antibody at a concentration of 5-10 mg/ml was modified using 10-15 fold molar excess of the commercially available heterobifunctional linker with -(CH 2 )- n alkyl groups (such as SPDB, SPP, SPDP) in aqueous buffer at pH 6.5-8 for 0.25-3 h at ambient temperature and then purified by gel filtration (using, for example, Sephadex G25 chromatography) to obtain antibody modified with an average 8-12 linker groups per antibody molecule in high yields (typically 80-90% yields).
- the commercially available heterobifunctional linker with -(CH 2 )- n alkyl groups such as SPDB, SPP, SPDP
- DTT 1 ,4-dithiothreitol
- the numbers of incorporated maytansinoid per antibody molecule were much lower (-5.2-5.5 average maytansinoid molecules per antibody molecule) than expected based on the much greater average number of initial reactive linker groups incorporated per antibody molecule (-8-12 reactive linker groups per antibody molecule) suggesting precipitation of the higher maytansinoid-bearing antibody conjugates.
- a humanized antibody was first modified with the SPDB heterobifunctional linker to incorporate 1 1 pyridyldithio groups per antibody molecule, which upon a second reaction with 1.7 fold molar excess of DM4 maytansinoid thiol showed significant precipitation in the reaction mixture resulting in a very poor recovery of ⁇ 30% antibody- maytansinoid conjugate.
- Using commercially available heterobifunctional linkers such as SPDB or SPDP with aliphatic spacers it is typically difficult to incorporate greater than 4 or 5 maytansinoid molecules per antibody at high conjugation yields for antibody- maytansinoid conjugate concentrations of 1 mg/ml or higher concentrations.
- hydrophilic spacers such as polyethylene oxide (PEG n , or (-CH 2 -
- a humanized antibody at a concentration of 8 mg/ml was modified with the PySS-PEG 4 -NHS reagent at several fold molar excess over antibody concentration in pH 8 buffer for 1 h at 3O 0 C and then purified by gel filtration.
- the linked dithiopyridyl groups per antibody molecule were estimated to be -4-16 by 2-thiopyridone release assay of aliquots using excess dithiothereitol, based upon which a 1.4-fold molar excess of DM4 maytansinoid thiol was added to each dithiopyridyl-PEG n -linker modified antibody solution for the conjugation step at pH 6.5, overnight at 25°C, and then the conjugate was purified by gel filtration ( Figure 12).
- the final incorporated maytansinoid per antibody values for the different conjugation mixtures with different initial linker incorporations ranged from 3 to 9 average maytansinoid per antibody molecule, with no observed precipitation, >70% yields and very high monomer (>90% monomer based on size-exclusion TSK-GEL G3000 HPLC using 20% isopropanol or 0.4 M sodium perchlorate).
- the unconjugated drug in the final conjugates was determined to be less than 0.6% by HiSep Mixed-Mode chromatography (HiSep column, Supelco) indicating that maytansinoids were covalently linked to antibody.
- a humanized antibody at a concentration of 8 mg/ml was modified with PySS-PEG 4 -NHS reagent at several fold molar excess over antibody concentration in pH 6.5 buffer for 1.5 h at 25 0 C and then purified by gel filtration.
- the dithiopyridyl-PEG n -bearing linker groups on antibody samples were estimated as 6-18 per antibody molecule, which were then reacted with 1.3-1.7-fold molar excess of DM4 maytansinoid thiol at pH 6.5, 25 0 C overnight, and then purified by gel filtration.
- PEG n hydrophilic polyethyleneoxide spacers
- iV-hydroxysuccinimide esters of maytansinoids with traditional aliphatic linkers such as alkyl linkers derived from SPP (described in W.C. Widdison et al., J Med. Chem., 2006, 49, 4392-4408) were used initially to conjugate antibodies in a 1-step method.
- DMl-MaI-PEG 2 -NHS reagent was used to obtain high numbers of conjugated maytansinoids linked per antibody molecule via thioether bonds.
- a murine IgGj antibody was conjugated at 4 mg/ml with 10- and 20-fold molar excess OfDMl-MaI-PEG 4 -NHS reagent in pH 8 buffer for 2 h at 30 0 C followed by gel filtration to obtain antibody-maytansinoid conjugates at ⁇ 1 mg/ml concentration with 4.1 and 7.8 covalently conjugated maytansinoid molecules per antibody molecule (98% monomer) with undetectable levels of unconjugated drug (HiSep HPLC assay).
- a humanized antibody was conjugated with excess DMl-MaI-PEG 4 -NHS reagent to obtain average 10.7 linked maytansinoid molecules per antibody (99% monomer; 1.1 mg/ml concentration).
- the PEG 4 -I inked thioether conjugates were also prepared from antibodies using a two-step conjugation procedure outlined in Figure 8 and Figure 10. Therefore large number of maytansinoid molecules can be introduced per antibody molecule by the use of hydrophilic linkers such as PEG n or (-CH 2 -CH 2 -O) n (see, for example, Figures 1, 2, 4, 5, 7, 8, 9, 10, 13, 14, 15, 16, 17, 18, 19, 20, and 21).
- N 2 -Deacetyl-7V 2 -(3-mercapto- 1 -oxopropyl)-maytansine (DM 1 , 13.4 mg, 0.0182 mmol) was prepared in 0.70 mL of THF and succinimidyl-[(N- maleimidopropionamido)-diethyleneglycol] ester (NHS-PEG 2 -Maleimide, Quanta Biodesign, 1 1.6 mg, 0.0273 mmol) was added in 1.5 mL of 2:1 (v/v) mixture of aqueous potassium phosphate buffer (50 mM, pH 6) and THF.
- CanAg, and CD56 by flow cytometry were incubated with conjugates or unmodified antibodies at 4°C, then with a secondary antibody-FITC conjugate at 4 0 C, fixed with formaldehyde (1% in PBS) and analyzed by flow cytometry. No significant difference was observed between the binding of the conjugate versus that of the unmodified antibody for all the conjugates evaluated.
- An example is shown in Figure 24, where a 10.7 maytansinoid bearing Ab-PEG 4 -MaI-DMl conjugate bound to antigen-positive cells with a high affinity similar to that of the unmodified antibody.
- the cytotoxic effects of the antibody-maytansinoid conjugates with thioether and disulfide linkers containing PEG n spacers were typically evaluated using a WST-8 cell- viability assay after a 4-5 day continuous incubation of the cancer cells with the conjugates.
- the antigen-expressing cancer cells ( ⁇ 1000-5000 cells per well) were incubated in 96-well plates in regular growth medium containing fetal bovine serum with various concentrations of the antibody-maytansinoid conjugates for about 5 days.
- the WST-8 reagent was then added and the plate absorbance was measured at 450 nm after —2-5 h.
- FIG. 25 shows the enhancement in potency of anti -EpCAM Ab-maytansinoid conjugates with increased drug load for the PEG 4 linked thioether conjugate (Ab-PEG 4 - MaI-DMl), which also shows greater potency than the thioether-linked SMCC-DMl and disulfide-linked SPDB-DM4 conjugates at similar drug loads of about 4 maytansinoid per antibody toward EpCAM antigen-positive COLO205 -multi drug resistant cells (COLO205-MDR cells).
- the potency of the thioether-linked anti-EpCAM Ab-PEG 4 - MaI-DMl conjugate at maytansinoid loads of 4.1 and 7.8 is novel and potentially very promising for therapeutic applications.
- Figure 26 shows the cytotoxic activities of anti-CanAg Ab-maytansinoid conjugates against CanAg antigen-positive COLO205-MDR cells.
- the thioether- linked Ab-PEG 4 -MaI-DMl and Ab-PEG 2 -MaI-DMl conjugates showed greater potency compared to the thioether-linked Ab-SMCC-DMl conjugate with similar maytansinoid loads.
- Figure 27 shows the cytotoxic activities of the anti-CD56 antibody-maytansinoid conjugates with PEG-containing thioether and disulfide linkers on CD56-expressing Molp-8 multiple myeloma cells.
- Figure 28 shows the enhancement in potency of anti-EpCAM Ab-maytansinoid conjugates bearing a PEG 4 linked thioether conjugate (Ab-PEG 4 -MaI-DMl), over the conventional thioether-linked SMCC-DMl at similar drug loads of about 4 maytansinoid per antibody toward EpCAM-positive multi drug resistant HCTl 5 cells.
- the high potency of the thioether-linked anti-EpCAM Ab-PEG 4 -MaI-DMl conjugate is a novel finding and potentially very promising for therapeutic applications.
- Figure 29 shows the enhancement in potency of anti-EpCAM Ab-maytansinoid conjugates bearing a PEG 4 linked thioether conjugate (Ab-PEG 4 -MaI-DMl), over the conventional thioether-linked SMCC-DMl at similar drug loads of about 4 maytansinoid per antibody toward EpCAM-positive multi drug resistant COLO 205 cells.
- the enhanced potency of the thioether-linked anti-EpCAM Ab-PEG 4 -MaI-DMl conjugate is a novel finding and potentially very promising for therapeutic applications.
- Figure 37 shows the potent enhancement in cytotoxicity of anti-EGFR Ab- Maytansinoid conjugate with the hydrophilic thioether-bonded PEG 4 linker (Ab-PEG 4 -MaI-DMl) compared to the non-hydrophilic SMCC-DMl conjugate with 3.7 maytansinoid/ Ab toward EGFR- positive UO-31 human renal carcinoma cells.
- the potency of the PEG 4 -MaI-DMl was about 10-fold greater than that of the SMCC-DMl conjugate with the traditional linker.
- the plasma samples were added to microtiter plates containing coated, immobilized goat-anti-human IgG (H+L) antibody, washed, and detected using horseradish peroxidase-conjugated goat- anti-human IgG (Fc ⁇ ) antibody.
- conjugate concentration the plasma samples were added to microtiter plates containing coated, immobilized goat-anti -human IgG (H+L) antibody, washed, and detected using biotinylated anti-maytansine antibody and alkaline phosphatase-conjugated streptavidin. Both antibody concentration and conjugate concentration ELISA results demonstrated that the Ab-PEG 4 -MaI-DMl conjugate with hydrophilic PEG 4 linker bearing the high 6.7 DMl /Ab load was well retained in plasma over the 4 week study period.
- Figure 38 A shows the in vivo pharmacokinetics of an Antibody- Maytansinoid conjugate using the PEG 4 linker with a high maytansinoid load (6.7 DM 1/Ab) compared to the standard linker conjugate bearing 4 DMl /Ab. Even with the high maytansinoid load, the PEG 4 linked thioether conjugate (Ab-PEG 4 -MaI-DMl) with 6.7 maytansinoid/ Ab has a longer half life than the standard conjugate.
- the plasma pharmacokinetics of a humanized C242 Ab-PEG 4 -MaI- 3 H-DMl conjugate with 3 H-labeled DMl was compared with unconjugated antibody and with Ab-SMCC- 3 H-DMl conjugate containing a traditional aliphatic carbon chain linker and bearing a similar 4.2 D/A load, in CD-I mice at 10-12 mg/kg i.v. dose ( Figure 38 B).
- the Ab-PEG 4 -MaI- 3 H-DMl conjugate showed higher plasma concentrations over 4 weeks compared to the traditional SMCC-linker conjugate with a similar maytansinoid load, as measured by both antibody concentrations (ELISA; Figure 38 B) and conjugate concentrations ( 3 H-label counts).
- the half life of the PEG 4 -MaI linked conjugate was 16 days compared to 12.6 days for the SMCC-linked conjugate and thus much improved over the SMCC conjugate ( Figure 38 B).
- HCTl 5 human colon carcinoma
- Tumor growth was monitored by measuring tumor size twice per week. Tumor size was calculated with the formula: length x width x height x !4
- tumors reached a tumor volume of 600 mm 3 by day 20, post cell inoculation.
- Treatment with muB38.1 -MCC-DMl resulted in tumor growth delay of 15 days.
- Treatment with muB38.1-PEG4-mal-DMl showed more anti-tumor effect with two of five animals having complete tumor regressions, lasting 44 days and three animals with a tumor growth delay of 32 days.
- muB38.1-PEG4-mal-DMl is significantly more efficacious than muB38.1 -MCC-DMl in this human colon cancer xenograft model.
- muB38.1 -MCC-DMl 20 mg conjugate protein/kg
- muB38.1-PEG4-mal-DMl antibody dose 20 mg/kg
- phosphate-buffered saline vehicle control
- Tumor growth was monitored by measuring tumor size twice per week. Tumor size was calculated with the formula: length x width x height x /4.
- muB38.1-PEG4-mal-DMl is significantly more efficacious than the conjugate muB38.1 -MCC-DMl, prepared with the previously described linker, in this human colon cancer xenograft model.
- the Ab-PEG n - MaI-DMx conjugates prepared with PEG 4 , PEG 8 , PEGj 2 , PEG 24 linkers were potent in cytotoxicity toward antigen-positive cells.
- Figure 35 demonstrates that the anti-CanAg antibody (huC242)-PEG n -Mal-DMl conjugates with 4 to 17 D/A killed the CanAg antigen-positive COLO205 cells with potent ICs 0 of about 0.1-0.5 nM upon incubation for 5 days.
- the pgp-expressing multi-drug resistant COLO205-MDR cells were killed by the huC242-PEG n -Mal-DMl conjugates bearing 4 to 17 D/A in a potent manner with IC 50 of about 0.05 to 0.5 nM ( Figure 36).
- the PEG 24 -MaI-DMl conjugate with high, 17.1 D/A was more potent in cytotoxicity than the PEG 24 -MaI-DMl conjugate with 4 D/A ( Figures 34, 36).
- a 10 mM stock (w/v) of DMl-SMCC reagent was prepared in DMA (10.7 mg/mL). The stock solution was diluted in EtOH and the absorbance was measured at 280 nm against a reagent blank of EtOH and DMA. The concentration of stock DMl - SMCC reagent was calculated by using an extinction coefficient of 5700 M "1 at 280 nm which is the extinction coefficient of DMl at this wavelength. Since the real extinction coefficient of DMl-SMCC has not been determined this is only an estimate of concentration.
- the antibody was conjugated with DMl-SMCC at 5 mg/mL using a 7-fold molar excess of the reagent. A titration of antibody with several excesses of DMl-SMCC was performed initially to determine the desired DMl :Ab ratio, typically this range is 6- 10- fold molar excess for human antibodies.
- the reaction was carried out in pH 7.5 buffer with DMA (5% v/v) for 90 minutes at room temperature. The reaction mixture was then kept at 4 0 C for 12-36 hours.
- the conjugate was then purified over a NAP-5 (Sephadex G25) column equilibrated in pH 5.5 citrate buffer, filtered and dialyzed against the pH 5.5 citrate buffer to remove any unreacted free drug.
- the number of DMl molecules per Ab antibody molecule (average) in the final conjugate was measured by determining absorbance of the conjugate at 252 and 280 nm and using known extinction coefficients for DMl and antibody at these two wavelengths.
- the final conjugate was also analyzed by size exclusion LC/MS.
- the conjugate made via the method described in this invention shows the desired MS spectrum of deglycoslyated conjugate containing only the expected distribution of peaks, Figure 45.
- the number of DM4 molecules linked per antibody molecule (average) in the final conjugate was measured by determining absorbance of the conjugate at 252 and 280 nm and using known extinction coefficients for DM4 and antibody at these two wavelengths. [204] Following purification, the conjugate had 3.7 DM4 molecules linked per molecule of antibody. SEC analysis was performed on the final conjugate to show that it was >95% monomelic however >5% free drug species was present on the final conjugate. Dialysis of the conjugate may have reduced the presence of undesired free drug species.
- the sulfhydryl-bearing maytansinoid DM4 was modified with a heterobifunctional, haloacetamide-bearing crosslinker to give an amine reactive maytansinoid bearing a thioacetamidyl moiety ( Figure 49).
- One-step conjugation of the isolated compound with antibody ( Figure 42) gave a non-cleavable thioacetamidyl- linked antibody maytansinoid conjugate ( Figure 40).
- a 10.8 mM stock (w/v) of DM 1 -SBA reagent was prepared in DMA.
- the antibody was conjugated with DMl-SBA at 2.5 mg/mL using a 10.2-fold molar excess of the reagent.
- the reaction was carried out in pH 7.5 buffer (100 mM potassium phosphate) with DMA (10% v/v) for 90 minutes at room temperature.
- the conjugate was then purified over a S300 gel filtration (Sephadex S300) column eluting with pH 6.5 buffer (10 mM potassium phosphate, 140 mM sodium chloride). Following purification, the conjugate had 3.7 DMl molecules linked per mole of antibody and -0.18% free drug
- Sulfhydryl-bearing maytansinoids such as DMl and DM4, may be modified to give non-cleavable thiosuccinimidyl-bearing carboxylic acid derivatives ( Figures 45, 46 and 48). These derivatives would be useful in the preparation of amine-reactive non- cleavable thiosuccinimidyl-linked maytansinoids described herein.
- Figures 45,46 and 48 show the formation of N-hydroxysuccinimide activated esters however it is obvious to one skilled in the art that several other activated esters could be formed, these include but are not limited to N-sulfosuccinimidyl esters, pentafluorophenol ester, tetrafluorosulfophenol, and nitrophenol ester.
- Sulfhydryl-bearing maytansinoids such as DMl and DM4
- the maleimide bearing maytansinoid containing a non-cleavable thiosuccinimidyl group was conjugated to a thiol containing antibody ( Figure 51).
- the maleimide bearing maytansinoid containing a non-cleavable thiosuccinimidyl group was prepared by coupling a thiol containing maytansinoid to a, bis-maleimide crosslinker
- huC242 (8 mg/mL) in 150 mM HEPES buffer, pH 8.0 containing 5% dimethyl acetamide was modified with 9 equivalents of SPDB for 1 hr at 30'C then eluted through a NAP5 sizing column using 50 mM phosphate, 50 mM NaCl, pH 7.5 buffer. To the recovered modified antibody was added 2 ⁇ L of 1 M dithiothreitol at 30'C. After 10 min the reaction was purified on a NAPlO column eluting with 50 mM phosphate, 50 mM NaCl, pH 6.5 buffer.
- DMl-mal-(CH 2 ) 6 -mal (1.7 mole equivalents) in dimethyl formamide was added to the fraction containing desired product to obtain 10% v/v dimethyl formamide/buffer. After 1 Hr the crude conjugate was purified on a NAP 25 column eluting with 10 mM citrate, 135 mM NaCl pH 5.5 buffer. Using the previously described binding assay, huC242-Mal-(CH 2 ) 6 -Mal-DMl was shown to bind to antigen-positive COLO205 cells to the same extent as the naked huC242 antibody ( Figure 53). The huC242-Mal-(CH 2 ) 6 -Mal-DMl conjugate was shown to be much more cytotoxic to antigen positive COLO205 cells than antigen negative Namalwa cells ( Figure 54).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Virology (AREA)
- Cell Biology (AREA)
- Transplantation (AREA)
- Tropical Medicine & Parasitology (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
Description


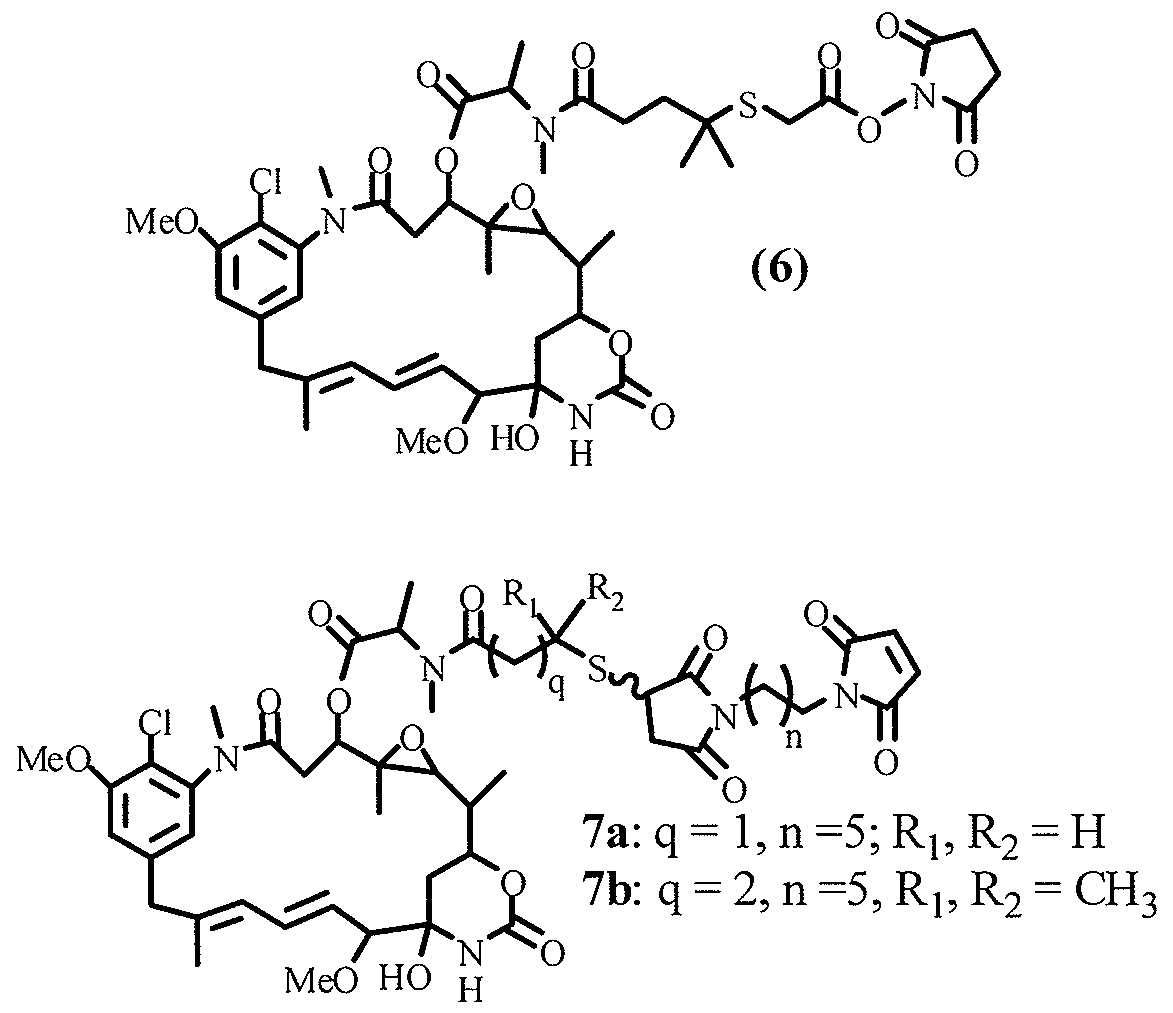
Claims





Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012533123A JP2013506709A (en) | 2009-10-06 | 2009-10-06 | Effective conjugates and hydrophilic linkers |
| CN2009801623736A CN102596922A (en) | 2009-10-06 | 2009-10-06 | Potent conjugates and hydrophilic linkers |
| EP09844200.7A EP2486023A4 (en) | 2009-10-06 | 2009-10-06 | Potent conjugates and hydrophilic linkers |
| KR1020127010084A KR20120080611A (en) | 2009-10-06 | 2009-10-06 | Potent conjugates and hydrophilic linkers |
| PCT/US2009/059620 WO2010126551A1 (en) | 2009-04-30 | 2009-10-06 | Potent conjugates and hydrophilic linkers |
| IN2780DEN2012 IN2012DN02780A (en) | 2009-10-06 | 2009-10-06 | |
| IL219020A IL219020A0 (en) | 2009-10-06 | 2012-04-03 | Maytansinoids, maytansinoid conjugates and processes for preparing the same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/433,668 | 2009-04-30 | ||
| PCT/US2009/059620 WO2010126551A1 (en) | 2009-04-30 | 2009-10-06 | Potent conjugates and hydrophilic linkers |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010126551A1 true WO2010126551A1 (en) | 2010-11-04 |
Family
ID=46467238
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2009/059620 WO2010126551A1 (en) | 2009-04-30 | 2009-10-06 | Potent conjugates and hydrophilic linkers |
Country Status (7)
| Country | Link |
|---|---|
| EP (1) | EP2486023A4 (en) |
| JP (1) | JP2013506709A (en) |
| KR (1) | KR20120080611A (en) |
| CN (1) | CN102596922A (en) |
| IL (1) | IL219020A0 (en) |
| IN (1) | IN2012DN02780A (en) |
| WO (1) | WO2010126551A1 (en) |
Cited By (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012074757A1 (en) * | 2010-11-17 | 2012-06-07 | Genentech, Inc. | Alaninyl maytansinol antibody conjugates |
| JP2014500273A (en) * | 2010-12-21 | 2014-01-09 | パナクセム カンパニー リミテッド | Conjugate for photodynamic diagnosis or treatment and method for producing the same |
| JP2014515742A (en) * | 2011-04-01 | 2014-07-03 | イミュノジェン, インコーポレイテッド | CD37 binding molecule and immune complex thereof |
| JP2014533954A (en) * | 2011-11-21 | 2014-12-18 | イミュノジェン, インコーポレイテッド | Method for treating tumors resistant to EGFR therapy with EGFR antibody cytotoxic drug conjugates |
| WO2014160160A3 (en) * | 2013-03-13 | 2015-01-08 | Novartis Ag | Antibody drug conjugates and corresponding antibodies |
| WO2015009740A2 (en) | 2013-07-15 | 2015-01-22 | Cell Signaling Technology, Inc. | Anti-mucin 1 binding agents and uses thereof |
| WO2015014879A1 (en) * | 2013-08-02 | 2015-02-05 | Sanofi | Use of anti-muc1 maytansinoid immunoconjugate antibody for the treatment of solid tumors |
| WO2015031396A1 (en) * | 2013-08-26 | 2015-03-05 | Regeneron Pharmaceuticals, Inc. | Pharmaceutical compositions comprising macrolide diastereomers, methods of their synthesis and therapeutic uses |
| WO2015118031A3 (en) * | 2014-02-06 | 2015-10-01 | Oncomatryx Biopharma, S.L. | Antibody-drug conjugates and immunotoxins |
| US9415118B2 (en) | 2013-03-13 | 2016-08-16 | Novartis Ag | Antibody drug conjugates |
| WO2016167798A1 (en) * | 2015-04-17 | 2016-10-20 | Northwestern University | Phosphatidylethanolamine-specific probes |
| US20160324980A1 (en) * | 2011-02-15 | 2016-11-10 | Immunogen, Inc. | Methods of preparation of conjugates |
| WO2016147031A3 (en) * | 2015-03-19 | 2016-11-10 | Hangzhou Dac Biotech Co., Ltd | Novel hydrophilic linkers and ligand-drug conjugates thereof |
| EP3125943A4 (en) * | 2014-04-04 | 2017-12-06 | Merck Sharp & Dohme Corp. | Phosphate based linkers for intracellular delivery of drug conjugates |
| US9950076B2 (en) | 2016-01-25 | 2018-04-24 | Regeneron Pharmaceuticals, Inc. | Maytansinoid derivatives, conjugates thereof, and methods of use |
| US10208127B2 (en) | 2009-02-05 | 2019-02-19 | Immunogen, Inc. | Benzodiazepine derivatives |
| US10570151B2 (en) | 2013-03-15 | 2020-02-25 | Regeneron Pharmaceuticals, Inc. | Biologically active molecules, conjugates thereof, and therapeutic uses |
| US10640508B2 (en) | 2017-10-13 | 2020-05-05 | Massachusetts Institute Of Technology | Diazene directed modular synthesis of compounds with quaternary carbon centers |
| US10646585B2 (en) | 2017-09-15 | 2020-05-12 | Hangzhou Dac Biotech Co., Ltd. | Hydrophilic linkers and ligand-drug conjugates thereof |
| US10654873B2 (en) | 2016-09-15 | 2020-05-19 | Polytherics Limited | Cytotoxic agents and conjugates thereof |
| US10899775B2 (en) | 2015-07-21 | 2021-01-26 | Immunogen, Inc. | Methods of preparing cytotoxic benzodiazepine derivatives |
| US10918735B2 (en) | 2012-12-04 | 2021-02-16 | Massachusetts Institute Of Technology | Substituted pyrazino[1′,2′:1,5]pyrrolo[2,3-b]indole-1,4-diones for cancer treatment |
| US10918627B2 (en) | 2016-05-11 | 2021-02-16 | Massachusetts Institute Of Technology | Convergent and enantioselective total synthesis of Communesin analogs |
| CN112543770A (en) * | 2018-06-26 | 2021-03-23 | 伊缪诺金公司 | Immunoconjugates targeting ADAM9 and methods of use thereof |
| US11103593B2 (en) | 2013-10-15 | 2021-08-31 | Seagen Inc. | Pegylated drug-linkers for improved ligand-drug conjugate pharmacokinetics |
| US11104740B2 (en) | 2015-08-28 | 2021-08-31 | Debiopharm International, S.A. | Antibodies and assays for detection of CD37 |
| US11229708B2 (en) | 2015-12-04 | 2022-01-25 | Seagen Inc. | Conjugates of quaternized tubulysin compounds |
| US11278629B2 (en) | 2016-11-02 | 2022-03-22 | Debiopharm International, S.A. | Methods for improving anti-CD37 immunoconjugate therapy |
| US11395796B2 (en) | 2015-06-08 | 2022-07-26 | Debiopharm International, S.A. | Anti-CD37 immunoconjugate and anti-CD20 antibody combinations |
| US11466095B2 (en) | 2010-03-12 | 2022-10-11 | Debiopharm International S.A. | CD37-binding molecules and immunoconjugates thereof |
| US11535634B2 (en) | 2019-06-05 | 2022-12-27 | Massachusetts Institute Of Technology | Compounds, conjugates, and compositions of epipolythiodiketopiperazines and polythiodiketopiperazines and uses thereof |
| US11730822B2 (en) | 2017-03-24 | 2023-08-22 | Seagen Inc. | Process for the preparation of glucuronide drug-linkers and intermediates thereof |
| US11793880B2 (en) | 2015-12-04 | 2023-10-24 | Seagen Inc. | Conjugates of quaternized tubulysin compounds |
| US11844839B2 (en) | 2016-03-25 | 2023-12-19 | Seagen Inc. | Process for the preparation of pegylated drug-linkers and intermediates thereof |
| US11932650B2 (en) | 2017-05-11 | 2024-03-19 | Massachusetts Institute Of Technology | Potent agelastatin derivatives as modulators for cancer invasion and metastasis |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103044677A (en) * | 2012-12-28 | 2013-04-17 | 上海景宇生物科技有限公司 | Heresy base telechelic polyethylene glycol and preparation method thereof |
| CN103083652B (en) * | 2013-02-06 | 2016-08-10 | 中国科学院过程工程研究所 | A kind of meningococcal polysaccharides combined vaccine with Heterobifunctional reagents as cross structure and preparation method thereof |
| CN103254311B (en) * | 2013-05-09 | 2015-05-13 | 齐鲁制药有限公司 | Method for preparing antibody-maytansine alkaloid medicine conjugate |
| CN103483357B (en) * | 2013-10-12 | 2015-11-18 | 齐鲁制药有限公司 | Intermediate new crystal of a kind of antibody-maytenin conjugate and preparation method thereof |
| RU2685728C2 (en) * | 2014-01-29 | 2019-04-23 | Шанхай Хэнжуй Фармасьютикал Ко., Лтд. | Ligand-cytotoxic drug conjugate, production method and use thereof |
| US10238748B2 (en) * | 2014-08-12 | 2019-03-26 | Novartis Ag | Anti-CDH6 antibody drug conjugates |
| CN105435241A (en) * | 2015-12-10 | 2016-03-30 | 浙江大学 | IRGD-anticancer drug conjugate, and preparation method and application thereof |
| CN110088138B (en) * | 2016-09-23 | 2023-08-25 | 瑞泽恩制药公司 | anti-STEAP 2 antibodies, antibody drug conjugates and bispecific antigen binding molecules that bind STEAP2 and CD3 and uses thereof |
| CR20200178A (en) * | 2017-11-01 | 2020-06-28 | Arrowhead Pharmaceuticals Inc | Integrin ligands and uses thereof |
Citations (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4361650A (en) | 1978-03-24 | 1982-11-30 | Takeda Chemical Industries, Ltd. | Fermentation process of preparing demethyl maytansinoids |
| US4563304A (en) | 1981-02-27 | 1986-01-07 | Pharmacia Fine Chemicals Ab | Pyridine compounds modifying proteins, polypeptides or polysaccharides |
| WO1992001047A1 (en) | 1990-07-10 | 1992-01-23 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| US5208020A (en) | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| US5475092A (en) | 1992-03-25 | 1995-12-12 | Immunogen Inc. | Cell binding agent conjugates of analogues and derivatives of CC-1065 |
| US5595499A (en) | 1993-10-06 | 1997-01-21 | The Whitaker Corporation | Coaxial connector having improved locking mechanism |
| US5635483A (en) | 1992-12-03 | 1997-06-03 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Tumor inhibiting tetrapeptide bearing modified phenethyl amides |
| US5639641A (en) | 1992-09-09 | 1997-06-17 | Immunogen Inc. | Resurfacing of rodent antibodies |
| US5665357A (en) | 1993-12-03 | 1997-09-09 | Zeneca Limited | Antibodies recognizing tumor associated antigen CA 55.1 |
| US5714586A (en) | 1995-06-07 | 1998-02-03 | American Cyanamid Company | Methods for the preparation of monomeric calicheamicin derivative/carrier conjugates |
| US5739116A (en) | 1994-06-03 | 1998-04-14 | American Cyanamid Company | Enediyne derivatives useful for the synthesis of conjugates of methyltrithio antitumor agents |
| WO1999006587A2 (en) | 1997-08-01 | 1999-02-11 | Morphosys Ag | Novel method and phage for the identification of nucleic acid sequences encoding members of a multimeric (poly)peptide complex |
| US5885793A (en) | 1991-12-02 | 1999-03-23 | Medical Research Council | Production of anti-self antibodies from antibody segment repertoires and displayed on phage |
| US6013748A (en) | 1997-05-08 | 2000-01-11 | Japan As Represented By Director General Of Agency Of Industrial Science And Technology | Aromatic ester compound and second-order non-linear optical material therewith |
| US6333410B1 (en) | 2000-08-18 | 2001-12-25 | Immunogen, Inc. | Process for the preparation and purification of thiol-containing maytansinoids |
| US6340701B1 (en) | 1999-11-24 | 2002-01-22 | Immunogen Inc | Cytotoxic agents comprising taxanes and their therapeutic use |
| WO2002016401A2 (en) | 2000-08-18 | 2002-02-28 | East Carolina University | Monoclonal antibody ds6, tumor-associated antigen ca6, and methods of use thereof |
| US6441163B1 (en) | 2001-05-31 | 2002-08-27 | Immunogen, Inc. | Methods for preparation of cytotoxic conjugates of maytansinoids and cell binding agents |
| US6534660B1 (en) | 2002-04-05 | 2003-03-18 | Immunogen, Inc. | CC-1065 analog synthesis |
| US6596757B1 (en) | 2002-05-14 | 2003-07-22 | Immunogen Inc. | Cytotoxic agents comprising polyethylene glycol-containing taxanes and their therapeutic use |
| US6630579B2 (en) | 1999-12-29 | 2003-10-07 | Immunogen Inc. | Cytotoxic agents comprising modified doxorubicins and daunorubicins and their therapeutic use |
| US20040001838A1 (en) * | 2001-12-21 | 2004-01-01 | Immunogen, Inc. | Cytotoxic agents bearing a reactive polyethylene glycol moiety, cytotoxic conjugates comprising polyethylene glycol linking groups, and methods of making and using the same |
| US6680311B1 (en) | 1996-08-30 | 2004-01-20 | Eli Lilly And Company | Cryptophycin compounds |
| US6747021B2 (en) | 2000-10-02 | 2004-06-08 | Eli Lilly And Company | Cryptophycin compound |
| US6756397B2 (en) | 2002-04-05 | 2004-06-29 | Immunogen, Inc. | Prodrugs of CC-1065 analogs |
| US20050107325A1 (en) | 2003-04-17 | 2005-05-19 | Muthiah Manoharan | Modified iRNA agents |
| US20050118183A1 (en) | 2002-11-07 | 2005-06-02 | Immunogen, Inc. | Anti-CD33 antibodies and method for treatment of acute myeloid leukemia using the same |
| US6913758B2 (en) | 2001-06-11 | 2005-07-05 | Playtex Products, Inc. | Antimicrobial glove and method of making same |
| US6913748B2 (en) | 2002-08-16 | 2005-07-05 | Immunogen, Inc. | Cross-linkers with high reactivity and solubility and their use in the preparation of conjugates for targeted delivery of small molecule drugs |
| US20050169933A1 (en) | 2003-10-10 | 2005-08-04 | Immunogen, Inc. | Method of targeting specific cell populations using cell-binding agent maytansinoid conjugates linked via a non-cleavable linker, said conjugates, and methods of making said conjugates |
| US20050176667A1 (en) | 2001-01-09 | 2005-08-11 | Alnylam Europe Ag | Compositions and methods for inhibiting expression of anti-apoptotic genes |
| US6956036B1 (en) | 2000-03-17 | 2005-10-18 | Alcon, Inc. | 6-hydroxy-indazole derivatives for treating glaucoma |
| US20050249740A1 (en) | 2002-07-09 | 2005-11-10 | R & D Biopharmaceuticals Gmbh | Tubulysin conjugates |
| US20050288244A1 (en) | 2004-04-30 | 2005-12-29 | Alnylam Pharmaceuticals, Inc. | Oligonucleotides comprising a C5-modified pyrimidine |
| US6989450B2 (en) | 2000-10-13 | 2006-01-24 | The University Of Mississippi | Synthesis of epothilones and related analogs |
| US20060022925A1 (en) | 2004-07-27 | 2006-02-02 | Seiko Epson Corporation | Grayscale voltage generation circuit, driver circuit, and electro-optical device |
| US20060035254A1 (en) | 2004-07-21 | 2006-02-16 | Alnylam Pharmaceuticals, Inc. | Oligonucleotides comprising a modified or non-natural nucleobase |
| US20060045877A1 (en) | 2004-08-30 | 2006-03-02 | Goldmakher Viktor S | Immunoconjugates targeting syndecan-1 expressing cells and use thereof |
| WO2006034488A2 (en) | 2004-09-23 | 2006-03-30 | Genentech, Inc. | Cysteine engineered antibodies and conjugates |
| US20060074008A1 (en) | 2002-07-31 | 2006-04-06 | Senter Peter D | Drug conjugates and their use for treating cancer, an autoimmune disease or an infectious disease |
| US20060127407A1 (en) | 2004-12-09 | 2006-06-15 | Qiming Chen | Anti-integrin immunoconjugates, methods and uses |
| US7101675B2 (en) | 1995-02-22 | 2006-09-05 | Yeda Research And Development Co. Ltd. | Modulators of regulatory proteins |
| US20060287260A1 (en) | 2004-06-30 | 2006-12-21 | Alnylam Pharmaceuticals, Inc. | Oligonucleotides comprising a non-phosphate backbone linkage |
| US20070054279A1 (en) | 2004-08-04 | 2007-03-08 | Alnylam Pharmaceuticals | Oligonucleotides comprising a ligand tethered to a modified or non-natural nucleobase |
| US20070082365A1 (en) | 1998-12-10 | 2007-04-12 | Adnexus Therapeutics, Inc. | Protein scaffolds for antibody mimics and other binding proteins |
| WO2007062466A1 (en) | 2005-11-29 | 2007-06-07 | The University Of Sydney | Demibodies: dimerisation-activated therapeutic agents |
| US20070161595A1 (en) | 2003-06-09 | 2007-07-12 | Mayo Foundation For Medical Education And Research | Method of treating neurodegenerative disease |
| US20070185050A1 (en) | 2002-01-22 | 2007-08-09 | Alnylam Europe Ag, A German Corporation | Double-stranded RNA (dsRNA) and method of use for inhibiting expression of a fusion gene |
| US20070213292A1 (en) | 2005-08-10 | 2007-09-13 | The Rockefeller University | Chemically modified oligonucleotides for use in modulating micro RNA and uses thereof |
| US7276497B2 (en) | 2003-05-20 | 2007-10-02 | Immunogen Inc. | Cytotoxic agents comprising new maytansinoids |
| US20070238667A1 (en) | 2006-03-03 | 2007-10-11 | Zongchao Jia | Compositions for treatment of cancer |
| US7301019B2 (en) | 2005-01-21 | 2007-11-27 | Immunogen, Inc. | Method for the preparation of maytansinoid esters |
| US20070275465A1 (en) | 2003-06-13 | 2007-11-29 | Alnylam Pharmaceuticals | Double-Stranded Ribonucleic Acid with Increased Effectiveness in an Organism |
| US7303749B1 (en) | 1999-10-01 | 2007-12-04 | Immunogen Inc. | Compositions and methods for treating cancer using immunoconjugates and chemotherapeutic agents |
| WO2007147213A1 (en) | 2006-06-22 | 2007-12-27 | Walter And Eliza Hall Institute Of Medical Research | Structure of the insulin receptor ectodomain |
| US20080008822A1 (en) | 2001-10-05 | 2008-01-10 | Cabot Corporation | Controlling ink migration during the formation of printable electronic features |
| US20080171040A1 (en) | 2004-06-01 | 2008-07-17 | Genentech, Inc. | Antibody-drug conjugates and methods |
| US20080305044A1 (en) | 2004-11-29 | 2008-12-11 | Seattle Genetics, Inc. | Engineered Antibodies and Immunoconjugates |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101186613B (en) * | 2003-05-20 | 2014-09-17 | 伊缪诺金公司 | Cytotoxic agents comprising new maytansinoids |
| RS53168B (en) * | 2006-05-30 | 2014-06-30 | Genentech Inc. | Antibodies and immunoconjugates and uses therefor |
| US9845355B2 (en) * | 2007-07-16 | 2017-12-19 | Genentech, Inc. | Humanized anti-CD79b antibodies and immunoconjugates and methods of use |
| RU2487877C2 (en) * | 2008-04-30 | 2013-07-20 | Иммьюноджен, Инк. | Potent conjugates and hydrophilic cross-linking agents (linkers) |
-
2009
- 2009-10-06 IN IN2780DEN2012 patent/IN2012DN02780A/en unknown
- 2009-10-06 EP EP09844200.7A patent/EP2486023A4/en not_active Withdrawn
- 2009-10-06 KR KR1020127010084A patent/KR20120080611A/en not_active Application Discontinuation
- 2009-10-06 CN CN2009801623736A patent/CN102596922A/en active Pending
- 2009-10-06 JP JP2012533123A patent/JP2013506709A/en active Pending
- 2009-10-06 WO PCT/US2009/059620 patent/WO2010126551A1/en active Application Filing
-
2012
- 2012-04-03 IL IL219020A patent/IL219020A0/en unknown
Patent Citations (74)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4361650A (en) | 1978-03-24 | 1982-11-30 | Takeda Chemical Industries, Ltd. | Fermentation process of preparing demethyl maytansinoids |
| US4563304A (en) | 1981-02-27 | 1986-01-07 | Pharmacia Fine Chemicals Ab | Pyridine compounds modifying proteins, polypeptides or polysaccharides |
| US5208020A (en) | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| US5416064A (en) | 1989-10-25 | 1995-05-16 | Immunogen, Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| WO1992001047A1 (en) | 1990-07-10 | 1992-01-23 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| US5885793A (en) | 1991-12-02 | 1999-03-23 | Medical Research Council | Production of anti-self antibodies from antibody segment repertoires and displayed on phage |
| US5585499A (en) | 1992-03-25 | 1996-12-17 | Immunogen Inc. | Cyclopropylbenzindole-containing cytotoxic drugs |
| US5846545A (en) | 1992-03-25 | 1998-12-08 | Immunogen, Inc. | Targeted delivery of cyclopropylbenzindole-containing cytotoxic drugs |
| US5475092A (en) | 1992-03-25 | 1995-12-12 | Immunogen Inc. | Cell binding agent conjugates of analogues and derivatives of CC-1065 |
| US5639641A (en) | 1992-09-09 | 1997-06-17 | Immunogen Inc. | Resurfacing of rodent antibodies |
| US5635483A (en) | 1992-12-03 | 1997-06-03 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Tumor inhibiting tetrapeptide bearing modified phenethyl amides |
| US5595499A (en) | 1993-10-06 | 1997-01-21 | The Whitaker Corporation | Coaxial connector having improved locking mechanism |
| US5665357A (en) | 1993-12-03 | 1997-09-09 | Zeneca Limited | Antibodies recognizing tumor associated antigen CA 55.1 |
| US5739116A (en) | 1994-06-03 | 1998-04-14 | American Cyanamid Company | Enediyne derivatives useful for the synthesis of conjugates of methyltrithio antitumor agents |
| US7101675B2 (en) | 1995-02-22 | 2006-09-05 | Yeda Research And Development Co. Ltd. | Modulators of regulatory proteins |
| US5714586A (en) | 1995-06-07 | 1998-02-03 | American Cyanamid Company | Methods for the preparation of monomeric calicheamicin derivative/carrier conjugates |
| US6680311B1 (en) | 1996-08-30 | 2004-01-20 | Eli Lilly And Company | Cryptophycin compounds |
| US6013748A (en) | 1997-05-08 | 2000-01-11 | Japan As Represented By Director General Of Agency Of Industrial Science And Technology | Aromatic ester compound and second-order non-linear optical material therewith |
| WO1999006587A2 (en) | 1997-08-01 | 1999-02-11 | Morphosys Ag | Novel method and phage for the identification of nucleic acid sequences encoding members of a multimeric (poly)peptide complex |
| US20070082365A1 (en) | 1998-12-10 | 2007-04-12 | Adnexus Therapeutics, Inc. | Protein scaffolds for antibody mimics and other binding proteins |
| US7303749B1 (en) | 1999-10-01 | 2007-12-04 | Immunogen Inc. | Compositions and methods for treating cancer using immunoconjugates and chemotherapeutic agents |
| US6706708B2 (en) | 1999-11-24 | 2004-03-16 | Immunogen, Inc. | Cytotoxic agents comprising taxanes and their therapeutic use |
| US7008942B2 (en) | 1999-11-24 | 2006-03-07 | Immunogen, Inc. | Cytotoxic agents comprising taxanes and their therapeutic use |
| US6372738B2 (en) | 1999-11-24 | 2002-04-16 | Immunogen Inc. | Cytotoxic agents comprising taxanes and their therapeutic use |
| US6436931B1 (en) | 1999-11-24 | 2002-08-20 | Immunogen Inc. | Cytotoxic agents comprising taxanes and their therapeutic use |
| US7217819B2 (en) | 1999-11-24 | 2007-05-15 | Immunogen, Inc. | Cytotoxic agents comprising taxanes and their therapeutic use |
| US6340701B1 (en) | 1999-11-24 | 2002-01-22 | Immunogen Inc | Cytotoxic agents comprising taxanes and their therapeutic use |
| US7276499B2 (en) | 1999-11-24 | 2007-10-02 | Immunogen Inc. | Cytotoxic agents comprising taxanes and their therapeutic use |
| US6630579B2 (en) | 1999-12-29 | 2003-10-07 | Immunogen Inc. | Cytotoxic agents comprising modified doxorubicins and daunorubicins and their therapeutic use |
| US6956036B1 (en) | 2000-03-17 | 2005-10-18 | Alcon, Inc. | 6-hydroxy-indazole derivatives for treating glaucoma |
| WO2002016401A2 (en) | 2000-08-18 | 2002-02-28 | East Carolina University | Monoclonal antibody ds6, tumor-associated antigen ca6, and methods of use thereof |
| USRE39151E1 (en) | 2000-08-18 | 2006-06-27 | Immunogen, Inc. | Process for the preparation and purification of thiol-containing maytansinoids |
| US6333410B1 (en) | 2000-08-18 | 2001-12-25 | Immunogen, Inc. | Process for the preparation and purification of thiol-containing maytansinoids |
| US6747021B2 (en) | 2000-10-02 | 2004-06-08 | Eli Lilly And Company | Cryptophycin compound |
| US6989450B2 (en) | 2000-10-13 | 2006-01-24 | The University Of Mississippi | Synthesis of epothilones and related analogs |
| US20050176667A1 (en) | 2001-01-09 | 2005-08-11 | Alnylam Europe Ag | Compositions and methods for inhibiting expression of anti-apoptotic genes |
| US6441163B1 (en) | 2001-05-31 | 2002-08-27 | Immunogen, Inc. | Methods for preparation of cytotoxic conjugates of maytansinoids and cell binding agents |
| US6913758B2 (en) | 2001-06-11 | 2005-07-05 | Playtex Products, Inc. | Antimicrobial glove and method of making same |
| US20080008822A1 (en) | 2001-10-05 | 2008-01-10 | Cabot Corporation | Controlling ink migration during the formation of printable electronic features |
| US6716821B2 (en) | 2001-12-21 | 2004-04-06 | Immunogen Inc. | Cytotoxic agents bearing a reactive polyethylene glycol moiety, cytotoxic conjugates comprising polyethylene glycol linking groups, and methods of making and using the same |
| US20040001838A1 (en) * | 2001-12-21 | 2004-01-01 | Immunogen, Inc. | Cytotoxic agents bearing a reactive polyethylene glycol moiety, cytotoxic conjugates comprising polyethylene glycol linking groups, and methods of making and using the same |
| US20070185050A1 (en) | 2002-01-22 | 2007-08-09 | Alnylam Europe Ag, A German Corporation | Double-stranded RNA (dsRNA) and method of use for inhibiting expression of a fusion gene |
| US6756397B2 (en) | 2002-04-05 | 2004-06-29 | Immunogen, Inc. | Prodrugs of CC-1065 analogs |
| US6586618B1 (en) | 2002-04-05 | 2003-07-01 | Immunogen Inc. | CC-1065 analog synthesis |
| US6534660B1 (en) | 2002-04-05 | 2003-03-18 | Immunogen, Inc. | CC-1065 analog synthesis |
| US7049316B2 (en) | 2002-04-05 | 2006-05-23 | Immunogen Inc. | Prodrugs of CC-1065 analogs |
| US6596757B1 (en) | 2002-05-14 | 2003-07-22 | Immunogen Inc. | Cytotoxic agents comprising polyethylene glycol-containing taxanes and their therapeutic use |
| US20050249740A1 (en) | 2002-07-09 | 2005-11-10 | R & D Biopharmaceuticals Gmbh | Tubulysin conjugates |
| US20060074008A1 (en) | 2002-07-31 | 2006-04-06 | Senter Peter D | Drug conjugates and their use for treating cancer, an autoimmune disease or an infectious disease |
| US6913748B2 (en) | 2002-08-16 | 2005-07-05 | Immunogen, Inc. | Cross-linkers with high reactivity and solubility and their use in the preparation of conjugates for targeted delivery of small molecule drugs |
| US7342110B2 (en) | 2002-11-07 | 2008-03-11 | Immunogen Inc. | Anti-CD33 antibodies and method for treatment of acute myeloid leukemia using the same |
| US20050118183A1 (en) | 2002-11-07 | 2005-06-02 | Immunogen, Inc. | Anti-CD33 antibodies and method for treatment of acute myeloid leukemia using the same |
| US20050107325A1 (en) | 2003-04-17 | 2005-05-19 | Muthiah Manoharan | Modified iRNA agents |
| US20070270585A1 (en) * | 2003-05-20 | 2007-11-22 | Immunogen, Inc. | Cytotoxic agents comprising new maytansinoids |
| US7276497B2 (en) | 2003-05-20 | 2007-10-02 | Immunogen Inc. | Cytotoxic agents comprising new maytansinoids |
| US20070161595A1 (en) | 2003-06-09 | 2007-07-12 | Mayo Foundation For Medical Education And Research | Method of treating neurodegenerative disease |
| US20070275465A1 (en) | 2003-06-13 | 2007-11-29 | Alnylam Pharmaceuticals | Double-Stranded Ribonucleic Acid with Increased Effectiveness in an Organism |
| US20080171865A1 (en) * | 2003-10-10 | 2008-07-17 | Immunogen, Inc. | Method of targeting specific cell populations using cell-binding agent maytansinoid conjugates linked via a non-cleavable linker, said conjugates and methods of making said conjugates |
| US20050169933A1 (en) | 2003-10-10 | 2005-08-04 | Immunogen, Inc. | Method of targeting specific cell populations using cell-binding agent maytansinoid conjugates linked via a non-cleavable linker, said conjugates, and methods of making said conjugates |
| US20050288244A1 (en) | 2004-04-30 | 2005-12-29 | Alnylam Pharmaceuticals, Inc. | Oligonucleotides comprising a C5-modified pyrimidine |
| US20080171040A1 (en) | 2004-06-01 | 2008-07-17 | Genentech, Inc. | Antibody-drug conjugates and methods |
| US20060287260A1 (en) | 2004-06-30 | 2006-12-21 | Alnylam Pharmaceuticals, Inc. | Oligonucleotides comprising a non-phosphate backbone linkage |
| US20060035254A1 (en) | 2004-07-21 | 2006-02-16 | Alnylam Pharmaceuticals, Inc. | Oligonucleotides comprising a modified or non-natural nucleobase |
| US20060022925A1 (en) | 2004-07-27 | 2006-02-02 | Seiko Epson Corporation | Grayscale voltage generation circuit, driver circuit, and electro-optical device |
| US20070054279A1 (en) | 2004-08-04 | 2007-03-08 | Alnylam Pharmaceuticals | Oligonucleotides comprising a ligand tethered to a modified or non-natural nucleobase |
| US20060045877A1 (en) | 2004-08-30 | 2006-03-02 | Goldmakher Viktor S | Immunoconjugates targeting syndecan-1 expressing cells and use thereof |
| WO2006034488A2 (en) | 2004-09-23 | 2006-03-30 | Genentech, Inc. | Cysteine engineered antibodies and conjugates |
| US20080305044A1 (en) | 2004-11-29 | 2008-12-11 | Seattle Genetics, Inc. | Engineered Antibodies and Immunoconjugates |
| US20060127407A1 (en) | 2004-12-09 | 2006-06-15 | Qiming Chen | Anti-integrin immunoconjugates, methods and uses |
| US7301019B2 (en) | 2005-01-21 | 2007-11-27 | Immunogen, Inc. | Method for the preparation of maytansinoid esters |
| US20070213292A1 (en) | 2005-08-10 | 2007-09-13 | The Rockefeller University | Chemically modified oligonucleotides for use in modulating micro RNA and uses thereof |
| WO2007062466A1 (en) | 2005-11-29 | 2007-06-07 | The University Of Sydney | Demibodies: dimerisation-activated therapeutic agents |
| US20070238667A1 (en) | 2006-03-03 | 2007-10-11 | Zongchao Jia | Compositions for treatment of cancer |
| WO2007147213A1 (en) | 2006-06-22 | 2007-12-27 | Walter And Eliza Hall Institute Of Medical Research | Structure of the insulin receptor ectodomain |
Non-Patent Citations (33)
| Title |
|---|
| "Physician's Desk Reference discloses that Taxotere", 2006, pages: 2947 |
| BINZ, H.K.; AMSTUTZ, P.; PLUCKTHUN, A., NATURE BIOTECHNOLOGY, vol. 23, 2005, pages 1257 - 1268 |
| BURGESS, IMMUNOLOGY TODAY, vol. 5, 1984, pages 155 - 158 |
| C. DRUMOND; B. I. SIKIC, J. CLIN. ONCOLOGY, vol. 17, 1999, pages 1061 - 1070 |
| CARLSSON ET AL., BIOCHEM. J., vol. 173, 1978, pages 723 - 737 |
| COLOMER ET AL., CANCER INVEST., vol. 19, 2001, pages 49 - 56 |
| D. G. GILLILAND ET AL., PROC. NATL. ACAD. SCI. USA., vol. 77, 1980, pages 4539 - 4543 |
| G, SZOKACS ET AL., NATURE REVIEWS, vol. 5, 2006, pages 219 - 234 |
| G. SZOKACS ET AL., NATURE REVIEWS, vol. 5, 2006, pages 219 - 235 |
| H. K. ERICKSON ET AL., CANCER RESEARCH, vol. 66, 2006, pages 4626 - 4433 |
| HEIDER, EUR. J. CANCER, vol. 31A, 1995, pages 2385 - 2391 |
| J. B. STIMMEL ET AL., J. BIOL. CHEM., vol. 275, 2000, pages 30445 - 30450 |
| LNF. J. ONCOL., vol. 15, 1999, pages 367 - 72 |
| MALONEY, BLOOD, vol. 90, 1997, pages 2188 - 2195 |
| MOLECULAR CANCER THERAPEUTICS, vol. 3, no. 8, 2004, pages 921 - 932 |
| NADLER ET AL., J. LMMUNOL., vol. 131, 1983, pages 244 - 250 |
| NISONOFF ET AL., ARCH. BIOCHEM. BIOPHYS., vol. 89, 1960, pages 230 - 244 |
| PARHAM, J. IMMUNOL., vol. 131, 1983, pages 2895 - 2902 |
| PEDERSEN ET AL., J. MOL. BIOL., vol. 235, 1994, pages 959 - 973 |
| R. JUE ET AL., BIOCHEMISTRY, vol. 17, 1978, pages 5399 - 5406 |
| R. SINGH ET AL., ANAL. BIOCHEM., vol. 304, 2002, pages 147 - 156 |
| RICHART, A. D.; TOLCHER, A. W., NATURE CLINICAL PRACTICE, vol. 4, 2007, pages 245 - 255 |
| ROGUSKA ET AL., PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES, vol. 91, 1994, pages 969 - 973 |
| ROY ET AL., BLOOD, vol. 77, 1991, pages 2404 - 2412 |
| ROY ET AL., J. NAT. CANCER INST., vol. 88, 1996, pages 1136 - 1145 |
| See also references of EP2486023A4 |
| SPRING ET AL., J. IMMUNOL., vol. 113, 1974, pages 470 - 478 |
| T. P. KING ET AL., BIOCHEMISTRY, vol. 17, 1978, pages 1499 - 1506 |
| W. C. WIDDISON ET AL., J. MED. CHEM., vol. 49, 2006, pages 4392 - 4408 |
| W.C. WIDDISON ET AL., J. MED. CHEM., vol. 49, 2006, pages 4392 - 4408 |
| WELT ET AL., J. CLIN. ONCOL., vol. 12, 1994, pages 1193 - 1203 |
| Y. V. KOVTUN ET AL., CANCER RESEARCH, vol. 66, 2006, pages 3214 - 3221 |
| ZAHND ET AL., J. BIOL. CHEM., vol. 281, no. 46, 2006, pages 35167 - 35175 |
Cited By (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11505617B2 (en) | 2009-02-05 | 2022-11-22 | Immunogen, Inc. | Benzodiazepine derivatives |
| US10947315B2 (en) | 2009-02-05 | 2021-03-16 | Immunogen, Inc. | Benzodiazepine derivatives |
| US10208127B2 (en) | 2009-02-05 | 2019-02-19 | Immunogen, Inc. | Benzodiazepine derivatives |
| US11466095B2 (en) | 2010-03-12 | 2022-10-11 | Debiopharm International S.A. | CD37-binding molecules and immunoconjugates thereof |
| CN103313990A (en) * | 2010-11-17 | 2013-09-18 | 基因泰克公司 | Alaninyl maytansinol antibody conjugates |
| JP2013544253A (en) * | 2010-11-17 | 2013-12-12 | ジェネンテック, インコーポレイテッド | Alaninyl maytansinol antibody conjugate |
| WO2012074757A1 (en) * | 2010-11-17 | 2012-06-07 | Genentech, Inc. | Alaninyl maytansinol antibody conjugates |
| CN103313990B (en) * | 2010-11-17 | 2016-07-20 | 基因泰克公司 | Alanyl maytansinol antibody coupling matter |
| JP2014500273A (en) * | 2010-12-21 | 2014-01-09 | パナクセム カンパニー リミテッド | Conjugate for photodynamic diagnosis or treatment and method for producing the same |
| US8946394B2 (en) | 2010-12-21 | 2015-02-03 | Panaxem Co., Ltd. | Conjugate for photodynamic diagnosis or therapy and method for preparing same |
| US9840564B2 (en) | 2011-02-15 | 2017-12-12 | Immunogen, Inc. | Cytotoxic benzodiazepine derivatives |
| USRE49918E1 (en) | 2011-02-15 | 2024-04-16 | Immunogen, Inc. | Cytotoxic benzodiazepine derivatives |
| US10570212B2 (en) | 2011-02-15 | 2020-02-25 | Immunogen, Inc. | Cytotoxic benzodiazepine derivatives |
| US9868791B2 (en) * | 2011-02-15 | 2018-01-16 | Immunogen, Inc. | Methods of preparation of conjugates |
| US10179818B2 (en) | 2011-02-15 | 2019-01-15 | Immunogen, Inc. | Cytotoxic benzodiazepine derivatives |
| US20160324980A1 (en) * | 2011-02-15 | 2016-11-10 | Immunogen, Inc. | Methods of preparation of conjugates |
| US10556958B2 (en) | 2011-04-01 | 2020-02-11 | Debiopharm International, S.A. | CD37-binding molecules and immunoconjugates thereof |
| US9447189B2 (en) | 2011-04-01 | 2016-09-20 | Immunogen, Inc. | CD37-binding molecules and immunoconjugates thereof |
| JP2014515742A (en) * | 2011-04-01 | 2014-07-03 | イミュノジェン, インコーポレイテッド | CD37 binding molecule and immune complex thereof |
| JP2014533954A (en) * | 2011-11-21 | 2014-12-18 | イミュノジェン, インコーポレイテッド | Method for treating tumors resistant to EGFR therapy with EGFR antibody cytotoxic drug conjugates |
| US10918735B2 (en) | 2012-12-04 | 2021-02-16 | Massachusetts Institute Of Technology | Substituted pyrazino[1′,2′:1,5]pyrrolo[2,3-b]indole-1,4-diones for cancer treatment |
| EP2968600A4 (en) * | 2013-03-13 | 2017-03-08 | Petropoulos, Konstantin | Antibody drug conjugates |
| US9498532B2 (en) | 2013-03-13 | 2016-11-22 | Novartis Ag | Antibody drug conjugates |
| US9415118B2 (en) | 2013-03-13 | 2016-08-16 | Novartis Ag | Antibody drug conjugates |
| WO2014160160A3 (en) * | 2013-03-13 | 2015-01-08 | Novartis Ag | Antibody drug conjugates and corresponding antibodies |
| US11345715B2 (en) | 2013-03-15 | 2022-05-31 | Regeneron Pharmaceuticals, Inc. | Biologically active molecules, conjugates thereof, and therapeutic uses |
| US10570151B2 (en) | 2013-03-15 | 2020-02-25 | Regeneron Pharmaceuticals, Inc. | Biologically active molecules, conjugates thereof, and therapeutic uses |
| EP3699200A1 (en) | 2013-07-15 | 2020-08-26 | Cell Signaling Technology, Inc. | Anti-mucin 1 binding agents and uses thereof |
| WO2015009740A2 (en) | 2013-07-15 | 2015-01-22 | Cell Signaling Technology, Inc. | Anti-mucin 1 binding agents and uses thereof |
| WO2015014879A1 (en) * | 2013-08-02 | 2015-02-05 | Sanofi | Use of anti-muc1 maytansinoid immunoconjugate antibody for the treatment of solid tumors |
| EA038192B1 (en) * | 2013-08-26 | 2021-07-21 | Регенерон Фармасьютикалз, Инк. | Pharmaceutical compositions comprising macrolide diastereomers, methods of their synthesis and therapeutic uses |
| CN105530942B (en) * | 2013-08-26 | 2019-10-11 | 瑞泽恩制药公司 | A kind of pharmaceutical composition comprising macrolides diastereomer, preparation method and use |
| WO2015031396A1 (en) * | 2013-08-26 | 2015-03-05 | Regeneron Pharmaceuticals, Inc. | Pharmaceutical compositions comprising macrolide diastereomers, methods of their synthesis and therapeutic uses |
| AU2014311361B2 (en) * | 2013-08-26 | 2018-11-29 | Regeneron Pharmaceuticals, Inc. | Pharmaceutical compositions comprising macrolide diastereomers, methods of their synthesis and therapeutic uses |
| KR102252925B1 (en) | 2013-08-26 | 2021-05-18 | 리제너론 파마슈티칼스 인코포레이티드 | Pharmaceutical compositions comprising macrolide diastereomers, methods of their synthesis and therapeutic uses |
| US11596635B2 (en) | 2013-08-26 | 2023-03-07 | Regeneron Pharmaceuticals, Inc. | Pharmaceutical compositions comprising macrolide diastereomers, methods of their synthesis and therapeutic uses |
| CN105530942A (en) * | 2013-08-26 | 2016-04-27 | 瑞泽恩制药公司 | Pharmaceutical compositions comprising macrolide diastereomers, methods of their synthesis and therapeutic uses |
| KR20160045146A (en) * | 2013-08-26 | 2016-04-26 | 리제너론 파마슈티칼스 인코포레이티드 | Pharmaceutical compositions comprising macrolide diastereomers, methods of their synthesis and therapeutic uses |
| US11103593B2 (en) | 2013-10-15 | 2021-08-31 | Seagen Inc. | Pegylated drug-linkers for improved ligand-drug conjugate pharmacokinetics |
| US10004812B2 (en) | 2014-02-06 | 2018-06-26 | Oncomatryx Biopharma, S.L. | Antibody-drug conjugates and immunotoxins |
| WO2015118031A3 (en) * | 2014-02-06 | 2015-10-01 | Oncomatryx Biopharma, S.L. | Antibody-drug conjugates and immunotoxins |
| EP3125943A4 (en) * | 2014-04-04 | 2017-12-06 | Merck Sharp & Dohme Corp. | Phosphate based linkers for intracellular delivery of drug conjugates |
| AU2019203316B2 (en) * | 2015-03-19 | 2020-05-14 | Hangzhou Dac Biotech Co., Ltd | Novel Hydrophilic Linkers and Ligand-Drug Conjugates Thereof |
| AU2020200566B2 (en) * | 2015-03-19 | 2020-05-21 | Hangzhou Dac Biotech Co., Ltd | Novel Hydrophilic Linkers and Ligand-Drug Conjugates Thereof |
| CN107614488A (en) * | 2015-03-19 | 2018-01-19 | 杭州多禧生物科技有限公司 | Novel hydrophilic connector and its application on ligand drug conjugation conjugate |
| US10456479B2 (en) | 2015-03-19 | 2019-10-29 | Hangzhou Dac Biotech Co., Ltd. | Hydrophilic linkers and ligand-drug conjugates thereof |
| WO2016147031A3 (en) * | 2015-03-19 | 2016-11-10 | Hangzhou Dac Biotech Co., Ltd | Novel hydrophilic linkers and ligand-drug conjugates thereof |
| WO2016167798A1 (en) * | 2015-04-17 | 2016-10-20 | Northwestern University | Phosphatidylethanolamine-specific probes |
| US11395796B2 (en) | 2015-06-08 | 2022-07-26 | Debiopharm International, S.A. | Anti-CD37 immunoconjugate and anti-CD20 antibody combinations |
| US10899775B2 (en) | 2015-07-21 | 2021-01-26 | Immunogen, Inc. | Methods of preparing cytotoxic benzodiazepine derivatives |
| US11104740B2 (en) | 2015-08-28 | 2021-08-31 | Debiopharm International, S.A. | Antibodies and assays for detection of CD37 |
| US11229708B2 (en) | 2015-12-04 | 2022-01-25 | Seagen Inc. | Conjugates of quaternized tubulysin compounds |
| US11793880B2 (en) | 2015-12-04 | 2023-10-24 | Seagen Inc. | Conjugates of quaternized tubulysin compounds |
| US9950076B2 (en) | 2016-01-25 | 2018-04-24 | Regeneron Pharmaceuticals, Inc. | Maytansinoid derivatives, conjugates thereof, and methods of use |
| US11446389B2 (en) | 2016-01-25 | 2022-09-20 | Regeneron Pharmaceuticals, Inc. | Maytansinoid derivatives, conjugates thereof, and methods of use |
| US10463749B2 (en) | 2016-01-25 | 2019-11-05 | Regeneron Pharmaceuticals, Inc. | Maytansinoid derivatives, conjugates thereof, and methods of use |
| US11844839B2 (en) | 2016-03-25 | 2023-12-19 | Seagen Inc. | Process for the preparation of pegylated drug-linkers and intermediates thereof |
| US10918627B2 (en) | 2016-05-11 | 2021-02-16 | Massachusetts Institute Of Technology | Convergent and enantioselective total synthesis of Communesin analogs |
| US10654873B2 (en) | 2016-09-15 | 2020-05-19 | Polytherics Limited | Cytotoxic agents and conjugates thereof |
| US11278629B2 (en) | 2016-11-02 | 2022-03-22 | Debiopharm International, S.A. | Methods for improving anti-CD37 immunoconjugate therapy |
| US11730822B2 (en) | 2017-03-24 | 2023-08-22 | Seagen Inc. | Process for the preparation of glucuronide drug-linkers and intermediates thereof |
| US11932650B2 (en) | 2017-05-11 | 2024-03-19 | Massachusetts Institute Of Technology | Potent agelastatin derivatives as modulators for cancer invasion and metastasis |
| US10646585B2 (en) | 2017-09-15 | 2020-05-12 | Hangzhou Dac Biotech Co., Ltd. | Hydrophilic linkers and ligand-drug conjugates thereof |
| US10640508B2 (en) | 2017-10-13 | 2020-05-05 | Massachusetts Institute Of Technology | Diazene directed modular synthesis of compounds with quaternary carbon centers |
| CN112543770A (en) * | 2018-06-26 | 2021-03-23 | 伊缪诺金公司 | Immunoconjugates targeting ADAM9 and methods of use thereof |
| US11535634B2 (en) | 2019-06-05 | 2022-12-27 | Massachusetts Institute Of Technology | Compounds, conjugates, and compositions of epipolythiodiketopiperazines and polythiodiketopiperazines and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102596922A (en) | 2012-07-18 |
| EP2486023A1 (en) | 2012-08-15 |
| KR20120080611A (en) | 2012-07-17 |
| IL219020A0 (en) | 2012-06-28 |
| EP2486023A4 (en) | 2014-05-07 |
| IN2012DN02780A (en) | 2015-09-18 |
| JP2013506709A (en) | 2013-02-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9150649B2 (en) | Potent conjugates and hydrophilic linkers | |
| EP2486023A1 (en) | Potent conjugates and hydrophilic linkers | |
| US20210261683A1 (en) | Method of targeting specific cell populations using cell-binding agent maytansinoid conjugates linked via a non-cleavable linker, said conjugates and methods of making said conjugates | |
| JP2013506709A5 (en) | ||
| AU2006236489B2 (en) | Elimination of heterogeneous or mixed cell population in tumors | |
| WO2014094353A1 (en) | Maytansinoid derivatives | |
| AU2012352210A1 (en) | Use of N-hydroxysuccinimide to improve conjugate stability |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980162373.6 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09844200 Country of ref document: EP Kind code of ref document: A1 |
|
| WPC | Withdrawal of priority claims after completion of the technical preparations for international publication |
Ref document number: 12/433,668 Country of ref document: US Date of ref document: 20110426 Free format text: WITHDRAWN AFTER TECHNICAL PREPARATION FINISHED |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2780/DELNP/2012 Country of ref document: IN Ref document number: 2009844200 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 219020 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012533123 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 20127010084 Country of ref document: KR Kind code of ref document: A |