WO2016033424A1 - Methods for the prevention and treatment of major adverse cardiovascular events using compounds that modulate apolipoprotein b - Google Patents
Methods for the prevention and treatment of major adverse cardiovascular events using compounds that modulate apolipoprotein b Download PDFInfo
- Publication number
- WO2016033424A1 WO2016033424A1 PCT/US2015/047372 US2015047372W WO2016033424A1 WO 2016033424 A1 WO2016033424 A1 WO 2016033424A1 US 2015047372 W US2015047372 W US 2015047372W WO 2016033424 A1 WO2016033424 A1 WO 2016033424A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mace
- antisense oligonucleotide
- patient
- certain embodiments
- oligonucleotide
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/11—Antisense
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/31—Chemical structure of the backbone
- C12N2310/315—Phosphorothioates
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/34—Spatial arrangement of the modifications
- C12N2310/341—Gapmers, i.e. of the type ===---===
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/34—Spatial arrangement of the modifications
- C12N2310/346—Spatial arrangement of the modifications having a combination of backbone and sugar modifications
Definitions
- a major adverse cardiovascular event in a subject with hypercholesterolemia, e.g., familial hypercholesterolemia or a condition or disease with an increased risk of MACE.
- ApoB Apolipoprotein B
- Coronary heart disease has been the leading cause of death in the United States for over a century, and complications from atherosclerosis are the most common causes of death in Western societies (Knopp, New Engl. J. Medicine, 1999, 341, 498-511; Davis and Hui, Arterioscler. Thromb. Vase. Biol, 2001, 21, 887-898; Bonow, Circulation, 2002, 106, 3140- 3141). Elevated low density lipoprotein-cholesterol (LDL-cholesterol) is widely recognized as a risk factor for CHD. However, despite pharmacologic intervention, many subjects are unable to lower LDL-cholesterol levels. Indeed, following acute coronary syndrome, the risk for future major cardiovascular events (MACE) is high and related to levels of LDL-C, even when the patients are on standard therapies (e.g., statins).
- MACE major cardiovascular events
- Low density lipoproteins are one of five broad classes of lipoproteins, which include the following: chylomicrons, responsible for the transport dietary lipids from intestine to tissues; very low density lipoproteins (VLDL); intermediate density lipoproteins (IDL); low density lipoproteins (LDL); all of which transport triacylglycerols and cholesterol from the liver to tissues; and high density lipoproteins (HDL), which transport endogenous cholesterol from tissues to the liver.
- VLDL very low density lipoproteins
- IDL intermediate density lipoproteins
- LDL low density lipoproteins
- HDL high density lipoproteins
- Lipoprotein particles undergo continuous metabolic processing and have variable properties and compositions.
- the protein components of lipoproteins are known as apolipoprotems. At least nine apolipoprotems, one of which is apolipoprotein B, are distributed in significant amounts among the various human lipoproteins.
- Apolipoprotein B (also known as ApoB, apolipoprotein B-100; ApoB-100, apolipoprotein B-48; ApoB-48 and Ag(x) antigen), is a large glycoprotein involved in the assembly and secretion of lipids and in the transport and receptor-mediated uptake and delivery of distinct classes of lipoproteins.
- Apolipoprotein B performs a variety of functions, including the absorption and processing of dietary lipids, as well as the regulation of circulating lipoprotein levels (Davidson and Shelness, Annu. Rev. Nutr., 2000, 20, 169-193).
- Apolipoprotein B is involved cholesterol homeostasis and its overproduction has been associated with various diseases, including familial hypercholesterolemia, familial defective ApoB and familial combined hypercholesterolemia (Kane and Havel, The Metabolic and Molecular Bases of Inherited Diseases, 2001, 8.sup.th edition, 2717-2751). Perturbations in the metabolism of ApoB that correspond with an increased risk of CHD are also observed in diabetes and obesity (Grundy, Am. J. Cardiol, 1998, 81, 18B-25B; Chan et al, Diabetes, 2002, 51, 2377-2386; Chan et al, Metabolism, 2002, 51, 1041-1046).
- LDL-C-lowering medications include statins, cholesterol absorption inhibitors, fibrates, niacin, and bile acid sequestrants.
- Statins are a commonly prescribed treatment for LDL-C lowering. While statins are potent apoB lowering agents, their efficacy in achieving therapeutic targets for LDL-C patients, for example in high-risk patients (FH) is limited. Resistance and intolerance to statins occur in a significant number of patients. Thus, there is a need for new lipid lowering therapies.
- a method of treating, preventing, or managing a major adverse cardiovascular event (MACE) in a hypercholesterolemia patient in need thereof comprising administering to the patient a therapeutically effect amount of an antisense olionucleotide complementary to a nucleic acid encoding human apolipoprotein B is described.
- MACE major adverse cardiovascular event
- a method of treating, preventing, or managing a major adverse cardiovascular event (MACE) in a patient comprising; selecting a patient having a disease or condition that increases the risk of MACE, and administering to the patient a therapeutically effect amount of an antisense olionucleotide complementary to a nucleic acid encoding human apolipoprotein B is described.
- MACE major adverse cardiovascular event
- the MACE is a myocardial infarction, reinfarction, stroke, unstable angina, cardiogenic shock, pulmonary edema, cardiac arrest, coronary revascularization, investigational angioplasty, interventional angioplasty, a percutaneous transluminal coronary angioplasty, percutaneous coronary intervention, a coronary artery bypass graft, or any combination thereof.
- the MACE is myocardial infarction.
- the patient is statin-resistant or statin intolerant.
- the oligonucleotide is administered for at least 12 months. In a fifth embodiment of the first or second aspect or any other embodiment, the oligonucleotide is administered for 12 to 24 months, or for a prolonged period of reduction of MACE events.
- the patient has an established cardiovascular disease.
- the administration results in reversed cardiac injury.
- the patient is homozygous for familial hypercholestrolemia.
- the patient is heterozygous for familial hypercholestrolemia.
- the patient is heterozygous for familial hypercholestrolemia with coronary artery disease.
- the patient has severe hypercholesterolemia.
- the patient has not previously been treated for MACE.
- the patient has been previously been treated for MACE.
- the method reduces the occurrence of or prevents MACE in a patient having an established CVD.
- the method reduces the occurrence of MACE in a patient at risk of CVD.
- the patient has a reduction of serum cholesterol, ApoB, serum low density lipoprotein (LDL), serum very low density lipoprotein (VLDL), serum triglycerides, serum apolipoprotein (a) and/or free fatty acids after administration of the antisence oligonucleotide.
- LDL serum low density lipoprotein
- VLDL serum very low density lipoprotein
- triglycerides serum triglycerides
- serum apolipoprotein (a) serum apolipoprotein (a) and/or free fatty acids after administration of the antisence oligonucleotide.
- the antisense oligonucleotide is 20 nucleobases in length.
- the antisence oligonucleotide is mipomersen.
- the antisense oligonucleotide has a nucleobase sequence consisting of the nucleobase sequence of SEQ ID NO: 247.
- the antisense oligonucleotide comprises a modified internucleoside linkage, a modified sugar moiety, a modified nucleobase, or a combination thereof.
- the modified sugar moiety is a 2'-0-methoxyethyl sugar moiety or a bicyclic sugar moiety.
- the modified internucleoside linkage is a phosphorothioate linkage.
- the modified nucleobase is a 5-methylcytosine.
- the antisense oligonucleotide is a chimeric oligonucleotide.
- the chimeric oligonucleotide comprises a gap segment often linked 2'-deoxynucleotides, wherein the gap segment is positioned between wing segments, wherein each nucleoside of each wing segment comprises a modified sugar moiety.
- the modified sugar moiety is a 2 '-O-methoxy ethyl sugar moiety.
- each wing segment comprises from 1 to 8 2'-0-methoxyethyl.
- each wing segment comprises 2'-methoxyethoxyl nucleotides.
- the antisense oligonucleotide is an antisense oligonucleotide 20 nucleotides in length having the nucleobase sequence of SEQ ID NO: 247, and comprising a 5-methylcytosine at nucleobases 2, 3, 5, 9, 12, 15, 17, 19, and 20, wherein every internucleoside linkage is a phosphorothioate linkage, nucleotides 1-5 and 16-20 can be 2 '-O-methoxy ethyl nucleotides, and nucleotides 6-15 can be 2'-deoxynucleotides, or wherein said antisense oligonucleotide is a pharmaceutically acceptable salt form thereof.
- the antisense oligonucleotide is administered in a dosage to achieve at least a 60% reduction in MACE.
- the antisense oligonucleotide is administered in a dosage to achieve at least a 65%, at least a 70%, at least a 75%, at least a 80%, at least a 85%, or at least a 90% reduction in MACE.
- the antisense oligonucleotide is administered at 200 mg per week.
- the administering comprises an induction phase, wherein a 210 mg dose of the antisense
- oligonucleotide per week is administered in two or more administrations for at least 13 weeks, followed by a maintenance phase, wherein a 210 mg dose of the antisense oligonucleotide per week is administered in two or more administrations.
- the administration of the antisense oligonucleotide cause reductions in atherogenic lipoproteins in plasma.
- the antisense oligonucleotide is administered with one or more additional compounds selected from the group consisting of angiotensin-converting- enzyme inhibitors, angiotensin receptor blockers, renin inhibitors, HMG CoA reductase inhibitors, dihydropyridine calcium channel blockers, antiarrhythmic agents, azetidinone-based cholesterol absorption inhibitors, PCSK9 inhibitors, niacin, niacin derivatives, PPAR agonists, PPAR antagonists, bile acid sequestrants; and antiplatelet drugs; or any pharmaceutically acceptable esters, derivatives, conjugates, precursors or salts thereof.
- the method reduces the occurrence of MACE as compared to the occurrence of the MACE prior to administration of the antisense oligonucleotide.
- the reduction in the occurrence of the MACE is as compared to the occurrence of the MACE in the 24 months prior to administration of the antisense oligonucleotide.
- Familial hypercholesterolemia is associated with a 10-20 fold increase in cardiovascular (CV) events and many of these patients are resistant or intolerant to statin therapy.
- CV cardiovascular
- MACE major adverse cardiovascular events
- the present application is based, in part, on the discovery that mipomersen significantly lowers levels of atherogenic lipoproteins in plasma; whereas previous safety analysis of all patients in phase 3 trials found no imbalance in CV events between placebo and mipomersen arms and the patients have a reduction in MACE events.
- a method for treating, preventing, or managing a MACE in one aspect, is a method for treating, preventing, or managing a MACE.
- the patient has a condition or disease that has an increase in the risk for MACE, e.g., familial hypercholesterolemia (FH).
- FH familial hypercholesterolemia
- the patent is resistant or intolerate to statins.
- the patient has familial hypercholesterolemia and is resistant or intolerate to statins.
- the method comprises administering an antisense oligonucleotide to the patient.
- Such methods can comprise the administration of a therapeutically effective amount of an antisense oligonucleotide.
- the antisense oligonucleotide targets ApoB.
- the method comprises administering to the patient a therapeutically effective amount of an antisense oligonucleotide comprising or consisting of a nucleobase sequence of SEQ ID NO: 247 (e.g., mipomersen).
- the MACE is a myocardial infarction, reinfarction, stroke, unstable angina, cardiogenic shock, pulmonary edema, cardiac arrest, atrial dysrhythmia, coronary revascularization, investigational angioplasty, interventional angioplasty, a
- the MACE is death.
- the MACE is a non-fatal myocardial infarction, stroke, unstable angina, or revascularization procedure (e.g., a percutaneous coronary intervention (PCI), coronary artery bypass graft (CABG) surgery), or any combination thereof.
- PCI percutaneous coronary intervention
- CABG coronary artery bypass graft
- the methods described sho a reversal of cardiac injury from the administration of mipomerson for at least 12 months.
- the patient is a mammal (e.g., a rodent, monkey), such as a human.
- the patient is homozygous for FH.
- the patient is heterozygous for FH.
- the patient has coronary artery disease, severe hypercholesterolemia, or a high risk of cardiovascular disease (CVD).
- CVD cardiovascular disease
- the patient was not previously treated for MACE. In other embodiments, the patient was previously treated for MACE. In some embodiments, the patient is a patient in need thereof. [0047] In certain embodiments, a method provided herein reduces the occurrence of or prevents MACE in a patient having established CVD. In some embodiments, a method provided herein reduces the occurrence of or prevents MACE in a patient at risk of CVD.
- the antisense oligonucleotide is an antisense oligonucleotide, which is targeted to a nucleic acid encoding ApoB. In certain embodiments, the antisense oligonucleotide is 20 nucleobases in length. In some embodiments, the antisense oligonucleotide is an antisense oligonucleotide 20 nucleobases in length. In other embodiments, the antisense oligonucleotide has a nucleobase sequence comprising of the nucleobase sequence of SEQ ID NO: 247. In yet other embodiments, the antisense oligonucleotide has a nucleobase sequence consisting of the nucleobase sequence on SEQ ID NO: 247.
- the antisense oligonucleotide includes a modified
- the modified sugar moiety is a 2'-0-methoxyethyl sugar moiety or a bicyclic sugar moiety.
- the modified internucleoside linkage is a phosphorothioate linkage.
- the modified nucleobase is a 5-methylcytosine.
- the antisense oligonucleotide is a chimeric oligonucleotide.
- the chimeric oligonucleotide can include a gap segment often linked 2'- deoxynucleotides.
- the gap segment is positioned between wing segments.
- each nucleoside of each wing segment includes a modified sugar moiety.
- the modified sugar moiety is a 2'-0-methoxyethyl sugar moiety.
- the gap segment is 10 2'-deoxynucleosides in length, and each wing segment includes from 1 to 8 2'-0-methoxyethyl.
- each wing segment includes 2'- methoxyethoxyl nucleotides.
- the antisense oligonucleotide is an antisense oligonucleotide 20 nucleotides in length having the nucleobase sequence of SEQ ID NO: 247, and can optionally include a 5-methylcytosine at nucleobases 2, 3, 5, 9, 12, 15, 17, 19, and 20.
- SEQ ID NO: 247 the nucleobase sequence of SEQ ID NO: 247
- every internucleoside linkage is a phosphorothioate linkage
- nucleotides 1-5 and 16-20 are 2'-0-methoxyethyl nucleotides
- nucleotides 6-15 are 2'-deoxynucleotides.
- the antisense oligonucleotide is a pharmaceutically acceptable salt form thereof.
- the antisense oligonucleotide is administered in a dosage to achieve at least a 60% reduction in MACE. In certain embodiments, the antisense oligonucleotide is administered in a dosage to achieve at least a 65%, at least a 70%, at least a 75%, at least an 80%, at least an 85%, or at least a 90% reduction in MACE.
- the antisense oligonucleotide is administered at 200 mg per day. In some embodiments, the antisense oligonucleotide is administered for at least 12 months.
- administration of the antisense oligonucleotide decreases total serum cholesterol, ApoB, serum low density lipoprotein (LDL), serum very low density lipoprotein (VLDL), serum triglycerides, serum apolipoprotein (a) and/or free fatty acids in the patient.
- LDL serum low density lipoprotein
- VLDL serum very low density lipoprotein
- a serum apolipoprotein
- administration of the antisense oligonucleotide decreases LDL cholesterol.
- the LDL level is reduced to about 100 mg/dl or lower, about 70 mg/dl or lower, or about 50 mg/dl or lower.
- administration of the antisense oligonucleotide causes reductions in atherogenic lipoproteins in plasma.
- the antisense oligonucleotide is administered with one or more additional oligonucleotide s selected from the group consisting of angiotensin-converting- enzyme inhibitors, angiotensin receptor blockers, renin inhibitors, HMG CoA reductase inhibitors, dihydropyridine calcium channel blockers, antiarrhythmic agents, azetidinone-based cholesterol absorption inhibitors, niacin, niacin derivatives, PPAR agonists, PPAR antagonists, bile acid sequestrants; and antiplatelet drugs; or any pharmaceutically acceptable esters, derivatives, conjugates, precursors or salts thereof.
- additional oligonucleotide s selected from the group consisting of angiotensin-converting- enzyme inhibitors, angiotensin receptor blockers, renin inhibitors, HMG CoA reductase inhibitors, dihydropyridine calcium channel blockers, antiarrhythmic agents, azetidinone-based cholesterol
- the method reduces the occurrence of MACE as compared to a patient that has not been administered the antisense oligonucleotide.
- the patient that has not received the antisense oligonucleotide is a patient that was administered a placebo.
- the method reduces the occurrence of MACE as compared to the occurrence of the MACE prior to administration of the antisense oligonucleotide. In some embodiments, the reduction in the occurrence of the MACE is compared to the occurrence of the MACE in the 24 months prior to administration of the antisense oligonucleotide.
- FIG. 1 depicts the general study design of Phase 3 randomized, placebo-controlled studies used for assessing MACE incidence in FH patients treated with mipomirsen for at least one year.
- FIG. 2 depicts that there is a significant reduction in MACE incidence in FH patients treated with mipomirsen for at least one year.
- the term “about” or “approximately” refers to within 20%, within 10%, within 5%, within 1% or less of a given range.
- acceptable safety profile means a pattern of side effects that is within clinically acceptable limits.
- active pharmaceutical ingredient means the substance in a pharmaceutical composition that provides a desired effect.
- ISIS 301012 SEQ ID NO:247; mipomersen
- saline is the active pharmaceutical ingredient in a pharmaceutical composition comprising ISIS 301012 and saline.
- active target region refers to a target region to which one or more active antisense oligonucleotides or compounds is targeted.
- active antisense compounds or “active antisense olionucleotides” refer to antisense compounds or oligonucelotides that reduce target nucleic acid levels.
- administering refers to providing a pharmaceutical agent to a subject, and includes, but is not limited to administering by a medical professional and self- administering.
- antisense refers to the modulation of function of a target nucleic acid by oligonucleotide s which specifically hybridize to it is.
- antisense oligonucleotide refers to an oligomeric oligonucleotide that is capable of undergoing hybridization to a target nucleic acid through hydrogen bonding.
- antisense inhibition refers to reduction of a target nucleic acid levels in the presence of an antisense oligonucleotide complementary to a target nucleic acid complementary to a target nucleic acid levels in the absence of the antisense oligonucleotide.
- antisense oligonucleotide refers to a single-stranded oligonucleotide having a nucleobase sequence that will permits hybridization to a corresponding region of a target nucleic acid. Such an antisense oligonucleotide is "targeted to" the nucleic acid.
- ApoB means apolipoprotein B-100 protein. Concentration of ApoB in serum (or plasma) is typically quantified in mg/dL or nmol/L.
- Sporum ApoB and plasma ApoB mean ApoB in the serum and plasma, respectively.
- ApoAl is apolipoprotein-Al protein in serum. Concentration of ApoAl in serum is typically quantified in mg/dL or nmol/L.
- ApoB ApoAl ratio
- ApoAl ratio is the ratio of ApoB concentration to ApoAl concentration.
- ApoB -containing lipoprotein refers to any lipoprotein that has ApoB as its protein component, and is understood to include LDL, VLDL, IDL, and lipoprotein (a).
- the term "atherosclerosis” refers to a hardening of the arteries affecting large and medium-sized arteries and is characterized by the presence of fatty deposits.
- the fatty deposits can be called “atheromas” or “plaques,” which consist mainly of cholesterol and other fats, calcium and scar tissue, and damage the lining of arteries.
- AUCt r ou gh or "plasma trough AUC” means the area under the concentration-time curve at a time when plasma pharmaceutical agent concentrations are in equilibrium with target tissue pharmaceutical agent concentrations.
- bicyclic sugar refers to a furosyl ring modified by the bridging of the two non-geminal ring atoms.
- a bicyclic sugar is a modified sugar.
- C h ou gh or "plasma trough concentration” means a minimum plasma concentration when plasma pharmaceutical agent concentrations are in equilibrium with target tissue pharmaceutical agent concentrations.
- a plasma trough concentration of ISIS 301012 is achieved when plasma ISIS 301012 concentrations are in equilibrium with liver tissue ISIS 301012 concentrations.
- cap structure or “terminal cap moiety” refers to chemical modifications, which have been incorporated at either terminus of an antisense oligonucleotide.
- cardiac injury refers to the disruption of normal cardiac myocyte membrane integrity resulting in the loss into the extracellular space (including blood) of intracellular constituents including detectable levels of a variety of biologically active cytosolic and structural proteins such as troponin, creatine kinase, myoglobin, heart-type fatty acid binding protein, and lactate dehydrogenase.
- a reversal of cardiac injury refers to an improvement of any of the biochemical characteristics of the injury, e.g., reduction of inflammation. Ischemia or infarction consequent to an imbalance between the supply and demand of oxygen (and nutrients) is the most common cause of cardiac injury. When a sufficient number of myocytes have died (myocyte necrosis) or lost function, acute clinical disease is apparent; examples include myocardial infarction (MI) or myocarditis.
- MI myocardial infarction
- cardiovascular outcome means the occurrence of major adverse cardiovascular events.
- CHD risk factors refers to CHD risk equivalents and major risk factors.
- CHD risk equivalents means indicators of clinical atherosclerotic disease that confer a high risk for coronary heart disease, and include clinical coronary heart disease, symptomatic carotid artery disease, peripheral arterial disease, and/or abdominal aortic aneurysm.
- chimeric antisense oligonucleotide s refers to an antisense oligonucleotide s that have at least 2 chemically distinct regions, each region having a plurality of subunits.
- cholesterol absorption inhibitor refers to a pharmaceutical agent that inhibits the absorption of exogenous cholesterol obtained from diet.
- cholesteryl ester content means the amount of cholesteryl ester present in liver tissue. In certain embodiments, serum cholesteryl ester concentration is used as an indicator of hepatic cholesteryl ester content.
- complementarity means the capacity for pairing between nucleobases of a first nucleic acid and a second nucleic acid.
- each nucleobase of an oligonucleotide is capable of precise base pairing with the corresponding nucleobases of a target nucleic acid.
- composition is intended to encompass a product containing the specified ingredients (e.g., an antisense oligonucleotide provided herein) and, optionally, in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in, optionally, the specified amounts.
- specified ingredients e.g., an antisense oligonucleotide provided herein
- co-administration refers to administration of two or more pharmaceutical agents to a subject.
- the two or more pharmaceutical agents can be in a single pharmaceutical composition, or can be in separate pharmaceutical compositions.
- Each of the two or more pharmaceutical agents can be administered through the same or different routes of administration.
- Co-administration encompasses administration in parallel or sequentially.
- the combination of therapies (e.g., use of therapeutic agents) which is more effective than the additive effects of any two or more single therapy.
- a synergistic effect of a combination of therapeutic agents permits the use of lower dosages of one or more of the agents and/or less frequent administration of the agents to a subject with a MACE.
- the ability to utilize lower dosages of prophylactic or therapeutic therapies and/or to administer the therapies less frequently reduces the toxicity associated with the administration of the therapies to a subject without reducing the efficacy of the therapies in the prevention,
- CHD coronary heart disease
- complementarity refers to the capacity for pairing between nucleobases of a first nucleic acid and a second nucleic acid.
- nucleobases refers to nucleobases immediately adjacent to each other.
- diabetes dyslipidemia or "Type II diabetes with dyslipidemia” means a condition characterized by Type II diabetes, reduced HDL-C, elevated serum
- the term "diluent” refers to an ingredient in a composition that lacks pharmacological activity, but is pharmaceutically necessary or desirable.
- the diluent can be a liquid, e.g., saline solution.
- a disease or condition that increases the risk of MACE including by not limited to FH, homozygous FH, severe heterozygous FH, heterozygous FH, diabetes, coronary heart disease, and obesity.
- the term "dose" refers a specified quantity of a pharmaceutical agent provided in a single administration.
- a dose can be administered in two or more boluses, tablets, or injections.
- the desired dose requires a volume not easily accommodated by a single injection.
- two or more injections can be used to achieve the desired dose.
- a dose can be administered in two or more injections to minimize injection site reaction in a subject.
- a dosage unit refers to a form in which a pharmaceutical agent is provided.
- a dosage unit is a vial containing lyophilized antisense oligonucleotide.
- a dosage unit is a vial containing reconstituted antisense oligonucleotide.
- a "dosing regimen” is a combination of doses designed to achieve one or more desired effects.
- a dose regimen is designed to provide a therapeutic effect quickly.
- a dose regimen is designed to reduce and undesired side effect, for example, liver toxicity.
- diabetes As used herein, the term "diabetic dyslipidemia" or "Type II diabetes with
- dyslipidemia refers to a condition characterized by Type II diabetes, reduced HDL-C, elevated serum triglycerides, and elevated small, dense LDL particles.
- the term “duration” refers to the period of time during which an activity or event continues.
- the duration of treatment is the period of time during which doses of a pharmaceutical agent can be administered.
- the duration of an induction phase is the period of time during which induction doses are
- the duration of a maintenance phase is the period of time during which maintenance doses are administered.
- efficacy refers to the ability to produce a desired effect.
- efficacy of a lipid-lowering therapy can be reduction in the concentration of one or more of LDL-C, VLDL-C, IDL-C, non-HDL-C, ApoB , lipoprotein (a), or triglycerides.
- the term "effective amount" as used herein refers to the amount of a therapy (e.g., a pharmaceutical composition provided herein) which is sufficient to reduce and/or prevent and/or manage a MACE or a symptom related thereto. This term also encompasses an amount necessary for the reduction or amelioration of the advancement a MACE or amelioration of the recurrence, or onset of a given MACE, and/or to improve or enhance the or therapeutic effect(s) of another therapy (e.g., a therapy other than the antisense oligonucleotide provided herein). In some embodiments, the effective amount of an antisense oligonucleotide provided herein is from about 200 mg/day to about 400 mg/day.
- an effective amount as used herein also refers to the amount of an antisense oligonucleotide provided herein to achieve a specified result (e.g., inhibition of ApoB synthesis).
- an effective amount is 200 mg/week.
- the 200 mg/week is adminitered one time per week.
- the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week.
- doses e.g., 2, 3, 4, 5, 6 or 7 doses
- efficacy of a lipid-lowering therapy may be reduction in MACE, or in the concentration of one or more of LDL-C, VLDL-C, IDL-C, non-HDL-C, ApoB, lipoprotein(a), or triglycerides.
- “elevated total cholesterol” means total cholesterol at a concentration in a subject at which lipid-lowering therapy is recommended, and includes, without limitation, elevated LDL-C", “elevated VLDL-C,” “elevated IDL-C,” and “elevated non-HDL-C.” In certain embodiments, total cholesterol concentrations of less than 200 mg/dL, 200-239 mg/dL, and greater than 240 mg/dL are considered desirable, borderline high, and high, respectively.
- LDL-C concentrations of 100 mg/dL, 100-129 mg/dL, 130-159 mg/dL, 160-189 mg/dL, and greater than 190 mg/dL are considered optimal, near optimal/above optimal, borderline high, high, and very high, respectively.
- “elevated triglyceride” means concentrations of triglyceride in the serum or liver at which lipid-lowering therapy is recommended, and includes “elevated serum triglyceride” and “elevated liver triglyceride.”
- serum triglyceride concentration of 150-199 mg/dL, 200-499 mg/dL, and greater than or equal to 500 mg/dL is considered borderline high, high, and very high, respectively.
- elevated small dense LDL particles means a concentration of small dense LDL particles in a subject at which lipid-lowering therapy is recommended.
- elevated lipoprotein(a) means a concentration of lipoprotein(a) in a subject at which lipid-lowering therapy is recommended.
- excipients refers to inert substances which can be commonly used as a diluent, vehicle, preservatives, binders, or stabilizing agent for drugs and includes, but not limited to, proteins (e.g., serum albumin, etc.), amino acids (e.g., aspartic acid, glutamic acid, lysine, arginine, glycine, histidine, etc.), fatty acids and phospholipids (e.g., alkyl sulfonates, caprylate, etc.), surfactants (e.g., SDS, polysorbate, nonionic surfactant, etc.), saccharides (e.g., sucrose, maltose, trehalose, etc.) and polyols (e.g., mannitol, sorbitol, etc.). See, also, Remington's Pharmaceutical Sciences (1990) Mack Publishing Co., Easton, PA, which is hereby incorporated by reference
- FH familial hypercholesterolemia
- a diagnosis of familial hypercholesterolemia is made when a subject meets one or more of the following criteria: genetic testing confirming 2 mutated LDL-receptor genes; genetic testing confirming one mutated LDL- receptor gene; document history of untreated serum LDL-cholesterol greater than 500 mg/dL; tendinous and/or cutaneous xanthoma prior to age 10 years; or, both parents have documented elevated serum LDL-cholesterol prior to lipid-lowering therapy consistent with heterozygous familial hypercholesterolemia.
- a first nucleic acid is an antisense oligonucleotide and a target nucleic acid is a second nucleic acid.
- an antisense oligonucleotide is a first nucleic acid and a target nucleic acid is a second nucleic acid.
- a "gapmer”, as described herein refers to an antisense oligonucleotide in which an internal position having a plurality of nucleotides that supports R aseH cleavage is positioned between external regions having one or more nucleotides that can be chemically distinct from the nucleosides of the internal region.
- gap segment refers to the plurality of nucleotides that make up the internal region of a gapmer that can support cleavage by the endonuclease RNaseH.
- gap-widened refers to an antisense oligonucleotide has a gap segment of 12 or more contiguous 2'-deoxyribonucleotides positioned between 5' and 3' wing segments having from one to six nucleotides having modified sugar moieties.
- HMG-CoA reductase inhibitor refers to a pharmaceutical agent that acts through the inhibition of the enzyme HMG-CoA reductase.
- homozygous familial hypercholesterolemia refers to a condition characterized by a mutation in both maternal and paternal LDL-R genes.
- heterozygous familial hypercholesterolemia refers to a condition characterized by a mutation in either the maternal or paternal LDL-R gene.
- high density lipoprotein-C means cholesterol associated with high density lipoprotein particles. Concentration of HDL-C in serum (or plasma) is typically quantified in mg/dL or nmol/L.
- serum HDL-C and “plasma HDL-C” mean HDL-C in the serum and plasma, respectively.
- history of coronary heart disease means the occurrence of clinically evident coronary heart disease in the medical history of a subject or a subject's family member.
- hypoglycemic or “hyperglycemia,” when used in reference to a condition of a subject refers to a transient or chronic abnormally high level of glucose present in the blood of a subject.
- the condition can be caused by a delay in glucose metabolism or absorption such that the subject exhibits glucose intolerance or a state of elevated glucose not typically found in normal subjects (e.g., in glucose-intolerant pre-diabetic subjects at risk of developing diabetes, or in diabetic subjects).
- Fasting plasma glucose levels for normoglycemia can be less than about 100 mg/dl, for impaired glucose metabolism, between about 100 and 126 mg/dl, and for diabetics greater than about 126 mg/dl.
- hypercholesterolemia refers to a condition characterized by elevated serum cholesterol.
- hypercholesterolemia includes, but is not limited to, polygenic hypercholesterolemia, heterozygous familial hypercholesterolemia, and a
- hypolipidemia refers to a condition characterized by elevated serum lipids.
- hypotriglyceridemia refers to a condition characterized by elevated triglyceride levels.
- hybridization refers to the annealing of complementary nucleic acid molecules.
- complementary nucleic acid molecules include, but are not limited to, an antisense oligonucleotide and a nucleic acid target.
- complementary nucleic acid molecules include, but are not limited to, an antisense oligonucleotide and a nucleic acid target
- the term "in combination" in the context of the administration of other therapies refers to the use of more than one therapy.
- the use of the term “in combination” does not restrict the order in which therapies can be administered to a subject with.
- a first therapy can be administered before (e.g., 1 minute, 45 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, 24 hours, 48 hours, 72 hours, 96 hours, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 8 weeks, or 12 weeks), concurrently, or after (e.g., 1 minute, 45 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, 24 hours, 48 hours, 72 hours, 96 hours, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 8 weeks, or 12 weeks) the administration of a second therapy to a subject which had, has, or is susceptible to a MACE.
- any additional therapy can be administered in any order with the other additional therapies.
- the antisense oligonucleotide s can be administered in combination with one or more therapies (e.g., therapies that are not the antisense oligonucleotide s provided herein) that can be currently administered to prevent, treat, manage, and/or ameliorate a MACE.
- the term "improved cardiovascular outcome” refers to a reduction in the occurrence of major adverse cardiovascular events, or the risk thereof.
- major adverse cardiovascular events include, without limitation, reinfarction, stroke, cardiogenic shock, pulmonary edema, cardiac arrest, and atrial dysrhythmia.
- the MACE is death.
- increased HDL-C means an increase in serum HDL-C in a subject over time.
- induction phase means a dosing phase during which administration is initiated and steady state concentrations of active pharmaceutical agent are achieved in a target tissue.
- an induction phase is a dosing phase during which steady state
- inflammation refers to a localized protective response elicited by injury or destruction of tissues, which serves to destroy, dilute, or wall off both the injurious agent and the injured tissue. Inflammation is a factor involved in initiation, progression and intability of atheroscherotic plaques. Infammation can be moditored by serum inflammatory biomarkers. Elevation of these biomarkers associate with an increased risk of experiencing a cardiovascular event. See Clin Biochem. 2013 Oct;46(15): 1353-71 for examples of biomarkers for evaluation of inflammation for use in the methods described herein.
- injection site reaction refers to inflammation or abnormal redness of skin at a site of injection in a subject.
- intermediate low density lipoprotein-cholesterol means cholesterol associated with intermediate density lipoprotein. Concentration of IDL-C in serum (or plasma) is typically quantified in mg/mL or nmol/L.
- Serum IDL-C and “plasma IDL-C” mean IDL-C in the serum or plasma, respectively.
- internucleoside linkage refers to the chemical bond or covalent linkage between adjacent nucleosides.
- intravenous administration refers to administration into a vein.
- ISIS 301012 and “mipomersen” mean a lipid-lowering agent that is an antisense oligonucleotide having the sequence "GCCTCAGTCTGCTTCGCACC” (SEQ ID NO:247), wherein each internucleoside linkage is a phosphorothioate internucleoside linkage, each cytosine is a 5-methylcytosine, nucleotides 6-15 are 2'-deoxynucleotides, and nucleotides 1-5 and 16-20 are 2'-0-methoxyethyl nucleotides.
- ISIS 301012 is complementary to nucleotides 3249-3268 of the sequence with GENBANK® Accession No. NM_000384.1.
- Mipomersen treatment in patients does not induce systemic inflammatory response (Flaim et al, J Am Heart Assoc. 2014; 3: e00056; originally published March 13, 2014).
- LDL apheresis means a form of apheresis by which LDL-C is removed from blood.
- a subject's blood is removed from a vein, and separated into red cells and plasma. LDL-C is filtered out of the plasma prior to return of the plasma and red blood cells to the subject.
- LDL-C target refers to an LDL-C level that is desired following lipid-lowering therapy.
- LDL/HDL ratio means the ratio of LDL-C to HDL-C.
- lipid-lowering therapy refers to a therapeutic regimen provided to a subject to reduce one or more lipids in a subject.
- a lipid-lowering therapy is provided to reduce one or more of ApoB total cholesterol, LDL-C, VLDL-C, IDL-C, non-HDL-C, triglycerides, small dense LDL particles, and Lp (a) in a subject.
- lipid-lowering refers to a reduction in one or more serum lipids in a subject over time.
- lipid-lowering agent means a pharmaceutical agent provided to a subject to achieve a lowering of lipids in the subject.
- a lipid-lowering agent is provided to a subject to reduce one or more of ApoB, LDL-C, total cholesterol, and triglyerides.
- lipid-lowering therapy means a therapeutic regimen provided to a subject to reduce one or more lipids in a subject.
- a lipid-lowering therapy is provide to reduce one or more of ApoB, total cholesterol, LDL-C, VLDL-C, IDL-C, non-HDL-C, triglycerides, small dense LDL particles, and Lp(a) in a subject.
- lipoprotein(a) or "Lp(a)” means a lipoprotein particle that is comprised of LDL-C, an apolipoprotein(a) particle, and an apolipoproteinB-100 particle.
- Elevated serum concentration is a risk factor of ACVD.
- Lp(a) levels are not modulated by diet, age, gender or physical activity.
- linked nucleoside refers to adjacent nucleosides which can be bonded together.
- linked deoxynucleoside refers to a nucleic acid base (A, G, C, T, U) substituted by deoxyribose linked by a phosphate ester to form a nucleotide.
- LDL-C low density lipoprotein-cholesterol
- lipid-lowering agent refers to a pharmaceutical agent provided to a subject to achieve a lowering of lipids in the subject.
- a lipid-lowering agent is provided to a subject to reduce one or more of ApoB , LDL-C, total cholesterol, and triglycerides.
- low LDL-receptor activity means LDL-receptor activity that is not sufficiently high to maintain clinically acceptable levels of LDL-C in the bloodstream.
- low HDL-C means a concentration of HDL-C in a subject at which lipid-lowering therapy is recommended. In certain embodiments lipid-lowering therapy is recommended when low HDL-C is accompanied by elevations in non-HDL-C and/or elevations in triglyceride. In certain embodiments, HDL-C concentrations of less than 40 mg/dL are considered low. In certain embodiments, HDL-C concentrations of less than 50 mg/dL are considered low.
- MACE major adverse cardiovascular event
- the terms “MACE” and “major adverse cardiovascular event” are used interchangeably and refers to, for example, a myocardial infarction, reinfarction, a nonfatal myocardial infarction, stroke, unstable angina, cardiogenic shock, pulmonary edema, cardiac arrest, atrial dysrhythmia, coronary revascularization, investigational angioplasty, interventional angioplasty, a percutaneous transluminal coronary angioplasty, percutaneous coronary intervention, a coronary artery bypass graft, or any combination thereof.
- the MACE is death.
- the MACE is a non- fatal myocardial infarction, stroke, unstable angina, or revascularization procedure (e.g., a percutaneous coronary intervention (PCI), coronary artery bypass graft (CABG) surgery), or any combination thereof.
- the MACE is a myocardial infarction.
- the MACE is a stroke.
- the MACE is an unstable angina.
- the MACE is a revascularization procedure.
- the revascularization procedure is a percutaneous coronary intervention (PCI).
- the revascularization procedure is a coronary artery bypass graft (CABG) surgery.
- the MACE is non-fatal.
- maintenance phase means a dosing phase after target tissue steady state concentrations of drug have been achieved.
- major risk factors refers to factors that contribute to a high risk for coronary heart disease, and include without limitation cigarette smoking, hypertension, low HDL-C, family history of coronary heart disease, diabetes, obesity and age.
- mixed dyslipidemia refers to a condition characterized by elevated serum cholesterol and elevated serum triglycerides.
- MTP inhibitor refers to a pharmaceutical agent that inhibits the enzyme microsomal triglyceride transfer protein.
- modified nucleotide refers to a nucleotide having, independently, a modified sugar moiety, modified internucleoside linkage, or modified nucleobase.
- modified nucleoside refers to a nucleotide having, independently, a modified sugar moiety or modified nucleobase.
- modified oligonucleotide refers to an oligonucleotide comprising a modified internucleoside linkage, a modified sugar, and/or a modified nucleobase.
- modified internucleoside linkage refers to substitution and/or any change from a naturally occurring internucleoside bond. In certain instances, the modified internucleoside linkage refers to a phosphodiester internucleoside bond.
- modified sugar moiety means a sugar moiety having a substitution and/or any change from a natural sugar moiety.
- metabolic syndrome refers to a condition characterized by a clustering of lipid and non-lipid cardiovascular risk factors of metabolic origin.
- metabolic syndrome is identified by the presence of any 3 of the following factors: waist circumference of greater than 102 cm in men or greater than 88 cm in women; serum triglyceride of at least 150 mg/dL; HDL-C less than 40 mg/dL in men or less than 50 mg/dL in women; blood pressure of at least 130/85 mmHg; and fasting glucose of at least 1 10 mg/dL.
- maintenance dose means a dose administered at a single administration during the maintenance phase.
- induction dose means a dose administered at a single administration during the induction phase.
- the terms “manage,” “managing,” and “management” refer to the beneficial effects that a subject derives from a therapy (e.g., a therapeutic agent), which does not result in a cure of a the disease, disorder or symptom thereof, such as MACE.
- a subject is administered one or more therapies (e.g., therapeutic agents, such as an antisense oligonucleotide provided herein) to "manage” a MACE (e.g., myocardial infarction, stroke, unstable angina, and the like), or one or more symptoms thereof, so as to prevent the progression or worsening of the MACE or symptom thereof.
- therapies e.g., therapeutic agents, such as an antisense oligonucleotide provided herein
- modified sugar moiety refers to substitution and/or any change from a natural sugar moiety.
- a “natural sugar moiety” is a sugar moiety found in DNA (2'-H) or RNA (2'-OH).
- modified sugar refers to a substitution and/or any change from a natural sugar.
- modified nucleobase refers to any nucleobase other than adenine, cytosine, guanine, thymidine, or uracil.
- An “unmodified nucleobase” refers to the purine bases adenine (A) and guanine (G), and the pyrimidine bases thymine (T), cytosine (C) and uracil (U).
- modified sugar moiety refers to a sugar moiety having a substitution and/or any change from a natural sugar moiety.
- motif refers to the pattern of unmodified and modified nucleosides in an antisense oligonucleotide.
- modulation refers to either an increase (stimulation) or a decrease (inhibition) in the expression of a gene.
- myocardial infarction refers the loss of living heart muscle as a result of coronary artery occlusion (e.g., heart attack).
- Myocardial infarction or its related syndromes usually occurs when an atheromatous plaque in a coronary artery ruptures, and the resulting clot obstructs the injured blood vessel. Perfusion of the muscular tissue that lies downstream from the blocked artery is lost. If blood flow is not restored within a few hours, the heart muscle dies.
- the term "myocardial infarction” focuses on the myocardium (the heart muscle) and the changes that occur in it due to the sudden deprivation of circulating blood. The main change is necrosis (death) of myocardial tissue.
- naturally occurring internucleoside linkage means a 3' to 5' phosphodiester linkage.
- Naturally occurring sugar moiety refers to a sugar moiety found in DNA (2'-H) or RNA (2'-OH).
- nucleic acid encoding human ApoB means DNA encoding ApoB, or RNA transcribed from DNA encoding ApoB.
- nucleobase means a heterocyclic base moiety
- nucleoside means a base-sugar combination.
- nucleotide nucleic acid
- nucleic acid molecule nucleic acid molecule
- nucleotide means a nucleoside having a phosphate group covalently linked to the sugar portion of the nucleoside.
- non-alcoholic fatty liver disease refers to a condition characterized by fatty inflammation of the liver that is not due to excessive alcohol use (for example, alcohol consumption of over 20 g/day).
- nonalcoholic fatty liver disease is related to insulin resistance and the metabolic syndrome.
- non-complementary nucleobase refers to a nucleobase of first nucleic acid that is not capable of pairing with the corresponding nucleobase of a target nucleic acid.
- non-familial hypercholesterolemia means a condition characterized by elevated cholesterol that is not the result of a single gene mutation.
- non-high density lipoprotein-cholesterol (Non-HDL-C) means cholesterol associated with lipoproteins other than high density lipoproteins, and includes, without limitation, LDL-C, VLDL-C, and IDL-C.
- nucleoside refers to a nucleobase linked to a sugar.
- nucleobase refers to a heterocyclic base moiety capable of pairing with a base of another nucleic acid.
- naturally occurring internucleoside linkage refers to a 3 'to 5'phosphodiester linkage.
- nucleotide refers to a nucleoside having a phosphate group covalently linked to the sugar portion of the nucleoside.
- nucleobase sequence refers to the order of contiguous nucleobases independent of any sugar, linkage, and/or nucleobase modification.
- oligomeric oligonucleotide refers to a polymer of linked monomeric subunits which is capable of hybridizing to at least a region of an RNA molecule
- oligonucleotide refers to a polymer of linked nucleosides each of which can be modified or unmodified, independent one from another.
- oligonucleoside refers to an oligonucleotide in which the internucleoside linkages do not contain a phosphorus atom.
- Oxidized-LDL or “Ox-LDL-C” means LDL-C that is oxidized following exposure to free radicals.
- parenteral administration refers to administration through injection or infusion.
- Parenteral administration includes, but is not limited to, subcutaneous administration, intravenous administration, or intramuscular administration
- the term “pharmaceutical agent” refers to a substance that provides a therapeutic benefit when administered to a subject.
- an antisense oligonucleotide targeted to ApoB is a pharmaceutical agent.
- the term “pharmaceutical composition” refers a mixture of substances suitable for administering to a subject.
- a pharmaceutical composition can comprise an antisense oligonucleotide and a sterile aqueous solution.
- pharmaceutically acceptable refers to being approved by a regulatory agency of the Federal or a state government, or listed in the U.S. Pharmacopeia, European Pharmacopeia or other generally recognized Pharmacopeia for use in animals, and more particularly in humans.
- the term "pharmaceutically acceptable carrier” refers to a medium or diluent that does not interfere with the structure of the oligonucleotide. Certain, of such carries enable pharmaceutical compositions to be formulated as, for example, tablets, pills, dragees, capsules, liquids, gels, syrups, slurries, suspension and lozenges for the oral ingestion by a subject.
- pharmaceutically acceptable salts means physiologically and pharmaceutically acceptable salts of antisense oligonucleotide s, i.e., salts that retain the desired biological activity of the parent oligonucleotide and do not impart undesired
- phosphorothioate linkage refers to a linkage between nucleosides where the phosphodiester bond is modified by replacing one of the non-bridging oxygen atoms with a sulfur atom.
- a phosphorothioate linkage is a modified internucleoside linkage.
- polygenic hypercholesterolemia means a condition characterized by elevated cholesterol that results from the influence of a variety of genetic factors. In certain embodiments, polygenic hypercholesterolemia may be exacerbated by dietary intake of lipids.
- portion refers to a defined number of contiguous ⁇ i.e., linked) nucleobases of a target nucleic acid. In certain embodiments, provided herein a portion is a defined number of contiguous nucleobases of a target nucleic acid. In certain embodiments, provided herein a portion is a defined number of contiguous nucleobases of an antisense oligonucleotide.
- prevention refers to the total or partial development, and recurrence of a disease or disorder, such as a MACE, and/or symptoms related thereto, resulting from the administration of a therapy or combination of therapies provided herein (e.g., a combination of therapeutic agents, such as an antisense oligonucleotide provided herein). Prevention can be, for example, in subjects predisposed to having a particular disorder(s).
- prodrug refers to a therapeutic agent that is prepared in an inactive form that is converted to an active form (i.e., drug) within the body or cells thereof by the action of endogenous enzymes or other chemicals and/or conditions.
- reduced coronary heart disease risk refers to a reduction in the likelihood that a subject will develop CHD.
- a reduction in CHD risk is measured by an improvement in one or more CHD risk factors, for example, a decrease in LDL-C levels.
- salts refers to physiologically and pharmaceutically acceptable salts of antisense oligonucleotide s, i.e., salts that retain the desired biological activity of the parent oligonucleotide and do not impart undesired toxico logical effects thereto.
- serum lipids include, but are not limited to, serum cholesterol and serum triglycerides.
- side effects encompasses unwanted and adverse effects of a therapy (e.g., a therapeutic agent). Unwanted effects are not necessarily adverse. An adverse effect from a therapy (e.g., therapeutic agent) might be harmful or uncomfortable or risky.
- a therapy e.g., therapeutic agent
- side effects include, diarrhea, cough, gastroenteritis, wheezing, nausea, vomiting, anorexia, abdominal cramping, fever, pain, loss of body weight, dehydration, alopecia, dyspenea, insomnia, dizziness, mucositis, nerve and muscle effects, fatigue, dry mouth, and loss of appetite, rashes or swellings at the site of administration, flu-like symptoms such as fever, chills and fatigue, digestive tract problems and allergic reactions.
- side effects include, without limitation, injection site reactions, liver function test abnormalities, renal function abnormalities, liver toxicity, renal toxicity, central nervous system abnormalities, and myopathies. For example, increased aminotransferase levels in serum can indicate liver toxicity or liver function abnormality.
- single-stranded modified oligonucleotide refers to a modified oligonucleotide which is not hybridized to a complementary strand.
- slows progression refers to decrease in the development of the said disease.
- small dense LDL particles and “small LDL particles” are used interchangeably herein and refer to a subclass of LDL particles characterized by a smaller, denser size compared to other LDL particles.
- intermediate LDL particles can be 23-27 nm in diameter.
- large LDL particles can be 21. 2-23 nm in diameter.
- small LDL particles can be 18-21. 2 nm in diameter.
- particle size is measured by nuclear magnetic resonance analysis.
- small VLDL particle refers to a subclass of VLDL particles characterized by a smaller, denser size compared to other VLDL particles.
- large VLDL particles can be greater than 60 nm in diameter.
- medium VLDL particles can be 35-60 nm in diameter.
- small VLDL particles can be 27-35 nm in diameter.
- particle size is measured by nuclear magnetic resonance analysis.
- oligonucleotide that hybridizes to a target nucleic acid to induce a desired effect, while exhibiting minimal or no effects on non-target nucleic acids.
- hybridizable and “complementary” can be used to indicate a sufficient degree of
- statin refers to a pharmaceutical agent that inhibits the activity of HMG-CoA reductase. Statins typically lover serum LDL-C by 30-50%.
- statin-resistant means a subject who is currently on statin therapy but is not meting the target in LDL levels.
- statin intolerant subject means a subject who as a result of statin therapy experiences one or more of creatine kinase increases, liver function test abnormalities, muscle aches, or central nervous system side effects.
- stringent hybridization conditions refers to conditions under which a nucleic acid molecule, such as an antisense oligonucleotide, will hybridize to a target nucleic acid sequence, but to a minimal number of other sequences.
- subcutaneous administration refers to administration just below the skin.
- a subject can be a mammal such as a non-primate (e.g., cows, pigs, horses, cats, dogs, rats, etc.) or a primate (e.g., monkey and human).
- the subject is a human.
- the subject is a mammal (e.g., a human) with FH having a MACE.
- the subject is a mammal (e.g., a human) with FH at risk of developing a MACE.
- subject compliance means adherence to a recommended or prescribed therapy by a subject.
- a subject having elevated LDL-C levels refers to a subject who has been identified by a medical professional (e.g., a physician) as having LDL-C levels near or above the level at which therapeutic intervention is recommended, according to guidelines recognized by medical professionals. Such a subject can also be considered “in need of treatment” to decrease LDL-C levels.
- a subject having elevated ApoB -100 levels refers to a subject who has been identified as having ApoB -100 levels near or below the level at which therapeutic intervention is recommended, according to guidelines recognized by medical professionals. Such a subject can also be considered “in need of treatment” to decrease ApoB - 100 levels.
- surrogate markers of cardiovascular outcome means indirect indicators of cardiovascular events, or the risk thereof.
- surrogate markers of cardiovascular outcome include carotid intimal media thickness (CIMT).
- CIMT carotid intimal media thickness
- IVUS intravascular ultrasound
- target nucleic acid and “nucleic acid encoding ApoB” encompass DNA encoding ApoB, RNA (including pre-mRNA and mRNA) transcribed from such DNA, and also cDNA derived from such RNA.
- targeting refers to the process of design and selection of an antisense oligonucleotide that will specifically hybridize to a target nucleic acid and induce a desired effect.
- target nucleic acid As used herein, the terms “target nucleic acid,” “target RNA,” “target RNA transcript” and “nucleic acid target” all mean any nucleic acid capable of being targeted by antisense oligonucleotides.
- ApoB target nucleic acid and “nucleic acid encoding ApoB” encompass nucleic acid, including, for example, DNA (including, for example, cDNA), RNA (including, for example pre-mRNA, and mRNA) transcribed from DNA encoding ApoB, and also cDNA derived from such RNA.
- an ApoB target nucleic acid is the sequence of GENBANK® Accession No. NM_000384.1 , first deposited with GENBANK® on March 24, 1999.
- targeted refers to having a nucleobase sequence that will allow specific hybridization of an antisense oligonucleotide to a target nucleic acid to induce a desired effect.
- a desired effect is reduction of a target nucleic acid.
- a desired effect is reduction of ApoB .
- target region refers to a fragment of a target nucleic acid to which an antisense oligonucleotide is targeted.
- target segment refers to the sequence of nucleotides of a target nucleic acid to which an antisense oligonucleotide is targeted.
- 5 ' target site refers to the 5 '-most nucleotide of a target segment.
- 3 ' target site refers to the 3 '-most nucleotide of a target segment.
- the term “therapy” refers to any protocol, method and/or agent that can be used in the prevention, management, treatment and/or amelioration of a MACE (e.g., myocardial infarction).
- the terms “therapies” and “therapy” refer to a biological therapy, supportive therapy, and/or other therapies useful in the prevention, management, treatment and/or amelioration of a MACE known to one of skill in the art such as medical personnel.
- the term “therapeutic agent” refers to any agent that can be used in the prevention, treatment, management a MACE and/or a symptom related thereto.
- the term “therapeutic agent” refers to an antisense oligonucleotide provided herein.
- the term “therapeutic agent” refers to an agent other than an antisense oligonucleotide provided herein.
- a therapeutic agent is an agent that is known to be useful for, or has been or is currently being used for the prevention, treatment, management of a MACE or one or more symptoms related thereto.
- the therapeutic agent is antisense oligonucleotide with having a nucleobase sequence of SEQ ID NO: 247.
- therapeutic lifestyle change refers to dietary and lifestyle changes intended to lower cholesterol and reduce the risk of developing heart disease, and includes recommendations for dietary intake of total daily calories, total fat, saturated fat, polyunsaturated fat, monounsaturated fat, carbohydrate, protein, cholesterol, insoluble fiber, as well as recommendations for physical activity.
- a "therapeutically effective amount” means an amount of a pharmaceutical agent that provides a therapeutic benefit to a subject.
- a pharmaceutical agent that provides a therapeutic benefit to a subject.
- therapeutically effective amount of an antisense oligonucleotide complementary to a nucleic acid encoding human ApoB is an amount that results, for example, in reduced LDL-C or a reduced incident of MACE.
- total cholesterol refers to all types of cholesterol, including, but not limited to, LDL-C, HDL-C, IDL-C and VLDL-C. Concentration of total cholesterol in serum (or plasma) is typically quantified in mg/dL or nmol/L.
- the terms “treat,” “treatment” and “treating” refers to the reduction or of a disorder or condition (such as a MACE), or a symptom thereof, resulting from the administration of one or more therapies (including, but not limited to, the administration of one or more therapeutic agents, such as an antisense oligonucleotide provided herein).
- the therapeutic agent is an antisense oligonucleotide having a nucleobase sequence of SEQ ID NO: 247.
- triglycerides means lipids that are the triesters of glycerol.
- serum triglycerides mean triglycerides present in serum.
- Liver triglycerides mean triglycerides present in liver tissue.
- nucleobases mean the purine bases adenine (A) and guanine (G), and the pyrimidine bases thymine (T), cytosine (C) and uracil (U).
- unmodified nucleotide refers to a nucleotide composed of naturally occurring nucleobases, sugar moieties and internucleoside linkages.
- an unmodified nucleotide is an R A nucleotide (i.e., ⁇ -D- ribonucleosides) or a DNA nucleotide (i.e., ⁇ -D-deoxyribonucleoside).
- VLDL-C very low density lipoprotein-cholesterol
- wing segment refers to the external region of a gapmer.
- 2'-0-methoxyethyl sugar moiety refers to a 2 '-substituted furosyl ring having a 2'-0(CH2)2-OCH3 (2'-0-methoxyethyl or 2'-MOE) substituent group.
- 2'-0-methoxyethyl nucleotide refers to a nucleotide comprising a 2'-0-methoxyethyl modified sugar moiety.
- 2'-0-methoxyethyl refers to an O-methoxy-ethyl modification of the 2' position of a furosyl ring.
- a 2'-0-methoxyethyl modified sugar is a modified sugar.
- 5-methylcytosine refers to a cytosine modified with a methyl group attached to the 5' position.
- a 5-methylcytosine is a modified nucleobase.
- Hypercholesterolemia and in particular, an elevated level of serum (or plasma) low density lipoprotein cholesterol (LDL-C), is associated with an increased risk of adverse cardiovascular events.
- LDL-C low density lipoprotein cholesterol
- Lipid lowering drug therapy particularly with statins, is indicated to decrease the risk of cardiovascular events in most individuals with established atherosclerotic cardiovascular disease and in many who are at high risk.
- statin-resistant Many patients treated with statins are considered statin-resistant because they fail to achieve adequate reduction of low density lipoprotein cholesterol (LDL-C) levels.
- LDL-C low density lipoprotein cholesterol
- Some patients are statin-intolerant because they are unable to tolerate statin therapy at all or to tolerate a full therapeutic statin dose because of adverse effects, particularly myopathy and increased activity of liver enzymes.
- statins are the treatment of choice for lowering LDL-C in the majority of patients, including those with Type II diabeties, many patients retain a high CVD risk despite achieving the recommended LDL-C targets with statins.
- apolipoprotein B Two forms of apolipoprotein B exist in mammals.
- ApoB-100 represents the full- length protein containing 4536 amino acid residues, synthesized primarily in the human liver (Davidson and Shelness, Annu. Rev. Nutr., 2000, 20, 169-193).
- a truncated form known as apoB-48 is co linear with the amino terminal 2152 residues and is synthesized in the small intestine of all mammals (Davidson and Shelness, Annu. Rev. Nutr., 2000, 20, 169-193).
- apoB-48 circulates in association with chylomicrons and chylomicron remnants and these particles are cleared by a distinct receptor known as the LDL-receptor-related protein (Davidson and Shelness, Annu. Rev. Nutr., 2000, 20, 169-193).
- ApoB-48 can be viewed as an adaptation by which dietary lipid is delivered from the small intestine to the liver, while apoB- 100 participates in the transport and delivery of cholesterol (Davidson and Shelness, Annu. Rev. Nutr., 2000, 20, 169-193).
- ApoB is the major protein component of LDL and contains the domain required for interaction of this lipoprotein species with the LDL receptor.
- ApoB contains an unpaired cysteine residue which mediates an interaction with
- apolipoprotein(a) and generates lipoprotein(a) or Lp(a), another distinct lipoprotein with atherogenic potential (Davidson and Shelness, Annu. Rev. Nutr., 2000, 20, 169-193). Elevated plasma levels of the ApoB-containing lipoprotein Lp(a) are associated with increased risk for atherosclerosis and its manifestations, which may include hypercholesterolemia (Seed et al, N. Engl. J. Med., 1990, 322, 1494-1499), myocardial infarction (Sandkamp et al, Clin. Chem., 1990, 36, 20-23), and thrombosis (Nowak-Gottl et al, Pediatrics, 1997, 99, El l).
- Elevated plasma apoB for example, as seen with familial hypercholesterolaemia (FH), is associated with an increased risk of theroclerotic cardiovascular disease (ACVD).
- ACVD theroclerotic cardiovascular disease
- hypobetalipoproteinemia patients with the genetic disorder, hypobetalipoproteinemia, are protected against ACVD due to diminished levels of apoB and LDL-cholesterol.
- the methods described herein provide benefits for patients in need of treatment for MACE, these benefits include but are not limited to administering a compound targeted to ApoB that does not produce proinflammatory events and reduces other lipoproteins, for example Lp (a), is advantageous in patient populations where conventional lipid lowering strategies have not hit the target, helps reverse cardiac injury, long term use is therapeutically beneficial (e.g., over 12 months) and can be used in conjuction with other lipid lowering therapies.
- a lipoproteins
- a method of treating, preventing, or managing a major adverse cardiovascular event (MACE) in a hypercholesterolemia patient in need thereof comprises administering to the patient a therapeutically effect amount of an antisense olionucleotide complementary to a nucleic acid encoding human apolipoprotein B.
- MACE major adverse cardiovascular event
- MACE is MACE is a myocardial infarction, reinfarction, stroke, unstable angina, cardiogenic shock, pulmonary edema, cardiac arrest, coronary revascularization, investigational angioplasty, interventional angioplasty, a percutaneous transluminal coronary angioplasty, percutaneous coronary intervention, a coronary artery bypass graft, or any combination thereof.
- the antisense olionucleotide is mipomersen.
- a method for treating, preventing, or managing MACE in a patient with familial hypercholesterolemia (FH) in need thereof includes administering to the patient a therapeutically effective amount of an antisense oligonucleotide having a nucleobase sequence of SEQ ID NO: 247 (e.g., mipomersen).
- the MACE is a myocardial infarction, reinfarction, stroke, unstable angina, cardiogenic shock, pulmonary edema, cardiac arrest, atrial dysrhythmia, coronary revascularization, investigational angioplasty, interventional angioplasty, a
- the MACE is death.
- the MACE is a non-fatal myocardial infarction, stroke, unstable angina, or revascularization procedure (e.g., a percutaneous coronary intervention (PCI), coronary artery bypass graft (CABG) surgery), or any combination thereof.
- the MACE is a myocardial infarction.
- the MACE is a stroke.
- the MACE is an unstable angina.
- the MACE is a revascularization procedure.
- the revascularization procedure is a percutaneous coronary intervention (PCI).
- PCI percutaneous coronary intervention
- revascularization procedure is a coronary artery bypass graft (CABG) surgery.
- CABG coronary artery bypass graft
- MACE non-fatal.
- the patient is a mammal (e.g., a rodent, monkey), such as a human.
- the patient is homozygous for FH.
- the patient is heterozygous for FH.
- the patient has coronary artery disease, severe hypercholesterolemia, or a high risk of CVD.
- the patient was not previously treated for MACE. In other embodiments, the patient was previously treated for MACE.
- a method provided herein reduces the occurrence of or prevents MACE in a patient having established CVD. In some embodiments, a method provided herein reduces the occurrence or prevents MACE in a patient at risk of CVD.
- the antisense oligonucleotide is an antisense oligonucleotide, which is targeted to a nucleic acid encoding apolipoprotein B. In certain embodiments, the antisense oligonucleotide is 20 nucleobases in length. In some embodiments, the antisense oligonucleotide is an antisense oligonucleotide 20 nucleobases in length. In other embodiments, the antisense oligonucleotide has a nucleobase sequence comprising or consisting of the nucleobase sequence of SEQ ID NO: 247.
- the antisense oligonucleotide includes a modified
- the modified sugar moiety is a 2'-0-methoxyethyl sugar moiety or a bicyclic sugar moiety.
- the modified internucleoside linkage is a phosphorothioate linkage.
- the modified nucleobase is a 5-methylcytosine.
- the antisense oligonucleotide is a chimeric oligonucleotide.
- the chimeric oligonucleotide can include a gap segment often linked 2'- deoxynucleotides.
- the gap segment is positioned between wing segments.
- each nucleoside of each wing segment includes a modified sugar moiety.
- the modified sugar moiety is a 2'-0-methoxyethyl sugar moiety.
- the gap segment is 10 2'-deoxynucleosides in length, and each wing segment includes from 1 to 8 2'-0-methoxyethyl.
- each wing segment includes 2'- methoxyethoxyl nucleotides.
- the antisense oligonucleotide is an antisense oligonucleotide 20 nucleotides in length having the nucleobase sequence of SEQ ID NO: 247, and can optionally include a 5-methylcytosine at nucleobases 2, 3, 5, 9, 12, 15, 17, 19, and 20.
- SEQ ID NO: 247 the nucleobase sequence of SEQ ID NO: 247
- 5-methylcytosine at nucleobases 2, 3, 5, 9, 12, 15, 17, 19, and 20.
- every internucleoside linkage is a phosphorothioate linkage
- nucleotides 1-5 and 16-20 are 2'-0-methoxyethyl nucleotides
- nucleotides 6-15 are 2'-deoxynucleotides.
- the antisense oligonucleotide is a pharmaceutically acceptable salt form thereof.
- the antisense oligonucleotide is administered in a dosage to achieve at least a 60% reduction in MACE. In certain embodiments, the antisense
- oligonucleotide is administered in a dosage to achieve at least a 65%, at least a 70%, at least a 75%, at least an 80%, at least an 85%, or at least a 90% reduction in MACE.
- the antisense oligonucleotide is administered at 200 mg per day. In some embodiments, the antisense oligonucleotide is administered for at least 12 months.
- the antisense oligonucleotide is administered at 200 mg/week. In some embodiments, the 200 mg/week is adminitered one time per week. In some embodiments, the subject is administered a single 200 mg/dose per week. In other embodiments, the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week. In some embodiments, the antisense oligonucleotide is administered for at least 12 months. In one embodiment, the antisense oligonucleotide is administered subcutaneously (s.c).
- administration of the antisense oligonucleotide decreases total serum cholesterol, ApoB, serum LDL, serum VLDL, serum triglycerides, serum
- apolipoprotein (a) and/or free fatty acids in the patient are examples of apolipoprotein (a) and/or free fatty acids in the patient.
- administration of the antisense oligonucleotide decreases LDL cholesterol.
- the LDL level is reduced to about 100 mg/dl or lower, about 70 mg/dl or lower, or about 50 mg/dl or lower.
- administration of the antisense oligonucleotide causes reductions in atherogenic lipoproteins in plasma.
- the antisense oligonucleotide is administered with one or more additional oligonucleotide s selected from the group consisting of angiotensin-converting- enzyme inhibitors, angiotensin receptor blockers, renin inhibitors, HMG CoA reductase inhibitors, dihydropyridine calcium channel blockers, antiarrhythmic agents, azetidinone-based cholesterol absorption inhibitors, niacin, niacin derivatives, PPAR agonists, PPAR antagonists, bile acid sequestrants; and antiplatelet drugs; or any pharmaceutically acceptable esters, derivatives, conjugates, precursors or salts thereof.
- additional oligonucleotide s selected from the group consisting of angiotensin-converting- enzyme inhibitors, angiotensin receptor blockers, renin inhibitors, HMG CoA reductase inhibitors, dihydropyridine calcium channel blockers, antiarrhythmic agents, azetidinone-based cholesterol
- the method reduces the occurrence of MACE as compared to a patient that has not been administered the antisense oligonucleotide.
- the patient that has not received the antisense oligonucleotide is a patient that was administered a placebo.
- the method reduces the occurrence of MACE as compared to the occurrence of the MACE prior to administration of the antisense oligonucleotide. In some embodiments, the reduction in the occurrence of the MACE is compared to the occurrence of the MACE in the 24 months prior to administration of the antisense oligonucleotide.
- oligonucleotide and uses thereof for treating, reducing the occurrence of and/or preventing MACE.
- the patient has FH.
- MACE includes, but is not limited to, cardiac death, hospitalization for unstable angina, stroke, transient ischemic attack and hospitalization for peripheral artery disease. Additional oligonucleotide s useful in treating, reducing the occurrence of, and/or preventing CVD or the underlying risk factors associated with CVD can also be beneficially co-administered with antisense compositions and pharmaceutical formulations provided herein.
- the subject has been identified as in need of lipid-lowering therapy. In some embodiments, the subject has been identified as in need of lipid-lowering therapy according to the guidelines established in 2001 by ATP III of the NCEP, and updated in 2004 (Grundy et al., Circulation, 2004, 110, 227-239). In other embodiments, the subject in need of lipid-lowering therapy has LDL-C above 190 mg/dL. In certain embodiments, the subject in need of lipid-lowering therapy has LDL-C above 160 mg/dL. In some embodiments, the subject in need of lipid-lowering therapy has LDL-C above 130 mg/dL.
- the subject in need of lipid-lowering therapy has LDL-C above 100 mg/dL. In some embodiments, the subject in need of lipid-lowering therapy should maintain LDL-C below 160 mg/dL. In certain embodiments, the subject in need of lipid- lowering therapy should maintain LDL-C below 130 mg/dL. In other embodiments, the subject in need of lipid-lowering therapy should maintain LDL-C below 100 mg/dL. In some embodiments, the subject should maintain LDL-C below 70 mg/dL or even below 50 mg/dL.
- provided herein are methods for reducing ApoB in a subject. In other embodiments, provided herein are methods for reducing ApoB -containing lipoprotein in a subject. In certain embodiments, provided herein are methods for reducing LDL-C in a subject. In other embodiments, provided herein are methods for reducing VLDL-C in a subject. In certain embodiments, provided herein are methods for reducing LDL-C in a subject. In other
- provided herein are methods for reducing non-HDL-C in a subject. In some embodiments, provided herein are methods for reducing Lp(a) in a subject. In other words, provided herein are methods for reducing non-HDL-C in a subject. In some embodiments, provided herein are methods for reducing Lp(a) in a subject. In other words, provided herein are methods for reducing non-HDL-C in a subject. In some embodiments, provided herein are methods for reducing Lp(a) in a subject. In other
- provided herein are methods for reducing serum triglyceride in a subject. In certain embodiments, provided herein are methods for reducing liver triglyceride in a subject. In some embodiments, provided herein are methods for reducing Ox-LDL-C in a subject. In other embodiments, provided herein are methods for reducing small LDL particles in a subject. In certain embodiments, provided herein are methods for reducing small VLDL particles in a subject. In certain embodiments, provided herein are methods for reducing phospholipids in a subject. In other embodiments, provided herein are methods for reducing oxidized phospholipids in a subject. Any combination of the two, three four or more of the foregoing methods is also contemplated herein.
- compositions comprising an antisense oligonucleotide for use, e.g., in the prevention, management, treatment and/or amelioration of a MACE.
- the MACE is a myocardial infarction.
- the MACE is a reinfarction.
- the MACE is a stroke.
- the MACE is a myocardial infarction. In other embodiments, the MACE is a reinfarction. In yet other embodiments, the MACE is a stroke. In certain embodiments, the MACE is unstable angina. In some embodiments, the MACE is cardiogenic shock. In other embodiments, the MACE is pulmonary edema. In other embodiments, the MACE is cardiac arrest. In certain embodiments, the MACE is atrial dysrhythmia. In some embodiments, the MACE is coronary revascularization. In other embodiments, the MACE is investigational angioplasty. In certain embodiments, the MACE is interventional angioplasty. In other embodiments, the MACE is percutaneous transluminal coronary angioplasty. In certain embodiments, the MACE is percutaneous coronary intervention. In some embodiments, the MACE is coronary artery bypass graft. In other embodiments, the MACE is death.
- the MACE is a non-fatal myocardial infarction, stroke, unstable angina, or revascularization procedure (e.g., a percutaneous coronary intervention (PCI), coronary artery bypass graft (CABG) surgery), or any combination thereof.
- the MACE is a myocardial infarction.
- the MACE is a stroke.
- the MACE is an unstable angina.
- the MACE is a revascularization procedure.
- the revascularization procedure is a percutaneous coronary intervention (PCI).
- PCI percutaneous coronary intervention
- revascularization procedure is a coronary artery bypass graft (CABG) surgery.
- CABG coronary artery bypass graft
- MACE non-fatal.
- MACE can range from mild to severe.
- Exemplary risk factors for MACE include hypercholesterolemia, mixed dyslipidemia, atherosclerosis, coronary heart disease, a history of coronary heart disease, early onset coronary heart disease, acute coronary syndrome, one or more risk factors for coronary heart disease, type II diabetes, type II diabetes with dyslipidemia, dyslipidemia, hypertriglyceridemia,
- hyperlipidemia hyperfattyacidemia, hepatic steatosis, nonalcoholic steatohepatitis, or nonalcoholic fatty liver disease.
- Other symptoms can include obesity, unhealthy diet, and harmful use of alcohol, smoking, age, or family history.
- compositions comprising an antisense oligonucleotide for use in the prevention, management, treatment and/or amelioration of a MACE, such as, but not limited to, a myocardial infarction, or a risk factor thereof.
- compositions and methods of administering and dosing are also useful in the other methods provided herein.
- a composition provided herein can be used either alone or in combination with other oligonucleotide s or compositions.
- kits for treating, preventing, or managing MACE in a subject comprising administering to the subject an effective amount of an antisense oligonucleotide.
- levels of atherogenic lipoproteins in plasma can also be decreased in the subject.
- the subject has FH.
- the subject administered an antisense oligonucleotide is, in certain embodiments, a mammal such as non-primate (e.g., cows, pigs, horses, cats, dogs, rats, etc.) or a primate (e.g., a monkey, such as a cynomolgus monkey, or a human). In a specific embodiment, the subject is a human.
- non-primate e.g., cows, pigs, horses, cats, dogs, rats, etc.
- a primate e.g., a monkey, such as a cynomolgus monkey, or a human.
- the subject is a human.
- the subject is a human with FH. In another embodiment, the subject has heterozygous FH. In another embodiment, the subject has homozygous FH.
- the subject has a premature CVD.
- the subject has heterozygous FH, and premature CVD.
- the subject has homozygous FH and can have premature CVD.
- the subject has severe hypercholesterolemia. In another embodiment, the subject has a high risk of CVD. In a specific embodiment, the subject has not previously been treated for MACE. In another embodiment, the subject has been treated for MACE.
- the methods provided herein can be useful in the treatment of MACE in patients with FH having established CVD.
- the patient has not had a prior myocardial infarction, and/or does not have any of the underlying risk factors or diseases that cause CVD. These can include, but are not limited to, hypertension, dyslipidemia, obesity and/or diabetes.
- the subject is susceptible or at risk of MACE. In another embodiment, the subject has a heightened risk of MACE.
- the subject has a body mass index of approximately 30.4+ 4 6 kg/m 2 .
- the subject has one or more risk factors, including but not limited to, hypercholesterolemia, mixed dyslipidemia, atherosclerosis, coronary heart disease, a history of coronary heart disease, early onset coronary heart disease, acute coronary syndrome, one or more risk factors for coronary heart disease, type II diabetes, type II diabetes with dyslipidemia, dyslipidemia, hypertriglyceridemia, hyperlipidemia, hyperfattyacidemia, hepatic steatosis, nonalcoholic steatohepatitis, or non-alcoholic fatty liver disease.
- Other symptoms can include obesity, unhealthy diet, and harmful use of alcohol, smoking, age, or family history.
- the subject has symptoms of FH, including, but not limited to xanthomas, xanthelasmas, or corneal arcus.
- the subject has an LDL-C level above 100 mg/dL, above 130 mg/dL, above 160 mg/dL, or above 190 mg/dL.
- the subject is a subject with FH receiving a lipid- lowering medication. In certain embodiments, the subject has FH.
- the subject that has not previously received an antisense provided herein.
- the patient previously received a placebo.
- the subject is administered a placebo for 6 months, followed by administration of the antisense oligonucleotide for at least one year.
- the subject is first administered blinded, an antisense oligonucleotide, followed by open labeled treatment with the antisense oligonucleotide.
- the subject is administered an antisense oligonucleotide concurrently with implementing lifestyle changes, including but not limited to, regular exercise, or reduced fat intake diet.
- methods for reducing the occurrence of MACE comprising administering the antisense inhibitor (e.g., mipomersen) to a subject.
- the subject has FH.
- methods for reducing the occurrence of or preventing MACE in a subject having established CVD are provided herein.
- methods for reducing the occurrence of MACE is a subject at risk of CVD.
- administering reduces the occurrence of MACE as compared to a patient that has not been administered with the antisense oligonucleotide.
- the method further reduces the occurrence of MACE as compared to the occurrence of MACE prior to
- the reduction in the occurrence of MACE is compared to the occurrence of the MACE in the 24 months prior to the administration of the antisense oligonucleotide.
- administering the antisense oligonucleotide or a composition thereof reduces the risk that a subject will develop MACE by at least about 10%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 50%, at least about 60%>, at least about 70%>, at least about 80%>, at least about 90%>, or more, compared to the risk of developing MACE in the absence of such treatment.
- administering the antisense oligonucleotide or a composition thereof reduces the risk of MACE proportionally to the reduction in LDL cholesterol levels.
- a method of decreasing LDL-C levels comprises selecting a subject in need of a decrease in LDL-C levels, and administering to the subject a therapeutically effective amount of an antisense oligonucleotide.
- a method of reducing coronary heart disease risk includes selecting a subject having elevated LDL-C levels and one or more additional indicators of coronary heart disease risk, and administering to the subject a therapeutically effective amount of an antisense oligonucleotide.
- a method for reducing MACE includes selecting a subject having elevated LDL-C levels and one or more MACE prior treatment.
- a method for reducing MACE includes selecting a subject having elevated LDL-C levels and no previous MACE. In a further embodiment, a method for reducing MACE includes selecting a subject having risk factors associated with MACE, as described herein.
- the LDL- C level is from 100-129 mg/dL, from 130-159 mg/dL, from 160-189 mg/dL, or greater than or equal to 190 mg/dL.
- administration of a therapeutically effective amount of an antisense oligonucleotide can be accompanied by monitoring of LDL-C levels in the serum of a subject, to determine a subject's response to administration of the antisense oligonucleotide.
- a subject's response to administration of the antisense oligonucleotide is used by a physician to determine the amount and duration of therapeutic intervention.
- administering results in LDL-C levels below 190 mg/dL, below 160 mg/dL, below 130 mg/dL, below 100 mg/dL, below 70 mg/dL, or below 50 mg/dL.
- administration of an antisense oligonucleotide results in LDL-C levels below 190 mg/dL, below 160 mg/dL, below 130 mg/dL, below 100 mg/dL, below 70 mg/dL, or below 50 mg/dL.
- administration of an antisense oligonucleotide results in LDL-C levels below 190 mg/dL, below 160 mg/dL, below 130 mg/dL, below 100 mg/dL, below 70 mg/dL, or below 50 mg/dL.
- oligonucleotide decreases LDL-C by at least 15%, by at least 25%, by at least 50%, by at least 60%, by at least 70%, by at least 75%, by at least 80%, by at least 85%, by at least 90%, or by at least 95%.
- a subject having elevated LDL-C levels can also exhibit reduced HDL-C levels and/or elevated total cholesterol levels. Accordingly, in one embodiment a therapeutically effective amount of an antisense oligonucleotide is administered to a subject having elevated LDL-C levels, which also has reduced HDL-C levels and/or elevated total cholesterol levels.
- Subjects having elevated LDL-C levels can also exhibit elevated triglyceride levels. Accordingly, in one embodiment a therapeutically effective amount of an antisense
- oligonucleotide is administered to a subject having elevated LDL-C levels, and also having elevated triglyceride levels.
- Measurements of cholesterol, lipoproteins and triglycerides can be obtained using serum or plasma collected from a subject. Methods of obtaining serum or plasma samples can be routine, as are methods of preparation of the serum samples for analysis of cholesterol, triglycerides, and other serum markers. A physician can determine the need for therapeutic intervention in cases where more or less aggressive LDL-lowering therapy is needed. The practice of the methods herein can be applied to any altered guidelines provided by the NCEP, or other entities that establish guidelines for physicians used in treating any of the diseases or conditions listed herein, for determining coronary heart disease risk and diagnosing metabolic syndrome.
- a pharmaceutical composition including an antisense oligonucleotide is for use in therapy.
- the therapy is the reduction of LDL-C, ApoB, VLDL-C, IDL-C, non-HDL-C, Lp(a) , serum triglyceride, liver triglyceride, Ox-LDL-C, small LDL particles, small VLDL, phospholipids, or oxidized phospholipids in a subject.
- the therapy is the treatment of
- hypercholesterolemia mixed dyslipidemia, atherosclerosis, a risk of developing atherosclerosis, coronary heart disease, acute coronary syndrome, a history of coronary heart disease, early onset coronary heart disease, one or more risk factors for coronary heart disease, type II diabetes, type II diabetes with dyslipidemia, dyslipidemia, hypertriglyceridemia, hyperlipidemia,
- the therapy is the reduction of CHD risk.
- the therapy is prevention of atherosclerosis.
- the therapy is the prevention of coronary heart disease.
- Measurements of cholesterol, lipoproteins and triglycerides can be obtained using serum or plasma collected from a subject. Methods of obtaining serum or plasma samples can be routine, as are methods of preparation of the serum samples for analysis of cholesterol, triglycerides, and other serum markers. A physician can determine the need for therapeutic intervention for subjects in cases where more or less aggressive LDL-lowering therapy is needed.
- the practice of the methods herein can be applied to any altered guidelines provided by the NCEP, or other entities that establish guidelines for physicians used in treating any of the diseases or conditions listed herein, for determining coronary heart disease risk and diagnosing metabolic syndrome.
- the therapy is the treatment, prevention, or management of MACE.
- the method comprises measuring the levels of one or more of certain analytes, such as proteins and metabolites, in a plasma or serum sample derived from a blood sample from that subject and then administering an antisense oligonucleotide in a subject likely to experience a MACE.
- certain analytes such as proteins and metabolites
- the antisense oligonucleotide is administered at 200 mg/week.
- the 200 mg/week is adminitered one time per week.
- the subject is administered a single 200 mg/dose per week.
- the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week.
- the antisense oligonucleotide is administered for at least 12 months.
- the antisense oligonucleotide is administered subcutaneously (s.c).
- Also provided herein are methods comprising administering to a subject a
- composition comprising an antisense oligonucleotide complementary to a nucleic acid encoding human apolipoprotein B-100, wherein the administering comprises an induction phase, wherein a dose of the antisense oligonucleotide ranging from 100-300 mg is administered once per week for at least 13 weeks.
- the induction phase is followed by a maintenance phase, wherein a dose of the antisense oligonucleotide ranging from 80-200 mg is administered once per week or once every two weeks for as long as needed, effective, and/or tolerated.
- the antisense oligonucleotide is administered at 200 mg/week.
- the 200 mg/week is adminitered one time per week.
- the subject is administered a single 200 mg/dose per week.
- the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week.
- the antisense oligonucleotide is administered for at least 12 months.
- the antisense oligonucleotide is administered subcutaneously (s.c).
- the dose administered in the induction phase is a 100 mg dose and the dose administered in the maintenance phase is a 200 mg dose administered once per week. In certain embodiments, the dose administered in the induction phase is a 200 mg dose, and the dose administered in the maintenance phase is a 300 mg dose administered once per week. In certain embodiments, the dose administered in the induction phase is a 100 mg dose and the dose administered in the maintenance phase is a 200 mg dose administered once per week, and wherein the tolerability or the effectiveness of the antisense oligonucleotide are assessed during or at the end of the induction period, or a portion thereof once per week during the maintenance phase. In certain embodiments, the dose administered in the induction phase is a 200 mg dose and the dose administered in the maintenance phase is a 300 mg dose
- the dose administered in the induction phase is a 100 mg dose and the dose administered in the maintenance phase is a 100 mg dose administered once every two weeks, and wherein the tolerability or the effectiveness of the antisense
- the dose administered in the induction phase is a 200 mg dose and the dose administered in the maintenance phase is a 200 mg dose administered once every two weeks, and wherein the tolerability or the effectiveness of the antisense oligonucleotide are assessed during or at the end of the induction period, or a portion thereof.
- the dose administered in the induction phase is from 100 mg to 200mg and the dose administered in the maintenance phase is from 200 mg to 300 mg and is administered once per week.
- said administering comprises parenteral administration.
- said parenteral administration comprises subcutaneous administration.
- each induction dose and each maintenance dose comprises a single injection.
- each induction dose and each maintenance dose independently comprise two or more injections.
- the methods further comprise assessing the tolerability or effectiveness of the antisense oligonucleotide during or at the end of the induction period, or a portion thereof. In certain embodiments, the tolerability and the effectiveness of the antisense oligonucleotide are assessed
- the tolerability of the antisense oligonucleotide is assessed by monitoring a rate of decrease of ApoB concentration in the plasma of said subject. In certain embodiments, the tolerability of the antisense oligonucleotide is assessed by monitoring ApoB concentration in the plasma of said subject. In certain embodiments, the tolerability of the antisense oligonucleotide is assessed by monitoring a rate of decrease of ApoB concentration and ApoB concentration in the plasma of said subject. In certain embodiments, the tolerability of the antisense oligonucleotide is assessed by monitoring ALT concentrations in the liver of the subject.
- the tolerability of the antisense oligonucleotide is assessed by monitoring ANT concentrations in the liver of said subject. In certain embodiments, the tolerability of the antisense oligonucleotide is assessed by monitoring bilirubin concentrations in the plasma of the subject.
- a rate of decrease in the ApoB concentration greater than about 30 mg/dL*day indicates that the subject is not tolerating administration of the antisense oligonucleotide.
- an ApoB concentration less than about 60 mg/dL indicates that the subject is not tolerating administration of the antisense oligonucleotide.
- a rate of decrease in the ApoB concentration greater than about 30 mg/dL*day and an ApoB concentration less than about 60 mg/dL indicates that the subject is not tolerating administration of the antisense oligonucleotide.
- the dose of antisense oligonucleotide is reduced following an indication that administration of said antisense oligonucleotide is not tolerated. In certain embodiments, the frequency of administration of antisense oligonucleotide is reduced following an indication that administration of said antisense oligonucleotide is not tolerated. In certain embodiments, the dose of antisense oligonucleotide is increased following an indication that administration of said antisense oligonucleotide is tolerated. In certain embodiments, the frequency of administration of antisense oligonucleotide is increased following an indication that administration of said antisense oligonucleotide is tolerated.
- the effectiveness of the antisense oligonucleotide is assessed by monitoring ApoB, LDL-C, VLDL-C, IDL-C, non-HDL-C, serum triglycerides, liver triglycerides, Lp(a), Ox-LDL-C, or small dense LDL particle concentration in the plasma of said subject.
- a reduction of ApoB, LDL-C, VLDL-C, IDL-C, non-HDL-C, serum triglycerides, liver triglycerides, Lp(a), Ox-LDL-C, or small dense LDL particle concentration indicates that the antisense oligonucleotide is effective.
- the effectiveness of the antisense oligonucleotide is assessed by monitoring MACE in said subject.
- a reduction of in MACE indicates that the antisense oligonucleotide is effective.
- the dose of antisense oligonucleotide is reduced following an indication that administration of said antisense oligonucleotide is effective.
- the dose of antisense oligonucleotide is increased following an indication that administration of said antisense oligonucleotide is not effective.
- the frequency of administration of antisense oligonucleotide is reduced following an indication that administration of said antisense oligonucleotide is effective.
- the frequency of administration of antisense oligonucleotide is increased following an indication that administration of said antisense oligonucleotide is not effective.
- said subject has elevated ApoB prior to said administering.
- said subject has elevated cholesterol prior to said administering.
- said elevated cholesterol is selected from elevated total cholesterol, elevated LDL-cholesterol, elevated VLDL-cholesterol, elevated IDL-cholesterol, or elevated non-HDL cholesterol prior to said administering.
- said subject has elevated Lp(a) prior to said administering.
- said subject has elevated serum triglycerides prior to said administering.
- said subject has elevated liver triglycerides prior to said administering.
- said subject has elevated small dense LDL particles prior to said administering.
- said subject has MACE prior to said administrating.
- said subject is at risk of developing MACE prior to said administrating.
- said subject has hypercholesterolemia.
- said subject has polygenic hypercholesterolemia. In certain embodiments, said subject has familial hypercholesterolemia. In certain embodiments, said subject has homozygous familial hypercholesterolemia. In certain embodiments, said subject has heterozygous familial hypercholesterolemia. In certain embodiments, said subject has mixed dyslipidemia. In certain embodiments, said subject has a history of coronary heart disease.
- said subject has one or more risk factors for coronary heart disease.
- said one or more risk factors is selected from age, smoking, hypertension, low HDL-cholesterol, and a family history of early coronary heart disease.
- said subject has type II diabetes with dyslipidemia.
- said subject has been treated by a statin. In certain embodiments, said subject failed to meet LDL-cholesterol target on statin therapy. In certain embodiments, said subject did not comply with recommended therapy. In certain embodiments, said subject experienced side effects of stain therapy. In certain embodiments, said subject has low LDL-receptor activity. In certain embodiments, said subject failed to meet LDL-cholesterol target on lipid-lowering therapy prior to said administering.
- said maintenance phase comprises administering said pharmaceutical composition throughout the lifetime of the subject.
- the duration of said maintenance phase is one year, 2 years, 3 years, 4 years, 5 years, 6 years, 7 years, 8 years, 9 years, 10 years, 1 1 years, 12 years, 13 years, 14 years, 15 years, 16 years, 17 years, 18 years, 19 years, or 20 years.
- the duration of said maintenance phase is from one week to twenty years.
- the induction dose is 100 mg. In certain embodiments, the induction dose is 200 mg. In certain embodiments, the induction dose is 300 mg. In certain embodiments, the maintenance dose is 100 mg. In certain embodiments, the maintenance dose is 200 mg.
- the antisense oligonucleotide is administered at 200 mg/week. In some embodiments, the 200 mg/week is adminitered one time per week. In some embodiments, the subject is administered a single 200 mg/dose per week. In other embodiments, the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week. In some embodiments, the antisense oligonucleotide is administered for at least 12 months. In one embodiment, the antisense oligonucleotide is administered subcutaneously (s.c).
- said administering of said pharmaceutical composition results in antisense oligonucleotide plasma trough levels between 5 and 100 ng/mL. In certain embodiments, said administering of said pharmaceutical composition results in antisense oligonucleotide plasma trough levels between 5 and 50 ng/mL. In certain embodiments, said administering of said pharmaceutical composition results in antisense oligonucleotide plasma trough levels between 10 and 40 ng/mL. In certain embodiments, said administering of said pharmaceutical composition results in antisense oligonucleotide plasma trough levels between 15 and 35 ng/mL. In certain embodiments, said administering of said pharmaceutical composition results in antisense oligonucleotide plasma trough levels between 20 and 30 ng/mL.
- said administering of said pharmaceutical composition results in ApoB reduction of at least 10%.
- said ApoB reduction is at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%), at least 90%>, at least 95%, or at least 100%).
- said ApoB reduction is between 10%> and 80%>, between 20%> and 70%>, between 30%> and 60%>, or between 30% and 70%.
- said administering of said pharmaceutical composition results in a LDL-cholesterol reduction of at least 10%.
- said LDL- cholesterol reduction is at least 15%, at least 20%>, at least 25%, at least 30%>, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100%.
- said administering of said pharmaceutical composition results in a LDL- cholesterol reduction between 10% and 100%.
- said administering of said pharmaceutical composition results in a VLDL-cholesterol reduction of at least 10%.
- said VLDL-cholesterol reduction is at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100%.
- said administering of said pharmaceutical composition results in a VLDL-cholesterol reduction bwtween 10% to 100%.
- said administering of said pharmaceutical composition results in Lp(a) reduction of at least 10%.
- said Lp(a) reduction is at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%o, at least 90%, at least 95%, or at least 100%).
- said administering of said pharmaceutical composition results in Lp(a) reduction between 10% and 100%.
- said administering of said pharmaceutical composition results in a small LDL-particle reduction of at least 10%.
- said small LDL-particle reduction is at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%), at least 50%>, at least 55%, at least 60%>, at least 65%, at least 70%>, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100%.
- said administering of said pharmaceutical composition results in a small LDL-particle reduction between 10% and 100%.
- said administering of said pharmaceutical composition results in a small LDL-particle reduction of at least 10%.
- said non- HDL-cholesterol reduction is at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100%.
- said administering of said pharmaceutical composition results in a small LDL- particle reduction between 10%> and 100%.
- said administering of said pharmaceutical composition results in reduced coronary heart disease risk in the subject. In certain embodiments, said administering of said pharmaceutical composition slows or stops the progression of
- said administering of said pharmaceutical composition reduces the risk of developing atherosclerosis in the subject.
- said administering of said pharmaceutical composition results in improved cardiovascular outcome the subject.
- said improved cardiovascular outcome is a reduced risk of major cardiovascular adverse events in the subject.
- said improved cardiovascular outcome is improved carotid intimal media thickness.
- said improved cardiovascular outcome is improved atheroma thickness.
- said improved cardiovascular outcome is increased HDL- cholesterol.
- said administering results in lipid lowering. In certain embodiments, said administering results in reductions in LDL-cholesterol, triglycerides, or small LDL particles, or a combination thereof. In certain embodiments, said administering results in an improved LDL/HDL ratio. In certain embodiments, said administering results in an HDL- cholesterol level increase of at least 10%.
- said HDL-cholesterol level increase is 15%, at least 20%>, at least 25%, at least 30%>, at least 35%, at least 40%>, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100%.
- said HDL-cholesterol level increase is 15%, at least 20%>, at least 25%, at least 30%>, at least 35%, at least 40%>, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100%.
- administering of said pharmaceutical composition results in a liver triglyceride level decrease of at least 10%.
- said liver triglyceride level decrease is at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%), at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100%.
- said administering of said pharmaceutical composition results in a hepatic cholesterol ester reduction of at least 10%.
- said reduced hepatic cholesterol ester reduction 15% at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100%.
- the methods further comprise co-administration of said pharmaceutical composition and at least one additional therapy.
- said co-administration is simultaneous.
- said pharmaceutical composition is administered prior to administration of said additional therapy.
- the method of claim 100 wherein said pharmaceutical composition is administered after administration of said additional therapy.
- the interval between administration of said pharmaceutical composition and said additional therapy is about one hour, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 9 hours, about 10 hours, about 11 hours, or about 12 hours.
- the interval between administration of said pharmaceutical composition and said additional therapy is about 1 day, about 1 week, about 2 weeks, about 3 weeks, about 4 weeks, about 5 weeks, about 6 weeks, about 7 weeks, about 8 weeks, about 9 weeks, about 10 weeks, about 11 weeks, or about 12 weeks. In certain embodiments, the interval between administration of said pharmaceutical composition and said additional therapy is about 1 month, about 2 months, about 3 months, about 4 months, about 5 months, or about 6 months. [00332] In certain embodiments, the methods further comprise administering a single additional therapy. In certain embodiments, the methods further comprise administering at 2 or more additional therapies. In certain embodiments, said additional therapy is a lipid-lowering therapy. In certain embodiments, said additional lipid-lowering therapy is therapeutic lifestyle change.
- said additional lipid-lowering therapy is an HMG-CoA reductase inhibitor.
- the HMG-CoA reductase inhibitor is selected from atorvastatin, rosuvastatin, or simvastatin.
- said additional lipid-lowering therapy is a cholesterol absorption inhibitor.
- the cholesterol absorption inhibitor is ezetimibe.
- said 2 or more additional therapies comprises an HMG-CoA reductase inhibitor and a cholesterol absorption inhibitor.
- said HMG-CoA reductase inhibitors is simvastatin and said cholesterol absorption inhibitor is ezetimibe.
- said additional lipid-lowering therapy is LDL apheresis. In certain embodiments, said administering of said additional therapy comprises intravenous administration. In certain embodiments, said additional lipid-lowering therapy is an MTP inhibitor. In certain embodiments, the additional lipid-lowering therapy is a PCSK9 inhibitor.
- said pharmaceutical composition comprises a
- the dose of the antisense oligonucleotide concentration is administered at a concentration of about 50 mg/ml, about 75 mg/ml, about 100 mg/ml, about 125 mg/ml, about 150 mg/ml, about 175 mg/ml, about 200 mg/ml, about 225 mg/ml, or about 250 mg/ml. In a specifc embodiment, the dose of the antisense oligonucleotide concentration is administered at a concentration of about 200 mg/ml.
- the antisense oligonucleotide comprises at least one modified sugar moiety.
- the modified sugar moiety comprises a 2'-methoxyethyl sugar moiety.
- the modified sugar moiety comprises a bicyclic nucleic acid sugar moiety.
- the antisense oligonucleotide comprises a 2'- deoxynucleotide gap segment positioned between wing segments, wherein each nucleotide of the wing segments comprises a modified sugar moiety.
- each nucleotide of the wing segment comprises a 2'-0-methoxyethyl sugar moiety.
- each nucleotide of the wing segment comprises a bicyclic nucleic acid sugar moiety.
- the gap segment comprises ten nucleotides and each wing segment comprises five nucleotides.
- At least one internucleoside linkage is a phosphorothioate internucleoside linkage. In certain embodiments, each internucleoside linkage is a
- At least one cytosine is a 5-methylcytosine. In certain embodiments, each cytosine is a 5-methylcytosine.
- the antisense oligonucleotide is at least 90% complementary to a nucleic acid encoding human ApoB. In certain embodiments, the antisense oligonucleotide is at least 95% complementary to a nucleic acid encoding human ApoB. In certain
- the antisense oligonucleotide is 100% complementary to a nucleic acid encoding human ApoB.
- the nucleic acid encoding human ApoB comprises a sequence identified by Accession number NM 000384.1.
- the antisense oligonucleotide comprises 12 to 30 nucleotides. In certain embodiments, the antisense oligonucleotide comprises 15 to 25 nucleotides. In certain embodiments, the antisense oligonucleotide comprises 17 to 23 nucleotides. In certain embodiments, the antisense oligonucleotide comprises 18 to 22 nucleotides. In certain embodiments, the antisense oligonucleotide comprises 19 to 21 nucleotides. In certain embodiments, the antisense oligonucleotide comprises 20 nucleotides.
- the antisense nucleotide comprises the nucleobase sequence of SEQ ID NO: 247. In some embodiments, the antisense nucleotide consists of the nucleobase sequence of SEQ ID NO: 247. In certain embodiments, the antisense oligonucleotide is ISIS 301012 (mipomersen). In some embodiments, the antisense nucleotide comprises the nucleobase sequence selected from the group of SEQ ID NOS: 124-515. In some embodiments, the antisense nucleotide consists of the nucleobase sequence selected from the group of SEQ ID NOS: 124-515. In some embodiments, the antisense nucleotide targets a region on ApoB that comprises a nucleobase sequence selected from the group of SEQ ID NOS: 516-804.
- kits comprising administering to a subject a pharmaceutical composition comprising an antisense oligonucleotide complementary to a nucleic acid encoding human ApoB, wherein the administering comprises an induction phase comprising at least one induction dose and a maintenance phase comprising at least one maintenance dose, wherein the duration of the induction phase is greater than five weeks.
- kits comprising administering to a subject a pharmaceutical composition comprising an antisense oligonucleotide complementary to a nucleic acid encoding human ApoB, wherein the administering comprises an induction phase comprising at least one induction dose and a maintenance phase comprising at least one maintenance dose, wherein an induction dose is less than a maintenance dose.
- kits comprising administering to a subject a pharmaceutical composition comprising an antisense oligonucleotide complementary to a nucleic acid encoding human ApoB, wherein the administering comprises an induction phase comprising at least one induction dose.
- kits comprising administering to a subject having familial hypercholesterolemia a pharmaceutical composition comprising an antisense oligonucleotide complementary to a nucleic acid encoding human ApoB, wherein the administering comprises an induction phase comprising at least one induction dose and a maintenance phase comprising at least one maintenance dose, wherein the induction phase is at least 8 weeks.
- kits including administering to a subject a pharmaceutical composition comprising an antisense oligonucleotide complementary to a nucleic acid encoding human apolipoprotein B-100, wherein the administering comprises a maintenance phase comprising at least one maintenance dose.
- Oligomeric compounds can be used in the various methods provided herein for modulating the function of nucleic acid molecules encoding ApoB, ultimately modulating the amount of ApoB produced. This is accomplished by providing antisense compounds which specifically hybridize with one or more nucleic acids encoding ApoB. The specific hybridization of an oligomeric compound with its target nucleic acid interferes with the normal function of the nucleic acid. The functions of DNA to be interfered with include replication and transcription.
- RNA to be interfered with include all vital functions such as, for example, translocation of the RNA to the site of protein translation, translation of protein from the RNA, splicing of the RNA to yield one or more mRNA species, and catalytic activity which can be engaged in or facilitated by the R A.
- the overall effect of such interference with target nucleic acid function is modulation of the expression of ApoB.
- inhibition is the form of modulation of gene expression and mRNA is a target.
- Targeting an antisense compound to a particular nucleic acid, is a multistep process. The process usually begins with the identification of a nucleic acid sequence whose function is to be modulated. This can be, for example, a cellular gene (or mRNA transcribed from the gene) whose expression is associated with a particular disorder or disease state, or a nucleic acid molecule from an infectious agent.
- the target is a nucleic acid molecule encoding ApoB.
- the targeting process also includes determination of a site or sites within this gene for the antisense interaction to occur such that the desired effect, e.g., detection or modulation of expression of the protein, will result.
- an intragenic site is the region encompassing the translation initiation or termination codon of the open reading frame (ORF) of the gene. Since, as is known in the art, the translation initiation codon is typically 5 '-AUG (in transcribed mRNA molecules; 5'-ATG in the corresponding DNA molecule), the translation initiation codon is also referred to as the "AUG codon,” the "start codon” or the "AUG start codon".
- translation initiation codon having the RNA sequence 5'-GUG, 5'-UUG or 5'-CUG, and 5'-AUA, 5'-ACG and 5'-CUG have been shown to function in vivo.
- the terms "translation initiation codon” and "start codon” can encompass many codon sequences, even though the initiator amino acid in each instance is typically methionine (in eukaryotes) or formylmethionine (in prokaryotes). It is also known in the art that eukaryotic and prokaryotic genes can have two or more alternative start codons, any one of which can be preferentially utilized for translation initiation in a particular cell type or tissue, or under a particular set of conditions.
- start codon and “translation initiation codon” refer to the codon or codons that can be used in vivo to initiate translation of an mRNA molecule transcribed from a gene encoding ApoB, regardless of the sequence(s) of such codons.
- a translation termination codon (or "stop codon”) of a gene can have one of three sequences, i.e., 5'-UAA, 5'-UAG and 5'-UGA (the corresponding DNA sequences can be 5'-TAA, 5 '-TAG and 5'-TGA, respectively).
- start codon region and “translation initiation codon region” refer to a portion of such an mRNA or gene that encompasses from about 25 to about 50 contiguous nucleotides in either direction ⁇ i.e., 5' or 3') from a translation initiation codon.
- stop codon region and “translation termination codon region” refer to a portion of such an mRNA or gene that encompasses from about 25 to about 50 contiguous nucleotides in either direction (i.e., 5' or 3') from a translation termination codon.
- Other target regions include the 5 ' untranslated region (5'UTR), known in the art to refer to the portion of an mRNA in the 5' direction from the translation initiation codon, and thus including nucleotides between the 5 ' cap site and the translation initiation codon of an mRNA or corresponding nucleotides on the gene, and the 3' untranslated region (3 'UTR), known in the art to refer to the portion of an mRNA in the 3 ' direction from the translation termination codon, and thus including nucleotides between the translation termination codon and 3' end of an mRNA or corresponding nucleotides on the gene.
- 5'UTR 5 ' untranslated region
- 3'UTR known in the art to refer to the portion of an mRNA in the 3 ' direction from the translation termination codon
- the 5' cap of an mRNA comprises an N7-methylated guanosine residue joined to the 5 '-most residue of the mRNA via a 5 '-5' triphosphate linkage.
- the 5' cap region of an mRNA is considered to include the 5' cap structure itself as well as the first 50 nucleotides adjacent to the cap.
- the 5' cap region can also be a target region.
- mRNA splice sites i.e., intron-exon junctions
- intron-exon junctions can also be target regions, and can be particularly useful in situations where aberrant splicing is implicated in disease, or where an overproduction of a particular mRNA splice product is implicated in disease. Aberrant fusion junctions due to rearrangements or deletions are also targets. It has also been found that introns can also be effective, and therefore , target regions for antisense compounds targeted, for example, to DNA or pre -mRNA.
- oligonucleotides can be chosen which can be sufficiently complementary to the target, i.e., hybridize sufficiently well and with sufficient specificity, to give the desired effect.
- Hybridization refers to hydrogen bonding, which can be Watson-Crick, Hoogsteen or reversed Hoogsteen hydrogen bonding, between complementary nucleoside and nucleotide bases.
- adenine and thymine can be complementary nucleobases which pair through the formation of hydrogen bonds.
- “Complementary,” as used herein, refers to the capacity for precise pairing between two nucleotides.
- oligonucleotide and the DNA or RNA can be considered to be complementary to each other at that position.
- the oligonucleotide and the DNA or RNA can be complementary to each other when a sufficient number of corresponding positions in each molecule can be occupied by nucleotides which can hydrogen bond with each other. It is understood in the art that the sequence of an antisense compound need not be 100%
- An antisense compound is specifically hybridizable when binding of the compound to the target DNA or RNA molecule interferes with the normal function of the target DNA or RNA to cause a loss of utility, and there is a sufficient degree of complementarity to avoid non-specific binding of the antisense compound to non-target sequences under conditions in which specific binding is desired, i.e., under physiological conditions in the case of in vivo assays or therapeutic treatment, and in the case of in vitro assays, under conditions in which the assays can be performed.
- Antisense and other compounds which hybridize to the target and inhibit expression of the target can be identified through experimentation, and the sequences of these compounds can be herein below identified as certain embodiments.
- the target sites to which these sequences can be complementary can be herein below referred to as "active sites” and can be therefore sites for targeting. Therefore another embodiment encompasses compounds which hybridize to these active sites.
- Antisense compounds can be commonly used as research reagents and diagnostics. For example, antisense oligonucleotides, which are able to inhibit gene expression with seventeen specificity, can be often used by those of ordinary skill to elucidate the function of particular genes. Antisense compounds can also be used, for example, to distinguish between functions of various members of a biological pathway. Antisense modulation has, therefore, been harnessed for research use.
- the antisense compounds provided herein can be used as tools in differential and/or combinatorial analyses to elucidate expression patterns of a portion or the entire complement of genes expressed within cells and tissues.
- Expression patterns within cells or tissues treated with an antisense compound can be compared to control cells or tissues not treated with antisense compounds and the patterns produced can be analyzed for differential levels of gene expression as they pertain, for example, to disease association, signaling pathway, cellular localization, expression level, size, structure or function of the genes examined. These analyses can be performed on stimulated or unstimulated cells and in the presence or absence of other compounds which affect expression patterns.
- Examples of methods of gene expression analysis known in the art include DNA arrays or microarrays (Brazma and Vilo, FEBS Lett., 2000, 480, 17-24; Celis, et al, FEBS Lett., 2000, 480, 2-16), SAGE (serial analysis of gene expression) (Madden, et al., Drug Discov.
- Antisense oligonucleotides have been employed as therapeutic moieties in the treatment of disease states in animals and man.
- Antisense oligonucleotide drugs including ribozymes, have been safely and effectively administered to humans and numerous clinical trials can be presently underway. It is thus established that oligonucleotides can be useful therapeutic modalities that can be configured to be useful in treatment regimes for treatment of cells, tissues and animals, especially humans.
- oligonucleotide refers to an oligomer or polymer of ribonucleic acid (R A) or deoxyribonucleic acid (DNA) or mimetics thereof.
- R A ribonucleic acid
- DNA deoxyribonucleic acid
- this term includes oligonucleotides composed of naturally-occurring nucleobases, sugars and covalent internucleoside (backbone) linkages (RNA and DNA) as well as oligonucleotides having non- naturally-occurring portions which function similarly (oligonucleotide mimetics).
- Oligonucleotide mimetics can be often over native forms because of desirable properties such as, for example, enhanced cellular uptake, enhanced affinity for nucleic acid target and increased stability in the presence of nucleases.
- antisense oligonucleotides can be an exemplary form of an antisense compound provided herein, other oligomeric antisense compounds, including but not limited to oligonucleotide mimetics such as those described below can also be contemplated.
- the antisense compounds provided herein can comprise from about 8 to about 50 nucleobases (i.e., from about 8 to about 50 linked nucleosides).
- Antisense compounds can be antisense oligonucleotides, even more are those comprising from about 12, about 14, about 20 to about 30 nucleobases.
- Antisense compounds include ribozymes, external guide sequence (EGS) oligonucleotides (oligozymes), and other short catalytic RNAs or catalytic oligonucleotides which hybridize to the target nucleic acid and modulate its expression.
- the antisense compound is non- catalytic oligonucleotide, i.e., is not dependent on a catalytic property of the oligonucleotide for its modulating activity.
- Antisense compounds can include double-stranded molecules wherein a first strand is stably hybridized to a second strand.
- compositions including one or more different oligonucleotides.
- those pharmaceutically acceptable salts including one or more different oligonucleotides.
- compositions comprise an antisense oligonucleotide complementary to a nucleic acid encoding human ApoB.
- such pharmaceutical compositions comprise ISIS 301012.
- ISIS 301012 is a pharmaceutical agent that, when administered to a subject, results in dose-dependent reductions of ApoB, ApoB-containing lipoproteins, including but not limited to LDL-C, triglycerides, and Lp(a). ISIS 301012 results in efficacy when administered alone, and also results in efficacy when
- compositions comprise an oligonucleotide having complementary to a target nucleic acid.
- a sufficient number of nucleobases of the oligonucleotide can undergo hydrogen bonding with corresponding nucleobases in a target nucleic acid such that a desired effect occurs.
- a desired effect is antisense inhibition of a target nucleic acid.
- a desired effect is antisense inhibition of ApoB.
- oligonucleotides are fully complementary (i.e., 100% complementary) to a target nucleic acid. In certain such embodiments, oligonucleotides are fully complementary to a nucleic acid encoding ApoB.
- a nucleic acid encoding human ApoB is ApoB mRNA.
- ApoB mRNA may or may not include some or all exons.
- oligonucleotides are 12 to 30 nucleotides in length, i.e., the oligonucleotides are from 12 to 30 linked nucleotides. In certain such embodiments,
- oligonucleotides are 15 to 25 nucleotides in length. In certain such embodiments,
- oligonucleotides are 17 to 23 nucleotides in length. In certain such embodiments,
- oligonucleotides are 18 to 22 nucleotides in length. In certain such embodiments,
- oligonucleotides are 19 to 21 nucleotides in length. In certain such embodiments,
- oligonucleotides are 20 nucleotides in length.
- oligonucleotides comprise a percent identity to a particular nucleotide sequence.
- An oligonucleotide has identity to another oligonucleotide if the nucleobases of each oligonucleotide have the same nucleobase pairing ability.
- an oligonucleotide has 90% identity to another oligonucleotide.
- an oligonucleotide has 95% identity to another oligonucleotide.
- an oligonucleotide has 100% identity to another oligonucleotide.
- the identity is over the full-length of the oligonucleotide.
- the identity is to a portion of an oligonucleotide.
- oligonucleotides comprise chemical modifications.
- mmodifications to oligonucleotides encompass substitutions or changes to internucleoside linkages, sugar moieties, or nucleobases.
- Modified oligonucleotides are often preferred over native forms because of desirable properties such as, for example, enhanced cellular uptake, enhanced affinity for nucleic acid target, increased stability in the presence of nucleases, or increased inhibitory activity.
- chemically modified nucleosides may also be employed to increase the binding affinity of a shortened or truncated antisense oligonucleotide for its target nucleic acid.
- nucleoside is a base-sugar combination.
- the base portion of the nucleoside is normally a heterocyclic base.
- heterocyclic bases can be the purines and the pyrimidines.
- Nucleotides can be nucleosides that further include a phosphate group covalently linked to the sugar portion of the nucleoside.
- the phosphate group can be linked to either the 2', 3' or 5' hydroxyl moiety of the sugar.
- the phosphate groups covalently link adjacent nucleosides to one another to form a linear polymeric compound. In turn the respective ends of this linear polymeric structure can be further joined to form a circular structure; however, open linear structures can be generally.
- the phosphate groups can be commonly referred to as forming the internucleoside backbone of the oligonucleotide.
- the normal linkage or backbone of RNA and DNA is a 3' to 5' phosphodiester linkage.
- antisense compounds include oligonucleotides containing modified backbones or non-natural internucleoside linkages.
- oligonucleotides having modified backbones include those that retain a phosphorus atom in the backbone and those that do not have a phosphorus atom in the backbone.
- modified oligonucleotides that do not have a phosphorus atom in their internucleoside backbone can also be considered to be
- oligonucleotide backbones include, for example,
- phosphorothioates chiral phosphorothioates, phosphorodithioates, phosphotriesters, aminoalkyl- phosphotriesters, methyl and other alkyl phosphonates including 3-alkylene phosphonates, 5'- alkylene phosphonates and chiral phosphonates, phosphinates, phosphoramidates including 3'- amino phosphoramidate and aminoalkylphosphoramidates, thionophosphoramidates,
- oligonucleotides having inverted polarity comprise a single 3' to 3' linkage at the 3 '-most internucleotide linkage i.e., a single inverted nucleoside residue which can be basic (the nucleobase is missing or has a hydroxyl group in place thereof).
- Various salts, mixed salts and free acid forms can also be included.
- oligonucleotides comprise one or more modified, i.e. non- naturally occurring, internucleoside linkages.
- oligonucleotides having modified internucleoside linkages include internucleoside linkages that retain a phosphorus atom as well as internucleoside linkages that do not have a phosphorus atom.
- Representative phosphorus containing internucleoside linkages include, but are not limited to, phosphodiesters, phosphotriesters, methylphosphonates, phosphoramidate, and
- Modified oligonucleotide backbones that do not include a phosphorus atom therein can have backbones that can be formed by short chain alkyl or cycloalkyl internucleoside linkages, mixed heteroatom and alkyl or cycloalkyl internucleoside linkages, or one or more short chain heteroatomic or heterocyclic internucleoside linkages.
- oligonucleosides include those having morpholino linkages (formed in part from the sugar portion of a nucleoside); siloxane backbones; sulfide, sulfoxide and sulfone backbones; formacetyl and thioformacetyl backbones; methylene formacetyl and thioformacetyl backbones; riboacetyl backbones; alkene containing backbones; sulfamate backbones; methyleneimino and methylenehydrazino backbones; sulfonate and sulfonamide backbones; amide backbones; and others having mixed N, O, S and CH 2 component parts.
- Representative United States patents that teach the preparation of the above oligonucleosides include, but are not limited to, U.S. Pat. Nos.: 5,034,506; 5,166,315;
- oligonucleotides comprise one or more nucleotides comprising modified sugar moieties.
- the furanosyl sugar ring of a nucleoside is modified in a number of ways including, but not limited to: addition of a substituent group, particularly at the 2' position; bridging of two non-geminal ring atoms to form a bicyclic nucleic acid (BNA); and substitution of an atom or group such as -S-, -N(R)- or - C(Ri)(R 2 ) for the ring oxygen at the 4'-position.
- substituted sugars especially 2 '-substituted sugars having a 2'-F, 2'-OCH 2 (2'-OMe) or a 2'-0(CH 2 ) 2 -OCH 3 (2'-0-methoxyethyl or 2'-MOE) substituent group
- BNAs bicyclic modified sugars
- both the sugar and the internucleoside linkage, i.e., the backbone, of the nucleotide units can be replaced with novel groups.
- the base units can be maintained for hybridization with an appropriate nucleic acid target compound.
- One such oligomeric compound, an oligonucleotide mimetic that has been shown to have excellent hybridization properties is referred to as a peptide nucleic acid (PNA).
- PNA peptide nucleic acid
- the sugar-backbone of an oligonucleotide is replaced with an amide containing backbone, in particular an aminoethylglycine backbone.
- the nucleobases can be retained and can be bound directly or indirectly to aza nitrogen atoms of the amide portion of the backbone.
- Representative United States patents that teach the preparation of PNA compounds include, but are not limited to, U.S. Pat. Nos. : 5,539,082; 5,714,331; and 5,719,262, each of which is herein incorporated by reference. Further teaching of PNA compounds can be found in Nielsen et al., Science, 1991, 254, 1497-1500.
- oligonucleotides with phosphorothioate backbones and oligonucleosides with heteroatom backbones and in particular -CH 2 -NH-0-CH 2 -, -CH 2 -N(CH 3 )-0-CH 2 - [known as a methylene(methylimino) or MMI backbone], -CH 2 -0- N(CH 3 )-CH 2 -, -CH 2 -N(CH 3 )-N(CH 3 )-CH 2 - and -0-N(CH 3 )-CH 2 -CH 2 - [wherein the native phosphodiester backbone is represented as -O-P-O-C H 2 -] of the above referenced U.S.
- oligonucleotides include those having morpholino backbone structures of the above- referenced U.S. Pat. No. 5,034,506.
- Modified oligonucleotides can also contain one or more substituted sugar moieties.
- Certain oligonucleotides comprise one of the following at the 2' position: OH; F; 0-, S-, or N- alkyl; 0-, S-, or N-alkenyl; 0-, S- or N-alkynyl; or O-alkyl-O-alkyl, wherein the alkyl, alkenyl and alkynyl can be substituted or unsubstituted CI to CIO alkyl or C2 to CIO alkenyl and alkynyl.
- Exemplary oligonucleotides have 0[(CH 2 )nO]mCH 3 , 0(CH 2 ) n OCH 3 , 0(CH 2 ) n NH 2 , 0(CH 2 ) n CH 3 , 0(CH 2 ) n ONH 2 , and 0(CH 2 ) n ON[(CH 2 ) n CH 3 )] 2 , where n and m can be from 1 to about 10.
- oligonucleotides can comprise one of the following at the 2' position: CI to CIO lower alkyl, substituted lower alkyl, alkenyl, alkynyl, alkaryl, aralkyl, O-alkaryl or O-aralkyl, SH, SCH 3 , OCN, CI, Br, CN, CF 3 , OCF 3 , SOCH 3 , S0 2 CH 3 , ON0 2 , N0 2 , N 3 , NH 2 ,
- heterocycloalkyl heterocycloalkaryl, aminoalkylamino, polyalkylamino, substituted silyl, an RNA cleaving group, a reporter group, an intercalator, a group for improving the
- One modification includes 2'-methoxyethoxy (2'-0-CH 2 CH 2 OCH 3 , also known as 2'-0-(2-methoxyethyl) or 2'-MOE) (Martin et al, Helv. Chim. Acta, 1995, 78, 486-504) i.e., an alkoxyalkoxy group.
- Another modification includes 2'-dimethylaminooxyethoxy, i.e., an 0(CH 2 ) 2 ON(CH 3 ) 2 group, also known as 2'-DMAOE, as described in examples herein below, and 2'-dimethylamino-ethoxyethoxy (also known in the art as 2'-0-dimethylamino-ethoxyethyl or 2'-DMAEOE), i.e., 2'-0-CH 2 -0-CH 2 -N(CH 2 ) 2 , also described in examples herein below.
- 2'-dimethylaminooxyethoxy i.e., an 0(CH 2 ) 2 ON(CH 3 ) 2 group
- 2'-DMAOE 2'-dimethylamino-ethoxyethoxy
- 2'-DMAEOE 2'-dimethylamino-ethoxyethoxy
- LNAs Locked Nucleic Acids
- the linkage is can a methylene (-CH 2 -) n group bridging the 2' oxygen atom and the 4' carbon atom wherein n is 1 or 2.
- LNAs and preparation thereof can be described in WO 98/39352 and WO 99/14226.
- the 2 '-modification can be in the arabino (up) position or ribo (down) position.
- a 2'-arabino modification can be 2'-F.
- oligonucleotide Similar modifications can also be made at other positions on the oligonucleotide, such as the 3' position of the sugar on the 3' terminal nucleotide or in 2'- 5' linked oligonucleotides and the 5' position of 5' terminal nucleotide. Oligonucleotides can also have sugar mimetics such as cyclobutyl moieties in place of the pentofuranosyl sugar. Representative United States patents that teach the preparation of such modified sugar structures include, but are not limited to, U.S. Pat. Nos.
- Oligonucleotides can also include nucleobase (often referred to in the art simply as “base”) modifications or substitutions.
- nucleobases include the purine bases adenine (A) and guanine (G), and the pyrimidine bases thymine (T), cytosine. (C) and uracil (U).
- Modified nucleobases include other synthetic and natural nucleobases such as 5-methylcytosine (5-me-C or 5-meC), 5-hydroxymethyl cytosine, xanthine, hypoxanthine, 2-aminoadenine, 6-methyl and other alkyl derivatives of adenine and guanine, 2- propyl and other alkyl derivatives of adenine and guanine, 2-thiouracil, 2-thiothymine and 2- thiocytosine, 5-halouracil and cytosine, 5-propynyl (-C ⁇ C-CH3) uracil and cytosine and other alkynyl derivatives of pyrimidine bases, 6-azo uracil, cytosine and thymine, 5-uracil
- nucleobases include tricyclic pyrimidines such as phenoxazine cytidine(lH-pyrimido[5,4-b][l,4]benzoxazin-2(3H)-one), phenothiazine cytidine (lH-pyrimido[5,4-b][l,4]benzothiazin-2(3H)-one), G-clamps such as a substituted phenoxazine cytidine (e.g.,9-(2-aminoethoxy)-H-pyrimido[5,4-b][l,4]benzoxazin-2(3H)-one), carbazole cytidine (2H-pyrimido[4,5-b]indol-2-one), pyridoindole cytidine (H- pyrido[3',2' :4,5]pyrrolo[2,3-d]pyrimidin-2-one).
- Modified nucleobases can also include those in which the purine or pyrimidine base is replaced with other heterocycles, for example 7-deaza- adenine, 7-deazaguanosine, 2-aminopyridine and 2-pyridone.
- Further nucleobases include those disclosed in U.S. Pat. No. 3,687,808, those disclosed in The Concise Encyclopedia Of Polymer Science And Engineering, pages 858-859, Kroschwitz, J. I., ed. John Wiley & Sons, 1990, those disclosed by Englisch et al., Angewandte Chemie, International Edition, 1991, 30, 613, and those disclosed by Sanghvi, Y.
- nucleobases can be useful for increasing the binding affinity of the oligomeric compounds.
- These include 5 -substituted pyrimidines, 6-azapyrimidines and N-2, N-6 and 0-6 substituted purines, including 2- aminopropyladenine, 5-propynyluracil and 5-propynylcytosine. 5-methylcytosine substitutions have been shown to increase nucleic acid duplex stability by 0. 6-1. 2° C. (Sanghvi, Y. S., Crooke, S. T.
- Another modification of the oligonucleotides can involve chemically linking to the oligonucleotide one or more moieties or conjugates which enhance the activity, cellular distribution or cellular uptake of the oligonucleotide.
- the compounds can include conjugate groups covalently bound to functional groups such as primary or secondary hydroxyl groups.
- Conjugate groups include intercalators, reporter molecules, polyamines, polyamides, poly ethylene glycols, polyethers, groups that enhance the pharmacodynamic properties of oligomers, and groups that enhance the pharmacokinetic properties of oligomers.
- Conjugates groups include cholesterols, lipids, phospholipids, biotin, phenazine, folate, phenanthridine, anthraquinone, acridine, fluoresceins, rhodamines, coumarins, and dyes. Groups that enhance the
- pharmacodynamic properties include groups that improve oligomer uptake, enhance oligomer resistance to degradation, and/or strengthen sequence-specific hybridization with RNA.
- Groups that enhance the pharmacokinetic properties include groups that improve oligomer uptake, distribution, metabolism or excretion.
- Representative conjugate groups can be disclosed in International Patent Application PCT/US92/09196, filed Oct. 23, 1992 the entire disclosure of which is incorporated herein by reference.
- Conjugate moieties can include but are not limited to lipid moieties, such as a cholesterol moiety (Letsinger et al, Proc. Natl. Acad. Sci. USA, 1989, 86, 6553-6556), cholic acid (Manoharan et al, Bioorg. Med. Chem.
- a thioether e.g., hexyl-S-tritylthiol (Manoharan et al, Ann. N. Y. Acad. Sci., 1992, 660, 306-309; Manoharan et al, Bioorg. Med. Chem. Let., 1993, 3, 2765-2770), a thiocholesterol (Oberhauser et. al, Nucl.
- Acids Res., 1990, 18, 3777-3783 a polyamine or a polyethylene glycol chain (Manoharan et al, Nucleosides & Nucleotides, 1995, 14, 969-973), or adamantane acetic acid (Manoharan et al, Tetrahedron Lett., 1995, 36, 3651-3654), a palmityl moiety (Mishra et al, Biochim. Biophys. Acta, 1995, 1264, 229-237), or an octadecylamine or hexylamino-carbonyl-oxy cholesterol moiety (Crooke et al, J. Pharmacol. Exp.
- Oligonucleotides can also be conjugated to active drug substances, for example, aspirin, warfarin, phenylbutazone, ibuprofen, suprofen, fenbufen, ketoprofen, (S)-(+)-pranoprofen, carprofen, dansylsarcosine, 2,3,5-triiodobenzoic acid, flufenamic acid, folinic acid, a benzothiadiazide, chlorothiazide, a diazepine, indomethicin, a barbiturate, a cephalosporin, a sulfa drug, an antidiabetic, an antibacterial or an antibiotic.
- active drug substances for example, aspirin, warfarin, phenylbutazone, ibuprofen, suprofen, fenbufen, ketoprofen, (S)-(+)-pranoprofen, carprofen, dansy
- oligonucleotide conjugates include, but are not limited to, U.S. Pat. Nos. : 4,828,979; 4,948,882; 5,218,105; 5,525,465; 5,541,313; 5,545,730; 5,552,538; 5,578,717, 5,580,731; 5,580,731;
- Antisense compounds which can be chimeric compounds are also provided herein. "Chimeric” antisense compounds or “chimeras,” can be antisense compounds, particularly oligonucleotides, which contain two or more chemically distinct regions, each made up of at least one monomer unit, i.e., a nucleotide in the case of an oligonucleotide compound.
- oligonucleotides can contain at least one region wherein the oligonucleotide is modified so as to confer upon the oligonucleotide increased resistance to nuclease degradation, increased cellular uptake, and/or increased binding affinity for the target nucleic acid.
- An additional region of the oligonucleotide can serve as a substrate for enzymes capable of cleaving R A:DNA or R A:R A hybrids.
- R ase H is a cellular endonuclease which cleaves the RNA strand of an R A:DNA duplex.
- RNA target Activation of RNase H, therefore, results in cleavage of the RNA target, thereby greatly enhancing the efficiency of oligonucleotide inhibition of gene expression. Consequently, comparable results can often be obtained with shorter oligonucleotides when chimeric oligonucleotides can be used, compared to phosphorothioate deoxyoligonucleotides hybridizing to the same target region.
- Cleavage of the RNA target can be routinely detected by gel electrophoresis and, if necessary, associated nucleic acid hybridization techniques known in the art.
- Chimeric antisense compounds can be formed as composite structures of two or more oligonucleotides, modified oligonucleotides, oligonucleosides and/or oligonucleotide mimetics as described above. Such compounds have also been referred to in the art as hybrids or
- the antisense compounds provided herein can be conveniently and routinely made through the well-known technique of solid phase synthesis.
- Equipment for such synthesis is sold by several vendors including, for example, Applied Biosy stems (Foster City, Calif. ). Any other refers to for such synthesis known in the art can additionally or alternatively be employed. It is well known to use similar techniques to prepare oligonucleotides such as the phosphorothioates and alkylated derivatives.
- the antisense compounds can be synthesized in vitro and do not include antisense compositions of biological origin, or genetic vector constructs designed to direct the in vivo synthesis of antisense molecules.
- the compounds can also be admixed, encapsulated, conjugated or otherwise associated with other molecules, molecule structures or mixtures of compounds, as for; example, liposomes, receptor targeted molecules, oral, rectal, topical or other formulations, for assisting in uptake, distribution and/or absorption.
- Representative United States patents that teach the preparation of such uptake, distribution and/or absorption assisting formulations include, but are not limited to, U.S. Pat. Nos. : 5,108,921; 5,354,844; 5,416,016; 5,459,127; 5,521,291;
- Antisense compounds provided herein encompass any pharmaceutically acceptable salts, esters, or salts of such esters, or any other compound which, upon administration to an animal including a human, is capable of providing (directly or indirectly) the biologically active metabolite or residue thereof. Accordingly, for example, the examples can also include prodrugs and pharmaceutically acceptable salts of the compounds, pharmaceutically acceptable salts of such prodrugs, and other bioequivalents.
- prodrug refers to a therapeutic agent that is prepared in an inactive form that is converted to an active form (i.e., drug) within the body or cells thereof by the action of endogenous enzymes or other chemicals and/or conditions.
- prodrug versions of the oligonucleotides can be prepared as SATE [(S-acetyl-2-thioethyl) phosphate] derivatives according to the methods disclosed in WO 93/24510 to Gosselin et al., published Dec. 9, 1993 or in WO 94/26764 and U.S. Pat. No. 5,770,713 to Imbach et al.
- pharmaceutically acceptable salts refers to physiologically and pharmaceutically acceptable salts of the compounds: i.e., salts that retain the desired biological activity of the parent compound and do not impart undesired toxicological effects thereto.
- Pharmaceutically acceptable base addition salts can be formed with metals or amines, such as alkali and alkaline earth metals or organic amines. Examples of metals that can be used as cations can be sodium, potassium, magnesium, calcium, and the like.
- Suitable amines can be ⁇ , ⁇ '-dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, dicyclohexylamine, ethylenediamine, N-methylglucamine, and procaine (see, for example, Berge et al., "Pharmaceutical Salts," J. of Pharma Sci., 1977, 66, 1-19).
- the base addition salts of said acidic compounds can be prepared by contacting the free acid form with a sufficient amount of the desired base to produce the salt in the conventional manner.
- the free acid form can be regenerated by contacting the salt form with an acid and isolating the free acid in the
- a "pharmaceutical addition salt” includes a pharmaceutically acceptable salt of an acid form of one of the components of the compositions. These include organic or inorganic acid salts of the amines. Exemplary acid salts can be the hydrochlorides, acetates, salicylates, nitrates and phosphates.
- Suitable pharmaceutically acceptable salts can be well known to those skilled in the art and include basic salts of a variety of inorganic and organic acids, such as, for example, with inorganic acids, such as for example hydrochloric acid, hydrobromic acid, sulfuric acid or phosphoric acid; with organic carboxylic, sulfonic, sulfo or phospho acids or N-substituted sulfamic acids, for example acetic acid, propionic acid, glycolic acid, succinic acid, maleic acid, hydroxymaleic acid, methylmaleic acid, fumaric acid, malic acid, tartaric acid, lactic acid, oxalic acid, gluconic acid, glucaric acid, glucuronic acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, salicylic acid, 4-aminosalicylic acid, 2-phenoxybenzoic acid, 2-acetoxybenzoic acid, embonic acid, nicotinic acid or isonico
- Pharmaceutically acceptable salts of compounds can also be prepared with a pharmaceutically acceptable cation.
- Suitable pharmaceutically acceptable cations can be well known to those skilled in the art and include alkaline, alkaline earth, ammonium and quaternary ammonium cations. Carbonates or hydrogen carbonates can also be possible.
- examples of pharmaceutically acceptable salts include but are not limited to (a) salts formed with cations such as sodium, potassium, ammonium, magnesium, calcium, polyamines' such as spermine and spermidine, etc.; (b) acid addition salts formed with inorganic acids, for example hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, nitric acid and the like; (c) salts formed with organic acids such as, for example, acetic acid, oxalic acid, tartaric acid, succinic acid, maleic acid, fumaric acid, gluconic acid, citric acid, malic acid, ascorbic acid, benzoic acid, tannic acid, palmitic acid, alginic acid, polyglutamic acid, naphthalenesulfonic acid, methanesulfonic acid, p-toluenesulfonic acid, naphthalenedisulfonic acid, poly
- the antisense compounds of provided herein can be utilized for diagnostics, therapeutics, prophylaxis and as research reagents and kits.
- an animal such as a human, suspected of having a disease or disorder which can be treated by modulating the expression of ApoB is treated by administering antisense compounds, provided herein.
- the compounds can be utilized in pharmaceutical compositions by adding an effective amount of an antisense compound to a suitable pharmaceutically acceptable diluent or carrier.
- the antisense compounds can be useful for research and diagnostics, because these compounds hybridize to nucleic acids encoding ApoB, enabling sandwich and other assays to easily be constructed to exploit this fact.
- Hybridization of the antisense oligonucleotides with a nucleic acid encoding ApoB can be detected by refers to known in the art. Such refers to can include conjugation of an enzyme to the oligonucleotide, radiolabelling of the oligonucleotide or any other suitable detection refers to. Kits using such detection refers to for detecting the level of ApoB in a sample can also be prepared.
- compositions and formulations which include an antisense compound.
- the pharmaceutical compositions provided herein can be administered in a number of ways depending upon whether local or systemic treatment is desired and upon the area to be treated. Administration can be topical (including ophthalmic and to mucous membranes including vaginal and rectal delivery), pulmonary, e.g., by inhalation or insufflation of powders or aerosols, including by nebulizer; intratracheal, intranasal, epidermal and transdermal), oral or parenteral.
- Parenteral administration includes intravenous, intra-arterial, subcutaneous, intraperitoneal or intramuscular injection or infusion; or intracranial, e.g., intratracheal or intraventricular, administration.
- Antisense compounds with at least one 2'-0-methoxyethyl modification can be believed to be particularly useful for oral administration.
- compositions and formulations for topical administration can include transdermal patches, ointments, lotions, creams, gels, drops, suppositories, sprays, liquids and powders.
- Conventional pharmaceutical carriers, aqueous, powder or oily bases, thickeners and the like can be necessary or desirable.
- Coated condoms, gloves and the like can also be useful.
- Topical formulations include those in which the antisense compounds provided herein, can be in admixture with a topical delivery agent such as lipids, liposomes, fatty acids, fatty acid esters, steroids, chelating agents and surfactants.
- Lipids and liposomes include neutral (e.g.,
- distearolyphosphatidyl choline) negative e.g., dimyristoylphosphatidyl glycerol DMPG
- cationic e.g., dioleoyltetramethylammopropyl DOTAP and dioleoylphosphatidyl ethanolamine DOTMA
- Antisense compounds can be encapsulated within liposomes or can form complexes thereto, in particular to cationic liposomes. Alternatively, antisense compounds can be complexed to lipids, in particular to cationic lipids.
- Fatty acids and esters include but are not limited arachidonic acid, oleic acid, eicosanoic acid, lauric acid, caprylic acid, capric acid, myristic acid, palmitic acid, stearic acid, linoleic acid, linolenic acid, dicaprate, tricaprate, monoolein, dilaurin, glyceryl 1-monocaprate, l-dodecylazacycloheptan-2-one, an acylcarnitine, an acylcholine, or a C 1-10 alkyl ester (e.g., isopropylmyristate IPM), monoglyceride, diglyceride or pharmaceutically acceptable salt thereof.
- Topical formulations can be described in detail in U.S. patent application Ser. No. 09/315,298 filed on Can 20, 1999, which is incorporated herein by reference in its entirety.
- compositions and formulations for oral administration include powders or granules, microparticulates, nanoparticulates, suspensions or solutions in water or non-aqueous media, capsules, gel capsules, sachets, tablets or minitablets. Thickeners, flavoring agents, diluents, emulsifiers, dispersing aids or binders can be desirable. Oral formulations can be those in which antisense compounds provided herein, can be administered in conjunction with one or more penetration enhancers surfactants and chelators.
- Surfactants include fatty acids and/or esters or salts thereof, bile acids and/or salts thereof.
- Bile acids/salts include chenodeoxycholic acid (CDCA) and ursodeoxychenodeoxycholic acid (UDCA), cholic acid, dehydrocholic acid, deoxycholic acid, glucholic acid, glycholic acid, glycodeoxycholic acid, taurocholic acid, taurodeoxycholic acid, sodium tauro-24,25-dihydro-fusidate, sodium glycodihydrofusidate.
- DCA chenodeoxycholic acid
- UDCA ursodeoxychenodeoxycholic acid
- cholic acid dehydrocholic acid
- deoxycholic acid deoxycholic acid
- glucholic acid glycholic acid
- glycodeoxycholic acid taurocholic acid
- taurodeoxycholic acid sodium tauro-24,25-dihydro-fusidate, sodium glycodihydrofusidate.
- Fatty acids include arachidonic acid, undecanoic acid, oleic acid, lauric acid, caprylic acid, capric acid, myristic acid, palmitic acid, stearic acid, linoleic acid, linolenic acid, dicaprate, tricaprate, monoolein, dilaurin, glyceryl 1-monocaprate, l-dodecylazacycloheptan-2-one, an acylcarnitine, an acylcholine, or a monoglyceride, a diglyceride or a pharmaceutically acceptable salt thereof (e.g., sodium).
- arachidonic acid arachidonic acid, undecanoic acid, oleic acid, lauric acid, caprylic acid, capric acid, myristic acid, palmitic acid, stearic acid, linoleic acid, linolenic acid, dicaprate, tricaprate, monoolein, dilaurin, gly
- penetration enhancers for example, fatty acids/salts in combination with bile acids/salts can also be used.
- An exemplary combination is the sodium salt of lauric acid, capric acid and UDCA.
- Further penetration enhancers include polyoxyethylene-9- lauryl ether, polyoxyethylene-20-cetyl ether.
- Antisense compounds provided herein can be delivered orally in granular form including sprayed dried particles, or complexed to form micro or nanoparticles.
- Antisense compound complexing agents include poly-amino acids; polyimines; polyacrylates; polyalkylacrylates, polyoxethanes, polyalkylcyanoacrylates; cationized gelatins, albumins, starches, acrylates, polyethyleneglycols (PEG) and starches; polyalkylcyanoacrylates; DEAE-derivatized polyimines, pollulans, celluloses and starches.
- Complexing agents include chitosan, N-trimethylchitosan, poly-L-lysine, polyhistidine, polyornithine, polyspermines, protamine, polyvinylpyridine, polythiodiethylamino-methylethylene P(TDAE),
- polyaminostyrene e.g.,p-amino
- Oral formulations for antisense compounds and their preparation can be described in detail in U.S. applications Ser.
- compositions and formulations for parenteral, intrathecal or intraventricular administration can include sterile aqueous solutions which can also contain buffers, diluents and other suitable additives such as, but not limited to, penetration enhancers, carrier compounds and other pharmaceutically acceptable carriers or excipients.
- compositions provided herein include, but are not limited to, solutions, emulsions, and liposome-containing formulations. These compositions can be generated from a variety of components that include, but are not limited to, preformed liquids, self-emulsifying solids and self-emulsifying semisolids.
- the pharmaceutical formulations provided herein can be prepared according to conventional techniques well known in the pharmaceutical industry. Such techniques include the step of bringing into association the active ingredients with the pharmaceutical carrier(s) or excipient(s).
- the formulations can be prepared by uniformly and intimately bringing into association the active ingredients with liquid carriers or finely divided solid carriers or both, and then, if necessary, shaping the product.
- compositions provided herein can be formulated into any of many possible dosage forms such as, but not limited to, tablets, capsules, gel capsules, liquid syrups, soft gels, suppositories, and enemas.
- the compositions provided herein can also be formulated as suspensions in aqueous, non-aqueous or mixed media.
- Aqueous suspensions can further contain substances, which increase the viscosity of the suspension including, for example, sodium carboxymethylcellulose, sorbitol and/or dextran.
- the suspension can also contain stabilizers.
- the pharmaceutical compositions can be formulated and used as foams.
- Pharmaceutical foams include formulations such as, but not limited to, emulsions, microemulsions, creams, jellies and liposomes. While basically similar in nature these formulations vary in the components and the consistency of the final product.
- the preparation of such compositions and formulations is generally known to those skilled in the pharmaceutical and formulation arts and can be applied to the formulation of the compositions provided herein.
- compositions comprise one or more oligonucleotides and one or more excipients.
- excipients are selected from water, salt solutions, alcohol, polyethylene glycols, gelatin, lactose, amylase, magnesium stearate, talc, silicic acid, viscous paraffin, hydroxymethylcellulosem and polyvinylpyrrolidone.
- a pharmaceutical composition is prepared using known techniques, including, but not limited to mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping or tabletting processes.
- a pharmaceutical composition is a liquid (e.g. , a suspension, elixir and/or solution).
- a liquid pharmaceutical composition is prepared using ingredients known in the art, including, but not limited to, water, glycols, oils, alcohols, flavoring agents, preservatives, and coloring agents.
- a pharmaceutical composition is a solid (e.g., a powder, tablet, and/or capsule).
- a solid pharmaceutical composition comprising one or more oligonucleotides is prepared using ingredients known in the art, including, but not limited to, starches, sugars, diluents, granulating agents, lubricants, binders, and disintegrating agents.
- a pharmaceutical composition is formulated as a depot preparation. Certain such depot preparations are typically longer acting than non-depot preparations. In certain embodiments, such preparations are administered by implantation (for example subcutaneously or intramuscularly) or by intramuscular injection. In certain
- depot preparations are prepared using suitable polymeric or hydrophobic materials (for example an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
- a pharmaceutical composition comprises a delivery system.
- delivery systems include, but are not limited to, liposomes and emulsions. Certain delivery systems are useful for preparing certain pharmaceutical compositions including those comprising hydrophobic compounds. In certain embodiments, certain organic solvents such as dimethylsulfoxide are used.
- a pharmaceutical composition comprises one or more tissue-specific delivery molecules designed to deliver the one or more pharmaceutical agents to specific tissues or cell types.
- pharmaceutical compositions include liposomes coated with a tissue-specific antibody.
- a pharmaceutical composition comprises a co-solvent system.
- co-solvent systems comprise, for example, benzyl alcohol, a nonpolar surfactant, a water-miscible organic polymer, and an aqueous phase.
- co-solvent systems are used for hydrophobic compounds.
- a non-limiting example of such a co-solvent system is the VPD co-solvent system, which is a solution of absolute ethanol comprising 3% w/v benzyl alcohol, 8% w/v of the nonpolar surfactant Polysorbate 80 TM , and 65% w/v polyethylene glycol 300.
- the proportions of such co-solvent systems may be varied considerably without significantly altering their solubility and toxicity characteristics.
- co-solvent components may be varied: for example, other surfactants may be used instead of Polysorbate 80TM; the fraction size of polyethylene glycol may be varied; other biocompatible polymers may replace polyethylene glycol, e.g., polyvinyl pyrrolidone; and other sugars or polysaccharides may substitute for dextrose.
- a pharmaceutical composition comprises a sustained-release system.
- a sustained-release system is a semi-permeable matrix of solid hydrophobic polymers.
- sustained-release systems may, depending on their chemical nature, release pharmaceutical agents over a period of hours, days, weeks or months.
- a pharmaceutical composition is prepared for oral administration.
- a pharmaceutical composition is formulated by combining one or more oligonucleotides with one or more pharmaceutically acceptable carriers.
- Certain of such carriers enable pharmaceutical compositions to be formulated as tablets, pills, dragees, capsules, liquids, gels, syrups, slurries, suspensions and the like, for oral ingestion by a subject.
- pharmaceutical compositions for oral use are obtained by mixing oligonucleotide and one or more solid excipient.
- Suitable excipients include, but are not limited to, fillers, such as sugars, including lactose, sucrose, mannitol, or sorbitol; cellulose preparations such as, for example, maize starch, wheat starch, rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose, hydroxypropylmethyl-cellulose, sodium
- compositions are formed to obtain tablets or dragee cores.
- disintegrating agents ⁇ e.g., cross-linked polyvinyl pyrrolidone, agar, or alginic acid or a salt thereof, such as sodium alginate) are added.
- dragee cores are provided with coatings.
- concentrated sugar solutions may be used, which may optionally contain gum arabic, talc, polyvinyl pyrrolidone, carbopol gel, polyethylene glycol, and/or titanium dioxide, lacquer solutions, and suitable organic solvents or solvent mixtures.
- Dyestuffs or pigments may be added to tablets or dragee coatings.
- compositions for oral administration are push-fit capsules made of gelatin.
- Certain of such push- fit capsules comprise one or more pharmaceutical agents in admixture with one or more filler such as lactose, binders such as starches, and/or lubricants such as talc or magnesium stearate and, optionally, stabilizers.
- pharmaceutical compositions for oral administration are soft, sealed capsules made of gelatin and a plasticizer, such as glycerol or sorbitol.
- one or more pharmaceutical agents are be dissolved or suspended in suitable liquids, such as fatty oils, liquid paraffin, or liquid polyethylene glycols.
- stabilizers may be added.
- compositions are prepared for buccal administration. Certain of such pharmaceutical compositions are tablets or lozenges formulated in conventional manner.
- a pharmaceutical composition is prepared for administration by injection (e.g., intravenous, subcutaneous, intramuscular, etc.).
- injection e.g., intravenous, subcutaneous, intramuscular, etc.
- a pharmaceutical composition comprises a carrier and is formulated in aqueous solution, such as water or physiologically compatible buffers such as Hanks's solution, Ringer's solution, or physiological saline buffer.
- aqueous solution such as water or physiologically compatible buffers such as Hanks's solution, Ringer's solution, or physiological saline buffer.
- other ingredients are included (e.g., ingredients that aid in solubility or serve as preservatives).
- injectable suspensions are prepared using appropriate liquid carriers, suspending agents and the like.
- Certain pharmaceutical compositions for injection are presented in unit dosage form, e.g., in ampoules or in multi-dose containers.
- Certain pharmaceutical compositions for injection are suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents.
- solvents suitable for use in pharmaceutical compositions for injection include, but are not limited to, lipophilic solvents and fatty oils, such as sesame oil, synthetic fatty acid esters, such as ethyl oleate or triglycerides, and liposomes.
- Aqueous injection suspensions may contain substances that increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol, or dextran.
- suspensions may also contain suitable stabilizers or agents that increase the solubility of the pharmaceutical agents to allow for the preparation of highly concentrated solutions.
- a pharmaceutical composition is prepared for transmucosal administration.
- penetrants appropriate to the barrier to be permeated are used in the formulation. Such penetrants are generally known in the art.
- a pharmaceutical composition is prepared for administration by inhalation.
- Certain of such pharmaceutical compositions for inhalation are prepared in the form of an aerosol spray in a pressurized pack or a nebulizer.
- Certain of such pharmaceutical compositions comprise a propellant, e.g. , dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas.
- the dosage unit may be determined with a valve that delivers a metered amount.
- capsules and cartridges for use in an inhaler or insufflator may be formulated.
- Certain of such formulations comprise a powder mixture of a pharmaceutical agent provided herein and a suitable powder base such as lactose or starch.
- a pharmaceutical composition is prepared for rectal administration, such as a suppositories or retention enema.
- Certain of such pharmaceutical compositions comprise known ingredients, such as cocoa butter and/or other glycerides.
- a pharmaceutical composition is prepared for topical administration.
- Certain of such pharmaceutical compositions comprise bland moisturizing bases, such as ointments or creams.
- suitable ointment bases include, but are not limited to, petrolatum, petrolatum plus volatile silicones, lanolin and water in oil emulsions such as
- EucerinTM available from Beiersdorf (Cincinnati, Ohio).
- suitable cream bases include, but are not limited to, NiveaTM Cream, available from Beiersdorf (Cincinnati, Ohio), cold cream (USP), Purpose CreamTM, available from Johnson & Johnson (New Brunswick, N. J.), hydrophilic ointment (USP) and LubridermTM, available from Pfizer (Morris Plains, N.J.).
- a pharmaceutical composition comprises an oligonucleotide in a therapeutically effective amount.
- the therapeutically effective amount is sufficient to prevent, alleviate or ameliorate symptoms of a disease or to prolong the survival of the subject being treated. Determination of a therapeutically effective amount is well within the capability of those skilled in the art.
- one or more oligonucleotides is formulated as a prodrug.
- a prodrug upon in vivo administration, is chemically converted to the biologically, pharmaceutically or therapeutically more active form of the oligonucleotide.
- prodrugs are useful because they are easier to administer than the corresponding active form. For example, in certain instances, a prodrug may be more
- a prodrug may have improved solubility compared to the corresponding active form.
- prodrugs are less water soluble than the corresponding active form.
- such prodrugs possess superior transmittal across cell membranes, where water solubility is detrimental to mobility.
- a prodrug is an ester.
- the ester is metabolically hydrolyzed to carboxylic acid upon administration.
- the carboxylic acid containing compound is the corresponding active form.
- a prodrug comprises a short peptide (polyaminoacid) bound to an acid group.
- the peptide is cleaved upon administration to form the corresponding active form.
- a prodrug is produced by modifying a pharmaceutically active compound such that the active compound will be regenerated upon in vivo administration.
- the prodrug can be designed to alter the metabolic stability or the transport characteristics of a drug, to mask side effects or toxicity, to improve the flavor of a drug or to alter other
- compositions including one or more pharmaceutical agents are useful for treating a conditions or disorders in a mammalian, and particularly in a human, subject.
- Suitable administration routes include, but are not limited to, oral, rectal, transmucosal, intestinal, enteral, topical, suppository, through inhalation, intrathecal, intraventricular, intraperitoneal, intranasal, intraocular and parenteral (e.g. , intravenous, intramuscular, intramedullary, and subcutaneous).
- pharmaceutical intrathecals are administered to achieve local rather than systemic exposures.
- pharmaceutical compositions may be injected directly in the area of desired effect (e.g., in the renal or cardiac area).
- a pharmaceutical composition is administered in the form of a dosage unit (e.g., tablet, capsule, bolus, etc.).
- such pharmaceutical compositions comprise an oligonucleotide in a dose selected from 25 mg, 30 mg, 35 mg, 40 mg, 45 mg, 50 mg, 55 mg, 60 mg, 65 mg, 70 mg, 75 mg, 80 mg, 85 mg, 90 mg, 95 mg, 100 mg, 105 mg, 110 mg, 115 mg, 120 mg, 125 mg, 130 mg, 135 mg, 140 mg, 145 mg, 150 mg, 155 mg, 160 mg, 165 mg, 170 mg, 175 mg, 180 mg, 185 mg, 190 mg, 195 mg, 200 mg, 205 mg, 210 mg, 215 mg, 220 mg, 225 mg, 230 mg, 235 mg, 240 mg, 245 mg, 250 mg, 255 mg, 260 mg, 265 mg, 270 mg, 270 mg, 280 mg, 285 mg, 290
- a pharmaceutical composition comprises a dose of oligonucleotide selected from 25 mg, 50 mg, 75 mg, 100 mg, 150 mg, 200 mg, 250 mg, 300 mg, 350 mg, 400 mg, 500 mg, 600 mg, 700 mg, and 800mg.
- a pharmaceutical composition is comprises a dose of oligonucleotide selected from 50 mg, 100 mg, 150 mg, 200 mg, 250 mg, 300 mg, and
- the dose is 200 mg. In an embodiment, the dose is 160 mg.
- a pharmaceutical agent is sterile lyophilized oligonucleotide that is reconstituted with a suitable diluent, e.g., sterile water for injection.
- a suitable diluent e.g., sterile water for injection.
- the reconstituted product is administered as a subcutaneous injection or as an intravenous infusion after dilution into saline.
- the lyophilized drug product consists of the oligonucleotide which has been prepared in water for injection, adjusted to pH 7.0-9.0 with acid or base during preparation, and then lyophilized.
- the lyophilized oligonucleotide may be 25-800 mg of the oligonucleotide.
- lyophilized oligonucleotide may be packaged in a 2 mL Type I, clear glass vial (ammonium sulfate-treated), stoppered with a bromobutyl rubber closure and sealed with an aluminum FLIP-OFF® overseal.
- the lyophilized pharmaceutical agent comprises ISIS 301012.
- compositions may additionally contain other adjunct components conventionally found in pharmaceutical compositions, at their art-established usage levels.
- the compositions may contain additional, compatible, pharmaceutically-active materials such as, for example, antipruritics, astringents, local anesthetics or anti-inflammatory agents, or may contain additional materials useful in physically formulating various dosage forms of the compositions provided herein, such as dyes, flavoring agents, preservatives, antioxidants, opacifiers, thickening agents and stabilizers.
- additional materials useful in physically formulating various dosage forms of the compositions provided herein such as dyes, flavoring agents, preservatives, antioxidants, opacifiers, thickening agents and stabilizers.
- such materials when added, should not unduly interfere with the biological activities of the components of the compositions provided herein.
- the formulations can be sterilized and, if desired, mixed with auxiliary agents, e.g., lubricants, preservatives, stabilizers, wetting agents, emulsifiers, salts for influencing osmotic pressure, buffers, colorings, flavorings and/or aromatic substances and the like which do not deleteriously interact with the oligonucleotide(s) of the formulation.
- auxiliary agents e.g., lubricants, preservatives, stabilizers, wetting agents, emulsifiers, salts for influencing osmotic pressure, buffers, colorings, flavorings and/or aromatic substances and the like which do not deleteriously interact with the oligonucleotide(s) of the formulation.
- compositions provided herein can be prepared and formulated as emulsions.
- Emulsions can be typically heterogeneous systems of one liquid dispersed in another in the form of droplets usually exceeding 0.1 ⁇ in diameter (Idson, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 199; Rosoff, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p.
- Emulsions can be often biphasic systems comprising of two immiscible liquid phases intimately mixed and dispersed with each other.
- emulsions can be either water-in-oil (w/o) or of the oil-in-water (o/w) variety.
- aqueous phase When an aqueous phase is finely divided into and dispersed as minute droplets into a bulk oily phase the resulting composition is called water-in-oil (w/o) emulsion.
- oily phase When an oily phase is finely divided into and dispersed as minute droplets into a bulk aqueous phase the resulting composition is called water-in-oil (w/o) emulsion.
- oily phase is finely divided into and dispersed as minute droplets into a bulk aqueous phase the resulting
- composition is called an oil-in-water (o/w) emulsion.
- Emulsions can contain additional components in addition to the dispersed phases and the active drug which can be present as a solution in either the aqueous phase, oily phase or itself as a separate phase.
- Pharmaceutical excipients such as emulsifiers, stabilizers, dyes, and anti-oxidants can also be present in emulsions as needed.
- Pharmaceutical emulsions can also be multiple emulsions that can be comprised of more than two phases such as, for example, in the case of oil-in-water-in-oil (o/w/o) and water-in-oil-in-water (w/o/w) emulsions.
- Such complex formulations often provide certain advantages that simple binary emulsions do not.
- Multiple emulsions in which oil droplets of an o/w emulsion enclose small water droplets constitute a w/o/w emulsion.
- a system of oil droplets enclosed in globules of water stabilized in an oily continuous provides an o/w/o emulsion.
- Emulsions can be characterized by little or no thermodynamic stability. Often, the dispersed or discontinuous phase of the emulsion is well dispersed into the external or continuous phase and maintained in this form through the means of emulsifiers or the viscosity of the formulation. Either of the phases of the emulsion can be a semisolid or a solid, as is the case of emulsion- style ointment bases and creams. Other means of stabilizing emulsions entail the use of emulsifiers that can be incorporated into either phase of the emulsion. Emulsifiers can broadly be classified into four categories: synthetic surfactants, naturally occurring emulsifiers, absorption bases, and finely dispersed solids (Idson, in Pharmaceutical Dosage Forms,
- Synthetic surfactants also known as surface active agents, have found wide applicability in the formulation of emulsions and have been reviewed in the literature (Rieger, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 285; Idson, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds. ), Marcel Dekker, Inc., New York, N.Y., 1988, volume 1, p. 199).
- Surfactants can be typically amphiphilic and comprise a hydrophilic and a hydrophobic portion.
- HLB hydrophile/lipophile balance
- surfactants can be classified into different classes based on the nature of the hydrophilic group: nonionic, anionic, cationic and amphoteric (Rieger, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 285).
- Naturally occurring emulsifiers used in emulsion formulations include lanolin, beeswax, phosphatides, lecithin and acacia.
- Absorption bases possess hydrophilic properties such that they can soak up water to form w/o emulsions yet retain their semisolid consistencies, such as anhydrous lanolin and hydrophilic petrolatum. Finely divided solids have also been used as good emulsifiers especially in combination with surfactants and in viscous preparations.
- polar inorganic solids such as heavy metal hydroxides, non-swelling clays such as bentonite, attapulgite, hectorite, kaolin, montmorillonite, colloidal aluminum silicate and colloidal magnesium aluminum silicate, pigments and nonpolar solids such as carbon or glyceryl tristearate.
- non-emulsifying materials can also be included in emulsion formulations and contribute to the properties of emulsions. These include fats, oils, waxes, fatty acids, fatty alcohols, fatty esters, humectants, hydrophilic colloids, preservatives and
- Hydrophilic colloids or hydrocolloids include naturally occurring gums and synthetic polymers such as polysaccharides (for example, acacia, agar, alginic acid, carrageenan, guar gum, karaya gum, and tragacanth), cellulose derivatives (for example, carboxymethylcellulose and carboxypropylcellulose), and synthetic polymers (for example, carbomers, cellulose ethers, and carboxyvinyl polymers). These disperse or swell in water to form colloidal solutions that stabilize emulsions by forming strong interfacial films around the dispersed-phase droplets and by increasing the viscosity of the external phase.
- polysaccharides for example, acacia, agar, alginic acid, carrageenan, guar gum, karaya gum, and tragacanth
- cellulose derivatives for example, carboxymethylcellulose and carboxypropylcellulose
- synthetic polymers for example, carbomers, cellulose ethers, and
- emulsions often contain a number of ingredients such as carbohydrates, proteins, sterols and phosphatides that can readily support the growth of microbes, these formulations often incorporate preservatives.
- preservatives included in emulsion formulations include methyl paraben, propyl paraben, quaternary ammonium salts,
- benzalkonium chloride esters of p-hydroxybenzoic acid, and boric acid.
- Antioxidants can also be commonly added to emulsion formulations to prevent deterioration of the formulation.
- Antioxidants used can be free radical scavengers such as tocopherols, alkyl gallates, butylated hydroxyanisole, butylated hydroxytoluene, or reducing agents such as ascorbic acid and sodium metabisulfite, and antioxidant synergists such as citric acid, tartaric acid, and lecithin.
- free radical scavengers such as tocopherols, alkyl gallates, butylated hydroxyanisole, butylated hydroxytoluene, or reducing agents such as ascorbic acid and sodium metabisulfite
- antioxidant synergists such as citric acid, tartaric acid, and lecithin.
- compositions of antisense compounds and nucleic acids can be formulated as microemulsions.
- a microemulsion can be defined as a system of water, oil and amphiphile which is a single optically isotropic and thermodynamically stable liquid solution (Rosoff, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 245).
- microemulsions can be systems that can be prepared by first dispersing an oil in an aqueous surfactant solution and then adding a sufficient amount of a fourth component, generally an intermediate chain-length alcohol to form a transparent system.
- microemulsions have also been described as thermodynamically stable, isotropically clear dispersions of two immiscible liquids that can be stabilized by interfacial films of surface-active molecules (Leung and Shah, in: Controlled Release of Drugs: Polymers and Aggregate Systems, Rosoff, M., Ed., 1989, VCH Publishers, New York, pages 185-215).
- Microemulsions commonly can be prepared via a combination of three to five components that include oil, water, surfactant, cosurfactant and electrolyte.
- microemulsion is of the water-in-oil (w/o) or an oil-in-water (o/w) type is dependent on the properties of the oil and surfactant used and on the structure and geometric packing of the polar heads and hydrocarbon tails of the surfactant molecules (Schott, in Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa., 1985, p. 271).
- microemulsions offer the advantage of solubilizing water-insoluble drugs in a formulation of thermodynamically stable droplets that can be formed spontaneously.
- Surfactants used in the preparation of microemulsions include, but are not limited to, ionic surfactants, non-ionic surfactants, Brij 96, polyoxyethylene oleyl ethers, polyglycerol fatty acid esters, tetraglycerol monolaurate (ML310), tetraglycerol monooleate (MO310),
- hexaglycerol monooleate PO310
- hexaglycerol pentaoleate PO500
- decaglycerol monocaprate MCA750
- decaglycerol monooleate MO750
- decaglycerol sequioleate SO750
- decaglycerol decaoleate DAO750
- cosurfactant usually a short-chain alcohol such as ethanol, 1-propanol, and 1-butanol, serves to increase the interfacial fluidity by penetrating into the surfactant film and consequently creating a disordered film because of the void space generated among surfactant molecules.
- Microemulsions can, however, be prepared without the use of cosurfactants and alcohol-free self-emulsifying microemulsion systems can be known in the art.
- the aqueous phase can typically be, but is not limited to, water, an aqueous solution of the drug, glycerol, PEG300, PEG400, polyglycerols, propylene glycols, and derivatives of ethylene glycol.
- the oil phase can include, but is not limited to, materials such as Captex 300, Captex 355, Capmul MCM, fatty acid esters, medium chain (C8-C12) mono, di, and tri-glycerides, polyoxyethylated glyceryl fatty acid esters, fatty alcohols, polyglycolized glycerides, saturated polyglycolized C8-C10 glycerides, vegetable oils and silicone oil.
- materials such as Captex 300, Captex 355, Capmul MCM, fatty acid esters, medium chain (C8-C12) mono, di, and tri-glycerides, polyoxyethylated glyceryl fatty acid esters, fatty alcohols, polyglycolized glycerides, saturated polyglycolized C8-C10 glycerides, vegetable oils and silicone oil.
- Microemulsions can be particularly of interest from the standpoint of drug
- Lipid based microemulsions both o/w and w/o have been proposed to enhance the oral bioavailability of drugs, including peptides
- Microemulsions afford advantages of improved drug solubilization, protection of drug from enzymatic hydrolysis, possible enhancement of drug absorption due to surfactant-induced alterations in membrane fluidity and permeability, ease of preparation, ease of oral administration over solid dosage forms, improved clinical potency, and decreased toxicity (Constantinides et al., Pharmaceutical Research, 1994, 11, 1385; Ho et al., J. Pharm. Sci., 1996, 85, 138-143). Often microemulsions can form spontaneously when their components can be brought together at ambient temperature. This can be particularly
- thermolabile drugs peptides or antisense compounds.
- Microemulsions have also been effective in the transdermal delivery of active components in both cosmetic and pharmaceutical applications. It is expected that the microemulsion
- compositions and formulations provided herein will facilitate the increased systemic absorption of antisense compounds and nucleic acids from the gastrointestinal tract, as well as improve the local cellular uptake of antisense compounds and nucleic acids within the gastrointestinal tract, vagina, buccal cavity and other can beas of administration.
- Microemulsions provided herein can also contain additional components and additives such as sorbitan monostearate (Grill 3), Labrasol, and penetration enhancers to improve the properties of the formulation and to enhance the absorption of the antisense compounds and nucleic acids provided herein.
- Penetration enhancers used in the microemulsions provided herein can be classified as belonging to one of five broad categories-surfactants, fatty acids, bile salts, chelating agents, and non-chelating non-surfactants (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p. 92). Each of these classes has been discussed above.
- lipids there are organized surfactant structures besides microemulsions that have been studied and used for the formulation of drugs. These include monolayers, micelles, bilayers and vesicles. Vesicles, such as liposomes, have attracted great interest because of their specificity and the duration of action they offer from the standpoint of drug delivery.
- liposome refers to a vesicle composed of amphiphilic lipids arranged in a spherical bilayer or bilayers.
- Liposomes can be unilamellar or multilamellar vesicles which have a membrane formed from a lipophilic material and an aqueous interior. The aqueous portion contains the composition to be delivered. Cationic liposomes possess the advantage of being able to fuse to the cell wall. Non-cationic liposomes, although not able to fuse as efficiently with the cell wall, can be taken up by macrophages in vivo. [00447] In order to cross intact mammalian skin, lipid vesicles must pass through a series of fine pores, each with a diameter less than 50 nm, under the influence of a suitable transdermal gradient. Therefore, it is desirable to use a liposome which is highly deformable and able to pass through such fine pores.
- liposomes obtained from natural phospholipids can be biocompatible and biodegradable; liposomes can incorporate a wide range of water and lipid soluble drugs; liposomes can protect encapsulated drugs in their internal compartments from metabolism and degradation (Rosoff, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 245).
- Important considerations in the preparation of liposome formulations can be the lipid surface charge, vesicle size and the aqueous volume of the liposomes.
- Liposomes can be useful for the transfer and delivery of active ingredients to the site of action. Because the liposomal membrane is structurally similar to biological membranes, when liposomes can be applied to a tissue, the liposomes start to merge with the cellular membranes. As the merging of the liposome and cell progresses, the liposomal contents can be emptied into the cell where the active agent can act.
- Liposomal formulations have been the focus of extensive investigation as the mode of delivery for many drugs. There is growing evidence that for topical administration, liposomes present several advantages over other formulations. Such advantages include reduced sided effects related to high systemic absorption of the administered drug, increased accumulation of the administered drug at the desired target, and the ability to administer a wide variety of drugs, both hydrophilic and hydrophobic, into the skin.
- liposomes to deliver agents including high- molecular weight DNA into the skin.
- Compounds including analgesics, antibodies, hormones and high-molecular weight DNAs have been administered to the skin. The majority of applications resulted in the targeting of the upper epidermis.
- Liposomes fall into two broad classes. Cationic liposomes can be positively charged liposomes which interact with the negatively charged DNA molecules to form a stable complex. The positively charged DNA/liposome complex binds to the negatively charged cell surface and is internalized in an endosome. Due to the acidic pH within the endosome, the liposomes can be ruptured, releasing their contents into the cell cytoplasm (Wang et al., Biochem. Biophys. Res. Commun., 1987, 147, 980-985).
- Liposomes which can be pH-sensitive or negatively-charged, entrap DNA rather than complex with it. Since both the DNA and the lipid can be similarly charged, repulsion rather than complex formation occurs. Nevertheless, some DNA is entrapped within the aqueous interior of these liposomes. pH-sensitive liposomes have been used to deliver DNA encoding the thymidine kinase gene to cell monolayers in culture. Expression of the exogenous gene was detected in the target cells (Zhou et al., Journal of Controlled Release, 1992, 19, 269-274).
- liposomal composition includes phospholipids other than naturally-derived phosphatidylcholine.
- Neutral liposome compositions for example, can be formed from dimyristoyl phosphatidylcholine (DMPC) or dipalmitoyl phosphatidylcholine (DPPC).
- Anionic liposome compositions generally can be formed from dimyristoyl
- phosphatidylglycerol while anionic-fusogenic liposomes can be formed primarily from dioleoyl phosphatidylethanolamine (DOPE).
- DOPE dioleoyl phosphatidylethanolamine
- Another type of liposomal composition is formed from phosphatidylcholine (PC) such as, for example, soybean PC, and egg PC.
- PC phosphatidylcholine
- Another type is formed from mixtures of phospholipid and/or phosphatidylcholine and/or cholesterol.
- Non-ionic liposomal systems have also been examined to determine their utility in the delivery of drugs to the skin, in particular systems comprising non-ionic surfactant and cholesterol.
- Non-ionic liposomal, formulations comprising NovasomeTM I (glyceryl)
- Liposomes also include "sterically stabilized" liposomes, a term which, as used herein, refers to liposomes comprising one or more specialized lipids that, when incorporated into liposomes, result in enhanced circulation lifetimes relative to liposomes lacking such specialized lipids.
- sterically stabilized liposomes can be those in which part of the vesicle-forming lipid portion of the liposome (A) comprises one or more glycolipids, such as monosialoganglioside GM1, or (B) is derivatized with one or more hydrophilic polymers, such as a polyethylene glycol (PEG) moiety.
- PEG polyethylene glycol
- Liposomes comprising sphingomyelin.
- Liposomes comprising 1 ,2-sn-dimyristoylphosphatidylcholine can be disclosed in WO 97/13499 (Lim et al. ).
- liposomes comprising lipids derivatized with one or more hydrophilic polymers, and methods of preparation thereof, can be known in the art.
- Sunamoto et al. (Bull. Chem. Soc. Jpn., 1980, 53, 2778) described liposomes comprising a nonionic detergent, 2C1215G, that contains a PEG moiety.
- Ilium et al. (FEBS Lett., 1984, 167, 79) noted that hydrophilic coating of polystyrene particles with polymeric glycols results in significantly enhanced blood half-lives.
- Synthetic phospholipids modified by the attachment of carboxylic groups of polyalkylene glycols ⁇ e.g., PEG) can be described by Sears ( U.S. Pat. Nos. 4,426,330 and 4,534,899). Klibanov et al. (FEBS Lett., 1990, 268, 235) described experiments
- DSPE distearoylphosphatidylethanolamine
- PEG distearoylphosphatidylethanolamine
- Liposomes having covalently bound PEG moieties on their external surface can be described in European Patent No. EP 0 445 131 Bl and WO 90/04384 to Fisher.
- Liposome compositions containing 1-20 mole percent of PE derivatized with PEG, and methods of use thereof, can be described by Woodle et al. ( U.S. Pat. Nos.
- Liposomes comprising a number of other lipid-polymer conjugates can be disclosed in WO 91/05545 and U.S. Pat. No. 5,225,212 (both to Martin et al. ) and in WO 94/20073 (Zalipsky et al. )
- Liposomes comprising PEG-modified ceramide lipids can be described in WO 96/10391 (Choi et al. ).
- U.S. Pat. No. 5,540,935 (Miyazaki et al. ) and U.S. Pat. No. 5,556,948 (Tagawa et al. ) describes PEG-containing liposomes that can be further derivatized with functional moieties on their surfaces.
- a limited number of liposomes comprising nucleic acids can be known in the art.
- WO 96/40062 to Thierry et al. discloses methods for encapsulating high molecular weight nucleic acids in liposomes.
- U.S. Pat. No. 5,264,221 to Tagawa et al. discloses protein-bonded liposomes and asserts that the contents of such liposomes can include an antisense RNA.
- U.S. Pat. No. 5,665,710 to Rahman et al. describes certain methods of encapsulating oligodeoxynucleotides in liposomes.
- WO 97/04787 to Love et al. discloses liposomes comprising antisense antisense compounds targeted to the raf gene.
- Transfersomes can be yet another type of liposomes, and can be highly deformable lipid aggregates which can be attractive candidates for drug delivery vehicles.
- Transfersomes can be described as lipid droplets which can be so highly, deformable that they can be easily able to penetrate through pores which can be smaller than the droplet.
- Transfersomes can be adaptable to the environment in which they can be used, e.g., they can be self-optimizing (adaptive to the shape of pores in the skin), self-repairing, frequently reach their targets without fragmenting, and often self-loading.
- surface edge-activators usually surfactants
- HLB hydrophile/lipophile balance
- Nonionic surfactants find wide application in pharmaceutical and cosmetic products and can be usable over a wide range of pH values. In general their HLB values range from 2 to about 18 depending on their structure.
- Nonionic surfactants include nonionic esters such as ethylene glycol esters, propylene glycol esters, glyceryl esters, polyglyceryl esters, sorbitan esters, sucrose esters, and ethoxylated esters.
- Nonionic alkanolamides and ethers such as fatty alcohol ethoxylates, propoxylated alcohols, and ethoxylated/propoxylated block polymers can also be included in this class.
- the polyoxyethylene surfactants can be the most popular members of the nonionic surfactant class.
- Anionic surfactants include carboxylates such as soaps, acyl lactylates, acyl amides of amino acids, esters of sulfuric acid such as alkyl sulfates and ethoxylated alkyl sulfates, sulfonates such as alkyl benzene sulfonates, acyl isethionates, acyl taurates and sulfosuccinates, and phosphates.
- the most important members of the anionic surfactant class can be the alkyl sulfates and the soaps.
- Cationic surfactants include quaternary ammonium salts and ethoxylated amines.
- the quaternary ammonium salts can be the most used members of this class.
- amphoteric surfactants include acrylic acid
- various penetration enhancers to effect the efficient delivery of nucleic acids, particularly antisense compounds, to the skin of animals.
- Most drugs can be present in solution in both ionized and nonionized forms. However, usually only lipid soluble or lipophilic drugs readily cross cell membranes. It has been discovered that even non-lipophilic drugs can cross cell membranes if the membrane to be crossed is treated with a penetration enhancer. In addition to aiding the diffusion of non-lipophilic drugs across cell membranes, penetration enhancers also enhance the permeability of lipophilic drugs.
- Penetration enhancers can be classified as belonging to one of five broad categories, i.e., surfactants, fatty acids, bile salts, chelating agents, and non-chelating non-surfactants (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p. 92). Each of the above mentioned classes of penetration enhancers can be described below in greater detail.
- surfactants can be chemical entities which, when dissolved in an aqueous solution, reduce the surface tension of the solution or the interfacial tension between the aqueous solution and another liquid, with the result that absorption of antisense compounds through the mucosa is enhanced.
- these penetration enhancers include, for example, sodium lauryl sulfate, polyoxyethylene-9-lauryl ether and polyoxyethylene-20-cetyl ether) (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p. 92); and perfluorochemical emulsions, such as FC-43. Takahashi et al., J. Pharm. Pharmacol, 1988, 40, 252).
- Various fatty acids and their derivatives which act as penetration enhancers include, for example, oleic acid, lauric acid, capric acid (n-decanoic acid), myristic acid, palmitic acid, stearic acid, linoleic acid; linolenic acid, dicaprate, tricaprate, monoolein (1-monooleoyl-rac- glycerol), dilaurin, caprylic acid, arachidonic acid, glycerol 1-monocaprate, 1- dodecylazacycloheptan-2-one, acylcarnitines, acylchoines, CI -10 alkyl esters thereof (e.g., methyl, isopropyl and t-butyl), and mono- and di-glycerides thereof (i.e., oleate, laurate, caprate, myristate, palmitate, stearate, linoleate, etc.) (Lee
- the physiological role of bile includes the facilitation of dispersion and absorption of lipids and fat-soluble vitamins (Brunton, Chapter 38 in: Goodman & Gilman's The
- bile salts includes any of the naturally occurring components of bile as well as any of their synthetic derivatives.
- the bile salts include, for example, cholic acid (or its pharmaceutically acceptable sodium salt, sodium cholate), dehydrocholic acid (sodium dehydrocholate), deoxycholic acid (sodium deoxycholate), glucholic acid (sodium glucholate), glycholic acid (sodium glycocholate), glycodeoxycholic acid (sodium glycodeoxycholate), taurocholic acid (sodium taurocholate), taurodeoxycholic acid (sodium taurodeoxycholate), chenodeoxycholic acid (sodium chenodeoxycholate), ursodeoxycholic acid (UDCA), sodium tauro-24,25-dihydro-fusidate (STDHF), sodium glycodihydrofusidate and polyoxyethylene-9- lauryl ether (POE) (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, page 92; Swinyard, Chapter 39 In: Remington's Pharmaceutical Sciences, 18th Ed
- Chelating agents can be defined as compounds that remove metallic ions from solution by forming complexes therewith, with the result that absorption of antisense compounds through the mucosa is enhanced.
- chelating agents have the added advantage of also serving as DNase inhibitors, as most characterized DNA nucleases require a divalent metal ion for catalysis and can be thus inhibited by chelating agents (Jarrett, J. Chromatogr., 1993, 618, 315-339).
- Chelating agents include but are not limited to disodium ethylenediaminetetraacetate (EDTA), citric acid, salicylates (e.g., sodium salicylate, 5 -methoxy salicylate and homovanilate), N-acyl derivatives of collagen, laureth-9 and N-amino acyl derivatives of beta-diketones (enamines) (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, page 92; Muranishi, Critical Reviews in Therapeutic Drug Carrier Systems, 1990, 7, 1-33; Buur et al., J. Control ReL, 1990, 14, 43-51).
- EDTA disodium ethylenediaminetetraacetate
- citric acid e.g., sodium salicylate, 5 -methoxy salicylate and homovanilate
- salicylates e.g., sodium salicylate, 5 -methoxy salicylate and homovanilate
- N-acyl derivatives of collagen e.g., sodium sal
- non-chelating non-surfactant penetration enhancing compounds can be defined as compounds that demonstrate insignificant activity as chelating agents or as surfactants but that nonetheless enhance absorption of antisense compounds through the alimentary mucosa (Muranishi, Critical Reviews in Therapeutic Drug Carrier Systems, 1990, 7, 1-33).
- This class of penetration enhancers includes, for example, unsaturated cyclic ureas, 1- alkyl- and 1-alkenylazacycloalkanone derivatives (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, page 92); and non-steroidal anti-inflammatory agents such as diclofenac sodium, indomethacin and phenylbutazone (Yamashita et al., J. Pharm. Pharmacol., 1987, 39, 621-626).
- Agents that enhance uptake of antisense compounds at the cellular level can also be added to the pharmaceutical and other compositions provided herein.
- cationic lipids such as lipofectin (Junichi et al, U.S. Pat. No. 5,705,188), cationic glycerol derivatives, and polycationic molecules, such as polylysine (Lollo et al., PCT Application WO 97/30731), can also be known to enhance the cellular uptake of antisense compounds.
- nucleic acids can be utilized to enhance the penetration of the administered nucleic acids, including glycols such as ethylene glycol and propylene glycol, pyrrols such as 2-pyrrol, azones, and terpenes such as limonene and menthone.
- glycols such as ethylene glycol and propylene glycol
- pyrrols such as 2-pyrrol
- azones such as 2-pyrrol
- terpenes such as limonene and menthone.
- compositions provided herein also incorporate carrier compounds in the formulation.
- carrier compound or “carrier” can refer to a nucleic acid, or analog thereof, which is inert (i.e., does not possess biological activity per se) but is recognized as a nucleic acid by in vivo processes that reduce the bioavailability of a nucleic acid having biological activity by, for example, degrading the biologically active nucleic acid or promoting its removal from circulation.
- a nucleic acid and a carrier compound can result in a substantial reduction of the amount of nucleic acid recovered in the liver, kidney or other extracirculatory reservoirs, presumably due to competition between the carrier compound and the nucleic acid for a common receptor.
- the recovery of a partially phosphorothioate antisense compound in hepatic tissue can be reduced when it is co administered with polyinosinic acid, dextran sulfate, polycytidic acid or 4-acetamido-4'isothiocyano-stilbene-2,2'-disulfonic acid (Miyao et al., Antisense Res. Dev., 1995, 5, 115-121; Takakura et al., Antisense & Nucl. Acid Drug Dev., 1996, 6, 177-183).
- a “pharmaceutical carrier” or “excipient” is a pharmaceutically acceptable solvent, suspending agent or any other pharmacologically inert vehicle for delivering one or more nucleic acids to an animal.
- the excipient can be liquid or solid and is selected, with the planned manner of administration in mind, so as to provide for the desired bulk, consistency, etc., when combined with a nucleic acid and the other components of a given pharmaceutical composition.
- Typical pharmaceutical carriers include, but are not limited to, binding agents (e.g., pregelatinized maize starch, polyvinylpyrrolidone or hydroxypropyl methylcellulose, etc.); fillers (e.g., lactose and other sugars, microcrystalline cellulose, pectin, gelatin, calcium sulfate, ethyl cellulose, polyacrylates or calcium hydrogen phosphate, etc.); lubricants (e.g., magnesium stearate, talc, silica, colloidal silicon dioxide, stearic acid, metallic stearates, hydrogenated vegetable oils, corn starch, polyethylene glycols, sodium benzoate, sodium acetate, etc.); disintegrants (e.g., starch, sodium starch glycolate, etc.); and wetting agents (e.g., sodium lauryl sulphate, etc.).
- binding agents e.g., pregelatinized maize starch, polyvinylpyrrolidone or hydroxyprop
- compositions provided herein can also be used to formulate the compositions provided herein.
- suitable pharmaceutically acceptable carriers include, but are not limited to, water, salt solutions, alcohols, polyethylene glycols, gelatin, lactose, amylose, magnesium stearate, talc, silicic acid, viscous paraffin,
- hydroxymethylcellulose polyvinylpyrrolidone and the like.
- Formulations for topical administration of nucleic acids can include sterile and non- sterile aqueous solutions, non-aqueous solutions in common solvents such as alcohols, or solutions of the nucleic acids in liquid or solid oil bases.
- the solutions can also contain buffers, diluents and other suitable additives.
- Pharmaceutically acceptable organic or inorganic excipients suitable for non-parenteral administration which do not deleteriously react with nucleic acids can be used.
- Suitable pharmaceutically acceptable excipients include, but are not limited to, water, salt solutions, alcohol, polyethylene glycols, gelatin, lactose, amylose, magnesium stearate, talc, silicic acid, viscous paraffin, hydroxymethylcellulose, polyvinylpyrrolidone and the like.
- the compounds provided herein can also be administered by pulsatile delivery.
- Pulsatile delivery refers to a pharmaceutical formulations that delivers a first pulse of drug combined with a penetration enhancer and a second pulse of penetration enhancer to promote absorption of drug which is not absorbed upon release with the first pulse of penetration enhancer.
- One embodiment provided herein is a delayed release oral formulation for enhanced intestinal drug absorption, comprising:
- a second population of carrier particles comprising a penetration enhancer and a delayed release coating or matrix, wherein the penetration enhancer is released at a second location in the intestine downstream from the first location, whereby absorption of the drug is enhanced when the drug reaches the second location.
- the penetration enhancer in (a) and (b) is different.
- This enhancement is obtained by encapsulating at least two populations of carrier particles.
- the first population of carrier particles comprises a biologically active substance and a penetration enhancer
- the second (and optionally additional) population of carrier particles comprises a penetration enhancer and a delayed release coating or matrix.
- a "first pass effect" that applies to orally administered drugs is degradation due to the action of gastric acid and various digestive enzymes.
- One means of ameliorating first pass clearance effects is to increase the dose of administered drug, thereby compensating for proportion of drug lost to first pass clearance.
- administration by, for example, simply providing more of the drug to an animal, other factors influence the bioavailability of drugs administered via non-parenteral refers to.
- a drug can be enzymatically or chemically degraded in the alimentary canal or blood stream and/or can be impermeable or semipermeable to various mucosal membranes.
- compositions can be capable of enhancing absorption of biologically active substances when administered via the rectal, vaginal, nasal or pulmonary routes. It is also contemplated that release of the biologically active substance can be achieved in any part of the gastrointestinal tract.
- Liquid pharmaceutical compositions of antisense compound can be prepared by combining the antisense compound with a suitable vehicle, for example sterile pyrogen free water, or saline solution. Other therapeutic compounds can optionally be included.
- a suitable vehicle for example sterile pyrogen free water, or saline solution.
- Other therapeutic compounds can optionally be included.
- compositions can comprise particles of antisense compound that can be of respirable size.
- particles can be prepared by, for example, grinding dry antisense compound by conventional refers to, fore example with a mortar and pestle, and then passing the resulting powder composition through a 400 mesh screen to segregate large particles and agglomerates.
- a solid particulate composition comprised of an active antisense compound can optionally contain a dispersant which serves to facilitate the formation of an aerosol, for example lactose.
- antisense compound compositions can be aerosolized. Aerosolization of liquid particles can be produced by any suitable refers to, such as with a nebulizer. See, for example, U.S. Pat. No. 4,501,729.
- Nebulizers can be commercially available devices which transform solutions or suspensions into a therapeutic aerosol mist either by means of acceleration of a compressed gas, typically air or oxygen, through a narrow venturi orifice or by means of ultrasonic agitation.
- Suitable nebulizers include those sold by Blairex® under the name PARI LC PLUS, PARI DURA-NEB 2000, PARI-BABY Size, PARI PRONEB Compressor with LC PLUS, PARI WALKHALER Compressor/Nebulizer System, PARI LC PLUS Reusable Nebulizer, and PARI LC Jet+ ⁇ Nebulizer.
- Exemplary formulations for use in nebulizers consist of an antisense compound in a liquid, such as sterile, pyragen free water, or saline solution, wherein the antisense compound comprises up to about 40% w/w of the formulation. Can, the antisense compound comprises less than 20%) w/w. If desired, further additives such as preservatives (for example, methyl hydroxybenzoate) antioxidants, and flavoring agents can be added to the composition.
- Solid particles comprising an antisense compound can also be aerosolized using any solid particulate medicament aerosol generator known in the art. Such aerosol generators produce respirable particles, as described above, and further produce reproducible metered dose per unit volume of aerosol. Suitable solid particulate aerosol generators include insufflators and metered dose inhalers. Metered dose inhalers can be used in the art and can be useful provided herein.
- Can, liquid or solid aerosols can be produced at a rate of from about 10 to 150 liters per minute, more can from about 30 to 150 liters per minute, and most can about 60 liters per minute.
- bioavailability refers to a measurement of what portion of an administered drug reaches the circulatory system when a non-parenteral mode of administration is used to introduce the drug into an animal.
- Penetration enhancers include, but are not limited to, members of molecular classes such as surfactants, fatty acids, bile salts, chelating agents, and non-chelating non-surfactant molecules. (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p. 92).
- Carriers can be inert molecules that can be included in the compositions provided herein to interfere with processes that lead to reduction in the levels of bioavailable drug.
- the formulations will comprise (or consist essentially of) an effective amount of a pharmaceutical composition along with suitable excipients that allow the formulations to be chewed by the patient, in additional embodiments, the formulations can further comprise one or more taste -masking or sweetening agents, such as those described herein.
- the formulations can further comprise one or more taste -masking or sweetening agents, such as those described herein.
- sucralose is used in the chewable formulations. Additional active agents, such as those described herein, can also optionally be added to the chewable formulations.
- a pharmaceutical composition other optional active agents and sweetening agents (e.g., sucralose) in the chewable formulations of provided herein, can be readily determinable by those of ordinary skill in the art, and include those amounts and combinations described herein.
- the chewable formulations of provided herein comprise (or consist essentially of) a pharmaceutical composition and about 0. 05% to about 0. 15% sucralose.
- Such chewable formulations can be especially useful in patient populations where compliance is an issue, such as children, the elderly, and patients who can have difficulty swallowing or using spray/inhalable formulations.
- the formulations can also contain colorants to improve the appearance of the chewable formulations, especially since an attractive coloration imparted by a colorant can improve patient compliance.
- the relative amounts of the colorants selected will vary depending upon the particular hue of the subject colorants and the resultant color desired.
- any standard pharmaceutically acceptable excipient can be used in the chewable tablet formulations which provides adequate compression such as diluents (e.g., mannitol, xylitol, maltitol, lactitol, sorbitol, lactose, sucrose, and compressible sugars such as DiPac® (dextrinized sucrose), available from Austin Products Inc. (Holmdel, N. J.), binders,
- diluents e.g., mannitol, xylitol, maltitol, lactitol, sorbitol, lactose, sucrose, and compressible sugars such as DiPac® (dextrinized sucrose), available from Austin Products Inc. (Holmdel, N. J.)
- binders e.g., mannitol, xylitol, maltitol, lactitol, sorbitol, lacto
- disintegrants e.g., polyvinyl polypyrrolidone, croscarmellose sodium (e.g., Ac-Di-Sol available from FMC BioPolymer, Philadelphia, Pa.), starches and derivatives, cellulose and derivatives, microcrystalline celluloses, such as AvicelTM PH 101 or AvicelTM CE- 15 (a microcrystalline modified with guar gum), both available from FMC
- splitting or swelling agents e.g., polyvinyl polypyrrolidone, croscarmellose sodium (e.g., Ac-Di-Sol available from FMC BioPolymer, Philadelphia, Pa.)
- starches and derivatives cellulose and derivatives, microcrystalline celluloses, such as AvicelTM PH 101 or AvicelTM CE- 15 (a microcrystalline modified with guar gum), both available from FMC
- BioPolymer (Philadelphia, Pa. ), lubricating agents (e.g., magnesium stearate), and flow agents (e.g., colloidal silicon dioxide, such as Cab-O-Sil M5® available from Cabot Corporation, Kokomo, Ind.).
- lubricating agents e.g., magnesium stearate
- flow agents e.g., colloidal silicon dioxide, such as Cab-O-Sil M5® available from Cabot Corporation, Kokomo, Ind.
- Suitable amounts of sweetener used in the chewable formulations, will be familiar to, and can be readily determined by, those skilled in the art.
- the sweetener is present in an amount from about 0. 05%> to about 5.
- 0%> e.g., about 0. 05%, about 0. 1%, about 0. 125%, about 0. 15%, about 0. 2%, about 0. 3%, about 0. 4%, about 0. 5%, about 0. 6%, about 0. 7%, about 0. 8%, about 0. 9%, about 1%, about 1. 25% about 1. 5%
- about 1. 75%) about 2%, about 2. 25%, about 2. 5%, about 2. 75%, about 3%, about 3. 25%, about 3. 5%, about 3.
- sweetener can vary depending on the strength of the particular sweetener used and the levels approved by the regulatory authorities for use in pharmaceutical products.
- Suitable cyclodextrins for use in the chewable formulations of provided herein include ⁇ , ⁇ , or ⁇ cyclodextrins, or an alkylated or hydroxyalkylated derivatives thereof, such as heptakis (2,6-di-o-methyl)-P-cyclodextrin (DIMEB), randomly methylated ⁇ -cyclodextrin (RAMEB), and hydroxypropyl ⁇ -cyclodextrin (HPpCD).
- DIMEB heptakis (2,6-di-o-methyl)-P-cyclodextrin
- RAMEB randomly methylated ⁇ -cyclodextrin
- HPpCD hydroxypropyl ⁇ -cyclodextrin
- a suitable cyclodextrin is ⁇ - cyclodextrin (available from Cerestar USA, Inc., Hammond, Ind. or from Roquette America, Inc., Keokuk.
- the complex of the active substance with cyclodextrin can be prepared in advance, for example, by malaxating or granulating a pharmaceutical composition and any additional active substance(s) and the cyclodextrin in the presence of water, or by preparing an aqueous solution containing a pharmaceutical composition and any additional active substance(s) and the cyclodextrin in the desired molar ratio.
- the pharmaceutical composition and any additional active substance(s) and the cyclodextrin can be simply mixed with other excipients and adjuvants.
- a typical manufacturing process for making either a single layer or bi-layer chewable tablet generally involves blending of the desired ingredients to form a uniform distribution of the pharmaceutical composition (and any other active agent(s)), excipients (e.g., colorants and flavoring agents as well as others).
- an inclusion complex of a pharmaceutical composition and any other active agent(s) and cyclodextrin e.g., ⁇ -cyclodextrin
- a pharmaceutical composition and any other active agent(s) and cyclodextrin can be formed prior to blending into the mixture by malaxating a pharmaceutical composition and any other active agent(s) and cyclodextrin in the presence of water in a planetary mixer for about 20 minutes. The mixture is then dried in a drying oven.
- the complex After drying, the complex is mixed with any color/flavoring blend.
- the blend is then compressed into a single layer or bi-layer tablet using standard methods well-known to those skilled in the art (e.g., Kilian T-100 tablet press or Courtoy 292/43 rotary bi-layer press).
- the colorants and flavoring agents can be added to both layers to form a uniform presentation of the tablet.
- Methods for preparation of chewable tablets and various components for use in the tablets can be found throughout the detailed description section and the Examples of U.S. Patent Publication No. 2003/0215503, the disclosure of which is incorporated by reference herein for all purposes. Additional chewable/orally dissolving tablets, and methods for their manufacture, can be disclosed in U.S. Patent Publication No. 2004/0265372 and U.S. Patent No. 6,270,790, the disclosures of each of which can be incorporated by reference herein for all purposes
- compositions provided herein can additionally contain other adjunct components conventionally found in pharmaceutical compositions, at their art-established usage levels.
- the compositions can contain additional, compatible, pharmaceutically-active materials such as, for example, antipruritics, astringents, local anesthetics or anti-inflammatory agents, or can contain additional materials useful in physically formulating various dosage forms of the compositions provided herein, such as dyes, flavoring agents, preservatives, antioxidants, opacifiers, thickening agents and stabilizers.
- additional materials useful in physically formulating various dosage forms of the compositions provided herein such as dyes, flavoring agents, preservatives, antioxidants, opacifiers, thickening agents and stabilizers.
- such materials when added, should not unduly interfere with the biological activities of the components of the compositions provided herein.
- the formulations can be sterilized and, if desired, mixed with auxiliary agents, e.g., lubricants, preservatives, stabilizers, wetting agents, emulsifiers, salts for influencing osmotic pressure, buffers, colorings, flavorings and/or aromatic substances and the like which do not deleteriously interact with the nucleic acid(s) of the formulation.
- auxiliary agents e.g., lubricants, preservatives, stabilizers, wetting agents, emulsifiers, salts for influencing osmotic pressure, buffers, colorings, flavorings and/or aromatic substances and the like which do not deleteriously interact with the nucleic acid(s) of the formulation.
- Aqueous suspensions can contain substances which increase the viscosity of the suspension including, for example, sodium carboxymethylcellulose, sorbitol and/or dextran.
- the suspension can also contain stabilizers.
- compositions containing (a) an antisense compound and (b) one or more other chemotherapeutic agents which function by a non-antisense mechanism.
- chemotherapeutic agents include but are not limited to daunorubicin, daunomycin, dactinomycin, doxorubicin, epirubicin, idarubicin, esorubicin, bleomycin, mafosfamide, ifosfamide, cytosine arabinoside, bis-chloroethylnitrosurea, busulfan, mitomycin C, actinomycin D, mithramycin, prednisone, hydroxyprogesterone, testosterone, tamoxifen, dacarbazine, procarbazine, hexamethylmelamine, pentamethylmelamine,
- mitoxantrone amsacrine, chlorambucil, methylcyclohexylnitrosurea, nitrogen mustards, melphalan, cyclophosphamide, 6-mercaptopurine, 6-thioguanine, cytarabine, 5-azacytidine, hydroxyurea, deoxycoformycin, 4-hydroxyperoxycyclophosphoramide, 5-fluorouracil (5-FU), 5- fluorodeoxyuridine (5-FUdR), methotrexate (MTX), colchicine, taxol, vincristine, vinblastine, etoposide (VP- 16), trimetrexate, irinotecan, topotecan, gemcitabine, teniposide, cisplatin and diethylstilbestrol (DES).
- 5-fluorouracil 5- fluorodeoxyuridine
- MTX methotrexate
- colchicine colchicine, taxol, vincristine, vinblastine, etoposide (
- chemotherapeutic agents can be used ly (e.g., 5-FU and antisense compound), sequentially (e.g., 5-FU and antisense compound for a period of time followed by MTX and antisense compound), or in combination with one or more other such chemotherapeutic agents (e.g., 5-FU, MTX and antisense compound, or 5-FU, radiotherapy and antisense compound).
- Anti-inflammatory drugs including but not limited to nonsteroidal anti -inflammatory drugs and corticosteroids
- antiviral drugs including but not limited to ribivirin, vidarabine, acyclovir and ganciclovir
- Other non-antisense chemotherapeutic agents can also be used. Two or more combined compounds can be used together or sequentially.
- compositions can contain an antisense compound, particularly antisense compounds, targeted to a first nucleic acid and one or more additional antisense compounds targeted to a second nucleic acid target.
- antisense compounds can be known in the art. Two or more combined compounds can be used together or sequentially.
- Optimal dosing schedules can be calculated from measurements of drug accumulation in the body of the patient. Persons of ordinary skill can easily determine optimum dosages, dosing methodologies and repetition rates. Optimum dosages can vary depending on the relative potency of antisense compounds, and can generally be estimated based on EC50s found to be effective in in vitro and in vivo animal models. In general, dosage is from 0. 01 ⁇ g to 100 g per kg of body weight, and can be given once or more daily, weekly, monthly or yearly, or even once every 2 to 20 years.
- administration of an antisense compound is parenteral administration.
- Parenteral administration can be intravenous or subcutaneous administration.
- administration of an antisense compound is intravenous or subcutaneous administration.
- Administration can include multiple doses of an antisense compound.
- the antisense compound is administered to a subject by, but not including oral, intradermal, intramuscular, intraperitoneal, intravenous, topical,
- subcutaneous, percutaneous, intranasal, and inhalation routes and via scarification (scratching the top layers of skin, e.g., using a bifurcated needle.
- subcutaneous or intravenous routes can be used.
- the preparation for use according to provided herein can be conveniently delivered in the form of an aerosol spray.
- compositions can be administered to the subject via oral administration.
- Methods of oral administration can be accomplished via liquid or solid form, and particularly in solid form such as in tablet or capsule form, using approaches and
- Suitable dosages ⁇ e.g., amounts, volumes, etc.) of the compositions will be apparent from the description herein, including the Examples below.
- pharmaceutical compositions for the treatment of MACE including the underlying causes, but not limited to hypercholesterolemia and hypertension.
- the compositions can be administered to the patient in a single dosage comprising a therapeutically effective amount of each of an antisense compounds.
- the compositions can be administered to the patient in a single does comprising a therapeutically effective amount of an antisense compound and, one or more pharmaceutical compositions as described herein, each in a therapeutically effective ⁇ i.e., MACE-treating or MACE-preventing amount).
- the compositions can be administered to the patient in a single, daily dosage form, once per day. In other embodiments, the compositions can be administered to the patient two or more ⁇ i.e., two, three, four or more) times per day, or as needed according to the particular treatment regiment designed by the patient's physician.
- the amount of the compositions administered each time throughout the treatment period can be the same; alternatively, the amount administered each time during the treatment period can vary ⁇ e.g., the amount administered at a given time can be more or less than the amount administered previously). For example, doses given during maintenance therapy can be lower than those administered during the acute phase of treatment. Appropriate dosing schedules depending on the specific circumstances will be apparent to persons of ordinary skill in the art.
- MACE rates can be remarkably lower following treatment with the antisense compound described herein.
- the antisense compound is administered in a dosage to achieve at least a 40% reduction, at least a 45% reduction, at least a 50% reduction, or at least a 55% reduction in MACE.
- the antisense compound is administered in a dosage to achieve at least a 60% reduction in MACE.
- the antisense compound is administered to in a dosage of at least a 65% reduction, at least a 70% reduction, at least a 75% reduction, at least a 75%) reduction, at least an 80%> reduction, at least an 85% reduction, 85% reduction, or at least a 90% reduction in MACE.
- the reduction in MACE correlates with a refers to reduction in LDL cholesterol. In a specific embodiment, the reduction in MACE correlates with a refers to reduction in LDL cholesterol of approximately -49 to -113 mg/dL.
- administration of the antisense compound decreases total serum cholesterol, ApoB , serum LDL, serum VLDL, serum triglycerides, serum apolipoprotein(a) and/or fatty free acids in the subject. In another embodiment administration of the antisense compound decreases LDL cholesterol. In more specific embodiments,
- administration of the antisense compound causes reductions in atherogenic lipoproteins in plasma.
- the antisense compound is administered to patients with a baseline LDL of approximately >100 mg/dl.
- subjects can be administered with at least 50 mg of the antisense compound, at least 100 mg of the antisense compound, at least 200 mg of the antisense compound or at least 300 mg of the antisense compound.
- at least 50 mg of the antisense compound at least 100 mg of the antisense compound, at least 200 mg of the antisense compound or at least 300 mg of the antisense compound.
- subjects can be administered with at least 50 mg per day, at least 100 mg per day, at least 200 mg per day, or at least 300 mg per day of the antisense compound.
- subjects receive subcutaneous antisense compound at least 50 mg per day, at least 100 mg per day, at least 200 mg per day, or at least 300 mg per day.
- the antisense compound is administered at 200 mg/week.
- the 200 mg/week is adminitered one time per week.
- the subject is administered a single 200 mg/dose per week.
- the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week.
- the antisense compound is administered weekly for at least 12 months.
- the antisense compound is administered subcutaneously (s.c).
- subjects receive 200 mg of the antisense compound on days 1 , 4, 8, and 1 1 , followed by once-weekly injections to a total of
- the antisense compound described herein is administered as a single dose once weekly for 26 weeks.
- subjects receive a placebo for the first 6 months followed by treatment with the antisense compound for at least 12 months.
- subjects receive blinded antisense compound for 6 months followed by at least 6 months of open labeled treatment.
- provided herein subjects can be administered with the antisense compound for at least 12 months.
- the dosage of the active ingredient depends upon the mode of administration as upon the subject, and their age, weight, condition, and the subject
- compositions are administered according to a dosing regimen.
- the dosing regimen comprises an induction phase and a maintenance phase.
- the induction phase includes one, two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, or more than twenty doses.
- the induction phase lasts from one day to six months. In certain embodiments an induction phase lasts from one week to five months as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts from one week to five months as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase.
- an induction phase lasts from two weeks to five months as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts from three weeks to four months as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts from five weeks to three months as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts five weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts six weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase.
- an induction phase lasts seven weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts eight weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts nine weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts ten weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts eleven weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase.
- an induction phase lasts twelve weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts thirteen weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts fourteen weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts fifteen weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts sixteen weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts seventeen weeks as measured from
- an induction phase lasts eighteen weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts nineteen weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts twenty weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts twenty-one weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase.
- an induction phase lasts twenty-two weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts twenty-three weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts twenty-four weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts twenty- five weeks as measured from
- the administration of the first dose of the induction phase to administration of the first dose of the maintenance phase is lower than the doses administered during the maintenance phase.
- the dose administered during the induction phase is lower than the dose administered during the maintenance phase to avoid undesired side effects.
- the undesired side effect is liver toxicity.
- the undesired side effect is increased ALT.
- the lower induction dose provides time for lipid metabolism in the liver to compensate for the decreased production of ApoB.
- mild increases in ALT reflect rapid lipid-lowering activity.
- the doses administered during the induction phase are all the same amount as one another. In certain embodiments, the doses administered during the induction phase are not all the same amount. In certain such embodiments, the doses increase over time. In certain embodiments, the doses decrease over time.
- an induction dose is administered by parenteral
- parenteral administration is subcutaneous administration. In certain such embodiments, the parenteral administration is intravenous infusion.
- the doses during the induction phase are selected from 25 mg, 30 mg, 35 mg, 40 mg, 45 mg, 50 mg, 55 mg, 60 mg, 65 mg, 70 mg, 75 mg, 80 mg, 85 mg, 90 mg, 95 mg, 100 mg, 105 mg, 110 mg, 115 mg, 120 mg, 125 mg, 130 mg, 135 mg, 140 mg,
- the doses during the induction phase are selected from 25 mg, 50 mg, 75 mg, 100 mg, 150 mg, 200 mg, 250 mg, 300 mg, 350 mg, 400 mg, 500 mg, 600 mg, 700 mg, and 800mg. In certain such embodiments, the doses during the induction phase are selected from 100 mg, 125 mg, 150 mg, 175 mg, 200 mg, 225 mg, 250 mg, 275 mg, 300 mg, 325 mg, 350 mg, 375 mg, and 400 mg.
- the doses during the induction phase are selected from 100 mg, 125 mg, 150 mg, 175 mg, 200 mg, 225 mg, and 250 mg. In certain embodiments, the dose administered during the induction phase is 100 mg. In certain embodiments, the dose administered during the induction phase is 125 mg. In certain
- the dose administered during the induction phase is 150 mg. In certain embodiments the dose administered during the induction phase is 150 mg. In certain
- the dose administered during the induction phase is 175 mg.
- the dose administered during the induction phase is 200 mg. In certain embodiments the dose administered during the induction phase is 200 mg. In certain
- the dose administered during the induction phase is 225 mg.
- the dose administered during the induction phase is 250 mg. In certain embodiments the dose administered during the induction phase is 250 mg. In certain
- the dose administered during the induction phase is 300 mg. In certain embodiments the dose administered during the induction phase is 300 mg. In certain
- the dose administered during the induction phase is 325 mg.
- the dose administered during the induction phase is 350 mg. In certain embodiments the dose administered during the induction phase is 350 mg. In certain
- the dose administered during the induction phase is 375 mg.
- the dose administered during the induction phase is 400 mg.
- an induction dose may be administered in two or more subcutaneous injections.
- two or more subcutaneous injections may be used to achieve the desired induction dose.
- two or more subcutaneous injections may be used to administer the desired induction dose and minimize or eliminate an injection site reaction in a subject.
- dose, dose frequency, and duration of the induction phase may be selected to achieve a desired effect.
- those variables are adjusted to result in a desired concentration of pharmaceutical agent in a subject.
- dose and dose frequency are adjusted to provide plasma concentration of a pharmaceutical agent at an amount sufficient to achieve a desired effect.
- the plasma concentration is maintained above the minimal effective concentration (MEC).
- pharmaceutical compositions are administered with a dosage regimen designed to maintain a concentration above the MEC for 10-90% of the time, between 30-90% of the time, or between 50-90% of the time.
- doses, dose frequency, and duration of the induction phase may be selected to achieve a desired plasma trough concentration of a pharmaceutical composition.
- the pharmaceutical composition is an oligonucleotide.
- the desired plasma trough concentration is from 5- 100 ng/mL. In certain such embodiments, the desired plasma trough concentration is from 5-50 ng/mL. In certain such embodiments, the desired plasma trough concentration is from 10-40 ng/mL. In certain such embodiments, the desired plasma trough concentration is from 15-35 ng/mL. In certain such embodiments, the desired plasma trough concentration is from 20-30 ng/mL.
- dose, dose frequency, and duration of the induction phase may be selected to achieve a desired effect within five to thirteen weeks.
- the dose is the same and the dose frequency is varied to achieve the desired effect within five to thirteen weeks.
- the dose increases over time and the dose frequency remains constant.
- doses and dose frequency are selected to achieve a desired effect within six to 13 weeks.
- doses and frequency are selected to achieve a desired effect within six weeks.
- doses and frequency are selected to achieve a desired effect within seven weeks.
- doses and frequency are selected to achieve a desired effect within eight weeks.
- doses and frequency are selected to achieve a desired effect within nine weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within ten weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within eleven weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within twelve weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within thirteen weeks. In certain such embodiments, one or more doses of the induction phase is greater than one or more doses of the maintenance phase. In certain such embodiments, each of the induction doses is greater than each of the maintenance doses.
- doses, dose frequency, and duration of the induction phase may be selected to achieve a desired effect within 13 to 25 weeks.
- the dose is the same and the dose frequency is varied to achieve the desired effect within 13 to 25 weeks.
- the dose increases over time and the dose frequency remains constant.
- doses and frequency are selected to achieve a desired effect within thirteen weeks.
- doses and frequency are selected to achieve a desired effect within fourteen weeks.
- doses and frequency are selected to achieve a desired effect within fifteen weeks.
- doses and frequency are selected to achieve a desired effect within sixteen weeks.
- doses and frequency are selected to achieve a desired effect within seventeen weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within eighteen weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within nineteen weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within twenty weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within twenty-one weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within twenty-two weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within twenty-three weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within twenty-four weeks.
- doses and frequency are selected to achieve a desired effect within twenty- five weeks.
- one or more doses of the induction phase is less than one or more doses of the maintenance phase.
- each dose of the induction phase is less than each dose of the maintenance phase.
- an induction phase with a high dose and/or high dose frequency may be desirable.
- Such embodiments may include administration to subjects with very high cholesterol concentrations.
- an induction phase with a low dose and/or low dose frequency and/or long duration may be desirable.
- a long induction phase, with relatively low doses may result in better tolerance of the pharmaceutical agent.
- Certain such embodiments result in physiological changes that result in reduced overall side effects.
- such a dose regimen results in reduced liver toxicity when compared to higher initial doses and/or frequency. Such embodiments may include gradual increases of dose over time.
- the dosage regimen is selected to achieve a desired local concentration of a
- doses, dose frequency, and duration of the induction phase may be selected to achieve an acceptable safety profile.
- such variables may be selected to mitigate toxicity of the pharmaceutical composition.
- such variables are selected to mitigate liver toxicity.
- such variables are selected to mitigate renal toxicity.
- doses increase over time.
- one or more doses of the induction phase is lower than one or more doses of the maintenance phase.
- a safety profile is not acceptable when ALT is 5-10 times the upper limit of normal.
- a safety profile is not acceptable when ALT is 5-10 times the upper limit of normal, and bilirubin is elevated two or more times the upper limit of normal.
- an acceptable safety profile comprises ALT elevations that are above three times the upper limit of normal, but do not exceed five times the upper limit of normal.
- acceptable safety profile comprises ALT elevations that are above three times the upper limit of normal, but do not exceed five times the upper limit of normal, and bilirubin elevations that do not exceed two times the upper limit of normal.
- the dose and/or dose frequency is adjusted to mitigate the ALT elevation.
- the dose and/or dose frequency is adjusted to mitigate the ALT elevation and bilirubin elevation. In certain such embodiments, the dose and/or dose frequency is adjusted to mitigate the bilirubin elevation alone.
- the maintenance phase includes one, two three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, or more than twenty doses.
- the maintenance phase lasts from one day to the lifetime of the subject. In certain embodiments the maintenance phase lasts from one week to twenty years as measured from administration of the last dose of the induction phase to administration of the last dose of the maintenance phase. In certain embodiments the maintenance phase lasts from two weeks to fifteen years as measured from administration of the last dose of the induction phase to administration of the last dose of the maintenance phase. In certain embodiments the maintenance phase lasts three weeks to ten years as measured from administration of the last dose of the induction phase to administration of the last dose of the maintenance phase. In certain embodiments the maintenance phase lasts from four weeks to ten years as measured from administration of the last dose of the induction phase to administration of the last dose of the maintenance phase. In certain embodiments the maintenance phase lasts as long as the dose continues to be needed, effective, and tolerated.
- the doses administered during the maintenance phase are all the same as one another. In certain embodiments, the doses administered during the maintenance phase are not all the same. In certain such embodiments, the doses increase over time. In certain embodiments, the doses decrease over time.
- a maintenance dose is administered by parenteral administration.
- the parenteral administration is subcutaneous administration.
- the parenteral administration is intravenous infusion.
- the doses during the maintenance phase are selected from 25 mg, 30 mg, 35 mg, 40 mg, 45 mg, 50 mg, 55 mg, 60 mg, 65 mg, 70 mg, 75 mg, 80 mg, 85 mg, 90 mg, 95 mg, 100 mg, 105 mg, 110 mg, 115 mg, 120 mg, 125 mg, 130 mg, 135 mg, 140 mg,
- the doses during the maintenance phase are selected from 25 mg, 50 mg, 75 mg, 100 mg, 150 mg, 200 mg, 250 mg, 300 mg, 350 mg, 400 mg, 500 mg, 600 mg, 700 mg, and 800mg. In certain such embodiments, the doses during the maintenance phase are selected from 100 mg, 125 mg, 150 mg, 175 mg, 200 mg, 225 mg, 250 mg, 275 mg, 300 mg, 325 mg, 350 mg, 375 mg, and 400 mg.
- the doses during the maintenance phase are selected from 100 mg, 125 mg, 150 mg, 175 mg, 200 mg, 225 mg, and 250 mg. In certain embodiments, the dose administered during the maintenance phase is 100 mg. In certain embodiments, the dose administered during the maintenance phase is 125 mg. In certain embodiments the dose administered during the maintenance phase is 150 mg. In certain embodiments the dose administered during the maintenance phase is 175 mg. In certain embodiments the dose administered during the maintenance phase is 200 mg. In certain embodiments the dose administered during the maintenance phase is 225 mg. In certain embodiments the dose administered during the maintenance phase is 250 mg. In certain embodiments the dose administered during the maintenance phase is 275 mg. In certain embodiments the dose administered during the maintenance phase is 300 mg.
- a maintenance dose may be administered in two or more subcutaneous injections.
- two or more subcutaneous injections may be used to achieve the desired maintenance dose.
- two or more subcutaneous injections may be used to administer the desired maintenance dose and minimize or eliminate an injection site reaction in a subject.
- doses, dose frequency, and duration of the maintenance phase may be selected to achieve a desired effect.
- those variables are adjusted to result in a desired concentration of pharmaceutical agent in a subject.
- dose and dose frequency are adjusted to provide plasma concentration of a pharmaceutical agent at an amount sufficient to achieve a desired effect.
- the plasma concentration is maintained above the minimal effective concentration (MEC).
- pharmaceutical compositions are administered with a dosage regimen designed to maintain a concentration above the MEC for 10-90% of the time, between 30-90% of the time, or between 50-90% of the time.
- doses, dose frequency, and duration of the maintenance phase may be selected to achieve a desired plasma trough concentration of a pharmaceutical composition.
- the pharmaceutical composition is an
- the desired plasma trough concentration is from 5- 100 ng/mL. In certain such embodiments, the desired plasma trough concentration is from 5-50 ng/mL. In certain such embodiments, the desired plasma trough concentration is from 10-40 ng/mL. In certain such embodiments, the desired plasma trough concentration is from 15-35 ng/mL. In certain such embodiments, the desired plasma trough concentration is from 20-30 ng/mL.
- doses, dose frequency, and duration of the maintenance phase may be selected to achieve a desired safety profile.
- such variables may be selected to mitigate toxicity of the pharmaceutical composition.
- such variables are selected to mitigate liver toxicity.
- such variables are selected to mitigate renal toxicity.
- doses increase over time.
- doses, dose frequency, and duration of the maintenance phase may be adjusted from time to time to achieve a desired effect.
- subjects are monitored for effects (therapeutic and/or toxic effects) and doses, dose frequency, and/or duration of the maintenance phase may be adjusted based on the results of such monitoring.
- dosage regimens listed in Tables A-F below.
- Tables A-F are dosage regimens listed in Tables A-F, below.
- Tables A-F are dosage regimens listed in Tables A-F, below.
- the variables in the table can be selected and combined independently.
- the table is included solely to illustrate how the variables may be combined and is does not limit the invention.
- the present invention is not limited to the variables listed on the table.
- the antisense compound is administered at 200 mg/week. In some embodiments, the 200 mg/week is adminitered one time per week. In some embodiments, the subject is administered a single 200 mg/dose per week. In other embodiments, the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week. In some embodiments, the antisense compound is administered for at least 12 months. In one embodiment, the antisense compound is administered subcutaneously (s.c).
- the antisense compound is administered at 200 mg/week during the induction phase.
- the 200 mg/week is adminitered one time per week.
- the subject is administered a single 200 mg/dose per week.
- the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week.
- the antisense compound is administered subcutaneously (s.c).
- the antisense compound is administered at 200 mg/week during the maintenance phase.
- the 200 mg/week is adminitered one time per week.
- the subject is administered a single 200 mg/dose per week.
- the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week.
- the antisense compound is administered subcutaneously (s.c).
- the antisense compound is administered at 200 mg/week during the induction phase and during the maintenance phase.
- the 200 mg/week is adminitered one time per week.
- the subject is administered a single 200 mg/dose per week.
- the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week.
- the antisense compound is administered subcutaneously (s.c).
- a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein an induction dose of 200-400 mg is
- the induction dose is administered once per week for at least 8 weeks, followed by a maintenance phase, wherein a maintenance dose of 100-300 mg is administered at intervals ranging from one per week to once per three months, for as long as needed to sustain the desired effect.
- the induction dose is administered once per week for 8-20 weeks. In certain embodiments the induction dose is administered once per week for 10-15 weeks. In certain embodiments, the induction dose is administered once per week for at least 12 weeks. In certain embodiments the induction dose is administered once per week for at least 14 weeks. In certain embodiments the induction dose is administered once per week for at least 16 weeks. In certain such embodiments, the induction dose is 200 mg. In certain such embodiments, the induction dose is 300 mg. In certain such embodiments, the induction dose is 400 mg.
- the maintenance dose ranges from 200-300 mg. In certain such embodiments, the maintenance dose is 150 mg. In certain such embodiments, the maintenance dose is 200 mg. In certain such embodiments, the maintenance dose is 250 mg. In certain such embodiments, the maintenance dose is 300 mg. In certain such embodiments, the maintenance dose is administered once per week. In certain such embodiments, the maintenance dose is administered once per month. In certain such embodiments, the maintenance dose is administered once per three months. In certain such embodiments, the maintenance dose is administered for at least 6 months. In certain such embodiments, the maintenance dose is administered for at least one year. In certain such embodiments, the maintenance dose is administered for up to five years. In certain such embodiments the maintenance dose is administered for up to ten years.
- the maintenance dose is administered for as long as is necessary to sustain the desired effect.
- the frequency of administration of the maintenance dose is adjusted to achieved desired efficacy and/or desired safety profile.
- the frequency of the maintenance dose is adjusted to achieve a desired plasma trough concentration of oligonucleotide.
- the plasma trough concentration of the administered antisense oligonucleotide is 15-40 ng/mL.
- the plasma trough concentration of the administered antisense oligonucleotide is 20-30 ng/mL.
- the desired effect is selected from reduced ApoB, reduced LDL-C, reduced VLDL-C, reduced IDL-C, reduced non-HDL-C, reduced serum triglycerides, reduced liver triglycerides, reduced Lp(a), reduced Ox-LDL-C, and reduced small dense LDL particles.
- the subject has polygenic
- the subject has familial
- the subject has homozygous familial hypercholesterolemia. In certain such embodiments, the subject has heterozygous familial hypercholesterolemia.
- the pharmaceutical composition is coadministered with a statin. In certain such embodiments, the subject is intolerant to statins. In certain such embodiments, the subject is not meeting LDL-C target on current therapy. Non- limiting examples of certain dosing regimens are illustrated in Table A.
- a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein a 200 mg dose is administered once per week for 13 weeks, followed by a maintenance phase, wherein a dose ranging from 80-200 mg is administered at intervals ranging from once per week to once per three months, for as long as needed to sustain the desired effect.
- the maintenance dose ranges from 100-150 mg. In certain such embodiments, the maintenance dose is 100 mg. In certain such embodiments, the maintenance dose is 125 mg. In certain such embodiments, the maintenance dose is 140 mg. In certain such embodiments, the maintenance dose is 150 mg. In certain such embodiments, the maintenance dose is 175 mg. In certain such embodiments, the maintenance dose is 180 mg. In certain such embodiments, the maintenance dose is 200 mg. In certain such embodiments, the maintenance dose is administered once per week. In certain such such
- the maintenance dose is administered once per month. In certain such embodiments,
- the maintenance dose is administered once per three months. In certain such embodiments, the maintenance dose is administered for at least 6 months. In certain such embodiments, the maintenance dose is administered for at least one year. In certain such embodiments, the maintenance dose is administered for up to five years. In certain such embodiments the maintenance dose is administered for up to ten years. In certain such embodiments the maintenance dose is administered for as long as is necessary to sustain the desired effect. In certain such embodiments, the frequency of administration of the maintenance dose is adjusted to achieved desired efficacy and/or desired safety profile. In certain such embodiments, the frequency of the maintenance dose is adjusted to achieve a desired plasma trough concentration of oligonucleotide.
- the plasma trough concentration of the administered antisense oligonucleotide is 15-40 ng/mL. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 20-30 ng/mL. In certain such embodiments, the desired effect is selected from reduced ApoB, reduced LDL-C, reduced VLDL-C, reduced IDL-C, reduced non-HDL-C, reduced serum triglycerides, reduced liver triglycerides, reduced Lp(a), reduced Ox-LDL-C, and reduced small dense LDL particles. In certain such embodiments, the subject has polygenic hypercholesterolemia. In certain such embodiments, the subject has familial
- the pharmaceutical composition is coadministered with a statin.
- the subject is intolerant to statins.
- the subject is not meeting LDL-C target on current therapy.
- Non- limiting examples of certain dosing regimens are illustrated in Table B.
- a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein a 300 mg dose is administered once per week for 13 weeks, followed by a maintenance phase, wherein a dose ranging from 100-250 mg is administered at intervals ranging from once per week to once per three months, for as long as needed to sustain the desired effect.
- the maintenance dose is 100 mg.
- the maintenance dose is 125 mg.
- the maintenance dose is 150 mg.
- the maintenance dose is 175 mg.
- the maintenance dose is 200 mg.
- the maintenance dose is 250 mg.
- the maintenance dose is administered once per week.
- the maintenance dose is administered once per month. In certain such embodiments, the maintenance dose is administered once per three months. In certain such embodiments, the maintenance dose is administered for at least 6 months. In certain such embodiments, the maintenance dose is administered for at least one year. In certain such embodiments, the maintenance dose is administered for up to five years. In certain such embodiments the maintenance dose is administered for up to ten years. In certain such embodiments the maintenance dose is administered for as long as is necessary to sustain the desired effect. In certain such embodiments, the frequency of administration of the maintenance dose is adjusted to achieved desired efficacy and/or desired safety profile. In certain such embodiments, the frequency of the maintenance dose is adjusted to achieve a desired plasma trough concentration of oligonucleotide.
- the plasma trough concentration of the administered antisense oligonucleotide is 15-40 ng/mL. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 20-30 ng/mL. In certain such embodiments, plasma trough concentration of the administered antisense oligonucleotide is 15-40 ng/mL. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 20-30 ng/mL.
- the desired effect is selected from reduced ApoB, reduced LDL-C, reduced VLDL-C, reduced IDL-C, reduced non-HDL-C, reduced serum triglycerides, reduced liver triglycerides, reduced Lp(a), reduced Ox-LDL-C, and reduced small dense LDL particles.
- the subject has polygenic hypercholesterolemia.
- the subject has familial hypercholesterolemia.
- the pharmaceutical composition is co-administered with a statin.
- the subject is intolerant to statins.
- the subject is not meeting LDL-C target on current therapy.
- Non-limiting examples of certain dosing regimens are illustrated in Table C.
- a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein a 100 mg dose is administered once per week for 13 weeks, followed by a maintenance phase, wherein a dose ranging from 100-300 mg is administered at intervals ranging from once per week to once per three months, for as long as needed to sustain the desired effect.
- the dose ranges from 150-250 mg.
- the maintenance dose is 100 mg.
- the maintenance dose is 125 mg.
- the maintenance dose is 150 mg.
- the maintenance dose is 175 mg.
- the maintenance dose is 200 mg. In certain such embodiments, the maintenance dose is 225 mg.
- the maintenance dose is 250 mg. In certain such embodiments, the maintenance dose is 275 mg. In certain such embodiments, the maintenance dose is 300 mg. In certain such embodiments, the maintenance dose is administered once per week. In certain such embodiments, the maintenance dose is administered once per month. In certain such
- the maintenance dose is administered once per three months. In certain such embodiments, the maintenance dose is administered for at least 6 months. In certain such embodiments, the maintenance dose is administered for at least one year. In certain such embodiments, the maintenance dose is administered for up to five years. In certain such embodiments the maintenance dose is administered for up to ten years. In certain such embodiments the maintenance dose is administered for as long as is necessary to sustain the desired effect. In certain such embodiments, the frequency of administration of the maintenance dose is adjusted to achieved desired efficacy and/or desired safety profile. In certain such embodiments, the frequency of the maintenance dose is adjusted to achieve a desired plasma trough concentration of oligonucleotide.
- the plasma trough concentration of the administered antisense oligonucleotide is 15-40 ng/mL. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 20-30 ng/mL. In certain such embodiments, the desired effect is selected from reduced ApoB, reduced LDL-C, reduced VLDL-C, reduced IDL-C, reduced non-HDL-C, reduced serum triglycerides, reduced liver triglycerides, reduced Lp(a), reduced Ox-LDL-C, and reduced small dense LDL particles. In certain such embodiments, the subject has polygenic
- the subject has familial
- the pharmaceutical composition is coadministered with a statin.
- the subject is intolerant to statins.
- the subject is not meeting LDL-C target on current therapy.
- Non- limiting examples of certain dosing regimens are illustrated in Table D. Table D
- a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein a dose ranging from 100-200 mg is administered once per week for 13 weeks, followed by a maintenance phase, wherein a dose ranging from 100-300 mg is administered at intervals ranging from once per week to once per three months, for as long as needed to sustain the desired effect.
- the induction dose is 100 mg.
- the induction dose is 125 mg.
- the induction dose is 150 mg.
- the induction dose is 175 mg.
- the induction dose is 200 mg. In certain such
- an during an induction phase four doses of 100 mg are followed by five doses of 150 mg which are followed by four doses of 200 mg.
- four doses of 100 mg are followed by four doses of 150 mg which are followed by five doses of 200 mg.
- five doses of 100 mg are followed by four doses of 150 mg which are followed by four doses of 200 mg.
- the maintenance dose is higher than the induction dose.
- the maintenance dose is 100 mg.
- the maintenance dose is 125 mg.
- the maintenance dose is 150 mg.
- the maintenance dose is 175 mg.
- the maintenance dose is 200 mg.
- the maintenance dose is 225 mg. In certain such embodiments, the maintenance dose is 250 mg. In certain such embodiments, the maintenance dose is 275 mg. In certain such embodiments, the maintenance dose is 300 mg. In certain such embodiments, the maintenance dose is administered once per week. In certain such embodiments, the maintenance dose is administered once per month. In certain such embodiments, the maintenance dose is administered once per three months. In certain such embodiments, the maintenance dose is administered for at least 6 months. In certain such embodiments, the maintenance dose is administered for at least one year. In certain such embodiments, the maintenance dose is administered for up to five years. In certain such embodiments the maintenance dose is administered for up to ten years. In certain such embodiments the maintenance dose is administered for as long as is necessary to sustain the desired effect. In certain such such
- the amount or frequency of the induction dose is adjusted to achieve desired efficacy and/or desired safety profile. In certain such embodiments, the amount or frequency of the induction dose is adjusted to achieve a desired plasma trough concentration of antisense oligonucleotide. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 15-40 ng/mL. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 20-30 ng/mL. In certain such embodiments, the amount or frequency of administration of the maintenance dose is adjusted to achieved desired efficacy and/or desired safety profile. In certain such embodiments, the frequency of the maintenance dose is adjusted to achieve a desired plasma trough
- the plasma trough concentration of the administered antisense oligonucleotide is 15-40 ng/mL. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 20-30 ng/mL.
- the desired effect is selected from reduced ApoB, reduced LDL-C, reduced VLDL-C, reduced IDL-C, reduced non-HDL-C, reduced serum triglycerides, reduced liver triglycerides, reduced Lp(a), reduced Ox-LDL-C, and reduced small dense LDL particles. In certain such embodiments, the subject has polygenic hypercholesterolemia.
- the subject has familial hypercholesterolemia.
- the pharmaceutical composition is co-administered with a statin.
- the subject is intolerant to statins.
- the subject is not meeting LDL-C target on current therapy.
- Non-limiting examples of certain dosing regimens are illustrated in Table E.
- a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein a dose of 100 mg is administered once per week for 14-20 weeks, followed by a maintenance phase, wherein a dose ranging from 100-300 mg is administered at a frequency ranging from once per week to once per three months, for as long as needed to sustain the desired effect.
- the induction phase is 16-20 weeks.
- the duration of the induction phase is 14 weeks.
- the duration of the induction phase is 15 weeks.
- the duration of the induction phase is 16 weeks. In certain such embodiments, the duration of the induction phase is 17 weeks. In certain such embodiments, the duration of the induction phase is 18 weeks. In certain such embodiments, the duration of the induction phase is 19 weeks. In certain such embodiments, the duration of the induction phase is 20 weeks. In certain such embodiments, the maintenance dose is higher than the induction dose. In certain such embodiments, the maintenance dose ranges from 100-300 mg. In certain such embodiments, the maintenance dose ranges from 100-300 mg. In certain such embodiments,
- the maintenance dose ranges from 100-200 mg. In certain such embodiments, the maintenance dose is 100 mg. In certain such embodiments, the maintenance dose is 125 mg. In certain such embodiments, the maintenance dose is 150 mg. In certain such embodiments, the maintenance dose is 175 mg. In certain such embodiments, the maintenance dose is 200 mg. In certain such embodiments, the maintenance dose is 225 mg. In certain such embodiments, the maintenance dose is 250 mg. In certain such embodiments, the maintenance dose is 275 mg. In certain such embodiments, the maintenance dose is 300 mg. In certain such embodiments, the maintenance dose is administered once per week. In certain such embodiments, the maintenance dose is administered once per month. In certain such embodiments, the maintenance dose is administered once per three months.
- the maintenance dose is administered for at least 6 months. In certain such embodiments, the maintenance dose is administered for at least one year. In certain such embodiments, the maintenance dose is administered for up to five years. In certain such embodiments the maintenance dose is administered for up to ten years. In certain such embodiments the maintenance dose is administered for as long as is necessary to sustain the desired effect. In certain such embodiments
- the frequency of administration of the maintenance dose is adjusted to achieved desired efficacy and/or desired safety profile.
- the desired effect is selected from reduced ApoB, reduced LDL-C, reduced VLDL-C, reduced IDL-C, reduced non- HDL-C, reduced serum triglycerides, reduced liver triglycerides, reduced Lp(a), reduced Ox- LDL-C, and reduced small dense LDL particles.
- the subject has polygenic hypercholesterolemia.
- the subject has familial hypercholesterolemia.
- the pharmaceutical composition is coadministered with a statin.
- the subject is intolerant to statins.
- the subject is not meeting LDL-C target on current therapy.
- Non- limiting examples of certain dosing regimens are illustrated in Table F.
- a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein a dose ranging from 100-300 mg is administered once per week for 13 weeks, followed by a maintenance phase, wherein a dose ranging from 80-200 mg is administered once per week for as long as needed, effective, and/or tolerated.
- the pharmaceutical composition is administered subcutaneously during the induction phase and/or the maintenance phase.
- the subject is afflicted with familial hypercholesterolemia (either heterozygous or homozygous), non-familial hypercholesterolemia, or polygenic hypercholesterolemia.
- the maintenance phase lasts from one day to the end of the subject's lifetime or any fraction thereof as discussed above.
- the induction dose is 100 mg, and the maintenance dose is 80 mg, 100 mg, 140 mg, 180 mg, or 200 mg. In certain of such embodiments, the induction dose is 200 mg, and the maintenance dose is 80 mg, 100 mg, 140 mg, 180 mg, or 200 mg. In certain of such embodiments, the induction dose is 300 mg, and the maintenance dose is 80 mg, 100 mg, 140 mg, 180 mg, or 200 mg.
- the administration at the end of the induction phase achieves a reduction in plasma concentration of ApoB of from about -28% to -65%.
- the administration after 13 weeks of the maintenance phase achieves a reduction in plasma concentration of ApoB of from about -32% to -48%, from about -35% to about -52%o, from about -40%> to about -60%>, from about -43% to about -65%, or from about - 45% to about -67%.
- the administration at the end of the induction phase achieves a reduction in plasma concentration of LDL-Col from about -26% to - 60%.
- the administration after 13 weeks of the maintenance phase achieves a reduction in plasma concentration of LDL-Col from about -29% to -44%, from about -32%o to about -48%, from about -37% to about -55%, from about -40% to about -61%, or from about -42% to about -63%.
- the administration at the end of the induction phase achieves a plasma trough concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 11 to 38 ng/mL.
- the administration after 13 weeks of the maintenance phase achieves a plasma trough concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 7 to 27 ng/mL, from about 8 to 31 ng/mL, from about 11 to 38 ng/mL, from about 13 to 46 ng/mL, or from about 14 to 50 ng/mL.
- the administration at the end of the induction phase achieves a liver concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 55 to 190 ⁇ g/G. In certain of such embodiments, the administration after 13 weeks of the maintenance phase achieves a liver concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 38 to 133 ⁇ , from about 44 to 152 ⁇ , from about 55 to 190 ⁇ , from about 66 to 228 ⁇ , or from about 7 to 247 ⁇ . [00567] In certain of such embodiments, the administration at the end of the induction phase achieves a reduction in plasma concentration of ApoB of from about -34% to -77%.
- the administration after 13 weeks of the maintenance phase achieves a reduction in plasma concentration of ApoB of from about -38% to -58%, from about -43% to about -65%, from about -47% to about -70%, or from about -49% to about -74%.
- the administration at the end of the induction phase achieves a reduction in plasma concentration of LDL-Col from about -31% to -73%.
- the administration after 13 weeks of the maintenance phase achieves a reduction in plasma concentration of LDL-Col from about -35% to -54%, from about -40% to about -61%, from about -44% to about -66%, or from about -46% to about -70%.
- the administration at the end of the induction phase achieves a plasma trough concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 16 to 57 ng/mL.
- the administration after 13 weeks of the maintenance phase achieves a plasma trough concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 10 to 37 ng/mL, from about 13 to 46 ng/mL, from about 16 to 55 ng/mL, or from about 18 to 65 ng/mL.
- the administration at the end of the induction phase achieves a liver concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 82 to 285 ⁇ g/G. In certain of such embodiments, the administration after 13 weeks of the maintenance phase achieves a liver concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 52 to 181 ⁇ g/G, from about 66 to 228 ⁇ , from about 80 to 276 ⁇ , or from about 94 to 323 ⁇ g/G.
- a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein a dose ranging from 100-300 mg is administered once per week for 13 weeks, followed by a maintenance phase, wherein a dose ranging from 100-200 mg is administered once per week for as long as needed, effective, and/or tolerated, wherein the efficacy and/or the tolerability of the antisense oligonucleotide is monitored during the induction phase, the maintenance phase, or both, or any portion thereof.
- the pharmaceutical composition is administered subcutaneously during the induction phase and/or the maintenance phase.
- the subject is afflicted with familial hypercholesterolemia (either heterozygous or homozygous), non- familial hypercholesterolemia, or polygenic hypercholesterolemia.
- the maintenance phase lasts from one day to the end of the subject's lifetime or any fraction thereof as discussed above.
- the rate of reduction in the plasma concentration of ApoB is monitored during the induction and/or maintenance phases. In certain of such embodiments, the plasma concentration of ApoB is monitored during the induction and/or maintenance phases. In certain embodiments, if the rate of reduction in the plasma concentration of ApoB exceeds 30 mg/dL*day, the dose of pharmaceutical composition is altered, e.g., reduced. In certain embodiments, if the rate of reduction in the plasma concentration of ApoB exceeds 30 mg/dL* day, the frequency of administration of pharmaceutical composition is altered, e.g., reduced. In certain embodiments, if the plasma concentration of ApoB falls below about 50 mg/dL, the dose of pharmaceutical composition is altered, e.g., reduced.
- the frequency of administration of pharmaceutical composition is altered, e.g., reduced. In certain embodiments, if the plasma concentration of ApoB falls below about 60 mg/dL, the dose of pharmaceutical composition is altered, e.g. , reduced. In certain embodiments, if the plasma concentration of ApoB falls below about 60 mg/dL, the frequency of administration of pharmaceutical composition is altered, e.g. , reduced.
- the dose of pharmaceutical composition is altered, e.g., reduced.
- the rate of reduction in the plasma concentration of ApoB exceeds 30 mg/dL*day and the plasma concentration of ApoB falls below about 50 mg/dL
- the frequency of administration of pharmaceutical composition is altered, e.g., reduced.
- the rate of reduction in the plasma concentration of ApoB exceeds 30 mg/dL* day and the plasma concentration of ApoB falls below about 60 mg/dL
- the dose of pharmaceutical composition is altered, e.g., reduced.
- the rate of reduction in the plasma concentration of ApoB exceeds 30 mg/dL* day and the plasma concentration of ApoB falls below about 60 mg/dL
- a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein a dose ranging from 100 to 200 mg is administered once per week for 13 weeks, and a maintenance phase, wherein a dose ranging from 200 to 300 mg is administered once per week for as long as needed, effective, and/or tolerated, wherein the tolerability and/or efficacy of the pharmaceutical composition is assessed during or at the end of the induction phase, or any portion thereof.
- the dose of the maintenance phase is increased relative to the dose of the maintenance phase if the dose of the induction phase is well-tolerated and treatment goals are not met.
- the pharmaceutical composition is administered subcutaneously during the induction phase and/or the maintenance phase.
- the subject is afflicted with familial hypercholesterolemia (either heterozygous or homozygous), non-familial hypercholesterolemia, or polygenic hypercholesterolemia.
- the maintenance phase lasts from one day to the end of the subject's lifetime or any fraction thereof as discussed above.
- the induction dose is 100 mg
- the maintenance dose is 200 mg.
- the induction dose is 200 mg, and the maintenance dose is 300 mg.
- the treatment goals are assessed by monitoring plasma concentration of ApoB, LDL-C, VLDL-C, non-HDL-C, HDL-C, ApoAl, total cholesterol, triglycerides, and Lp(a).
- tolerability is assessed by monitoring ALT activity, AST activity, and plasma bilirubin concentrations.
- the administration at the end of the induction phase achieves a reduction in plasma concentration of ApoB of from about -17% to -40%. In certain of such embodiments, the administration after 26 weeks of the maintenance phase achieves a reduction in plasma concentration of ApoB of from about -42% to -63%. In certain of such embodiments, the administration at the end of the induction phase achieves a reduction in plasma concentration of LDL-Col from about -14% to -35%. In certain of such embodiments, the administration after 13 weeks of the maintenance phase achieves a reduction in plasma concentration of LDL-Col from about -39% to -60%.
- the administration at the end of the induction phase achieves a plasma trough concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 5 to 19 ng/mL. In certain of such embodiments, the administration after 26 weeks of the maintenance phase achieves a plasma trough concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 12 to 44 ng/mL. In certain of such embodiments, the administration at the end of the induction phase achieves a liver concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 27 to 95 ⁇ g/G. In certain of such embodiments, the administration after 26 weeks of the maintenance phase achieves a liver concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 63 to 220 ⁇ g/G.
- a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein a dose ranging from 100 to 200 mg is administered once per week for 13 weeks, and a maintenance phase, wherein a dose ranging from 200 to 300 mg is administered once every one or two weeks for as long as needed, effective, and/or tolerated, wherein the tolerability and/or efficacy of the pharmaceutical composition is assessed during or at the end of the induction phase, or any portion thereof.
- the frequency of administration of dose during the maintenance phase is reduced relative if the dose of the induction phase is not well-tolerated and/or treatment goals are met.
- the pharmaceutical composition is administered subcutaneously during the induction phase and/or the maintenance phase.
- the subject is afflicted with familial hypercholesterolemia (either heterozygous or homozygous), non-familial hypercholesterolemia, or polygenic hypercholesterolemia.
- the maintenance phase lasts from one day to the end of the subject's lifetime or any fraction thereof as discussed above.
- the induction dose is 100 mg, and the maintenance dose is 200 mg. In certain of such embodiments, the induction dose is 200 mg, and the maintenance dose is 300 mg.
- the treatment goals are assessed by monitoring plasma concentration of ApoB, LDL-C, VLDL-C, non-HDL-C, HDL-C, ApoAl, total cholesterol, triglycerides, and Lp(a).
- tolerability is assessed by monitoring ALT levels, AST levels, plasma bilirubin concentrations or total bilirubin.
- pharmaceutical agents is designed to treat the same disease or condition as the one or more antisense compound. In some embodiments, such one or more other pharmaceutical agents is designed to treat a different disease or condition as the one or more antisense compound. In other embodiments, such one or more other pharmaceutical agents is designed to treat an undesired effect of the one or more antisense compound. In certain embodiments, the one or more antisense compound is co-administered with another pharmaceutical agent to treat an undesired effect of that other pharmaceutical agent. In some embodiments, the one or more antisense compound, and one or more other pharmaceutical agents are administered at the same time. In other embodiments, the one or more antisense compound, and one or more other pharmaceutical agents are administered at different times.
- one or more pharmaceutical compositions described herein, and one or more other pharmaceutical agents can be prepared together in a single formulation. In some embodiments, one or more pharmaceutical compositions described herein, and one or more other pharmaceutical agents are prepared separately.
- a composition can comprise a pharmaceutical agent for separate, sequential, or simultaneous administration with an antisense compound.
- pharmaceutical agents that can be co-administered with a pharmaceutical composition described herein include lipid- lowering agents or LXR agonists.
- pharmaceutical agents that can be co-administered with a pharmaceutical composition described herein include, but are not limited to atorvastatin, simvastatin, rosuvastatin, and ezetimibe.
- the lipid-lowering agent is administered prior to administration of a pharmaceutical composition described herein.
- the lipid-lowering agent is administered following administration of a pharmaceutical composition described herein.
- the lipid-lowering agent is administered at the same time as a pharmaceutical composition described herein.
- the dose of a coadministered lipid-lowering agent is the same as the dose that would be administered if the lipid- lowering agent was administered alone. In some embodiments, the dose of a co-administered lipid-lowering agent is lower than the dose that would be administered if the lipid-lowering agent was administered alone. In certain embodiments, the dose of a co-administered lipid-lowering agent is greater than the dose that would be administered if the lipid-lowering agent was administered alone.
- a co-administered lipid-lowering agent is a HMG-CoA reductase inhibitor.
- the HMG-CoA reductase inhibitor is a statin.
- the statin is selected from, for example, atorvastatin, simvastatin, pravastatin, fluvastatin, and rosuvastatin.
- a co-administered lipid-lowering agent is a cholesterol absorption inhibitor.
- cholesterol absorption inhibitor is ezetimibe.
- a co-administered lipid-lowering agent is a co-formulated HMG-CoA reductase inhibitor and cholesterol absorption inhibitor.
- the co-formulated lipid-lowering agent is ezetimibe/simvastatin.
- a co-administered lipid-lowering agent is a microsomal triglyceride transfer protein inhibitor (MTP inhibitor).
- MTP inhibitor microsomal triglyceride transfer protein inhibitor
- a co-administered lipid-lowering agent is an oligonucleotide selected from an oligonucleotide targeted to PCSK9, an oligonucleotide targeted to ACAT-2, an oligonucleotide targeted to endothelial lipase, and an oligonucleotide targeted to CETP.
- a co-administered lipid-lowering agent is an oligonucleotide targeted to ApoB.
- a co-administered pharmaceutical agent is a bile acid sequestrant.
- the bile acid sequestrant is selected from, for example, cholestyramine, colestipol, and colesevelam.
- a co-administered pharmaceutical agent is a nicotinic acid.
- the nicotinic acid is selected from immediate release nicotinic acid, extended release nicotinic acid, and sustained release nicotinic acid.
- a co-administered pharmaceutical agent is a fibric acid.
- a fibric acid is selected from gemfibrozil, fenofibrate, clofibrate, bezafibrate, and ciprofibrate.
- a co-administered pharmaceutical agent is an
- the antihypertensive agent is selected from, for example, angiotensin-converting enzyme (ACE) inhibitors, beta and alpha adrenergic blockers, calcium-channel blockers, renin inhibitors, aldosterone receptor antagonists, and angiotensin- receptor blockers.
- ACE angiotensin-converting enzyme
- the antihypertensive agent is administered at the same time as a pharmaceutical composition described herein.
- the dose of a coadministered antihypertensive agent is the same as the dose that would be administered if the antihypertensive agent was administered alone.
- the angiotensin-converting enzyme inhibitor is selected from, for example, captopril, enalapril, fosinopril, lisinopril, perindopril, quinapril, and ramipril.
- the beta adrenergic blocker is selected from, for example, atenolol, metoprolol, propranolol, timolol, and oxprenolol.
- the alpha adrengergic blocker is selected from, for example, doxazosin, indoramin, prozosin, terazosin, tolazoline, and phentolamine.
- the calcium-channel blocker is selected from, for example, dihydropyridines.
- the dihydropyridine is selected from, for example, amlodipine, cinidipine, felodipine, isradipine, mad nimodipine.
- the calcium-channel blocker is selected from, for example, non-dihydropyridines.
- the non-dihydropyridine is selected from, for example, diltiazem, or verapamil.
- the renin inhibitor is selected from for example, Aliskiren.
- the aldosterone receptor antagonist is selected from for example, elerenone or spironolactone.
- the angiotensin-receptor blocker is selected from for example, azilsartan, candesartan eprosartan, irbesartan, losartan, olmesartan, telmisartan, and valsartan.
- a co-administered pharmaceutical agent is a thrombolytic agent.
- the thrombolytic agent is administered at the same time as a pharmaceutical composition described herein.
- the dose of a coadministered thrombolytic agent is the same as the dose that would be administered if the thrombolytic agent was administered alone.
- the dose of a co-administered thrombolytic agent is lower than the dose that would be administered if the thrombolytic agent was administered alone.
- the dose of a co-administered thrombolytic agent is greater than the dose that would be administered if the thrombolytic agent was administered alone.
- the thrombolytic agent is selected from, for example, eminase, retavase, streptase, t-PA, TNKase, abbokinase, and kinlytic.
- a co-administered pharmaceutical agent is an antiplatelet agent.
- the antiplatelet agent is administered at the same time as a pharmaceutical composition described herein.
- the dose of a coadministered antiplatelet agent is the same as the dose that would be administered if the antiplatelet agent was administered alone.
- the dose of a co-administered antiplatelet agent is lower than the dose that would be administered if the antiplatelet agent was administered alone.
- the dose of a co-administered antiplatelet agent is greater than the dose that would be administered if the antiplatelet agent was administered alone.
- the antiplatelet agent is selected from, for example, cyclooxygenase inhibitors, adenosine diphosphate (ADP) receptor inhibitors, phosphodiesterase inhibitors, glycoprotein IIB/IIIA inhibitors, adenosine reuptake inhibitors, and thromboxane inhibitors.
- cyclooxygenase inhibitors adenosine diphosphate (ADP) receptor inhibitors
- phosphodiesterase inhibitors phosphodiesterase inhibitors
- glycoprotein IIB/IIIA inhibitors glycoprotein IIB/IIIA inhibitors
- adenosine reuptake inhibitors adenosine reuptake inhibitors
- thromboxane inhibitors thromboxane inhibitors
- the cyclooxygenase inhibitor is selected from, for example aspirin, or triflusal.
- the adenosine diphosphate (ADP) receptor inhibitor is selected from, for example, clopidogrel, prasugrel, ticagrelor, ticlopidine, phosphodiesterase inhibitors, and cilostazol.
- the Glycoprotein IIB/IIIA inhibitor is selected from, for example, abciximab, eptifibatide, and tirofiban.
- the adenosine reuptake inhibitor is selected from, for example, dipyridamole.
- the thromboxane inhibitor is selected from, for example, thromboxane synthase inhibitors and thromboxane receptor antagonists.
- compositions described herein include, but are not limited to, antiarrhythmic agents; azetidinone-based cholesterol absorption inhibitors; niacin; niacin derivatives; PPAR agonists; PPAR antagonists; antiplatelet drugs; corticosteroids, including but not limited to prednisone; LXR agonists; immunoglobulins, including, but not limited to intravenous immunoglobulin (IVIg); analgesics (e.g., acetaminophen); anti-inflammatory agents, including, but not limited to non-steroidal anti-inflammatory drugs (e.g., ibuprofen, COX-1 inhibitors, and COX-2, inhibitors); salicylates; antibiotics; antivirals; antifungal agents; antidiabetic agents (e.g., biguanides, glucosidase inhibitors, insulins, sulfony
- osteoporosis agents e.g., biphosphonates, calcitonin, and estrogens
- prostaglandins e.g., biphosphonates, calcitonin, and estrogens
- antineoplastic agents include psychotherapeutic agents; sedatives; poison oak or poison sumac products; antibodies; and vaccines.
- the pharmaceutical compositions described herein are administered in conjunction with a lipid-lowering therapy.
- a lipid-lowering therapy is therapeutic lifestyle change.
- a lipid-lowering therapy is LDL apheresis.
- the pharmaceutical compositions described herein and one or more other pharmaceutical agents as described herein provides use in the manufacture of a medicament for the treatment, prevention, or management of a disease or conditions described herein.
- the antisense compound is targeted to a specific tissue, organ or location in the body.
- exemplary targets include liver, lung, kidney, heart, and atherosclerotic plaques within a blood vessel. Methods of targeting compounds can be well known in the art.
- the compound is targeted by direct or local administration.
- the compound when targeting a blood vessel, the compound is administered directly to the relevant portion of the vessel from inside the lumen of the vessel, e.g., single balloon or double balloon catheter, or through the adventitia with material aiding slow release of the compound, e.g., a pluronic gel system as described by Simons et al., Nature 359: 67-70 (1992).
- Other slow release techniques for local delivery of the compound to a vessel include coating a stent with the compound.
- the compound when targeting a particular tissue or organ, can be administered in or around that tissue or organ.
- U.S. Pat. No. 6,547,787 incorporated herein by reference in its entirety, discloses methods and devices for targeting therapeutic agents to the heart.
- administration occurs by direct injection or by injection into a blood vessel associated with the tissue or organ.
- the compound when targeting the liver, can be administered by injection or infusion through the portal vein.
- methods of targeting a compound include associating the compound with an agent that directs uptake of the compound by one or more cell types.
- agents include lipids and lipid-based structures such as liposomes generally in combination with an organ- or tissue-specific targeting moiety such as, for example, an antibody, a cell surface receptor, a ligand for a cell surface receptor, a polysaccharide, a drug, a hormone, a hapten, a special lipid and a nucleic acid as described in U.S. Pat. No. 6,495,532, the disclosure of which is incorporated herein by reference in its entirety.
- targeting agents include porous polymeric microspheres, which can be derived from copolymeric, and homopolymeric polyesters containing hydrolyzable ester linkages, which can be biodegradable, as described in U.S. Pat. No. 4,818,542, the disclosure of which is incorporated herein by reference in its entirety.
- Typical polyesters include polyglycolic (PGA) and polylactic (PLA) acids, and copolymers of glycolide and L(-lactide) (PGL), which can be particularly suited for the methods and compositions provided herein in that they exhibit low human toxicity and can be biodegradable.
- polyester or other polymer, oligomer, or copolymer utilized as the microspheric polymer matrix is not critical and a variety of polymers can be utilized depending on desired porosity, consistency, and shape and size distribution.
- Other biodegradable or bioerodable polymers or copolymers include, for example, gelatin, agar, starch, arabinogalactan, albumin, collagen, natural and synthetic materials or polymers, such as, poly(8-caprolactone), poly(8-caprolactone-CO-lactic acid), poly(8- caprolactone-CO-glycolic acid), poly(P-hydroxy butyric acid), polyethylene oxide, polyethylene, poly(alkyl-2-cyanoacrylate), (e.g., methyl, ethyl, butyl), hydrogels such as poly(hydroxyethyl methacrylate), polyamides (e.g., polyacrylamide), poly(amino acids) (i.e., L-leucine, L-aspart
- condensation polymerization reactions from the suitable monomers or, comonomers or oligomers from the suitable monomers or, comonomers or oligomers.
- U.S. Pat. No. 6,562,864 the disclosure of which is incorporated herein by reference in its entirety, describes catechins, including epi and other carbo-cationic isomers and derivatives thereof, which as monomers, dimers and higher multimers can form complexes with nucleophilic and cationic bioactive agents for use as delivery agents.
- Catechin multimers have a strong affinity for polar proteins, such as those residing in the vascular endothelium, and on cell/organelle membranes and can be particularly useful for targeted delivery of bioactive agents to select sites in vivo.
- delivery agents include substituted catechin multimers, including amidated catechin multimers which can be formed from reaction between catechin and nitrogen containing moities such as ammonia.
- ADEPT antibody- directed enzyme prodrug therapy
- GDEPT gene-directed EPT
- VDEPT virus-directed EPT
- kits can be used for targeted delivery, wherein the device is, for example, a syringe, stent, or catheter.
- Kits include a device for administering a compound and a container comprising an antisense compound provided herein.
- the compound is preloaded into the device.
- the kit provides instructions for methods of administering the compound and dosages.
- U.S. patents describing medical devices and kits for delivering an antisense compound include U.S. Pat. Nos. 6,368,356; 6,344,035; 6,344,028; 6,287,285; 6,200,304; 5,824,049; 5,749,915; 5,674,242;
- provided herein are methods of treating a subject comprising administering one or more pharmaceutical agents provided herein.
- such subject has hypercholesterolemia, hyperlipidemia, non-familial hypercholesterolemia, familial hypercholesterolemia, heterozygous familial hypercholesterolemia, homozygous familial hypercholesterolemia, coronary heart disease, atherosclerosis, mixed dyslipidemia, diabetic dyslipidemia.
- such subject has been identified as having one or more CHD risk equivalents.
- such subject has been identified has having major risk factors for coronary heart disease.
- such subject has been identified as having one or more CHD risk factors.
- such subject has been identified being at risk for atherosclerosis. In certain embodiments, such subject has been identified as having a history of coronary heart disease. In certain embodiments, such subject has been identified as having a family history of early onset coronary heart disease.
- the subject has been identified as having elevated
Abstract
Provided herein, for example, are methods generally relating to preventing, treating and/or managing a major adverse cardiovascular event in a subject with a disease or condition at risk for a major adverse cardiovascular event, e.g., familial hypercholesterolemia. Also provided herein are methods relating to administering to the patient a therapeutically effective amount of an antisense oligonucleotide having a nucleobase SEQ ID NO: 247 (e.g., mipomersen).
Description
METHODS FOR THE PREVENTION AND TREATMENT OF MAJOR ADVERSE CARDIOVASCULAR EVENTS USING COMPOUNDS THAT MODULATE
APOLIPOPROTEIN B
CROSS REFERENCE TO RELATED APPLICATION
[0001] This application claims the benefit of priority to U.S. Provisional Application No. 62/043,989 filed August 29, 2014 and U.S. Provisional Application No. 62/087,712 filed
December 4, 2014, the disclosures of which are incorporated herein in their entirety.
FIELD
[0002] Provided herein, for example, are methods generally relating to preventing, treating and/or managing a major adverse cardiovascular event in a subject with hypercholesterolemia, e.g., familial hypercholesterolemia or a condition or disease with an increased risk of MACE. Also provided herein are methods relating to administering to the patient a therapeutically effective amount of an antisense compound that targets Apolipoprotein B (ApoB).
BACKGROUND
[0003] Coronary heart disease (CHD) has been the leading cause of death in the United States for over a century, and complications from atherosclerosis are the most common causes of death in Western societies (Knopp, New Engl. J. Medicine, 1999, 341, 498-511; Davis and Hui, Arterioscler. Thromb. Vase. Biol, 2001, 21, 887-898; Bonow, Circulation, 2002, 106, 3140- 3141). Elevated low density lipoprotein-cholesterol (LDL-cholesterol) is widely recognized as a risk factor for CHD. However, despite pharmacologic intervention, many subjects are unable to lower LDL-cholesterol levels. Indeed, following acute coronary syndrome, the risk for future major cardiovascular events (MACE) is high and related to levels of LDL-C, even when the patients are on standard therapies (e.g., statins).
[0004] Low density lipoproteins are one of five broad classes of lipoproteins, which include the following: chylomicrons, responsible for the transport dietary lipids from intestine to tissues; very low density lipoproteins (VLDL); intermediate density lipoproteins (IDL); low density lipoproteins (LDL); all of which transport triacylglycerols and cholesterol from the liver to tissues; and high density lipoproteins (HDL), which transport endogenous cholesterol from tissues to the liver. Lipoprotein particles undergo continuous metabolic processing and have variable properties and compositions. The protein components of lipoproteins are known as
apolipoprotems. At least nine apolipoprotems, one of which is apolipoprotein B, are distributed in significant amounts among the various human lipoproteins.
[0005] Apolipoprotein B (also known as ApoB, apolipoprotein B-100; ApoB-100, apolipoprotein B-48; ApoB-48 and Ag(x) antigen), is a large glycoprotein involved in the assembly and secretion of lipids and in the transport and receptor-mediated uptake and delivery of distinct classes of lipoproteins. Apolipoprotein B performs a variety of functions, including the absorption and processing of dietary lipids, as well as the regulation of circulating lipoprotein levels (Davidson and Shelness, Annu. Rev. Nutr., 2000, 20, 169-193).
[0006] Apolipoprotein B is involved cholesterol homeostasis and its overproduction has been associated with various diseases, including familial hypercholesterolemia, familial defective ApoB and familial combined hypercholesterolemia (Kane and Havel, The Metabolic and Molecular Bases of Inherited Diseases, 2001, 8.sup.th edition, 2717-2751). Perturbations in the metabolism of ApoB that correspond with an increased risk of CHD are also observed in diabetes and obesity (Grundy, Am. J. Cardiol, 1998, 81, 18B-25B; Chan et al, Diabetes, 2002, 51, 2377-2386; Chan et al, Metabolism, 2002, 51, 1041-1046). Furthermore, genetic studies in mouse models have demonstrated a correlation between elevated apolipoprotein B, elevated cholesterol levels and atherosclerosis (Kim and Young, J. Lipid Res., 1998, 39, 703-723; Nishina et al, J. Lipid Res., 1990, 31, 859-869).
[0007] In studies of patients with familial hypobetalipoproteinemia (FHBL), these patients exhibit lowered serum apolipoprotein B levels, lowered serum LDL-cholesterol levels and a reduced incidence of coronary artery disease (Schonfeld et al, J. Lipid Res., 2003, 44, 878-883). Murine studies have demonstrated that mice having heterozygous deficiencies in apolipoprotein B exhibit reduced serum LDL-cholesterol and apolipoprotein B levels, and, furthermore, are protected from diet-induced hypercholesterolemia. (Farese et al, Proc. Natl. Acad. Sci. U.S.A., 1995, 92, 1774-1778).
[0008] Current LDL-C-lowering medications include statins, cholesterol absorption inhibitors, fibrates, niacin, and bile acid sequestrants. Statins are a commonly prescribed treatment for LDL-C lowering. While statins are potent apoB lowering agents, their efficacy in achieving therapeutic targets for LDL-C patients, for example in high-risk patients (FH) is limited. Resistance and intolerance to statins occur in a significant number of patients. Thus, there is a need for new lipid lowering therapies.
SUMMARY
[0009] In a first aspect, a method of treating, preventing, or managing a major adverse cardiovascular event (MACE) in a hypercholesterolemia patient in need thereof, comprising administering to the patient a therapeutically effect amount of an antisense olionucleotide complementary to a nucleic acid encoding human apolipoprotein B is described.
[0010] In a second aspect, a method of treating, preventing, or managing a major adverse cardiovascular event (MACE) in a patient comprising; selecting a patient having a disease or condition that increases the risk of MACE, and administering to the patient a therapeutically effect amount of an antisense olionucleotide complementary to a nucleic acid encoding human apolipoprotein B is described.
[0011] In a first embodiment of the first or second aspect, the MACE is a myocardial infarction, reinfarction, stroke, unstable angina, cardiogenic shock, pulmonary edema, cardiac arrest, coronary revascularization, investigational angioplasty, interventional angioplasty, a percutaneous transluminal coronary angioplasty, percutaneous coronary intervention, a coronary artery bypass graft, or any combination thereof. In a second embodiment of the first or second aspect, the MACE is myocardial infarction. In a third embodiment of the first or second aspect or any embodiment, the patient is statin-resistant or statin intolerant. In the fourth embodiment of the first aspect or second or any other embodiment, the oligonucleotide is administered for at least 12 months. In a fifth embodiment of the first or second aspect or any other embodiment, the oligonucleotide is administered for 12 to 24 months, or for a prolonged period of reduction of MACE events.
[0012] In a sixth embodiment of the first or second aspect or any other embodiment, the patient has an established cardiovascular disease.
[0013] In a seventh embodiment of the first or second aspect or any other embodiment, the administration results in reversed cardiac injury.
[0014] In an eighth embodiment of the first or second aspect or any other embodiment, the patient is homozygous for familial hypercholestrolemia.
[0015] In a ninth embodiment of the first or second aspect or any other embodiment, the patient is heterozygous for familial hypercholestrolemia.
[0016] In a tenth embodiment of the first or second aspect or any other embodiment, the patient is heterozygous for familial hypercholestrolemia with coronary artery disease.
[0017] In an eleventh embodiment of the first or second aspect or any other embodiment, the patient has severe hypercholesterolemia.
[0018] In a twelveth embodiment of the first or second aspect or any other embodiment, the patient has not previously been treated for MACE.
[0019] In a thirteenth embodiment of the first or second aspect or any other embodiment, the patient has been previously been treated for MACE.
[0020] In a fourteenth embodiment of the first or second aspect or any other embodiment, the method reduces the occurrence of or prevents MACE in a patient having an established CVD.
[0021] In a fifteenth embodiment of the first or second aspect or any other embodiment, the method reduces the occurrence of MACE in a patient at risk of CVD.
[0022] In a sixteenth of the first or second aspect or any other embodiment, the patient has a reduction of serum cholesterol, ApoB, serum low density lipoprotein (LDL), serum very low density lipoprotein (VLDL), serum triglycerides, serum apolipoprotein (a) and/or free fatty acids after administration of the antisence oligonucleotide.
[0023] In a seventeen embodiment of the first or second aspect or any other embodiment, the antisense oligonucleotide is 20 nucleobases in length.
[0024] In an eighteenth embodiment of the first or second aspect or any other embodiment, the antisence oligonucleotide is mipomersen.
[0025] In a ninteenth embodiment of the first or second aspect or any other embodiment, the antisense oligonucleotide has a nucleobase sequence consisting of the nucleobase sequence of SEQ ID NO: 247.
[0026] In a twentieth embodiment of the first or second aspect or any other embodiment, wherein the antisense oligonucleotide comprises a modified internucleoside linkage, a modified sugar moiety, a modified nucleobase, or a combination thereof.
[0027] In a twenty-first embodiment, of the first or second aspect, or any other embodiment, the modified sugar moiety is a 2'-0-methoxyethyl sugar moiety or a bicyclic sugar moiety.
[0028] In a twenty-second embodiment of the first or second aspect or any other
embodiment, the modified internucleoside linkage is a phosphorothioate linkage.
[0029] In a twenty-third embodiment of the first or second aspect of any other embodiment, the modified nucleobase is a 5-methylcytosine.
[0030] In a twenty-second embodiment of the first or second aspect or any other
embodiment, the antisense oligonucleotide is a chimeric oligonucleotide.
[0031] In a twenty-third embodiment of the first or second aspect or any other embodiment the chimeric oligonucleotide comprises a gap segment often linked 2'-deoxynucleotides, wherein the gap segment is positioned between wing segments, wherein each nucleoside of each wing segment comprises a modified sugar moiety.
[0032] In a twenty-fourth embodiment of the first or second aspect or any other embodiment the modified sugar moiety is a 2 '-O-methoxy ethyl sugar moiety.
[0033] In a twenty-fifth embodiment of the first or second aspect or any other embodiment wherein the gap segment is 10 2'-deoxynucleosides in length, and each wing segment comprises from 1 to 8 2'-0-methoxyethyl.
[0034] In a twenty-sixth embodiment of the first or second aspect or any other embodiment each wing segment comprises 2'-methoxyethoxyl nucleotides.
[0035] In a twenty-seventh embodiment of the first or second aspect or any other embodiment the antisense oligonucleotide is an antisense oligonucleotide 20 nucleotides in length having the nucleobase sequence of SEQ ID NO: 247, and comprising a 5-methylcytosine at nucleobases 2, 3, 5, 9, 12, 15, 17, 19, and 20, wherein every internucleoside linkage is a phosphorothioate linkage, nucleotides 1-5 and 16-20 can be 2 '-O-methoxy ethyl nucleotides, and nucleotides 6-15 can be 2'-deoxynucleotides, or wherein said antisense oligonucleotide is a pharmaceutically acceptable salt form thereof.
[0036] In a twenty-eighth embodiment of the first or second aspect or any other embodiment the antisense oligonucleotide is administered in a dosage to achieve at least a 60% reduction in MACE.
[0037] In a twenty-ninth embodiment of the first or second aspect or any other embodiment the antisense oligonucleotide is administered in a dosage to achieve at least a 65%, at least a 70%, at least a 75%, at least a 80%, at least a 85%, or at least a 90% reduction in MACE.
[0038] In a thirtieth embodiment of the first or second aspect or any other embodiment the antisense oligonucleotide is administered at 200 mg per week.
[0039] In a thirty-first embodiment of the first or second aspect or any other embodiment the administering comprises an induction phase, wherein a 210 mg dose of the antisense
oligonucleotide per week is administered in two or more administrations for at least 13 weeks, followed by a maintenance phase, wherein a 210 mg dose of the antisense oligonucleotide per week is administered in two or more administrations.
[0040] In a thirty-second embodiment of the first or second aspect or any of the
embodiments of these aspects, the administration of the antisense oligonucleotide cause reductions in atherogenic lipoproteins in plasma. In a thirty-third embodiment of the first or second aspect or any of the embodiments, the antisense oligonucleotide is administered with one or more additional compounds selected from the group consisting of angiotensin-converting- enzyme inhibitors, angiotensin receptor blockers, renin inhibitors, HMG CoA reductase inhibitors, dihydropyridine calcium channel blockers, antiarrhythmic agents, azetidinone-based cholesterol absorption inhibitors, PCSK9 inhibitors, niacin, niacin derivatives, PPAR agonists, PPAR antagonists, bile acid sequestrants; and antiplatelet drugs; or any pharmaceutically acceptable esters, derivatives, conjugates, precursors or salts thereof. In a thirty-fourth embodiment of the first or second aspect or any of the embodiments reduces the occurrence of MACE as compared to a patient that has not been administered the antisense oligonucleotide. In thirty- fifth embodiment of the first or second aspect or any of the embodiments the patient that has not received the antisense oligonucleotide is a patient that has been administered a placebo. In thirty-sixth embodiment of the first or second aspect or any of the embodiments the method reduces the occurrence of MACE as compared to the occurrence of the MACE prior to administration of the antisense oligonucleotide. In a thirty-seventh embodiment of the first or second aspect or any of the embodiments the reduction in the occurrence of the MACE is as compared to the occurrence of the MACE in the 24 months prior to administration of the antisense oligonucleotide.
[0041] Familial hypercholesterolemia (FH) is associated with a 10-20 fold increase in cardiovascular (CV) events and many of these patients are resistant or intolerant to statin therapy. Thus, there remains a need for additional therapeutic methods for the treatment, prevention and management of CV events, including major adverse cardiovascular events (MACE), for example, to improve patient outcomes. The present application is based, in part, on the discovery that mipomersen significantly lowers levels of atherogenic lipoproteins in plasma;
whereas previous safety analysis of all patients in phase 3 trials found no imbalance in CV events between placebo and mipomersen arms and the patients have a reduction in MACE events.
[0042] Accordingly, in one aspect, provided herein is a method for treating, preventing, or managing a MACE. In certain embodiments, the patient has a condition or disease that has an increase in the risk for MACE, e.g., familial hypercholesterolemia (FH). In certain
embodiments, the patent is resistant or intolerate to statins. In certain embodiments, the patient has familial hypercholesterolemia and is resistant or intolerate to statins. In a specific embodiment, the method comprises administering an antisense oligonucleotide to the patient. Such methods can comprise the administration of a therapeutically effective amount of an antisense oligonucleotide. In some embodiments, the antisense oligonucleotide targets ApoB. In certain embodiments, the method comprises administering to the patient a therapeutically effective amount of an antisense oligonucleotide comprising or consisting of a nucleobase sequence of SEQ ID NO: 247 (e.g., mipomersen).
[0043] In some embodiments, the MACE is a myocardial infarction, reinfarction, stroke, unstable angina, cardiogenic shock, pulmonary edema, cardiac arrest, atrial dysrhythmia, coronary revascularization, investigational angioplasty, interventional angioplasty, a
percutaneous transluminal coronary angioplasty, percutaneous coronary intervention, a coronary artery bypass graft, or any combination thereof. In certain of the methods of preventing a MACE provided herein, the MACE is death.
[0044] In some embodiments, the MACE is a non-fatal myocardial infarction, stroke, unstable angina, or revascularization procedure (e.g., a percutaneous coronary intervention (PCI), coronary artery bypass graft (CABG) surgery), or any combination thereof.
[0045] In certain embodiments, the methods described sho a reversal of cardiac injury from the administration of mipomerson for at least 12 months.
[0046] In certain embodiments, the patient is a mammal (e.g., a rodent, monkey), such as a human. In some embodiments, the patient is homozygous for FH. In some embodiments, the patient is heterozygous for FH. In other embodiments, the patient has coronary artery disease, severe hypercholesterolemia, or a high risk of cardiovascular disease (CVD). In some
embodiments, the patient was not previously treated for MACE. In other embodiments, the patient was previously treated for MACE. In some embodiments, the patient is a patient in need thereof.
[0047] In certain embodiments, a method provided herein reduces the occurrence of or prevents MACE in a patient having established CVD. In some embodiments, a method provided herein reduces the occurrence of or prevents MACE in a patient at risk of CVD.
[0048] In certain embodiments, the antisense oligonucleotide is an antisense oligonucleotide, which is targeted to a nucleic acid encoding ApoB. In certain embodiments, the antisense oligonucleotide is 20 nucleobases in length. In some embodiments, the antisense oligonucleotide is an antisense oligonucleotide 20 nucleobases in length. In other embodiments, the antisense oligonucleotide has a nucleobase sequence comprising of the nucleobase sequence of SEQ ID NO: 247. In yet other embodiments, the antisense oligonucleotide has a nucleobase sequence consisting of the nucleobase sequence on SEQ ID NO: 247.
[0049] In certain embodiments, the antisense oligonucleotide includes a modified
internucleoside linkage, a modified sugar moiety, a modified nucleobase, or a combination thereof. In some embodiments, the modified sugar moiety is a 2'-0-methoxyethyl sugar moiety or a bicyclic sugar moiety. In certain embodiments, the modified internucleoside linkage is a phosphorothioate linkage. In some embodiments, the modified nucleobase is a 5-methylcytosine. In certain embodiments, the antisense oligonucleotide is a chimeric oligonucleotide.
[0050] The chimeric oligonucleotide can include a gap segment often linked 2'- deoxynucleotides. In some embodiments, the gap segment is positioned between wing segments. In other embodiments, each nucleoside of each wing segment includes a modified sugar moiety. In some embodiments, the modified sugar moiety is a 2'-0-methoxyethyl sugar moiety. In other embodiments, the gap segment is 10 2'-deoxynucleosides in length, and each wing segment includes from 1 to 8 2'-0-methoxyethyl. In other embodiments, each wing segment includes 2'- methoxyethoxyl nucleotides.
[0051] In certain embodiments, the antisense oligonucleotide is an antisense oligonucleotide 20 nucleotides in length having the nucleobase sequence of SEQ ID NO: 247, and can optionally include a 5-methylcytosine at nucleobases 2, 3, 5, 9, 12, 15, 17, 19, and 20. In some
embodiments, every internucleoside linkage is a phosphorothioate linkage, nucleotides 1-5 and 16-20 are 2'-0-methoxyethyl nucleotides, and nucleotides 6-15 are 2'-deoxynucleotides. In other embodiments, the antisense oligonucleotide is a pharmaceutically acceptable salt form thereof.
[0052] In certain embodiments, the antisense oligonucleotide is administered in a dosage to achieve at least a 60% reduction in MACE. In certain embodiments, the antisense
oligonucleotide is administered in a dosage to achieve at least a 65%, at least a 70%, at least a 75%, at least an 80%, at least an 85%, or at least a 90% reduction in MACE.
[0053] In certain embodiments, the antisense oligonucleotide is administered at 200 mg per day. In some embodiments, the antisense oligonucleotide is administered for at least 12 months.
[0054] In certain embodiments, administration of the antisense oligonucleotide decreases total serum cholesterol, ApoB, serum low density lipoprotein (LDL), serum very low density lipoprotein (VLDL), serum triglycerides, serum apolipoprotein (a) and/or free fatty acids in the patient.
[0055] In certain embodiments, administration of the antisense oligonucleotide decreases LDL cholesterol. In some embodiments, the LDL level is reduced to about 100 mg/dl or lower, about 70 mg/dl or lower, or about 50 mg/dl or lower. In some embodiments, administration of the antisense oligonucleotide causes reductions in atherogenic lipoproteins in plasma.
[0056] In certain embodiments, the antisense oligonucleotide is administered with one or more additional oligonucleotide s selected from the group consisting of angiotensin-converting- enzyme inhibitors, angiotensin receptor blockers, renin inhibitors, HMG CoA reductase inhibitors, dihydropyridine calcium channel blockers, antiarrhythmic agents, azetidinone-based cholesterol absorption inhibitors, niacin, niacin derivatives, PPAR agonists, PPAR antagonists, bile acid sequestrants; and antiplatelet drugs; or any pharmaceutically acceptable esters, derivatives, conjugates, precursors or salts thereof.
[0057] In certain embodiments, the method reduces the occurrence of MACE as compared to a patient that has not been administered the antisense oligonucleotide. In some embodiments, the patient that has not received the antisense oligonucleotide is a patient that was administered a placebo.
[0058] In certain embodiments, the method reduces the occurrence of MACE as compared to the occurrence of the MACE prior to administration of the antisense oligonucleotide. In some embodiments, the reduction in the occurrence of the MACE is compared to the occurrence of the MACE in the 24 months prior to administration of the antisense oligonucleotide.
BRIEF DESCRIPTION OF THE DRAWINGS
[0059] FIG. 1 depicts the general study design of Phase 3 randomized, placebo-controlled studies used for assessing MACE incidence in FH patients treated with mipomirsen for at least one year.
[0060] FIG. 2 depicts that there is a significant reduction in MACE incidence in FH patients treated with mipomirsen for at least one year.
DETAILED DESCRIPTION
[0061] The following description is merely intended to illustrate various embodiments. As such, the specific modifications discussed are not to be construed as limitations on the scope. It will be apparent to one skilled in the art that various equivalents, changes, and modifications can be made without departing from the scope described herein, and it is understood that such equivalent embodiments, can be to be included herein.
[0062] Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs. As used herein, the term "about," when used in reference to a particular recited numerical value, means that the value may vary from the recited value by no more than 1%. For example, as used herein, the expression "about 100" includes 99 and 101 and all values in between (e.g., 99.1 , 99.2, 99.3, 99.4, etc.).
[0063] Although any methods and materials similar or equivalent to those described herein can be used in the practice of the present invention, the preferred methods and materials are now described. All publications mentioned herein are incorporated herein by reference to describe in their entirety.
Definitions
[0064] Unless defined otherwise, all technical and scientific terms used herein have the same refers meaning as is commonly understood by one of skill in the art. Unless specific definitions can be provided, the nomenclature utilized in connection with, and the procedures and techniques of, analytical chemistry, synthetic organic chemistry, and medicinal and
pharmaceutical chemistry described herein can be those well known and commonly used in the art. Standard techniques can be used for chemical synthesis, chemical analysis, pharmaceutical preparation, formulation and delivery, and treatment of subjects. Some techniques and procedures can be found for example in "Antisense Drug Technology: Principles, Strategies, and Applications." by Stanley Crooke, Boca Raton: Taylor & Francis Group, 2008; "Carbohydrate Modifications in Antisense Research" Edited by Sangvi and Cook, American Chemical Society, Washington D. C, 1994; and "Remington's Pharmaceutical Sciences," Mack Publishing Co.,
Easton, Pa., 18th edition, 1990; and which is hereby incorporated by reference for any purpose. Where permitted, all patents, patent applications, published applications and publications, GENBANK® sequences, websites and other published materials referred to throughout the entire disclosure herein, unless noted otherwise, can be incorporated by reference in their entirety. All of the in GENBANK® Accession Nos. along with their associated sequence and structural data pertaining to such sequences including gene organization and structural elements and SNP information that can be found in sequence databases such as the National Center for Biotechnology Information (NCBI) can be incorporated herein by reference in their entirety. In the event that there is a plurality of definitions for terms herein, those in this section prevail. Where reference is made to a URL or other such identifier or address, it is understood that such identifiers can change and particular information on the internet can come and go, but equivalent information can be found by searching the internet. Reference thereto evidences the availability and public dissemination of such information.
[0065] As used herein, the term "about" or "approximately" refers to within 20%, within 10%, within 5%, within 1% or less of a given range.
[0066] As used herein, "acceptable safety profile" means a pattern of side effects that is within clinically acceptable limits.
[0067] As used herein, "active pharmaceutical ingredient" means the substance in a pharmaceutical composition that provides a desired effect. For example, ISIS 301012 (SEQ ID NO:247; mipomersen) is the active pharmaceutical ingredient in a pharmaceutical composition comprising ISIS 301012 and saline.
[0068] As used herein, the term "active target region" refers to a target region to which one or more active antisense oligonucleotides or compounds is targeted.
[0069] As used herein, the term "active antisense compounds " or "active antisense olionucleotides" refer to antisense compounds or oligonucelotides that reduce target nucleic acid levels.
[0070] As used herein, the term "administering" refers to providing a pharmaceutical agent to a subject, and includes, but is not limited to administering by a medical professional and self- administering.
[0071] As used herein, the term "antisense" refers to the modulation of function of a target nucleic acid by oligonucleotide s which specifically hybridize to it is.
[0072] As used herein, the term "antisense oligonucleotide" refers to an oligomeric oligonucleotide that is capable of undergoing hybridization to a target nucleic acid through hydrogen bonding.
[0073] As used herein, the term "antisense inhibition" refers to reduction of a target nucleic acid levels in the presence of an antisense oligonucleotide complementary to a target nucleic acid complementary to a target nucleic acid levels in the absence of the antisense oligonucleotide.
[0074] As used herein, the term "antisense oligonucleotide" refers to a single-stranded oligonucleotide having a nucleobase sequence that will permits hybridization to a corresponding region of a target nucleic acid. Such an antisense oligonucleotide is "targeted to" the nucleic acid.
[0075] As used herein, "ApoB" means apolipoprotein B-100 protein. Concentration of ApoB in serum (or plasma) is typically quantified in mg/dL or nmol/L. "Serum ApoB" and "plasma ApoB" mean ApoB in the serum and plasma, respectively.
[0076] As used herein, "ApoAl" is apolipoprotein-Al protein in serum. Concentration of ApoAl in serum is typically quantified in mg/dL or nmol/L.
[0077] As used herein, "ApoB: ApoAl ratio" is the ratio of ApoB concentration to ApoAl concentration.
[0078] As used herein, the term "ApoB -containing lipoprotein" refers to any lipoprotein that has ApoB as its protein component, and is understood to include LDL, VLDL, IDL, and lipoprotein (a).
[0079] As used herein, the term "atherosclerosis" refers to a hardening of the arteries affecting large and medium-sized arteries and is characterized by the presence of fatty deposits. The fatty deposits can be called "atheromas" or "plaques," which consist mainly of cholesterol and other fats, calcium and scar tissue, and damage the lining of arteries.
[0080] As used herein, "AUCtrough" or "plasma trough AUC" means the area under the concentration-time curve at a time when plasma pharmaceutical agent concentrations are in equilibrium with target tissue pharmaceutical agent concentrations.
[0081] As used herein, the term "bicyclic sugar" refers to a furosyl ring modified by the bridging of the two non-geminal ring atoms. A bicyclic sugar is a modified sugar.
[0082] As used herein, the term "bicyclic nucleic acid sugar moiety" refers to a furosyl ring modified by the bridging of two non-geminal ring atoms.
[0083] As used herein, "Chough" or "plasma trough concentration" means a minimum plasma concentration when plasma pharmaceutical agent concentrations are in equilibrium with target tissue pharmaceutical agent concentrations. For example, in certain embodiments, a plasma trough concentration of ISIS 301012 is achieved when plasma ISIS 301012 concentrations are in equilibrium with liver tissue ISIS 301012 concentrations.
[0084] As used herein, the term "cap structure" or "terminal cap moiety" refers to chemical modifications, which have been incorporated at either terminus of an antisense oligonucleotide.
[0085] As used herein, "cardiac injury" refers to the disruption of normal cardiac myocyte membrane integrity resulting in the loss into the extracellular space (including blood) of intracellular constituents including detectable levels of a variety of biologically active cytosolic and structural proteins such as troponin, creatine kinase, myoglobin, heart-type fatty acid binding protein, and lactate dehydrogenase. A reversal of cardiac injury refers to an improvement of any of the biochemical characteristics of the injury, e.g., reduction of inflammation. Ischemia or infarction consequent to an imbalance between the supply and demand of oxygen (and nutrients) is the most common cause of cardiac injury. When a sufficient number of myocytes have died (myocyte necrosis) or lost function, acute clinical disease is apparent; examples include myocardial infarction (MI) or myocarditis.
[0086] As used herein, "cardiovascular outcome" means the occurrence of major adverse cardiovascular events.
[0087] As used herein, the term "CHD risk factors" refers to CHD risk equivalents and major risk factors.
[0088] As used herein, "CHD risk equivalents," means indicators of clinical atherosclerotic disease that confer a high risk for coronary heart disease, and include clinical coronary heart disease, symptomatic carotid artery disease, peripheral arterial disease, and/or abdominal aortic aneurysm.
[0089] As used herein, the term "chimeric antisense oligonucleotide s" refers to an antisense oligonucleotide s that have at least 2 chemically distinct regions, each region having a plurality of subunits.
[0090] As used herein, the term "cholesterol absorption inhibitor" refers to a pharmaceutical agent that inhibits the absorption of exogenous cholesterol obtained from diet.
[0091] As used herein, "cholesteryl ester content" means the amount of cholesteryl ester present in liver tissue. In certain embodiments, serum cholesteryl ester concentration is used as an indicator of hepatic cholesteryl ester content.
[0092] As used herein, "complementarity" means the capacity for pairing between nucleobases of a first nucleic acid and a second nucleic acid. As used herein, "fully
complementary" means each nucleobase of an oligonucleotide is capable of precise base pairing with the corresponding nucleobases of a target nucleic acid.
[0093] As used herein, "comply" as used herein means the adherence to a recommended therapy by a subject.
[0094] As used herein, the term "composition" is intended to encompass a product containing the specified ingredients (e.g., an antisense oligonucleotide provided herein) and, optionally, in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in, optionally, the specified amounts.
[0095] As used herein, the term "co-administration" refers to administration of two or more pharmaceutical agents to a subject. The two or more pharmaceutical agents can be in a single pharmaceutical composition, or can be in separate pharmaceutical compositions. Each of the two or more pharmaceutical agents can be administered through the same or different routes of administration. Co-administration encompasses administration in parallel or sequentially.
[0096] As used herein, the combination of therapies (e.g., use of therapeutic agents) which is more effective than the additive effects of any two or more single therapy. For example, a synergistic effect of a combination of therapeutic agents permits the use of lower dosages of one or more of the agents and/or less frequent administration of the agents to a subject with a MACE. The ability to utilize lower dosages of prophylactic or therapeutic therapies and/or to administer the therapies less frequently reduces the toxicity associated with the administration of the therapies to a subject without reducing the efficacy of the therapies in the prevention,
management, treatment of a MACE. In addition, a synergistic effect can result in improved efficacy of therapies in the prevention, or in the management, treatment of a MACE. Finally, synergistic effect of a combination of therapies (e.g., therapeutic agents) can avoid or reduce adverse or unwanted side effects associated with the use of any single therapy.
[0097] As used herein, the term "coronary heart disease (CHD)" refers to a narrowing of the small blood vessels that supply blood and oxygen to the heart, which is often a result of atherosclerosis.
[0098] As used herein, the term "complementarity" refers to the capacity for pairing between nucleobases of a first nucleic acid and a second nucleic acid.
[0099] As used herein, the term "contiguous nucleobases" refers to nucleobases immediately adjacent to each other.
[00100] As used herein, "diabetic dyslipidemia" or "Type II diabetes with dyslipidemia" means a condition characterized by Type II diabetes, reduced HDL-C, elevated serum
triglycerides, and elevated small, dense LDL particles.
[00101] As used herein, the term "diluent" refers to an ingredient in a composition that lacks pharmacological activity, but is pharmaceutically necessary or desirable. For example, in drugs that can be injected the diluent can be a liquid, e.g., saline solution.
[00102] As used herein, a disease or condition that increases the risk of MACE including by not limited to FH, homozygous FH, severe heterozygous FH, heterozygous FH, diabetes, coronary heart disease, and obesity.
[00103] As used herein, the term "dose" refers a specified quantity of a pharmaceutical agent provided in a single administration. In certain embodiments, provided herein a dose can be administered in two or more boluses, tablets, or injections. For example, in certain embodiments, provided herein where subcutaneous administration is desired, the desired dose requires a volume not easily accommodated by a single injection. In such embodiments, two or more injections can be used to achieve the desired dose. In certain embodiments, provided herein a dose can be administered in two or more injections to minimize injection site reaction in a subject.
[00104] As used herein, the term "dosage unit" refers to a form in which a pharmaceutical agent is provided. In certain embodiments, provided herein a dosage unit is a vial containing lyophilized antisense oligonucleotide. In certain embodiments, provided herein a dosage unit is a vial containing reconstituted antisense oligonucleotide.
[00105] As used herein, a "dosing regimen" is a combination of doses designed to achieve one or more desired effects. In certain embodiments, a dose regimen is designed to provide a
therapeutic effect quickly. In certain embodiments a dose regimen is designed to reduce and undesired side effect, for example, liver toxicity.
[00106] As used herein, the term "diabetic dyslipidemia" or "Type II diabetes with
dyslipidemia" refers to a condition characterized by Type II diabetes, reduced HDL-C, elevated serum triglycerides, and elevated small, dense LDL particles.
[00107] As used herein, the term "duration" refers to the period of time during which an activity or event continues. In certain embodiments, the duration of treatment is the period of time during which doses of a pharmaceutical agent can be administered. For example, the duration of an induction phase is the period of time during which induction doses are
administered. For example, the duration of a maintenance phase is the period of time during which maintenance doses are administered.
[00108] As used herein, the term "early onset coronary heart disease" rafters to a diagnosis of coronary heart disease prior to age 50.
[00109] As used herein, the term "efficacy" refers to the ability to produce a desired effect. For example, efficacy of a lipid-lowering therapy can be reduction in the concentration of one or more of LDL-C, VLDL-C, IDL-C, non-HDL-C, ApoB , lipoprotein (a), or triglycerides.
[00110] The term "effective amount" as used herein refers to the amount of a therapy (e.g., a pharmaceutical composition provided herein) which is sufficient to reduce and/or prevent and/or manage a MACE or a symptom related thereto. This term also encompasses an amount necessary for the reduction or amelioration of the advancement a MACE or amelioration of the recurrence, or onset of a given MACE, and/or to improve or enhance the or therapeutic effect(s) of another therapy (e.g., a therapy other than the antisense oligonucleotide provided herein). In some embodiments, the effective amount of an antisense oligonucleotide provided herein is from about 200 mg/day to about 400 mg/day. In some embodiments, "effective amount" as used herein also refers to the amount of an antisense oligonucleotide provided herein to achieve a specified result (e.g., inhibition of ApoB synthesis). In certain embodiments, an effective amount is 200 mg/week. In some embodiments, the 200 mg/week is adminitered one time per week. In certain embodiments, the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week.
[00111] As used herein, "efficacy" means the ability to produce a desired effect. For example, efficacy of a lipid-lowering therapy may be reduction in MACE, or in the concentration of one or more of LDL-C, VLDL-C, IDL-C, non-HDL-C, ApoB, lipoprotein(a), or triglycerides.
[00112] As used herein, "elevated total cholesterol" means total cholesterol at a concentration in a subject at which lipid-lowering therapy is recommended, and includes, without limitation, elevated LDL-C", "elevated VLDL-C," "elevated IDL-C," and "elevated non-HDL-C." In certain embodiments, total cholesterol concentrations of less than 200 mg/dL, 200-239 mg/dL, and greater than 240 mg/dL are considered desirable, borderline high, and high, respectively. In certain embodiments, LDL-C concentrations of 100 mg/dL, 100-129 mg/dL, 130-159 mg/dL, 160-189 mg/dL, and greater than 190 mg/dL are considered optimal, near optimal/above optimal, borderline high, high, and very high, respectively.
[00113] As used herein, "elevated triglyceride" means concentrations of triglyceride in the serum or liver at which lipid-lowering therapy is recommended, and includes "elevated serum triglyceride" and "elevated liver triglyceride." In certain embodiments, serum triglyceride concentration of 150-199 mg/dL, 200-499 mg/dL, and greater than or equal to 500 mg/dL is considered borderline high, high, and very high, respectively.
[00114] As used herein, "elevated small dense LDL particles" means a concentration of small dense LDL particles in a subject at which lipid-lowering therapy is recommended.
[00115] As used herein, "elevated lipoprotein(a)" means a concentration of lipoprotein(a) in a subject at which lipid-lowering therapy is recommended.
[00116] The term "excipients" as used herein refers to inert substances which can be commonly used as a diluent, vehicle, preservatives, binders, or stabilizing agent for drugs and includes, but not limited to, proteins (e.g., serum albumin, etc.), amino acids (e.g., aspartic acid, glutamic acid, lysine, arginine, glycine, histidine, etc.), fatty acids and phospholipids (e.g., alkyl sulfonates, caprylate, etc.), surfactants (e.g., SDS, polysorbate, nonionic surfactant, etc.), saccharides (e.g., sucrose, maltose, trehalose, etc.) and polyols (e.g., mannitol, sorbitol, etc.). See, also, Remington's Pharmaceutical Sciences (1990) Mack Publishing Co., Easton, PA, which is hereby incorporated by reference in its entirety.
[00117] As used herein, the term "familial hypercholesterolemia (FH)" refers to an autosomal dominant metabolic disorder characterized by a mutation in the LDL-receptor (LDL-R) gene, markedly elevated LDL-C and premature onset of atherosclerosis. A diagnosis of familial
hypercholesterolemia is made when a subject meets one or more of the following criteria: genetic testing confirming 2 mutated LDL-receptor genes; genetic testing confirming one mutated LDL- receptor gene; document history of untreated serum LDL-cholesterol greater than 500 mg/dL; tendinous and/or cutaneous xanthoma prior to age 10 years; or, both parents have documented elevated serum LDL-cholesterol prior to lipid-lowering therapy consistent with heterozygous familial hypercholesterolemia.
[00118] As used herein, the term "fully complementary" refers to each nucleobase of a first nucleic acid is capable of pairing with each nucleobase of a second nucleic acid. In certain embodiments, provided herein a first nucleic acid is an antisense oligonucleotide and a target nucleic acid is a second nucleic acid. In some embodiments, an antisense oligonucleotide is a first nucleic acid and a target nucleic acid is a second nucleic acid.
[00119] A "gapmer", as described herein refers to an antisense oligonucleotide in which an internal position having a plurality of nucleotides that supports R aseH cleavage is positioned between external regions having one or more nucleotides that can be chemically distinct from the nucleosides of the internal region.
As used herein, the term "gap segment" refers to the plurality of nucleotides that make up the internal region of a gapmer that can support cleavage by the endonuclease RNaseH.
[00120] As used herein, the term "gap-widened" refers to an antisense oligonucleotide has a gap segment of 12 or more contiguous 2'-deoxyribonucleotides positioned between 5' and 3' wing segments having from one to six nucleotides having modified sugar moieties.
[00121] As used herein, the term "HMG-CoA reductase inhibitor" refers to a pharmaceutical agent that acts through the inhibition of the enzyme HMG-CoA reductase.
[00122] As used herein, the term "homozygous familial hypercholesterolemia" refers to a condition characterized by a mutation in both maternal and paternal LDL-R genes.
[00123] As used herein, the term, "heterozygous familial hypercholesterolemia" refers to a condition characterized by a mutation in either the maternal or paternal LDL-R gene.
[00124] As used herein, "high density lipoprotein-C (HDL-C)" means cholesterol associated with high density lipoprotein particles. Concentration of HDL-C in serum (or plasma) is typically quantified in mg/dL or nmol/L. "Serum HDL-C" and "plasma HDL-C" mean HDL-C in the serum and plasma, respectively.
[00125] As used herein, "history of coronary heart disease" means the occurrence of clinically evident coronary heart disease in the medical history of a subject or a subject's family member.
[00126] As used herein, the term "hyperglycemic" or "hyperglycemia," when used in reference to a condition of a subject refers to a transient or chronic abnormally high level of glucose present in the blood of a subject. The condition can be caused by a delay in glucose metabolism or absorption such that the subject exhibits glucose intolerance or a state of elevated glucose not typically found in normal subjects (e.g., in glucose-intolerant pre-diabetic subjects at risk of developing diabetes, or in diabetic subjects). Fasting plasma glucose levels for normoglycemia can be less than about 100 mg/dl, for impaired glucose metabolism, between about 100 and 126 mg/dl, and for diabetics greater than about 126 mg/dl.
As used herein, the term "hypercholesterolemia" refers to a condition characterized by elevated serum cholesterol. In certain embodiments, hypercholesterolemia includes, but is not limited to, polygenic hypercholesterolemia, heterozygous familial hypercholesterolemia, and a
homozgygous familial hypercholesterolemia.
[00127] As used herein, the term "hyperlipidemia" refers to a condition characterized by elevated serum lipids.
[00128] As used herein, the term "hypertriglyceridemia" refers to a condition characterized by elevated triglyceride levels.
[00129] As used herein, the term "hybridization" refers to the annealing of complementary nucleic acid molecules. In certain embodiments, provided herein complementary nucleic acid molecules include, but are not limited to, an antisense oligonucleotide and a nucleic acid target. In some embodiments, complementary nucleic acid molecules include, but are not limited to, an antisense oligonucleotide and a nucleic acid target
[00130] As used herein, the term "in combination" in the context of the administration of other therapies refers to the use of more than one therapy. The use of the term "in combination" does not restrict the order in which therapies can be administered to a subject with. A first therapy can be administered before (e.g., 1 minute, 45 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, 24 hours, 48 hours, 72 hours, 96 hours, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 8 weeks, or 12 weeks), concurrently, or after (e.g., 1 minute, 45 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, 24 hours, 48 hours, 72 hours, 96 hours, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 8 weeks, or 12
weeks) the administration of a second therapy to a subject which had, has, or is susceptible to a MACE. Any additional therapy can be administered in any order with the other additional therapies. In certain embodiments, the antisense oligonucleotide s can be administered in combination with one or more therapies (e.g., therapies that are not the antisense oligonucleotide s provided herein) that can be currently administered to prevent, treat, manage, and/or ameliorate a MACE.
[00131] As used herein, the term "improved cardiovascular outcome" refers to a reduction in the occurrence of major adverse cardiovascular events, or the risk thereof. Examples of major adverse cardiovascular events include, without limitation, reinfarction, stroke, cardiogenic shock, pulmonary edema, cardiac arrest, and atrial dysrhythmia. In certain of the methods of preventing a MACE provided herein, the MACE is death.
[00132] As used herein, "increased HDL-C" means an increase in serum HDL-C in a subject over time.
[00133] As used herein, "induction phase" means a dosing phase during which administration is initiated and steady state concentrations of active pharmaceutical agent are achieved in a target tissue. For example, an induction phase is a dosing phase during which steady state
concentrations of antisense oligonucleotide are achieved in liver
[00134] As used herein, "inflammation" refers to a localized protective response elicited by injury or destruction of tissues, which serves to destroy, dilute, or wall off both the injurious agent and the injured tissue. Inflammation is a factor involved in initiation, progression and intability of atheroscherotic plaques. Infammation can be moditored by serum inflammatory biomarkers. Elevation of these biomarkers associate with an increased risk of experiencing a cardiovascular event. See Clin Biochem. 2013 Oct;46(15): 1353-71 for examples of biomarkers for evaluation of inflammation for use in the methods described herein.
[00135] As used herein, the term "injection site reaction" refers to inflammation or abnormal redness of skin at a site of injection in a subject.
[00136] As used herein, "intermediate low density lipoprotein-cholesterol (IDL-C)" means cholesterol associated with intermediate density lipoprotein. Concentration of IDL-C in serum (or plasma) is typically quantified in mg/mL or nmol/L. "Serum IDL-C" and "plasma IDL-C" mean IDL-C in the serum or plasma, respectively.
[00137] As used herein, the term "internucleoside linkage" refers to the chemical bond or covalent linkage between adjacent nucleosides.
[00138] As used herein, the term "intravenous administration" refers to administration into a vein.
[00139] As used herein "ISIS 301012" and "mipomersen" mean a lipid-lowering agent that is an antisense oligonucleotide having the sequence "GCCTCAGTCTGCTTCGCACC" (SEQ ID NO:247), wherein each internucleoside linkage is a phosphorothioate internucleoside linkage, each cytosine is a 5-methylcytosine, nucleotides 6-15 are 2'-deoxynucleotides, and nucleotides 1-5 and 16-20 are 2'-0-methoxyethyl nucleotides. ISIS 301012 is complementary to nucleotides 3249-3268 of the sequence with GENBANK® Accession No. NM_000384.1.
Mipomersen treatment in patients does not induce systemic inflammatory response (Flaim et al, J Am Heart Assoc. 2014; 3: e00056; originally published March 13, 2014).
[00140] As used herein, "LDL apheresis" means a form of apheresis by which LDL-C is removed from blood. Typically, a subject's blood is removed from a vein, and separated into red cells and plasma. LDL-C is filtered out of the plasma prior to return of the plasma and red blood cells to the subject.
[00141] As used herein, the term "LDL-C target" refers to an LDL-C level that is desired following lipid-lowering therapy.
[00142] As used herein, "LDL/HDL ratio" means the ratio of LDL-C to HDL-C.
[00143] As used herein, the term "lipid-lowering therapy" refers to a therapeutic regimen provided to a subject to reduce one or more lipids in a subject. In certain embodiments, provided herein a lipid-lowering therapy is provided to reduce one or more of ApoB total cholesterol, LDL-C, VLDL-C, IDL-C, non-HDL-C, triglycerides, small dense LDL particles, and Lp (a) in a subject.
[00144] As used herein, the term, "lipid-lowering" refers to a reduction in one or more serum lipids in a subject over time.
[00145] As used herein, "lipid-lowering agent" means a pharmaceutical agent provided to a subject to achieve a lowering of lipids in the subject. For example, in certain embodiments, a lipid-lowering agent is provided to a subject to reduce one or more of ApoB, LDL-C, total cholesterol, and triglyerides.
[00146] As used herein, "lipid-lowering therapy" means a therapeutic regimen provided to a subject to reduce one or more lipids in a subject. In certain embodiments, a lipid-lowering therapy is provide to reduce one or more of ApoB, total cholesterol, LDL-C, VLDL-C, IDL-C, non-HDL-C, triglycerides, small dense LDL particles, and Lp(a) in a subject.
[00147] As used herein, "lipoprotein(a)" or "Lp(a)" means a lipoprotein particle that is comprised of LDL-C, an apolipoprotein(a) particle, and an apolipoproteinB-100 particle.
Elevated serum concentration is a risk factor of ACVD. Lp(a) levels are not modulated by diet, age, gender or physical activity.
[00148] As used herein, the term "linked nucleoside" refers to adjacent nucleosides which can be bonded together.
[00149] As used herein, the term "linked deoxynucleoside" refers to a nucleic acid base (A, G, C, T, U) substituted by deoxyribose linked by a phosphate ester to form a nucleotide.
[00150] As used herein, the term "low density lipoprotein-cholesterol (LDL-C)" refers to cholesterol associated with low density lipoprotein particles. Concentration of LDL-C in serum (or plasma) is typically quantified in mg/dL or nmol/L. "Serum LDL-C" and "plasma LDL-C" refers to LDL-C in the serum and plasma, respectively.
[00151] As used herein, the term "lipid-lowering agent" refers to a pharmaceutical agent provided to a subject to achieve a lowering of lipids in the subject. For example, in certain embodiments, provided herein a lipid-lowering agent is provided to a subject to reduce one or more of ApoB , LDL-C, total cholesterol, and triglycerides.
[00152] As used herein, "low LDL-receptor activity" means LDL-receptor activity that is not sufficiently high to maintain clinically acceptable levels of LDL-C in the bloodstream.
[00153] As used herein, "low HDL-C" means a concentration of HDL-C in a subject at which lipid-lowering therapy is recommended. In certain embodiments lipid-lowering therapy is recommended when low HDL-C is accompanied by elevations in non-HDL-C and/or elevations in triglyceride. In certain embodiments, HDL-C concentrations of less than 40 mg/dL are considered low. In certain embodiments, HDL-C concentrations of less than 50 mg/dL are considered low.
[00154] As used herein, the terms "MACE" and "major adverse cardiovascular event" are used interchangeably and refers to, for example, a myocardial infarction, reinfarction, a nonfatal myocardial infarction, stroke, unstable angina, cardiogenic shock, pulmonary edema,
cardiac arrest, atrial dysrhythmia, coronary revascularization, investigational angioplasty, interventional angioplasty, a percutaneous transluminal coronary angioplasty, percutaneous coronary intervention, a coronary artery bypass graft, or any combination thereof. In certain of the methods of preventing a MACE provided herein, the MACE is death. In some embodiments, the MACE is a non- fatal myocardial infarction, stroke, unstable angina, or revascularization procedure (e.g., a percutaneous coronary intervention (PCI), coronary artery bypass graft (CABG) surgery), or any combination thereof. In some embodiments, the MACE is a myocardial infarction. In some embodiments, the MACE is a stroke. In some embodiments, the MACE is an unstable angina. In some embodiments, the MACE is a revascularization procedure. In certain embodiments, the revascularization procedure is a percutaneous coronary intervention (PCI). In other embodiments, the revascularization procedure is a coronary artery bypass graft (CABG) surgery. In specific embodiments the MACE is non-fatal.
[00155] As used herein, "maintenance phase" means a dosing phase after target tissue steady state concentrations of drug have been achieved.
[00156] As used herein, the term "major risk factors" refers to factors that contribute to a high risk for coronary heart disease, and include without limitation cigarette smoking, hypertension, low HDL-C, family history of coronary heart disease, diabetes, obesity and age.
[00157] As used herein, the term "mixed dyslipidemia" refers to a condition characterized by elevated serum cholesterol and elevated serum triglycerides.
[00158] As used herein, the term "MTP inhibitor" refers to a pharmaceutical agent that inhibits the enzyme microsomal triglyceride transfer protein.
[00159] As used herein, the term "modified nucleotide" refers to a nucleotide having, independently, a modified sugar moiety, modified internucleoside linkage, or modified nucleobase.
[00160] As used herein, the term "modified nucleoside" refers to a nucleotide having, independently, a modified sugar moiety or modified nucleobase.
[00161] As used herein, the term "modified oligonucleotide" refers to an oligonucleotide comprising a modified internucleoside linkage, a modified sugar, and/or a modified nucleobase.
[00162] As used herein, the term "modified internucleoside linkage" refers to substitution and/or any change from a naturally occurring internucleoside bond. In certain instances, the modified internucleoside linkage refers to a phosphodiester internucleoside bond.
[00163] As used herein "modified sugar moiety" means a sugar moiety having a substitution and/or any change from a natural sugar moiety.
[00164] As used herein, the term "metabolic syndrome" refers to a condition characterized by a clustering of lipid and non-lipid cardiovascular risk factors of metabolic origin. In certain embodiments, provided herein metabolic syndrome is identified by the presence of any 3 of the following factors: waist circumference of greater than 102 cm in men or greater than 88 cm in women; serum triglyceride of at least 150 mg/dL; HDL-C less than 40 mg/dL in men or less than 50 mg/dL in women; blood pressure of at least 130/85 mmHg; and fasting glucose of at least 1 10 mg/dL.
[00165] As used herein, "maintenance dose" means a dose administered at a single administration during the maintenance phase. As used herein, "induction dose" means a dose administered at a single administration during the induction phase.
[00166] As used herein, the terms "manage," "managing," and "management" refer to the beneficial effects that a subject derives from a therapy (e.g., a therapeutic agent), which does not result in a cure of a the disease, disorder or symptom thereof, such as MACE. In certain embodiments, provided herein a subject is administered one or more therapies (e.g., therapeutic agents, such as an antisense oligonucleotide provided herein) to "manage" a MACE (e.g., myocardial infarction, stroke, unstable angina, and the like), or one or more symptoms thereof, so as to prevent the progression or worsening of the MACE or symptom thereof.
[00167] As used herein, the term "modified sugar moiety" refers to substitution and/or any change from a natural sugar moiety. For the purposes of this disclosure, a "natural sugar moiety" is a sugar moiety found in DNA (2'-H) or RNA (2'-OH).
[00168] As used herein, the term "modified sugar" refers to a substitution and/or any change from a natural sugar.
[00169] As used herein, the term "modified nucleobase" refers to any nucleobase other than adenine, cytosine, guanine, thymidine, or uracil. An "unmodified nucleobase" refers to the purine bases adenine (A) and guanine (G), and the pyrimidine bases thymine (T), cytosine (C) and uracil (U).
[00170] As used herein, the term "modified sugar moiety" refers to a sugar moiety having a substitution and/or any change from a natural sugar moiety.
[00171] As used herein, the term "motif refers to the pattern of unmodified and modified nucleosides in an antisense oligonucleotide.
[00172] As used herein, "modulation" refers to either an increase (stimulation) or a decrease (inhibition) in the expression of a gene.
[00173] As used herein, myocardial infarction" refers the loss of living heart muscle as a result of coronary artery occlusion (e.g., heart attack). Myocardial infarction or its related syndromes (acute coronary syndrome or unstable angina) usually occurs when an atheromatous plaque in a coronary artery ruptures, and the resulting clot obstructs the injured blood vessel. Perfusion of the muscular tissue that lies downstream from the blocked artery is lost. If blood flow is not restored within a few hours, the heart muscle dies. The term "myocardial infarction" focuses on the myocardium (the heart muscle) and the changes that occur in it due to the sudden deprivation of circulating blood. The main change is necrosis (death) of myocardial tissue.
[00174] As used herein "naturally occurring internucleoside linkage" means a 3' to 5' phosphodiester linkage.
[00175] As used herein, the term "natural sugar moiety" refers to a sugar moiety found in DNA (2'-H) or RNA (2'-OH).
[00176] As used herein, "a nucleic acid encoding human ApoB" means DNA encoding ApoB, or RNA transcribed from DNA encoding ApoB.
[00177] As used herein, "nucleobase" means a heterocyclic base moiety.
[00178] As used herein, "nucleoside" means a base-sugar combination.
[00179] As used herein, the term "nucleotide," nucleic acid" "nucleic acid molecule" and other similar terms can be used interchangeable and include DNA, RNA, mRNA and the like. As used herein, "nucleotide" means a nucleoside having a phosphate group covalently linked to the sugar portion of the nucleoside.
[00180] As used herein, the term "non-alcoholic fatty liver disease" refers to a condition characterized by fatty inflammation of the liver that is not due to excessive alcohol use (for example, alcohol consumption of over 20 g/day). In certain embodiments, provided herein nonalcoholic fatty liver disease is related to insulin resistance and the metabolic syndrome.
[00181] As used herein, the term "non-complementary nucleobase" refers to a nucleobase of first nucleic acid that is not capable of pairing with the corresponding nucleobase of a target nucleic acid.
[00182] As used herein, "non-familial hypercholesterolemia" means a condition characterized by elevated cholesterol that is not the result of a single gene mutation.
[00183] As used herein, "non-high density lipoprotein-cholesterol (Non-HDL-C)" means cholesterol associated with lipoproteins other than high density lipoproteins, and includes, without limitation, LDL-C, VLDL-C, and IDL-C.
[00184] As used herein, the term "nucleoside" refers to a nucleobase linked to a sugar.
[00185] As used herein, the term "nucleobase" refers to a heterocyclic base moiety capable of pairing with a base of another nucleic acid.
[00186] As used herein, the term "naturally occurring internucleoside linkage" refers to a 3 'to 5'phosphodiester linkage.
[00187] As used herein, the term "nucleotide" refers to a nucleoside having a phosphate group covalently linked to the sugar portion of the nucleoside.
[00188] As used herein, the term "nucleobase sequence" refers to the order of contiguous nucleobases independent of any sugar, linkage, and/or nucleobase modification.
[00189] As used herein, the term "oligomeric oligonucleotide" refers to a polymer of linked monomeric subunits which is capable of hybridizing to at least a region of an RNA molecule
[00190] The terms "optional" or "optionally" as used herein refers to that the subsequently described event or circumstance may or may not occur, and that the description includes, without limitation, instances where said event or circumstance occurs and instances in which it does not.
[00191] As used herein, the term "oligonucleotide" refers to a polymer of linked nucleosides each of which can be modified or unmodified, independent one from another.
[00192] As used herein, the term "oligonucleoside" refers to an oligonucleotide in which the internucleoside linkages do not contain a phosphorus atom.
[00193] As used herein, "Oxidized-LDL" or "Ox-LDL-C" means LDL-C that is oxidized following exposure to free radicals.
[00194] The term "parenteral administration," used herein refers to administration through injection or infusion. Parenteral administration includes, but is not limited to, subcutaneous administration, intravenous administration, or intramuscular administration
[00195] As used herein, the term "pharmaceutical agent" refers to a substance that provides a therapeutic benefit when administered to a subject. For example, in certain embodiments, provided herein an antisense oligonucleotide targeted to ApoB is a pharmaceutical agent.
[00196] As used herein, the term "pharmaceutical composition" refers a mixture of substances suitable for administering to a subject. For example, a pharmaceutical composition can comprise an antisense oligonucleotide and a sterile aqueous solution.
[00197] The term "pharmaceutically acceptable" as used herein refers to being approved by a regulatory agency of the Federal or a state government, or listed in the U.S. Pharmacopeia, European Pharmacopeia or other generally recognized Pharmacopeia for use in animals, and more particularly in humans.
[00198] As used herein, the term "pharmaceutically acceptable carrier" refers to a medium or diluent that does not interfere with the structure of the oligonucleotide. Certain, of such carries enable pharmaceutical compositions to be formulated as, for example, tablets, pills, dragees, capsules, liquids, gels, syrups, slurries, suspension and lozenges for the oral ingestion by a subject.
[00199] As used herein, the term "pharmaceutically acceptable salts" means physiologically and pharmaceutically acceptable salts of antisense oligonucleotide s, i.e., salts that retain the desired biological activity of the parent oligonucleotide and do not impart undesired
toxico logical effects thereto.
[00200] As used herein, the term "phosphorothioate linkage" refers to a linkage between nucleosides where the phosphodiester bond is modified by replacing one of the non-bridging oxygen atoms with a sulfur atom. A phosphorothioate linkage is a modified internucleoside linkage.
[00201] As used herein, "polygenic hypercholesterolemia" means a condition characterized by elevated cholesterol that results from the influence of a variety of genetic factors. In certain embodiments, polygenic hypercholesterolemia may be exacerbated by dietary intake of lipids.
[00202] As used herein, the term "portion" refers to a defined number of contiguous {i.e., linked) nucleobases of a target nucleic acid. In certain embodiments, provided herein a portion is a defined number of contiguous nucleobases of a target nucleic acid. In certain embodiments, provided herein a portion is a defined number of contiguous nucleobases of an antisense oligonucleotide.
[00203] As used herein, the terms "prevent," "preventing," and "prevention" refer to the total or partial development, and recurrence of a disease or disorder, such as a MACE, and/or symptoms related thereto, resulting from the administration of a therapy or combination of
therapies provided herein (e.g., a combination of therapeutic agents, such as an antisense oligonucleotide provided herein). Prevention can be, for example, in subjects predisposed to having a particular disorder(s).
[00204] As used herein, the term "prodrug" refers to a therapeutic agent that is prepared in an inactive form that is converted to an active form (i.e., drug) within the body or cells thereof by the action of endogenous enzymes or other chemicals and/or conditions.
[00205] As used herein, the term "reduced coronary heart disease risk" refers to a reduction in the likelihood that a subject will develop CHD. In certain embodiments, provided herein a reduction in CHD risk is measured by an improvement in one or more CHD risk factors, for example, a decrease in LDL-C levels.
[00206] As used herein, the term "salts" refers to physiologically and pharmaceutically acceptable salts of antisense oligonucleotide s, i.e., salts that retain the desired biological activity of the parent oligonucleotide and do not impart undesired toxico logical effects thereto.
[00207] As used herein, "serum lipids" include, but are not limited to, serum cholesterol and serum triglycerides.
[00208] As used herein, the term "side effects" encompasses unwanted and adverse effects of a therapy (e.g., a therapeutic agent). Unwanted effects are not necessarily adverse. An adverse effect from a therapy (e.g., therapeutic agent) might be harmful or uncomfortable or risky.
Examples of side effects include, diarrhea, cough, gastroenteritis, wheezing, nausea, vomiting, anorexia, abdominal cramping, fever, pain, loss of body weight, dehydration, alopecia, dyspenea, insomnia, dizziness, mucositis, nerve and muscle effects, fatigue, dry mouth, and loss of appetite, rashes or swellings at the site of administration, flu-like symptoms such as fever, chills and fatigue, digestive tract problems and allergic reactions. In certain embodiments, provided herein side effects include, without limitation, injection site reactions, liver function test abnormalities, renal function abnormalities, liver toxicity, renal toxicity, central nervous system abnormalities, and myopathies. For example, increased aminotransferase levels in serum can indicate liver toxicity or liver function abnormality. For example, increased bilirubin can indicate liver toxicity or liver function abnormality. Additional undesired effects experienced by patients can be numerous and known in the art. Many can be described in the Physician's Desk Reference (60th ed., 2006).
[00209] As used herein, the term "single-stranded modified oligonucleotide" refers to a modified oligonucleotide which is not hybridized to a complementary strand.
[00210] As used herein, the term "slows progression" refers to decrease in the development of the said disease.
[00211] As used herein, the terms "small dense LDL particles" and "small LDL particles" are used interchangeably herein and refer to a subclass of LDL particles characterized by a smaller, denser size compared to other LDL particles. In certain embodiments, provided herein intermediate LDL particles can be 23-27 nm in diameter. In certain embodiments, provided herein large LDL particles can be 21. 2-23 nm in diameter. In certain embodiments, provided herein small LDL particles can be 18-21. 2 nm in diameter. In certain embodiments, provided herein particle size is measured by nuclear magnetic resonance analysis.
[00212] As used herein, the term "small VLDL particle" refers to a subclass of VLDL particles characterized by a smaller, denser size compared to other VLDL particles. In certain embodiments, provided herein large VLDL particles can be greater than 60 nm in diameter. In certain embodiments, provided herein medium VLDL particles can be 35-60 nm in diameter. In certain embodiments, provided herein small VLDL particles can be 27-35 nm in diameter. In certain embodiments, provided herein particle size is measured by nuclear magnetic resonance analysis.
[00213] As used herein, the term "specifically hybridizable" refers to an antisense
oligonucleotide that hybridizes to a target nucleic acid to induce a desired effect, while exhibiting minimal or no effects on non-target nucleic acids. The terms "specifically
hybridizable" and "complementary" can be used to indicate a sufficient degree of
complementarity or precise pairing such that stable and specific binding occurs between the oligonucleotide and the DNA or R A target.
[00214] As used herein, the term "statin" refers to a pharmaceutical agent that inhibits the activity of HMG-CoA reductase. Statins typically lover serum LDL-C by 30-50%.
[00215] As used herein, the term "statin-resistant" means a subject who is currently on statin therapy but is not meting the target in LDL levels.
[00216] As used herein, "statin intolerant subject" means a subject who as a result of statin therapy experiences one or more of creatine kinase increases, liver function test abnormalities, muscle aches, or central nervous system side effects.
[00217] As used herein, the term "stringent hybridization conditions" refers to conditions under which a nucleic acid molecule, such as an antisense oligonucleotide, will hybridize to a target nucleic acid sequence, but to a minimal number of other sequences.
[00218] As used herein, the term "subcutaneous administration" refers to administration just below the skin.
[00219] As used herein, the terms "subject" and "patient" are used interchangeably. As used herein, a subject can be a mammal such as a non-primate (e.g., cows, pigs, horses, cats, dogs, rats, etc.) or a primate (e.g., monkey and human). In specific embodiments, the subject is a human. In one embodiment, the subject is a mammal (e.g., a human) with FH having a MACE. In another embodiment, the subject is a mammal (e.g., a human) with FH at risk of developing a MACE.
[00220] As used herein, "subject compliance" means adherence to a recommended or prescribed therapy by a subject.
[00221] As used herein, the term "a subject having elevated LDL-C levels" refers to a subject who has been identified by a medical professional (e.g., a physician) as having LDL-C levels near or above the level at which therapeutic intervention is recommended, according to guidelines recognized by medical professionals. Such a subject can also be considered "in need of treatment" to decrease LDL-C levels.
[00222] As used herein, the term "a subject having elevated ApoB -100 levels" refers to a subject who has been identified as having ApoB -100 levels near or below the level at which therapeutic intervention is recommended, according to guidelines recognized by medical professionals. Such a subject can also be considered "in need of treatment" to decrease ApoB - 100 levels.
[00223] As used herein, "surrogate markers of cardiovascular outcome" means indirect indicators of cardiovascular events, or the risk thereof. For example, surrogate markers of cardiovascular outcome include carotid intimal media thickness (CIMT). Another example of a surrogate marker of cardiovascular outcome includes atheroma size. Atheroma size may be determined by intravascular ultrasound (IVUS).
[00224] As used herein, the terms "target nucleic acid" and "nucleic acid encoding ApoB" encompass DNA encoding ApoB, RNA (including pre-mRNA and mRNA) transcribed from such DNA, and also cDNA derived from such RNA.
[00225] As used herein, the term "targeting" refers to the process of design and selection of an antisense oligonucleotide that will specifically hybridize to a target nucleic acid and induce a desired effect.
[00226] As used herein, the terms "target nucleic acid," "target RNA," "target RNA transcript" and "nucleic acid target" all mean any nucleic acid capable of being targeted by antisense oligonucleotides. As used herein, the terms "ApoB target nucleic acid" and "nucleic acid encoding ApoB" encompass nucleic acid, including, for example, DNA (including, for example, cDNA), RNA (including, for example pre-mRNA, and mRNA) transcribed from DNA encoding ApoB, and also cDNA derived from such RNA. In one embodiment, an ApoB target nucleic acid is the sequence of GENBANK® Accession No. NM_000384.1 , first deposited with GENBANK® on March 24, 1999.
[00227] As used herein, the term "targeted" refers to having a nucleobase sequence that will allow specific hybridization of an antisense oligonucleotide to a target nucleic acid to induce a desired effect. In certain embodiments, provided herein a desired effect is reduction of a target nucleic acid. In some embodiments, a desired effect is reduction of ApoB .
[00228] As used herein, the term "target region" refers to a fragment of a target nucleic acid to which an antisense oligonucleotide is targeted.
[00229] As used herein, the term "target segment" refers to the sequence of nucleotides of a target nucleic acid to which an antisense oligonucleotide is targeted. "5 ' target site" refers to the 5 '-most nucleotide of a target segment. "3 ' target site" refers to the 3 '-most nucleotide of a target segment.
[00230] As used herein, the term "therapy" refers to any protocol, method and/or agent that can be used in the prevention, management, treatment and/or amelioration of a MACE (e.g., myocardial infarction). In certain embodiments, the terms "therapies" and "therapy" refer to a biological therapy, supportive therapy, and/or other therapies useful in the prevention, management, treatment and/or amelioration of a MACE known to one of skill in the art such as medical personnel.
[00231] As used herein, the term "therapeutic agent" refers to any agent that can be used in the prevention, treatment, management a MACE and/or a symptom related thereto. In certain embodiments, the term "therapeutic agent" refers to an antisense oligonucleotide provided herein. In certain other embodiments, the term "therapeutic agent" refers to an agent other than
an antisense oligonucleotide provided herein. In certain embodiments, provided herein a therapeutic agent is an agent that is known to be useful for, or has been or is currently being used for the prevention, treatment, management of a MACE or one or more symptoms related thereto. In specific embodiments, the therapeutic agent is antisense oligonucleotide with having a nucleobase sequence of SEQ ID NO: 247.
[00232] As used herein, the term "therapeutic lifestyle change" refers to dietary and lifestyle changes intended to lower cholesterol and reduce the risk of developing heart disease, and includes recommendations for dietary intake of total daily calories, total fat, saturated fat, polyunsaturated fat, monounsaturated fat, carbohydrate, protein, cholesterol, insoluble fiber, as well as recommendations for physical activity.
[00233] As used herein, a "therapeutically effective amount" means an amount of a pharmaceutical agent that provides a therapeutic benefit to a subject. For example, a
therapeutically effective amount of an antisense oligonucleotide complementary to a nucleic acid encoding human ApoB is an amount that results, for example, in reduced LDL-C or a reduced incident of MACE.
[00234] As used herein, the term "total cholesterol" refers to all types of cholesterol, including, but not limited to, LDL-C, HDL-C, IDL-C and VLDL-C. Concentration of total cholesterol in serum (or plasma) is typically quantified in mg/dL or nmol/L.
[00235] As used herein, the terms "treat," "treatment" and "treating" refers to the reduction or of a disorder or condition (such as a MACE), or a symptom thereof, resulting from the administration of one or more therapies (including, but not limited to, the administration of one or more therapeutic agents, such as an antisense oligonucleotide provided herein). In specific embodiments, the therapeutic agent is an antisense oligonucleotide having a nucleobase sequence of SEQ ID NO: 247.
[00236] As used herein, "triglycerides" means lipids that are the triesters of glycerol. "Serum triglycerides" mean triglycerides present in serum. "Liver triglycerides" mean triglycerides present in liver tissue.
[00237] As used herein "unmodified" nucleobases mean the purine bases adenine (A) and guanine (G), and the pyrimidine bases thymine (T), cytosine (C) and uracil (U).
[00238] As used herein, the term "unmodified nucleotide" refers to a nucleotide composed of naturally occurring nucleobases, sugar moieties and internucleoside linkages. In certain
embodiments, provided herein an unmodified nucleotide is an R A nucleotide (i.e., β-D- ribonucleosides) or a DNA nucleotide (i.e., β-D-deoxyribonucleoside).
[00239] As used herein, the term "very low density lipoprotein-cholesterol (VLDL-C)" refers to cholesterol associated with very low density lipoprotein particles. Concentration of VLDL-C in serum (or plasma) is typically quantified in mg/dL or nmol/L. "Serum VLDL-C" and "plasma VLDL-C" refer to VLDL-C in the serum or plasma, respectively
[00240] As used herein, the term "wing segment" refers to the external region of a gapmer.
[00241] As used herein, the term "2'-0-methoxyethyl sugar moiety" refers to a 2 '-substituted furosyl ring having a 2'-0(CH2)2-OCH3 (2'-0-methoxyethyl or 2'-MOE) substituent group.
[00242] As used herein, the term "2'-0-methoxyethyl nucleotide" refers to a nucleotide comprising a 2'-0-methoxyethyl modified sugar moiety.
[00243] As used herein, the term "2'-0-methoxyethyl" refers to an O-methoxy-ethyl modification of the 2' position of a furosyl ring. A 2'-0-methoxyethyl modified sugar is a modified sugar.
[00244] As used herein, the term "5-methylcytosine" refers to a cytosine modified with a methyl group attached to the 5' position. A 5-methylcytosine is a modified nucleobase.
[00245] Hypercholesterolemia, and in particular, an elevated level of serum (or plasma) low density lipoprotein cholesterol (LDL-C), is associated with an increased risk of adverse cardiovascular events. Lipid lowering drug therapy, particularly with statins, is indicated to decrease the risk of cardiovascular events in most individuals with established atherosclerotic cardiovascular disease and in many who are at high risk.
[00246] Many patients treated with statins are considered statin-resistant because they fail to achieve adequate reduction of low density lipoprotein cholesterol (LDL-C) levels. Some patients are statin-intolerant because they are unable to tolerate statin therapy at all or to tolerate a full therapeutic statin dose because of adverse effects, particularly myopathy and increased activity of liver enzymes. (Nutr Metab Cardiovasc Dis. 2014 Oct; 24(10): 1057-66.)
[00247] While statins are the treatment of choice for lowering LDL-C in the majority of patients, including those with Type II diabeties, many patients retain a high CVD risk despite achieving the recommended LDL-C targets with statins.
[00248] Two forms of apolipoprotein B exist in mammals. ApoB-100 represents the full- length protein containing 4536 amino acid residues, synthesized primarily in the human liver
(Davidson and Shelness, Annu. Rev. Nutr., 2000, 20, 169-193). A truncated form known as apoB-48 is co linear with the amino terminal 2152 residues and is synthesized in the small intestine of all mammals (Davidson and Shelness, Annu. Rev. Nutr., 2000, 20, 169-193). In humans, apoB-48 circulates in association with chylomicrons and chylomicron remnants and these particles are cleared by a distinct receptor known as the LDL-receptor-related protein (Davidson and Shelness, Annu. Rev. Nutr., 2000, 20, 169-193). ApoB-48 can be viewed as an adaptation by which dietary lipid is delivered from the small intestine to the liver, while apoB- 100 participates in the transport and delivery of cholesterol (Davidson and Shelness, Annu. Rev. Nutr., 2000, 20, 169-193). ApoB is the major protein component of LDL and contains the domain required for interaction of this lipoprotein species with the LDL receptor. In addition, ApoB contains an unpaired cysteine residue which mediates an interaction with
apolipoprotein(a) and generates lipoprotein(a) or Lp(a), another distinct lipoprotein with atherogenic potential (Davidson and Shelness, Annu. Rev. Nutr., 2000, 20, 169-193). Elevated plasma levels of the ApoB-containing lipoprotein Lp(a) are associated with increased risk for atherosclerosis and its manifestations, which may include hypercholesterolemia (Seed et al, N. Engl. J. Med., 1990, 322, 1494-1499), myocardial infarction (Sandkamp et al, Clin. Chem., 1990, 36, 20-23), and thrombosis (Nowak-Gottl et al, Pediatrics, 1997, 99, El l).
[00249] Elevated plasma apoB, for example, as seen with familial hypercholesterolaemia (FH), is associated with an increased risk of theroclerotic cardiovascular disease (ACVD). In contrast, patients with the genetic disorder, hypobetalipoproteinemia, are protected against ACVD due to diminished levels of apoB and LDL-cholesterol. (Sahebjar and Watts,
Cardiovascular Drugs Ther. (2013) 27:559-567).
[00250] The methods described herein provide benefits for patients in need of treatment for MACE, these benefits include but are not limited to administering a compound targeted to ApoB that does not produce proinflammatory events and reduces other lipoproteins, for example Lp (a), is advantageous in patient populations where conventional lipid lowering strategies have not hit the target, helps reverse cardiac injury, long term use is therapeutically beneficial (e.g., over 12 months) and can be used in conjuction with other lipid lowering therapies.
1.1 Methods of Use
[00251] In one aspect, provided herein is a method of treating, preventing, or managing a major adverse cardiovascular event (MACE) in a hypercholesterolemia patient in need thereof, wherein the method comprises administering to the patient a therapeutically effect amount of an antisense olionucleotide complementary to a nucleic acid encoding human apolipoprotein B. MACE is MACE is a myocardial infarction, reinfarction, stroke, unstable angina, cardiogenic shock, pulmonary edema, cardiac arrest, coronary revascularization, investigational angioplasty, interventional angioplasty, a percutaneous transluminal coronary angioplasty, percutaneous coronary intervention, a coronary artery bypass graft, or any combination thereof. In a certain embodiment of this aspect, the antisense olionucleotide is mipomersen.
[00252] In another aspect, provided herein is a method for treating, preventing, or managing MACE in a patient with familial hypercholesterolemia (FH) in need thereof. In certain embodiments, the method includes administering to the patient a therapeutically effective amount of an antisense oligonucleotide having a nucleobase sequence of SEQ ID NO: 247 (e.g., mipomersen).
[00253] In some embodiments, the MACE is a myocardial infarction, reinfarction, stroke, unstable angina, cardiogenic shock, pulmonary edema, cardiac arrest, atrial dysrhythmia, coronary revascularization, investigational angioplasty, interventional angioplasty, a
percutaneous transluminal coronary angioplasty, percutaneous coronary intervention, a coronary artery bypass graft, or any combination thereof. In certain of the methods of preventing a MACE provided herein, the MACE is death.
[00254] In some embodiments, the MACE is a non-fatal myocardial infarction, stroke, unstable angina, or revascularization procedure (e.g., a percutaneous coronary intervention (PCI), coronary artery bypass graft (CABG) surgery), or any combination thereof. In some embodiments, the MACE is a myocardial infarction. In some embodiments, the MACE is a stroke. In some embodiments, the MACE is an unstable angina. IN some embodiments, the MACE is a revascularization procedure. In certain embodiments, the revascularization procedure is a percutaneous coronary intervention (PCI). In other embodiments, the
revascularization procedure is a coronary artery bypass graft (CABG) surgery. In specific embodiments the MACE is non-fatal.
[00255] In certain embodiments, the patient is a mammal (e.g., a rodent, monkey), such as a human. In some embodiments, the patient is homozygous for FH. In some embodiments, the
patient is heterozygous for FH. In other embodiments, the patient has coronary artery disease, severe hypercholesterolemia, or a high risk of CVD. In some embodiments, the patient was not previously treated for MACE. In other embodiments, the patient was previously treated for MACE.
[00256] In certain embodiments, a method provided herein reduces the occurrence of or prevents MACE in a patient having established CVD. In some embodiments, a method provided herein reduces the occurrence or prevents MACE in a patient at risk of CVD.
[00257] In certain embodiments, the antisense oligonucleotide is an antisense oligonucleotide, which is targeted to a nucleic acid encoding apolipoprotein B. In certain embodiments, the antisense oligonucleotide is 20 nucleobases in length. In some embodiments, the antisense oligonucleotide is an antisense oligonucleotide 20 nucleobases in length. In other embodiments, the antisense oligonucleotide has a nucleobase sequence comprising or consisting of the nucleobase sequence of SEQ ID NO: 247.
[00258] In certain embodiments, the antisense oligonucleotide includes a modified
internucleoside linkage, a modified sugar moiety, a modified nucleobase, or a combination thereof. In some embodiments, the modified sugar moiety is a 2'-0-methoxyethyl sugar moiety or a bicyclic sugar moiety. In certain embodiments, the modified internucleoside linkage is a phosphorothioate linkage. In some embodiments, the modified nucleobase is a 5-methylcytosine. In certain embodiments, the antisense oligonucleotide is a chimeric oligonucleotide.
[00259] The chimeric oligonucleotide can include a gap segment often linked 2'- deoxynucleotides. In some embodiments, the gap segment is positioned between wing segments. In other embodiments, each nucleoside of each wing segment includes a modified sugar moiety. In some embodiments, the modified sugar moiety is a 2'-0-methoxyethyl sugar moiety. In other embodiments, the gap segment is 10 2'-deoxynucleosides in length, and each wing segment includes from 1 to 8 2'-0-methoxyethyl. In other embodiments, each wing segment includes 2'- methoxyethoxyl nucleotides.
[00260] In certain embodiments, the antisense oligonucleotide is an antisense oligonucleotide 20 nucleotides in length having the nucleobase sequence of SEQ ID NO: 247, and can optionally include a 5-methylcytosine at nucleobases 2, 3, 5, 9, 12, 15, 17, 19, and 20. In some
embodiments, every internucleoside linkage is a phosphorothioate linkage, nucleotides 1-5 and
16-20 are 2'-0-methoxyethyl nucleotides, and nucleotides 6-15 are 2'-deoxynucleotides. In other embodiments, the antisense oligonucleotide is a pharmaceutically acceptable salt form thereof.
[00261] In certain embodiments, the antisense oligonucleotide is administered in a dosage to achieve at least a 60% reduction in MACE. In certain embodiments, the antisense
oligonucleotide is administered in a dosage to achieve at least a 65%, at least a 70%, at least a 75%, at least an 80%, at least an 85%, or at least a 90% reduction in MACE.
[00262] In certain embodiments, the antisense oligonucleotide is administered at 200 mg per day. In some embodiments, the antisense oligonucleotide is administered for at least 12 months.
[00263] In certain embodiments, the antisense oligonucleotide is administered at 200 mg/week. In some embodiments, the 200 mg/week is adminitered one time per week. In some embodiments, the subject is administered a single 200 mg/dose per week. In other embodiments, the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week. In some embodiments, the antisense oligonucleotide is administered for at least 12 months. In one embodiment, the antisense oligonucleotide is administered subcutaneously (s.c).
[00264] In certain embodiments, administration of the antisense oligonucleotide decreases total serum cholesterol, ApoB, serum LDL, serum VLDL, serum triglycerides, serum
apolipoprotein (a) and/or free fatty acids in the patient.
[00265] In certain embodiments, administration of the antisense oligonucleotide decreases LDL cholesterol. In some embodiments, the LDL level is reduced to about 100 mg/dl or lower, about 70 mg/dl or lower, or about 50 mg/dl or lower. In some embodiments, administration of the antisense oligonucleotide causes reductions in atherogenic lipoproteins in plasma.
[00266] In certain embodiments, the antisense oligonucleotide is administered with one or more additional oligonucleotide s selected from the group consisting of angiotensin-converting- enzyme inhibitors, angiotensin receptor blockers, renin inhibitors, HMG CoA reductase inhibitors, dihydropyridine calcium channel blockers, antiarrhythmic agents, azetidinone-based cholesterol absorption inhibitors, niacin, niacin derivatives, PPAR agonists, PPAR antagonists, bile acid sequestrants; and antiplatelet drugs; or any pharmaceutically acceptable esters, derivatives, conjugates, precursors or salts thereof.
[00267] In certain embodiments, the method reduces the occurrence of MACE as compared to a patient that has not been administered the antisense oligonucleotide. In some embodiments, the
patient that has not received the antisense oligonucleotide is a patient that was administered a placebo.
[00268] In certain embodiments, the method reduces the occurrence of MACE as compared to the occurrence of the MACE prior to administration of the antisense oligonucleotide. In some embodiments, the reduction in the occurrence of the MACE is compared to the occurrence of the MACE in the 24 months prior to administration of the antisense oligonucleotide.
[00269] In one aspect, provided herein are compositions including an antisense
oligonucleotide, and uses thereof for treating, reducing the occurrence of and/or preventing MACE. In some embodiments, the patient has FH. In some embodiments, MACE includes, but is not limited to, cardiac death, hospitalization for unstable angina, stroke, transient ischemic attack and hospitalization for peripheral artery disease. Additional oligonucleotide s useful in treating, reducing the occurrence of, and/or preventing CVD or the underlying risk factors associated with CVD can also be beneficially co-administered with antisense compositions and pharmaceutical formulations provided herein.
[00270] Guidelines for lipid-lowering therapy were established in 2001 by Adult Treatment Panel III (ATP III) of the National Cholesterol Education Program (NCEP), and updated in 2004 (Grundy et al., Circulation, 2004, 110, 227-239). The guidelines include obtaining a complete lipoprotein profile, typically after a 9 to 12 hour fast, for determination of LDL-C, total cholesterol, and HDL-C levels. According to the most recently established guidelines, LDL-C levels of 130-159 mg/dL, 160-189 mg/dL, and greater than or equal to 190 mg/dL can be considered borderline high, high, and very high, respectively. Total cholesterol levels of 200-239 and greater than or equal to 240 mg/dL can be considered borderline high and high, respectively. HDL-C levels of less than 40 mg/dL can be considered low.
[00271] In certain embodiments, the subject has been identified as in need of lipid-lowering therapy. In some embodiments, the subject has been identified as in need of lipid- lowering therapy according to the guidelines established in 2001 by ATP III of the NCEP, and updated in 2004 (Grundy et al., Circulation, 2004, 110, 227-239). In other embodiments, the subject in need of lipid-lowering therapy has LDL-C above 190 mg/dL. In certain embodiments, the subject in need of lipid-lowering therapy has LDL-C above 160 mg/dL. In some embodiments, the subject in need of lipid-lowering therapy has LDL-C above 130 mg/dL. In other embodiments, the subject in need of lipid-lowering therapy has LDL-C above 100 mg/dL. In some embodiments,
the subject in need of lipid-lowering therapy should maintain LDL-C below 160 mg/dL. In certain embodiments, the subject in need of lipid- lowering therapy should maintain LDL-C below 130 mg/dL. In other embodiments, the subject in need of lipid-lowering therapy should maintain LDL-C below 100 mg/dL. In some embodiments, the subject should maintain LDL-C below 70 mg/dL or even below 50 mg/dL.
[00272] In some embodiments, provided herein are methods for reducing ApoB in a subject. In other embodiments, provided herein are methods for reducing ApoB -containing lipoprotein in a subject. In certain embodiments, provided herein are methods for reducing LDL-C in a subject. In other embodiments, provided herein are methods for reducing VLDL-C in a subject. In certain embodiments, provided herein are methods for reducing LDL-C in a subject. In other
embodiments, provided herein are methods for reducing non-HDL-C in a subject. In some embodiments, provided herein are methods for reducing Lp(a) in a subject. In other
embodiments, provided herein are methods for reducing serum triglyceride in a subject. In certain embodiments, provided herein are methods for reducing liver triglyceride in a subject. In some embodiments, provided herein are methods for reducing Ox-LDL-C in a subject. In other embodiments, provided herein are methods for reducing small LDL particles in a subject. In certain embodiments, provided herein are methods for reducing small VLDL particles in a subject. In certain embodiments, provided herein are methods for reducing phospholipids in a subject. In other embodiments, provided herein are methods for reducing oxidized phospholipids in a subject. Any combination of the two, three four or more of the foregoing methods is also contemplated herein.
[00273] In certain embodiments, provided herein can be compositions comprising an antisense oligonucleotide for use, e.g., in the prevention, management, treatment and/or amelioration of a MACE. In some embodiments, the MACE is a myocardial infarction. In other embodiments, the MACE is a reinfarction. In yet other embodiments, the MACE is a stroke.
[00274] In some embodiments, the MACE is a myocardial infarction. In other embodiments, the MACE is a reinfarction. In yet other embodiments, the MACE is a stroke. In certain embodiments, the MACE is unstable angina. In some embodiments, the MACE is cardiogenic shock. In other embodiments, the MACE is pulmonary edema. In other embodiments, the MACE is cardiac arrest. In certain embodiments, the MACE is atrial dysrhythmia. In some embodiments, the MACE is coronary revascularization. In other embodiments, the MACE is
investigational angioplasty. In certain embodiments, the MACE is interventional angioplasty. In other embodiments, the MACE is percutaneous transluminal coronary angioplasty. In certain embodiments, the MACE is percutaneous coronary intervention. In some embodiments, the MACE is coronary artery bypass graft. In other embodiments, the MACE is death.
[00275] In some embodiments, the MACE is a non-fatal myocardial infarction, stroke, unstable angina, or revascularization procedure (e.g., a percutaneous coronary intervention (PCI), coronary artery bypass graft (CABG) surgery), or any combination thereof. In some embodiments, the MACE is a myocardial infarction. In some embodiments, the MACE is a stroke. In some embodiments, the MACE is an unstable angina. IN some embodiments, the MACE is a revascularization procedure. In certain embodiments, the revascularization procedure is a percutaneous coronary intervention (PCI). In other embodiments, the
revascularization procedure is a coronary artery bypass graft (CABG) surgery. In specific embodiments the MACE is non-fatal.
[00276] In certain embodiments, provided herein MACE can range from mild to severe.
[00277] Exemplary risk factors for MACE include hypercholesterolemia, mixed dyslipidemia, atherosclerosis, coronary heart disease, a history of coronary heart disease, early onset coronary heart disease, acute coronary syndrome, one or more risk factors for coronary heart disease, type II diabetes, type II diabetes with dyslipidemia, dyslipidemia, hypertriglyceridemia,
hyperlipidemia, hyperfattyacidemia, hepatic steatosis, nonalcoholic steatohepatitis, or nonalcoholic fatty liver disease. Other symptoms can include obesity, unhealthy diet, and harmful use of alcohol, smoking, age, or family history.
[00278] In certain embodiments, provided herein are compositions comprising an antisense oligonucleotide for use in the prevention, management, treatment and/or amelioration of a MACE, such as, but not limited to, a myocardial infarction, or a risk factor thereof.
[00279] The compositions and methods of administering and dosing are also useful in the other methods provided herein. As discussed in more detail elsewhere herein, a composition provided herein can be used either alone or in combination with other oligonucleotide s or compositions.
[00280] In some embodiments, provided herein are methods for treating, preventing, or managing MACE in a subject, comprising administering to the subject an effective amount of an
antisense oligonucleotide. In some embodiments, levels of atherogenic lipoproteins in plasma can also be decreased in the subject. In certain embodiments, the subject has FH.
[00281] The subject administered an antisense oligonucleotide is, in certain embodiments, a mammal such as non-primate (e.g., cows, pigs, horses, cats, dogs, rats, etc.) or a primate (e.g., a monkey, such as a cynomolgus monkey, or a human). In a specific embodiment, the subject is a human.
[00282] In certain embodiments, the subject is a human with FH. In another embodiment, the subject has heterozygous FH. In another embodiment, the subject has homozygous FH.
[00283] In another embodiment, the subject has a premature CVD. In more specific embodiments, the subject has heterozygous FH, and premature CVD. In another embodiment, the subject has homozygous FH and can have premature CVD.
[00284] In certain embodiments, the subject has severe hypercholesterolemia. In another embodiment, the subject has a high risk of CVD. In a specific embodiment, the subject has not previously been treated for MACE. In another embodiment, the subject has been treated for MACE.
[00285] In another embodiment, the methods provided herein can be useful in the treatment of MACE in patients with FH having established CVD. In some embodiments, the patient has not had a prior myocardial infarction, and/or does not have any of the underlying risk factors or diseases that cause CVD. These can include, but are not limited to, hypertension, dyslipidemia, obesity and/or diabetes.
[00286] In another embodiment, the subject is susceptible or at risk of MACE. In another embodiment, the subject has a heightened risk of MACE.
[00287] In another embodiment, the subject has a body mass index of approximately 30.4+ 4 6 kg/m2.
[00288] In yet another embodiment, the subject has one or more risk factors, including but not limited to, hypercholesterolemia, mixed dyslipidemia, atherosclerosis, coronary heart disease, a history of coronary heart disease, early onset coronary heart disease, acute coronary syndrome, one or more risk factors for coronary heart disease, type II diabetes, type II diabetes with dyslipidemia, dyslipidemia, hypertriglyceridemia, hyperlipidemia, hyperfattyacidemia, hepatic steatosis, nonalcoholic steatohepatitis, or non-alcoholic fatty liver disease. Other symptoms can include obesity, unhealthy diet, and harmful use of alcohol, smoking, age, or family history.
[00289] In yet another specific embodiment, the subject has symptoms of FH, including, but not limited to xanthomas, xanthelasmas, or corneal arcus.
[00290] In another embodiment, the subject has an LDL-C level above 100 mg/dL, above 130 mg/dL, above 160 mg/dL, or above 190 mg/dL.
[00291] In another embodiment, the subject is a subject with FH receiving a lipid- lowering medication. In certain embodiments, the subject has FH.
[00292] In another embodiment, the subject that has not previously received an antisense provided herein. In other embodiments, the patient previously received a placebo. In another embodiment, the subject is administered a placebo for 6 months, followed by administration of the antisense oligonucleotide for at least one year.
[00293] In another embodiment, the subject is first administered blinded, an antisense oligonucleotide, followed by open labeled treatment with the antisense oligonucleotide.
[00294] In another embodiment, the subject is administered an antisense oligonucleotide concurrently with implementing lifestyle changes, including but not limited to, regular exercise, or reduced fat intake diet.
[00295] Also provided herein, are methods for reducing the occurrence of MACE, comprising administering the antisense inhibitor (e.g., mipomersen) to a subject. In some embodiments, the subject has FH. In another embodiment, provided herein are methods for reducing the occurrence of or preventing MACE in a subject having established CVD. In another embodiment, provided herein are methods for reducing the occurrence of MACE is a subject at risk of CVD.
[00296] In certain embodiments, of the methods provided herein, administration of the antisense oligonucleotide reduces the occurrence of MACE as compared to a patient that has not been administered with the antisense oligonucleotide. In other embodiments, the method further reduces the occurrence of MACE as compared to the occurrence of MACE prior to
administration of the antisense oligonucleotide. In another embodiment the reduction in the occurrence of MACE is compared to the occurrence of the MACE in the 24 months prior to the administration of the antisense oligonucleotide.
[00297] In another embodiment, administering the antisense oligonucleotide or a composition thereof reduces the risk that a subject will develop MACE by at least about 10%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least
about 50%, at least about 60%>, at least about 70%>, at least about 80%>, at least about 90%>, or more, compared to the risk of developing MACE in the absence of such treatment.
[00298] In yet another embodiment, administering the antisense oligonucleotide or a composition thereof reduces the risk of MACE proportionally to the reduction in LDL cholesterol levels.
[00299] In another embodiment, a method of decreasing LDL-C levels comprises selecting a subject in need of a decrease in LDL-C levels, and administering to the subject a therapeutically effective amount of an antisense oligonucleotide. In another embodiment, a method of reducing coronary heart disease risk includes selecting a subject having elevated LDL-C levels and one or more additional indicators of coronary heart disease risk, and administering to the subject a therapeutically effective amount of an antisense oligonucleotide. In other embodiments, a method for reducing MACE includes selecting a subject having elevated LDL-C levels and one or more MACE prior treatment. In a further embodiment, a method for reducing MACE includes selecting a subject having elevated LDL-C levels and no previous MACE. In a further embodiment, a method for reducing MACE includes selecting a subject having risk factors associated with MACE, as described herein.
[00300] In certain embodiments, of the various methods described provided herein, the LDL- C level is from 100-129 mg/dL, from 130-159 mg/dL, from 160-189 mg/dL, or greater than or equal to 190 mg/dL.
[00301] In one embodiment, administration of a therapeutically effective amount of an antisense oligonucleotide can be accompanied by monitoring of LDL-C levels in the serum of a subject, to determine a subject's response to administration of the antisense oligonucleotide. A subject's response to administration of the antisense oligonucleotide is used by a physician to determine the amount and duration of therapeutic intervention.
[00302] In one embodiment, administration of an antisense oligonucleotide results in LDL-C levels below 190 mg/dL, below 160 mg/dL, below 130 mg/dL, below 100 mg/dL, below 70 mg/dL, or below 50 mg/dL. In another embodiment, administration of an antisense
oligonucleotide decreases LDL-C by at least 15%, by at least 25%, by at least 50%, by at least 60%, by at least 70%, by at least 75%, by at least 80%, by at least 85%, by at least 90%, or by at least 95%.
[00303] In another embodiment, a subject having elevated LDL-C levels can also exhibit reduced HDL-C levels and/or elevated total cholesterol levels. Accordingly, in one embodiment a therapeutically effective amount of an antisense oligonucleotide is administered to a subject having elevated LDL-C levels, which also has reduced HDL-C levels and/or elevated total cholesterol levels.
[00304] Subjects having elevated LDL-C levels can also exhibit elevated triglyceride levels. Accordingly, in one embodiment a therapeutically effective amount of an antisense
oligonucleotide is administered to a subject having elevated LDL-C levels, and also having elevated triglyceride levels.
[00305] Measurements of cholesterol, lipoproteins and triglycerides can be obtained using serum or plasma collected from a subject. Methods of obtaining serum or plasma samples can be routine, as are methods of preparation of the serum samples for analysis of cholesterol, triglycerides, and other serum markers. A physician can determine the need for therapeutic intervention in cases where more or less aggressive LDL-lowering therapy is needed. The practice of the methods herein can be applied to any altered guidelines provided by the NCEP, or other entities that establish guidelines for physicians used in treating any of the diseases or conditions listed herein, for determining coronary heart disease risk and diagnosing metabolic syndrome.
[00306] In certain embodiments, provided herein does a pharmaceutical composition including an antisense oligonucleotide is for use in therapy. In certain embodiments, the therapy is the reduction of LDL-C, ApoB, VLDL-C, IDL-C, non-HDL-C, Lp(a) , serum triglyceride, liver triglyceride, Ox-LDL-C, small LDL particles, small VLDL, phospholipids, or oxidized phospholipids in a subject. In certain embodiments, the therapy is the treatment of
hypercholesterolemia, mixed dyslipidemia, atherosclerosis, a risk of developing atherosclerosis, coronary heart disease, acute coronary syndrome, a history of coronary heart disease, early onset coronary heart disease, one or more risk factors for coronary heart disease, type II diabetes, type II diabetes with dyslipidemia, dyslipidemia, hypertriglyceridemia, hyperlipidemia,
hyperfattyacidemia, hepatic steatosis, non-alcoholic steatohepatitis, or non-alcoholic fatty liver disease. In additional embodiments, the therapy is the reduction of CHD risk. In certain aspects, the therapy is prevention of atherosclerosis. In certain embodiments, the therapy is the prevention of coronary heart disease. Measurements of cholesterol, lipoproteins and triglycerides
can be obtained using serum or plasma collected from a subject. Methods of obtaining serum or plasma samples can be routine, as are methods of preparation of the serum samples for analysis of cholesterol, triglycerides, and other serum markers. A physician can determine the need for therapeutic intervention for subjects in cases where more or less aggressive LDL-lowering therapy is needed. The practice of the methods herein can be applied to any altered guidelines provided by the NCEP, or other entities that establish guidelines for physicians used in treating any of the diseases or conditions listed herein, for determining coronary heart disease risk and diagnosing metabolic syndrome. In some embodiments, the therapy is the treatment, prevention, or management of MACE.
[00307] In other embodiments of the various methods provided herein, the method comprises measuring the levels of one or more of certain analytes, such as proteins and metabolites, in a plasma or serum sample derived from a blood sample from that subject and then administering an antisense oligonucleotide in a subject likely to experience a MACE.
[00308] In certain embodiments of the various methods provided herein, the antisense oligonucleotide is administered at 200 mg/week. In some embodiments, the 200 mg/week is adminitered one time per week. In some embodiments, the subject is administered a single 200 mg/dose per week. In other embodiments, the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week. In some embodiments, the antisense oligonucleotide is administered for at least 12 months. In one embodiment, the antisense oligonucleotide is administered subcutaneously (s.c).
[00309] Also provided herein are methods comprising administering to a subject a
pharmaceutical composition comprising an antisense oligonucleotide complementary to a nucleic acid encoding human apolipoprotein B-100, wherein the administering comprises an induction phase, wherein a dose of the antisense oligonucleotide ranging from 100-300 mg is administered once per week for at least 13 weeks. In some embodiments, the induction phase is followed by a maintenance phase, wherein a dose of the antisense oligonucleotide ranging from 80-200 mg is administered once per week or once every two weeks for as long as needed, effective, and/or tolerated. See for example, Flaim et al., "A phase I study in Healthy Volunteers to Evaluate the Pharokinetics, Safety and th Tolerability of Mipomersen in Three dosing Regimens" Poster Presentation (201 1) and US Patent Appliction US20100297105.
[00310] In certain embodiments, the antisense oligonucleotide is administered at 200 mg/week. In some embodiments, the 200 mg/week is adminitered one time per week. In some embodiments, the subject is administered a single 200 mg/dose per week. In other embodiments, the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week. In some embodiments, the antisense oligonucleotide is administered for at least 12 months. In one embodiment, the antisense oligonucleotide is administered subcutaneously (s.c).
[00311] In certain embodiments, the dose administered in the induction phase is a 100 mg dose and the dose administered in the maintenance phase is a 200 mg dose administered once per week. In certain embodiments, the dose administered in the induction phase is a 200 mg dose, and the dose administered in the maintenance phase is a 300 mg dose administered once per week. In certain embodiments, the dose administered in the induction phase is a 100 mg dose and the dose administered in the maintenance phase is a 200 mg dose administered once per week, and wherein the tolerability or the effectiveness of the antisense oligonucleotide are assessed during or at the end of the induction period, or a portion thereof once per week during the maintenance phase. In certain embodiments, the dose administered in the induction phase is a 200 mg dose and the dose administered in the maintenance phase is a 300 mg dose
administered once per week, and wherein the tolerability or the effectiveness of the antisense oligonucleotide are assessed during or at the end of the induction period, or a portion thereof.
[00312] In certain embodiments, the dose administered in the induction phase is a 100 mg dose and the dose administered in the maintenance phase is a 100 mg dose administered once every two weeks, and wherein the tolerability or the effectiveness of the antisense
oligonucleotide are assessed during or at the end of the induction period, or a portion thereof. In certain embodiments, the dose administered in the induction phase is a 200 mg dose and the dose administered in the maintenance phase is a 200 mg dose administered once every two weeks, and wherein the tolerability or the effectiveness of the antisense oligonucleotide are assessed during or at the end of the induction period, or a portion thereof. In certain embodiments, the dose administered in the induction phase is from 100 mg to 200mg and the dose administered in the maintenance phase is from 200 mg to 300 mg and is administered once per week.
[00313] In certain embodiments, said administering comprises parenteral administration. In certain embodiments, said parenteral administration comprises subcutaneous administration. In certain embodiments, each induction dose and each maintenance dose comprises a single
injection. In certain embodiments, each induction dose and each maintenance dose independently comprise two or more injections. In certain embodiments, the methods further comprise assessing the tolerability or effectiveness of the antisense oligonucleotide during or at the end of the induction period, or a portion thereof. In certain embodiments, the tolerability and the effectiveness of the antisense oligonucleotide are assessed
[00314] In certain embodiments, the tolerability of the antisense oligonucleotide is assessed by monitoring a rate of decrease of ApoB concentration in the plasma of said subject. In certain embodiments, the tolerability of the antisense oligonucleotide is assessed by monitoring ApoB concentration in the plasma of said subject. In certain embodiments, the tolerability of the antisense oligonucleotide is assessed by monitoring a rate of decrease of ApoB concentration and ApoB concentration in the plasma of said subject. In certain embodiments, the tolerability of the antisense oligonucleotide is assessed by monitoring ALT concentrations in the liver of the subject. In certain embodiments, the tolerability of the antisense oligonucleotide is assessed by monitoring ANT concentrations in the liver of said subject. In certain embodiments, the tolerability of the antisense oligonucleotide is assessed by monitoring bilirubin concentrations in the plasma of the subject.
[00315] In certain embodiments, a rate of decrease in the ApoB concentration greater than about 30 mg/dL*day indicates that the subject is not tolerating administration of the antisense oligonucleotide. In certain embodiments, an ApoB concentration less than about 60 mg/dL indicates that the subject is not tolerating administration of the antisense oligonucleotide. In certain embodiments, a rate of decrease in the ApoB concentration greater than about 30 mg/dL*day and an ApoB concentration less than about 60 mg/dL indicates that the subject is not tolerating administration of the antisense oligonucleotide. In certain embodiments, the dose of antisense oligonucleotide is reduced following an indication that administration of said antisense oligonucleotide is not tolerated. In certain embodiments, the frequency of administration of antisense oligonucleotide is reduced following an indication that administration of said antisense oligonucleotide is not tolerated. In certain embodiments, the dose of antisense oligonucleotide is increased following an indication that administration of said antisense oligonucleotide is tolerated. In certain embodiments, the frequency of administration of antisense oligonucleotide is increased following an indication that administration of said antisense oligonucleotide is tolerated.
[00316] In certain embodiments, the effectiveness of the antisense oligonucleotide is assessed by monitoring ApoB, LDL-C, VLDL-C, IDL-C, non-HDL-C, serum triglycerides, liver triglycerides, Lp(a), Ox-LDL-C, or small dense LDL particle concentration in the plasma of said subject. In certain embodiments, a reduction of ApoB, LDL-C, VLDL-C, IDL-C, non-HDL-C, serum triglycerides, liver triglycerides, Lp(a), Ox-LDL-C, or small dense LDL particle concentration indicates that the antisense oligonucleotide is effective. In other embodiments, the effectiveness of the antisense oligonucleotide is assessed by monitoring MACE in said subject. In certain embodiments, a reduction of in MACE indicates that the antisense oligonucleotide is effective. In certain embodiments, the dose of antisense oligonucleotide is reduced following an indication that administration of said antisense oligonucleotide is effective. In certain embodiments, the dose of antisense oligonucleotide is increased following an indication that administration of said antisense oligonucleotide is not effective. In certain embodiments, the frequency of administration of antisense oligonucleotide is reduced following an indication that administration of said antisense oligonucleotide is effective. In certain embodiments, the frequency of administration of antisense oligonucleotide is increased following an indication that administration of said antisense oligonucleotide is not effective.
[00317] In certain embodiments, said subject has elevated ApoB prior to said administering. In certain embodiments, said subject has elevated cholesterol prior to said administering. In certain embodiments, said elevated cholesterol is selected from elevated total cholesterol, elevated LDL-cholesterol, elevated VLDL-cholesterol, elevated IDL-cholesterol, or elevated non-HDL cholesterol prior to said administering. In certain embodiments, said subject has elevated Lp(a) prior to said administering. In certain embodiments, said subject has elevated serum triglycerides prior to said administering. In certain embodiments, said subject has elevated liver triglycerides prior to said administering. In certain embodiments, said subject has elevated small dense LDL particles prior to said administering. In some embodiments, said subject has MACE prior to said administrating. In other embodiments, said subject is at risk of developing MACE prior to said administrating.
[00318] In certain embodiments, said subject has hypercholesterolemia. In certain
embodiments, said subject has polygenic hypercholesterolemia. In certain embodiments, said subject has familial hypercholesterolemia. In certain embodiments, said subject has homozygous familial hypercholesterolemia. In certain embodiments, said subject has heterozygous familial
hypercholesterolemia. In certain embodiments, said subject has mixed dyslipidemia. In certain embodiments, said subject has a history of coronary heart disease.
[00319] In certain embodiments, said subject has one or more risk factors for coronary heart disease. In certain embodiments, said one or more risk factors is selected from age, smoking, hypertension, low HDL-cholesterol, and a family history of early coronary heart disease. In certain embodiments, said subject has type II diabetes with dyslipidemia. In certain
embodiments, said subject has been treated by a statin. In certain embodiments, said subject failed to meet LDL-cholesterol target on statin therapy. In certain embodiments, said subject did not comply with recommended therapy. In certain embodiments, said subject experienced side effects of stain therapy. In certain embodiments, said subject has low LDL-receptor activity. In certain embodiments, said subject failed to meet LDL-cholesterol target on lipid-lowering therapy prior to said administering.
[00320] In certain embodiments, said maintenance phase comprises administering said pharmaceutical composition throughout the lifetime of the subject. In certain embodiments, the duration of said maintenance phase is one year, 2 years, 3 years, 4 years, 5 years, 6 years, 7 years, 8 years, 9 years, 10 years, 1 1 years, 12 years, 13 years, 14 years, 15 years, 16 years, 17 years, 18 years, 19 years, or 20 years. In certain embodiments, the duration of said maintenance phase is from one week to twenty years.
[00321] In certain embodiments, the induction dose is 100 mg. In certain embodiments, the induction dose is 200 mg. In certain embodiments, the induction dose is 300 mg. In certain embodiments, the maintenance dose is 100 mg. In certain embodiments, the maintenance dose is 200 mg.
[00322] In certain embodiments, the antisense oligonucleotide is administered at 200 mg/week. In some embodiments, the 200 mg/week is adminitered one time per week. In some embodiments, the subject is administered a single 200 mg/dose per week. In other embodiments, the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week. In some embodiments, the antisense oligonucleotide is administered for at least 12 months. In one embodiment, the antisense oligonucleotide is administered subcutaneously (s.c).
[00323] In certain embodiments, said administering of said pharmaceutical composition results in antisense oligonucleotide plasma trough levels between 5 and 100 ng/mL. In certain embodiments, said administering of said pharmaceutical composition results in antisense
oligonucleotide plasma trough levels between 5 and 50 ng/mL. In certain embodiments, said administering of said pharmaceutical composition results in antisense oligonucleotide plasma trough levels between 10 and 40 ng/mL. In certain embodiments, said administering of said pharmaceutical composition results in antisense oligonucleotide plasma trough levels between 15 and 35 ng/mL. In certain embodiments, said administering of said pharmaceutical composition results in antisense oligonucleotide plasma trough levels between 20 and 30 ng/mL.
[00324] In certain embodiments, said administering of said pharmaceutical composition results in ApoB reduction of at least 10%. In certain embodiments, said ApoB reduction is at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%), at least 90%>, at least 95%, or at least 100%). In certain embodiments, said ApoB reduction is between 10%> and 80%>, between 20%> and 70%>, between 30%> and 60%>, or between 30% and 70%.
[00325] In certain embodiments, said administering of said pharmaceutical composition results in a LDL-cholesterol reduction of at least 10%. In certain embodiments, said LDL- cholesterol reduction is at least 15%, at least 20%>, at least 25%, at least 30%>, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100%. In certain embodiments, said administering of said pharmaceutical composition results in a LDL- cholesterol reduction between 10% and 100%. In certain embodiments, said administering of said pharmaceutical composition results in a VLDL-cholesterol reduction of at least 10%. In certain embodiments, said VLDL-cholesterol reduction is at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100%. In certain embodiments, said administering of said pharmaceutical composition results in a VLDL-cholesterol reduction bwtween 10% to 100%.
[00326] In certain embodiments, said administering of said pharmaceutical composition results in Lp(a) reduction of at least 10%. In certain embodiments, said Lp(a) reduction is at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%o, at least 90%, at least 95%, or at least 100%). In certain embodiments, said
administering of said pharmaceutical composition results in Lp(a) reduction between 10% and 100%. In certain embodiments, said administering of said pharmaceutical composition results in a small LDL-particle reduction of at least 10%. In certain embodiments, said small LDL-particle reduction is at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%), at least 50%>, at least 55%, at least 60%>, at least 65%, at least 70%>, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100%. In certain embodiments, said administering of said pharmaceutical composition results in a small LDL-particle reduction between 10% and 100%.
[00327] In certain embodiments, said administering of said pharmaceutical composition results in a small LDL-particle reduction of at least 10%. In certain embodiments, said non- HDL-cholesterol reduction is at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100%. In certain embodiments, said administering of said pharmaceutical composition results in a small LDL- particle reduction between 10%> and 100%.
[00328] In certain embodiments, said administering of said pharmaceutical composition results in reduced coronary heart disease risk in the subject. In certain embodiments, said administering of said pharmaceutical composition slows or stops the progression of
atherosclerosis in the subject. In certain embodiments, said administering of said pharmaceutical composition reduces the risk of developing atherosclerosis in the subject. In certain
embodiments, said administering of said pharmaceutical composition results in improved cardiovascular outcome the subject. In certain embodiments, said improved cardiovascular outcome is a reduced risk of major cardiovascular adverse events in the subject. In certain embodiments, said improved cardiovascular outcome is improved carotid intimal media thickness. In certain embodiments, said improved cardiovascular outcome is improved atheroma thickness. In certain embodiments, said improved cardiovascular outcome is increased HDL- cholesterol.
[00329] In certain embodiments, said administering results in lipid lowering. In certain embodiments, said administering results in reductions in LDL-cholesterol, triglycerides, or small LDL particles, or a combination thereof. In certain embodiments, said administering results in an improved LDL/HDL ratio. In certain embodiments, said administering results in an HDL-
cholesterol level increase of at least 10%. In certain embodiments, said HDL-cholesterol level increase is 15%, at least 20%>, at least 25%, at least 30%>, at least 35%, at least 40%>, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100%. In certain embodiments, said
administering of said pharmaceutical composition results in a liver triglyceride level decrease of at least 10%.
[00330] In certain embodiments, said liver triglyceride level decrease is at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%), at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100%. In certain embodiments, said administering of said pharmaceutical composition results in a hepatic cholesterol ester reduction of at least 10%. In certain embodiments, said reduced hepatic cholesterol ester reduction 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100%.
[00331] In certain embodiments, the methods further comprise co-administration of said pharmaceutical composition and at least one additional therapy. In certain embodiments, said co-administration is simultaneous. In certain embodiments, said pharmaceutical composition is administered prior to administration of said additional therapy. The method of claim 100, wherein said pharmaceutical composition is administered after administration of said additional therapy. In certain embodiments, the interval between administration of said pharmaceutical composition and said additional therapy is about one hour, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 9 hours, about 10 hours, about 11 hours, or about 12 hours. In certain embodiments, the interval between administration of said pharmaceutical composition and said additional therapy is about 1 day, about 1 week, about 2 weeks, about 3 weeks, about 4 weeks, about 5 weeks, about 6 weeks, about 7 weeks, about 8 weeks, about 9 weeks, about 10 weeks, about 11 weeks, or about 12 weeks. In certain embodiments, the interval between administration of said pharmaceutical composition and said additional therapy is about 1 month, about 2 months, about 3 months, about 4 months, about 5 months, or about 6 months.
[00332] In certain embodiments, the methods further comprise administering a single additional therapy. In certain embodiments, the methods further comprise administering at 2 or more additional therapies. In certain embodiments, said additional therapy is a lipid-lowering therapy. In certain embodiments, said additional lipid-lowering therapy is therapeutic lifestyle change. In certain embodiments, said additional lipid-lowering therapy is an HMG-CoA reductase inhibitor. In certain embodiments, the HMG-CoA reductase inhibitor is selected from atorvastatin, rosuvastatin, or simvastatin. In certain embodiments, said additional lipid-lowering therapy is a cholesterol absorption inhibitor. In certain embodiments, the cholesterol absorption inhibitor is ezetimibe. In certain embodiments, said 2 or more additional therapies comprises an HMG-CoA reductase inhibitor and a cholesterol absorption inhibitor. In certain embodiments, said HMG-CoA reductase inhibitors is simvastatin and said cholesterol absorption inhibitor is ezetimibe. In certain embodiments, said additional lipid-lowering therapy is LDL apheresis. In certain embodiments, said administering of said additional therapy comprises intravenous administration. In certain embodiments, said additional lipid-lowering therapy is an MTP inhibitor. In certain embodiments, the additional lipid-lowering therapy is a PCSK9 inhibitor.
[00333] In certain embodiments, said pharmaceutical composition comprises a
pharmaceutically acceptable excipient. In certain embodiments, said pharmaceutically acceptable excipient is saline. In certain embodiments, the dose of the antisense oligonucleotide concentration is administered at a concentration of about 50 mg/ml, about 75 mg/ml, about 100 mg/ml, about 125 mg/ml, about 150 mg/ml, about 175 mg/ml, about 200 mg/ml, about 225 mg/ml, or about 250 mg/ml. In a specifc embodiment, the dose of the antisense oligonucleotide concentration is administered at a concentration of about 200 mg/ml.
[00334] In certain embodiments, the antisense oligonucleotide comprises at least one modified sugar moiety. In certain embodiments, the modified sugar moiety comprises a 2'-methoxyethyl sugar moiety. In certain embodiments, the modified sugar moiety comprises a bicyclic nucleic acid sugar moiety.
[00335] In certain embodiments, the antisense oligonucleotide comprises a 2'- deoxynucleotide gap segment positioned between wing segments, wherein each nucleotide of the wing segments comprises a modified sugar moiety. In certain embodiments, each nucleotide of the wing segment comprises a 2'-0-methoxyethyl sugar moiety. In certain embodiments, each nucleotide of the wing segment comprises a bicyclic nucleic acid sugar moiety. In certain
embodiments, the gap segment comprises ten nucleotides and each wing segment comprises five nucleotides.
[00336] In certain embodiments, at least one internucleoside linkage is a phosphorothioate internucleoside linkage. In certain embodiments, each internucleoside linkage is a
phosphorothioate internucleoside linkage.
[00337] In certain embodiments, at least one cytosine is a 5-methylcytosine. In certain embodiments, each cytosine is a 5-methylcytosine.
[00338] In certain embodiments, the antisense oligonucleotide is at least 90% complementary to a nucleic acid encoding human ApoB. In certain embodiments, the antisense oligonucleotide is at least 95% complementary to a nucleic acid encoding human ApoB. In certain
embodiments, the antisense oligonucleotide is 100% complementary to a nucleic acid encoding human ApoB.
[00339] In certain embodiments, the nucleic acid encoding human ApoB comprises a sequence identified by Accession number NM 000384.1.
[00340] In certain embodiments, the antisense oligonucleotide comprises 12 to 30 nucleotides. In certain embodiments, the antisense oligonucleotide comprises 15 to 25 nucleotides. In certain embodiments, the antisense oligonucleotide comprises 17 to 23 nucleotides. In certain embodiments, the antisense oligonucleotide comprises 18 to 22 nucleotides. In certain embodiments, the antisense oligonucleotide comprises 19 to 21 nucleotides. In certain embodiments, the antisense oligonucleotide comprises 20 nucleotides.
[00341] In some embodiments, the antisense nucleotide comprises the nucleobase sequence of SEQ ID NO: 247. In some embodiments, the antisense nucleotide consists of the nucleobase sequence of SEQ ID NO: 247. In certain embodiments, the antisense oligonucleotide is ISIS 301012 (mipomersen). In some embodiments, the antisense nucleotide comprises the nucleobase sequence selected from the group of SEQ ID NOS: 124-515. In some embodiments, the antisense nucleotide consists of the nucleobase sequence selected from the group of SEQ ID NOS: 124-515. In some embodiments, the antisense nucleotide targets a region on ApoB that comprises a nucleobase sequence selected from the group of SEQ ID NOS: 516-804.
[00342] In certain embodiments, provided are methods comprising administering to a subject a pharmaceutical composition comprising an antisense oligonucleotide complementary to a nucleic acid encoding human ApoB, wherein the administering comprises an induction phase
comprising at least one induction dose and a maintenance phase comprising at least one maintenance dose, wherein the duration of the induction phase is greater than five weeks.
[00343] In certain embodiments, provided are methods comprising administering to a subject a pharmaceutical composition comprising an antisense oligonucleotide complementary to a nucleic acid encoding human ApoB, wherein the administering comprises an induction phase comprising at least one induction dose and a maintenance phase comprising at least one maintenance dose, wherein an induction dose is less than a maintenance dose.
[00344] In certain embodiments, provided are methods comprising administering to a subject a pharmaceutical composition comprising an antisense oligonucleotide complementary to a nucleic acid encoding human ApoB, wherein the administering comprises an induction phase comprising at least one induction dose.
[00345] In certain embodiments, provided are methods comprising administering to a subject having familial hypercholesterolemia a pharmaceutical composition comprising an antisense oligonucleotide complementary to a nucleic acid encoding human ApoB, wherein the administering comprises an induction phase comprising at least one induction dose and a maintenance phase comprising at least one maintenance dose, wherein the induction phase is at least 8 weeks.
[00346] In certain embodiments, provided are methods including administering to a subject a pharmaceutical composition comprising an antisense oligonucleotide complementary to a nucleic acid encoding human apolipoprotein B-100, wherein the administering comprises a maintenance phase comprising at least one maintenance dose.
1.2 Antisense oligonucleotides or compounds
[00347] Oligomeric compounds, particularly antisense oligonucleotides, can be used in the various methods provided herein for modulating the function of nucleic acid molecules encoding ApoB, ultimately modulating the amount of ApoB produced. This is accomplished by providing antisense compounds which specifically hybridize with one or more nucleic acids encoding ApoB. The specific hybridization of an oligomeric compound with its target nucleic acid interferes with the normal function of the nucleic acid. The functions of DNA to be interfered with include replication and transcription. The functions of RNA to be interfered with include all vital functions such as, for example, translocation of the RNA to the site of protein translation, translation of protein from the RNA, splicing of the RNA to yield one or more mRNA species,
and catalytic activity which can be engaged in or facilitated by the R A. The overall effect of such interference with target nucleic acid function is modulation of the expression of ApoB. In this context, inhibition is the form of modulation of gene expression and mRNA is a target.
[00348] It is to target specific nucleic acids for antisense. "Targeting" an antisense compound to a particular nucleic acid, is a multistep process. The process usually begins with the identification of a nucleic acid sequence whose function is to be modulated. This can be, for example, a cellular gene (or mRNA transcribed from the gene) whose expression is associated with a particular disorder or disease state, or a nucleic acid molecule from an infectious agent. Provided herein, the target is a nucleic acid molecule encoding ApoB. The targeting process also includes determination of a site or sites within this gene for the antisense interaction to occur such that the desired effect, e.g., detection or modulation of expression of the protein, will result. Within this context, an intragenic site is the region encompassing the translation initiation or termination codon of the open reading frame (ORF) of the gene. Since, as is known in the art, the translation initiation codon is typically 5 '-AUG (in transcribed mRNA molecules; 5'-ATG in the corresponding DNA molecule), the translation initiation codon is also referred to as the "AUG codon," the "start codon" or the "AUG start codon". A minority of genes have a translation initiation codon having the RNA sequence 5'-GUG, 5'-UUG or 5'-CUG, and 5'-AUA, 5'-ACG and 5'-CUG have been shown to function in vivo. Thus, the terms "translation initiation codon" and "start codon" can encompass many codon sequences, even though the initiator amino acid in each instance is typically methionine (in eukaryotes) or formylmethionine (in prokaryotes). It is also known in the art that eukaryotic and prokaryotic genes can have two or more alternative start codons, any one of which can be preferentially utilized for translation initiation in a particular cell type or tissue, or under a particular set of conditions. In the context, "start codon" and "translation initiation codon" refer to the codon or codons that can be used in vivo to initiate translation of an mRNA molecule transcribed from a gene encoding ApoB, regardless of the sequence(s) of such codons.
[00349] It is also known in the art that a translation termination codon (or "stop codon") of a gene can have one of three sequences, i.e., 5'-UAA, 5'-UAG and 5'-UGA (the corresponding DNA sequences can be 5'-TAA, 5 '-TAG and 5'-TGA, respectively). The terms "start codon region" and "translation initiation codon region" refer to a portion of such an mRNA or gene that encompasses from about 25 to about 50 contiguous nucleotides in either direction {i.e., 5' or 3')
from a translation initiation codon. Similarly, the terms "stop codon region" and "translation termination codon region" refer to a portion of such an mRNA or gene that encompasses from about 25 to about 50 contiguous nucleotides in either direction (i.e., 5' or 3') from a translation termination codon.
[00350] The open reading frame (ORF) or "coding region," which is known in the art to refer to the region between the translation initiation codon and the translation termination codon, is also a region which can be targeted effectively. Other target regions include the 5 ' untranslated region (5'UTR), known in the art to refer to the portion of an mRNA in the 5' direction from the translation initiation codon, and thus including nucleotides between the 5 ' cap site and the translation initiation codon of an mRNA or corresponding nucleotides on the gene, and the 3' untranslated region (3 'UTR), known in the art to refer to the portion of an mRNA in the 3 ' direction from the translation termination codon, and thus including nucleotides between the translation termination codon and 3' end of an mRNA or corresponding nucleotides on the gene. The 5' cap of an mRNA comprises an N7-methylated guanosine residue joined to the 5 '-most residue of the mRNA via a 5 '-5' triphosphate linkage. The 5' cap region of an mRNA is considered to include the 5' cap structure itself as well as the first 50 nucleotides adjacent to the cap. The 5' cap region can also be a target region.
[00351] Although some eukaryotic mRNA transcripts can be directly translated, many contain one or more regions, known as "introns," which can be excised from a transcript before it is translated. The remaining (and therefore translated) regions can be known as "exons" and can be spliced together to form a continuous mRNA sequence. mRNA splice sites, i.e., intron-exon junctions, can also be target regions, and can be particularly useful in situations where aberrant splicing is implicated in disease, or where an overproduction of a particular mRNA splice product is implicated in disease. Aberrant fusion junctions due to rearrangements or deletions are also targets. It has also been found that introns can also be effective, and therefore , target regions for antisense compounds targeted, for example, to DNA or pre -mRNA.
[00352] Once one or more target sites have been identified, oligonucleotides can be chosen which can be sufficiently complementary to the target, i.e., hybridize sufficiently well and with sufficient specificity, to give the desired effect.
[00353] "Hybridization" refers to hydrogen bonding, which can be Watson-Crick, Hoogsteen or reversed Hoogsteen hydrogen bonding, between complementary nucleoside and nucleotide
bases. For example, adenine and thymine can be complementary nucleobases which pair through the formation of hydrogen bonds. "Complementary," as used herein, refers to the capacity for precise pairing between two nucleotides. For example, if a nucleotide at a certain position of an oligonucleotide is capable of hydrogen bonding with a nucleotide at the same position of a DNA or RNA molecule, then the oligonucleotide and the DNA or RNA can be considered to be complementary to each other at that position. The oligonucleotide and the DNA or RNA can be complementary to each other when a sufficient number of corresponding positions in each molecule can be occupied by nucleotides which can hydrogen bond with each other. It is understood in the art that the sequence of an antisense compound need not be 100%
complementary to that of its target nucleic acid to be specifically hybridizable. An antisense compound is specifically hybridizable when binding of the compound to the target DNA or RNA molecule interferes with the normal function of the target DNA or RNA to cause a loss of utility, and there is a sufficient degree of complementarity to avoid non-specific binding of the antisense compound to non-target sequences under conditions in which specific binding is desired, i.e., under physiological conditions in the case of in vivo assays or therapeutic treatment, and in the case of in vitro assays, under conditions in which the assays can be performed.
[00354] Antisense and other compounds which hybridize to the target and inhibit expression of the target can be identified through experimentation, and the sequences of these compounds can be herein below identified as certain embodiments. The target sites to which these sequences can be complementary can be herein below referred to as "active sites" and can be therefore sites for targeting. Therefore another embodiment encompasses compounds which hybridize to these active sites.
[00355] Antisense compounds can be commonly used as research reagents and diagnostics. For example, antisense oligonucleotides, which are able to inhibit gene expression with exquisite specificity, can be often used by those of ordinary skill to elucidate the function of particular genes. Antisense compounds can also be used, for example, to distinguish between functions of various members of a biological pathway. Antisense modulation has, therefore, been harnessed for research use.
[00356] For use in kits and diagnostics, the antisense compounds provided herein, either alone or in combination with other antisense compounds or therapeutics, can be used as tools in
differential and/or combinatorial analyses to elucidate expression patterns of a portion or the entire complement of genes expressed within cells and tissues.
[00357] Expression patterns within cells or tissues treated with an antisense compound can be compared to control cells or tissues not treated with antisense compounds and the patterns produced can be analyzed for differential levels of gene expression as they pertain, for example, to disease association, signaling pathway, cellular localization, expression level, size, structure or function of the genes examined. These analyses can be performed on stimulated or unstimulated cells and in the presence or absence of other compounds which affect expression patterns.
[00358] Examples of methods of gene expression analysis known in the art include DNA arrays or microarrays (Brazma and Vilo, FEBS Lett., 2000, 480, 17-24; Celis, et al, FEBS Lett., 2000, 480, 2-16), SAGE (serial analysis of gene expression) (Madden, et al., Drug Discov.
Today, 2000, 5, 415-425), READS (restriction enzyme amplification of digested cDNAs) (Prashar and Weissman, Methods EnzymoL, 1999, 303, 258-72), TOGA (total gene expression analysis) (Sutcliffe, et al., Proc. Natl. Acad. Sci. U.S. A., 2000, 97, 1976-81), protein arrays and proteomics (Celis, et al., FEBS Lett., 2000, 480, 2-16; Jungblut, et al., Electrophoresis, 1999, 20, 2100-10), expressed sequence tag (EST) sequencing (Celis, et al., FEBS Lett., 2000, 480, 2-16; Larsson, et al., J. BiotechnoL, 2000, 80, 143-57), subtractive RNA fingerprinting (SuRF) (Fuchs, et al, Anal. Biochem., 2000, 286, 91-98; Larson, et al, Cytometry, 2000, 41, 203-208), subtractive cloning, differential display (DD) (Jurecic and Belmont, Curr. Opin. Microbiol., 2000, 3, 316-21), comparative genomic hybridization (Carulli, et al, J. Cell Biochem. SuppL, 1998, 31, 286-96), FISH (fluorescent in situ hybridization) techniques (Going and Gusterson, Eur. J. Cancer, 1999, 35, 1895-904) and mass spectrometry methods (reviewed in (To, Comb. Chem. High Throughput Screen, 2000, 3, 235-41).
[00359] The specificity and sensitivity of antisense is also harnessed by those of skill in the art for therapeutic uses. Antisense oligonucleotides have been employed as therapeutic moieties in the treatment of disease states in animals and man. Antisense oligonucleotide drugs, including ribozymes, have been safely and effectively administered to humans and numerous clinical trials can be presently underway. It is thus established that oligonucleotides can be useful therapeutic modalities that can be configured to be useful in treatment regimes for treatment of cells, tissues and animals, especially humans.
[00360] As described herein, the term "oligonucleotide" refers to an oligomer or polymer of ribonucleic acid (R A) or deoxyribonucleic acid (DNA) or mimetics thereof. Thus, this term includes oligonucleotides composed of naturally-occurring nucleobases, sugars and covalent internucleoside (backbone) linkages (RNA and DNA) as well as oligonucleotides having non- naturally-occurring portions which function similarly (oligonucleotide mimetics).
Oligonucleotide mimetics can be often over native forms because of desirable properties such as, for example, enhanced cellular uptake, enhanced affinity for nucleic acid target and increased stability in the presence of nucleases.
[00361] While antisense oligonucleotides can be an exemplary form of an antisense compound provided herein, other oligomeric antisense compounds, including but not limited to oligonucleotide mimetics such as those described below can also be contemplated. The antisense compounds provided herein can comprise from about 8 to about 50 nucleobases (i.e., from about 8 to about 50 linked nucleosides). Antisense compounds can be antisense oligonucleotides, even more are those comprising from about 12, about 14, about 20 to about 30 nucleobases. Antisense compounds include ribozymes, external guide sequence (EGS) oligonucleotides (oligozymes), and other short catalytic RNAs or catalytic oligonucleotides which hybridize to the target nucleic acid and modulate its expression. In certain embodiments, the antisense compound is non- catalytic oligonucleotide, i.e., is not dependent on a catalytic property of the oligonucleotide for its modulating activity. Antisense compounds can include double-stranded molecules wherein a first strand is stably hybridized to a second strand.
[00362] In certain embodiments, provided are pharmaceutical compositions including one or more different oligonucleotides. In certain such embodiments, those pharmaceutical
compositions comprise an antisense oligonucleotide complementary to a nucleic acid encoding human ApoB. In certain embodiments, such pharmaceutical compositions comprise ISIS 301012. ISIS 301012 is a pharmaceutical agent that, when administered to a subject, results in dose-dependent reductions of ApoB, ApoB-containing lipoproteins, including but not limited to LDL-C, triglycerides, and Lp(a). ISIS 301012 results in efficacy when administered alone, and also results in efficacy when
[00363] In certain embodiments, pharmaceutical compositions comprise an oligonucleotide having complementary to a target nucleic acid. In certain such embodiments, a sufficient number of nucleobases of the oligonucleotide can undergo hydrogen bonding with corresponding
nucleobases in a target nucleic acid such that a desired effect occurs. In certain such embodiments, a desired effect is antisense inhibition of a target nucleic acid. In certain such embodiments, a desired effect is antisense inhibition of ApoB. In certain such embodiments, least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%), at least 98%> or at least 99% of the nucleobases of an oligonucleotide can undergo hydrogen bonding with a corresponding nucleobase of a target nucleic acid. In certain such embodiments, 100% of the nucleobases of an oligonucleotide can undergo hydrogen bonding with a corresponding nucleobase of a target nucleic acid. In these embodiments, oligonucleotides are fully complementary (i.e., 100% complementary) to a target nucleic acid. In certain such embodiments, oligonucleotides are fully complementary to a nucleic acid encoding ApoB.
[00364] In certain embodiments, a nucleic acid encoding human ApoB is ApoB mRNA. In certain embodiments, such ApoB mRNA may or may not include some or all exons.
[00365] In certain embodiments, oligonucleotides are 12 to 30 nucleotides in length, i.e., the oligonucleotides are from 12 to 30 linked nucleotides. In certain such embodiments,
oligonucleotides are 15 to 25 nucleotides in length. In certain such embodiments,
oligonucleotides are 17 to 23 nucleotides in length. In certain such embodiments,
oligonucleotides are 18 to 22 nucleotides in length. In certain such embodiments,
oligonucleotides are 19 to 21 nucleotides in length. In certain such embodiments,
oligonucleotides are 20 nucleotides in length.
[00366] In certain embodiments, oligonucleotides comprise a percent identity to a particular nucleotide sequence. An oligonucleotide has identity to another oligonucleotide if the nucleobases of each oligonucleotide have the same nucleobase pairing ability. In certain such embodiments, an oligonucleotide has 90% identity to another oligonucleotide. In certain such embodiments, an oligonucleotide has 95% identity to another oligonucleotide. In certain such embodiments, an oligonucleotide has 100% identity to another oligonucleotide. In certain such embodiments, the identity is over the full-length of the oligonucleotide. In certain such embodiments, the identity is to a portion of an oligonucleotide.
[00367] In certain embodiments, oligonucleotides comprise chemical modifications. In certain such embodiments, mmodifications to oligonucleotides encompass substitutions or changes to internucleoside linkages, sugar moieties, or nucleobases. Modified oligonucleotides are often preferred over native forms because of desirable properties such as, for example, enhanced
cellular uptake, enhanced affinity for nucleic acid target, increased stability in the presence of nucleases, or increased inhibitory activity.
[00368] In certain embodiments, chemically modified nucleosides may also be employed to increase the binding affinity of a shortened or truncated antisense oligonucleotide for its target nucleic acid.
[00369] As is known in the art, a nucleoside is a base-sugar combination. The base portion of the nucleoside is normally a heterocyclic base. The two most common classes of such
heterocyclic bases can be the purines and the pyrimidines. Nucleotides can be nucleosides that further include a phosphate group covalently linked to the sugar portion of the nucleoside. For those nucleosides that include a pentofuranosyl sugar, the phosphate group can be linked to either the 2', 3' or 5' hydroxyl moiety of the sugar. In forming oligonucleotides, the phosphate groups covalently link adjacent nucleosides to one another to form a linear polymeric compound. In turn the respective ends of this linear polymeric structure can be further joined to form a circular structure; however, open linear structures can be generally. Within the oligonucleotide structure, the phosphate groups can be commonly referred to as forming the internucleoside backbone of the oligonucleotide. The normal linkage or backbone of RNA and DNA is a 3' to 5' phosphodiester linkage.
[00370] Specific examples of antisense compounds include oligonucleotides containing modified backbones or non-natural internucleoside linkages. As defined in this specification, oligonucleotides having modified backbones include those that retain a phosphorus atom in the backbone and those that do not have a phosphorus atom in the backbone. For the purposes of this specification, and as sometimes referenced in the art, modified oligonucleotides that do not have a phosphorus atom in their internucleoside backbone can also be considered to be
oligonucleosides.
[00371] Exemplary modified oligonucleotide backbones include, for example,
phosphorothioates, chiral phosphorothioates, phosphorodithioates, phosphotriesters, aminoalkyl- phosphotriesters, methyl and other alkyl phosphonates including 3-alkylene phosphonates, 5'- alkylene phosphonates and chiral phosphonates, phosphinates, phosphoramidates including 3'- amino phosphoramidate and aminoalkylphosphoramidates, thionophosphoramidates,
thionoalkylphosphonates, thionoalkylphosphotriesters, selenophosphates and boranophosphates having normal 3 '-5' linkages, 2 '-5' linked analogs of these, and those having inverted polarity
wherein one or more internucleotide linkages is a 3' to 3', 5' to 5' or 2' to 2' linkage, oligonucleotides having inverted polarity comprise a single 3' to 3' linkage at the 3 '-most internucleotide linkage i.e., a single inverted nucleoside residue which can be basic (the nucleobase is missing or has a hydroxyl group in place thereof). Various salts, mixed salts and free acid forms can also be included.
[00372] In certain embodiments, oligonucleotides comprise one or more modified, i.e. non- naturally occurring, internucleoside linkages. In certain such embodiments, oligonucleotides having modified internucleoside linkages include internucleoside linkages that retain a phosphorus atom as well as internucleoside linkages that do not have a phosphorus atom.
Representative phosphorus containing internucleoside linkages include, but are not limited to, phosphodiesters, phosphotriesters, methylphosphonates, phosphoramidate, and
phosphorothioates. Methods of preparation of phosphorous-containing and non-phosphorous- containing linkages are well known.
[00373] Representative United States patents that teach the preparation of the above phosphorus-containing linkages include, but are not limited to, U.S. Pat. Nos.: 3,687,808; 4,469,863; 4,476,301; 5,023,243; 5,177,196; 5,188,897; 5,264,423; 5,276,019; 5,278,302;
5,286,717; 5,321,131; 5,399,676; 5,405,939; 5,453,496; 5,455,233; 5,466,677; 5,476,925;
5,519,126; 5,536,821; 5,541,306; 5,550,111 ; 5,563,253; 5,571,799; 5,587,361; 5,194,599;
5,565,555; 5,527,899; 5,721,218; 5,672,697 and 5,625,050, and each of which is herein incorporated by reference.
[00374] Modified oligonucleotide backbones that do not include a phosphorus atom therein can have backbones that can be formed by short chain alkyl or cycloalkyl internucleoside linkages, mixed heteroatom and alkyl or cycloalkyl internucleoside linkages, or one or more short chain heteroatomic or heterocyclic internucleoside linkages. These include those having morpholino linkages (formed in part from the sugar portion of a nucleoside); siloxane backbones; sulfide, sulfoxide and sulfone backbones; formacetyl and thioformacetyl backbones; methylene formacetyl and thioformacetyl backbones; riboacetyl backbones; alkene containing backbones; sulfamate backbones; methyleneimino and methylenehydrazino backbones; sulfonate and sulfonamide backbones; amide backbones; and others having mixed N, O, S and CH2 component parts.
[00375] Representative United States patents that teach the preparation of the above oligonucleosides include, but are not limited to, U.S. Pat. Nos.: 5,034,506; 5,166,315;
5,185,444; 5,214,134; 5,216,141; 5,235,033; 5,264,562; 5,264,564; 5,405,938; 5,434,257;
5,466,677; 5,470,967; 5,489,677; 5,541,307; 5,561,225; 5,596,086; 5,602,240; 5,610,289;
5,602,240; 5,608,046; 5,610,289; 5,618,704; 5,623,070; 5,663,312; 5,633,360; 5,677,437;
5,792,608; 5,646,269 and 5,677,439, and each of which is herein incorporated by reference.
[00376] In certain embodiments, oligonucleotides comprise one or more nucleotides comprising modified sugar moieties. In certain such embodiments, the furanosyl sugar ring of a nucleoside is modified in a number of ways including, but not limited to: addition of a substituent group, particularly at the 2' position; bridging of two non-geminal ring atoms to form a bicyclic nucleic acid (BNA); and substitution of an atom or group such as -S-, -N(R)- or - C(Ri)(R2) for the ring oxygen at the 4'-position. In certain such embodiments, modified sugars include, but are not limited to: substituted sugars, especially 2 '-substituted sugars having a 2'-F, 2'-OCH2 (2'-OMe) or a 2'-0(CH2)2-OCH3 (2'-0-methoxyethyl or 2'-MOE) substituent group; and bicyclic modified sugars (BNAs), having a 4'-(CH2)n-0-2' bridge, where n=l or n=2.
Methods for the preparations of modified sugars are well known to those skilled in the art.
[00377] In other exemplary oligonucleotide mimetics, both the sugar and the internucleoside linkage, i.e., the backbone, of the nucleotide units can be replaced with novel groups. The base units can be maintained for hybridization with an appropriate nucleic acid target compound. One such oligomeric compound, an oligonucleotide mimetic that has been shown to have excellent hybridization properties, is referred to as a peptide nucleic acid (PNA). In PNA compounds, the sugar-backbone of an oligonucleotide is replaced with an amide containing backbone, in particular an aminoethylglycine backbone. The nucleobases can be retained and can be bound directly or indirectly to aza nitrogen atoms of the amide portion of the backbone. Representative United States patents that teach the preparation of PNA compounds include, but are not limited to, U.S. Pat. Nos. : 5,539,082; 5,714,331; and 5,719,262, each of which is herein incorporated by reference. Further teaching of PNA compounds can be found in Nielsen et al., Science, 1991, 254, 1497-1500.
[00378] In certain embodiments, provided herein are oligonucleotides with phosphorothioate backbones and oligonucleosides with heteroatom backbones, and in particular -CH2-NH-0-CH2-, -CH2-N(CH3)-0-CH2- [known as a methylene(methylimino) or MMI backbone], -CH2-0-
N(CH3)-CH2-, -CH2-N(CH3)-N(CH3)-CH2- and -0-N(CH3)-CH2-CH2- [wherein the native phosphodiester backbone is represented as -O-P-O-C H2-] of the above referenced U.S. Pat. No. 5,489,677, and the amide backbones of the above referenced U.S. Pat. No. 5,602,240. Other exemplary oligonucleotides include those having morpholino backbone structures of the above- referenced U.S. Pat. No. 5,034,506.
[00379] Modified oligonucleotides can also contain one or more substituted sugar moieties. Certain oligonucleotides comprise one of the following at the 2' position: OH; F; 0-, S-, or N- alkyl; 0-, S-, or N-alkenyl; 0-, S- or N-alkynyl; or O-alkyl-O-alkyl, wherein the alkyl, alkenyl and alkynyl can be substituted or unsubstituted CI to CIO alkyl or C2 to CIO alkenyl and alkynyl. Exemplary oligonucleotides have 0[(CH2)nO]mCH3, 0(CH2)nOCH3, 0(CH2)nNH2, 0(CH2)nCH3, 0(CH2)nONH2, and 0(CH2)nON[(CH2)nCH3)]2, where n and m can be from 1 to about 10. Other oligonucleotides can comprise one of the following at the 2' position: CI to CIO lower alkyl, substituted lower alkyl, alkenyl, alkynyl, alkaryl, aralkyl, O-alkaryl or O-aralkyl, SH, SCH3, OCN, CI, Br, CN, CF3, OCF3, SOCH3, S02CH3, ON02, N02, N3, NH2,
heterocycloalkyl, heterocycloalkaryl, aminoalkylamino, polyalkylamino, substituted silyl, an RNA cleaving group, a reporter group, an intercalator, a group for improving the
pharmacokinetic properties of an oligonucleotide, or a group for improving the
pharmacodynamic properties of an oligonucleotide, and other substituents having similar properties. One modification includes 2'-methoxyethoxy (2'-0-CH2CH2OCH3, also known as 2'-0-(2-methoxyethyl) or 2'-MOE) (Martin et al, Helv. Chim. Acta, 1995, 78, 486-504) i.e., an alkoxyalkoxy group. Another modification includes 2'-dimethylaminooxyethoxy, i.e., an 0(CH2)2ON(CH3)2 group, also known as 2'-DMAOE, as described in examples herein below, and 2'-dimethylamino-ethoxyethoxy (also known in the art as 2'-0-dimethylamino-ethoxyethyl or 2'-DMAEOE), i.e., 2'-0-CH2-0-CH2-N(CH2)2, also described in examples herein below.
[00380] Another modification includes Locked Nucleic Acids (LNAs) in which the 2'- hydroxyl group is linked to the 3' or 4' carbon atom of the sugar ring thereby forming a bicyclic sugar moiety. The linkage is can a methylene (-CH2-) n group bridging the 2' oxygen atom and the 4' carbon atom wherein n is 1 or 2. LNAs and preparation thereof can be described in WO 98/39352 and WO 99/14226.
[00381] Other modifications include 2'-methoxy (2'-0-CH3), 2'-aminopropoxy (2'- OCH2CH2CH2NH2), 2'-allyl (2'-CH2-CH=CH2), 2'-0-allyl (2'-0-CH2-CH=CH2) and 2'-
fluoro(2'-F). The 2 '-modification can be in the arabino (up) position or ribo (down) position. A 2'-arabino modification can be 2'-F. Similar modifications can also be made at other positions on the oligonucleotide, such as the 3' position of the sugar on the 3' terminal nucleotide or in 2'- 5' linked oligonucleotides and the 5' position of 5' terminal nucleotide. Oligonucleotides can also have sugar mimetics such as cyclobutyl moieties in place of the pentofuranosyl sugar. Representative United States patents that teach the preparation of such modified sugar structures include, but are not limited to, U.S. Pat. Nos. : 4,981,957; 5,118,800; 5,319,080; 5,359,044; 5,393,878; 5,446,137; 5,466,786; 5,514,785; 5,519,134; 5,567,811; 5,576,427; 5,591,722;
5,597,909; 5,610,300; 5,627,053; 5,639,873; 5,646,265; 5,658,873; 5,670,633; 5,792,747; and 5,700,920, certain of which can be commonly owned with the instant application, and each of which is herein incorporated by reference in its entirety.
[00382] Oligonucleotides can also include nucleobase (often referred to in the art simply as "base") modifications or substitutions. As used herein, "unmodified" or "natural" nucleobases include the purine bases adenine (A) and guanine (G), and the pyrimidine bases thymine (T), cytosine. (C) and uracil (U). Modified nucleobases include other synthetic and natural nucleobases such as 5-methylcytosine (5-me-C or 5-meC), 5-hydroxymethyl cytosine, xanthine, hypoxanthine, 2-aminoadenine, 6-methyl and other alkyl derivatives of adenine and guanine, 2- propyl and other alkyl derivatives of adenine and guanine, 2-thiouracil, 2-thiothymine and 2- thiocytosine, 5-halouracil and cytosine, 5-propynyl (-C≡C-CH3) uracil and cytosine and other alkynyl derivatives of pyrimidine bases, 6-azo uracil, cytosine and thymine, 5-uracil
(pseudouracil), 4-thiouracil, 8-halo, 8-amino, 8-thiol, 8-thioalkyl, 8-hydroxyl and other 8- substituted adenines and guanines, 5-halo particularly 5-bromo, 5-trifluoromethyl and other 5- substituted uracils and cytosines, 7-methylguanine and 7-methyladenine, 2-F-adenine, 2-aminoadenine, 8-azaguanine and 8-azaadenine, 7-deazaguanine and 7-deazaadenine and 3- deazaguanine and 3-deazaadenine. Further modified nucleobases include tricyclic pyrimidines such as phenoxazine cytidine(lH-pyrimido[5,4-b][l,4]benzoxazin-2(3H)-one), phenothiazine cytidine (lH-pyrimido[5,4-b][l,4]benzothiazin-2(3H)-one), G-clamps such as a substituted phenoxazine cytidine (e.g.,9-(2-aminoethoxy)-H-pyrimido[5,4-b][l,4]benzoxazin-2(3H)-one), carbazole cytidine (2H-pyrimido[4,5-b]indol-2-one), pyridoindole cytidine (H- pyrido[3',2' :4,5]pyrrolo[2,3-d]pyrimidin-2-one). Modified nucleobases can also include those in which the purine or pyrimidine base is replaced with other heterocycles, for example 7-deaza-
adenine, 7-deazaguanosine, 2-aminopyridine and 2-pyridone. Further nucleobases include those disclosed in U.S. Pat. No. 3,687,808, those disclosed in The Concise Encyclopedia Of Polymer Science And Engineering, pages 858-859, Kroschwitz, J. I., ed. John Wiley & Sons, 1990, those disclosed by Englisch et al., Angewandte Chemie, International Edition, 1991, 30, 613, and those disclosed by Sanghvi, Y. S., Chapter 15, Antisense Research and Applications, pages 289-302, Crooke, S.T. and Lebleu, B., ed., CRC Press, 1993. Certain of these nucleobases can be useful for increasing the binding affinity of the oligomeric compounds. These include 5 -substituted pyrimidines, 6-azapyrimidines and N-2, N-6 and 0-6 substituted purines, including 2- aminopropyladenine, 5-propynyluracil and 5-propynylcytosine. 5-methylcytosine substitutions have been shown to increase nucleic acid duplex stability by 0. 6-1. 2° C. (Sanghvi, Y. S., Crooke, S. T. and Lebleu, B., eds., Antisense Research and Applications, CRC Press, Boca Raton, 1993, pp. 276-278) and can be exemplary base substitutions, such as particularly when combined with 2'-0-methoxyethyl sugar modifications.
[00383] Representative United States patents that teach the preparation of certain of the above noted modified nucleobases as well as other modified nucleobases include, but are not limited to, the above noted U.S. Pat No. 3,687,808, as well as U.S. Pat. Nos. : 4,845,205; 5,130,302;
5,134,066; 5,175,273; 5,367,066; 5,432,272; 5,457,187; 5,459,255; 5,484,908; 5,502,177;
5,525,711; 5,552,540; 5,587,469; 5,594,121 , 5,596,091; 5,614,617; 5,645,985; 5,830,653;
5,763,588; 6,005,096; 5,681,94; each of which is herein incorporated by reference.
[00384] Another modification of the oligonucleotides can involve chemically linking to the oligonucleotide one or more moieties or conjugates which enhance the activity, cellular distribution or cellular uptake of the oligonucleotide. The compounds can include conjugate groups covalently bound to functional groups such as primary or secondary hydroxyl groups. Conjugate groups include intercalators, reporter molecules, polyamines, polyamides, poly ethylene glycols, polyethers, groups that enhance the pharmacodynamic properties of oligomers, and groups that enhance the pharmacokinetic properties of oligomers. Conjugates groups include cholesterols, lipids, phospholipids, biotin, phenazine, folate, phenanthridine, anthraquinone, acridine, fluoresceins, rhodamines, coumarins, and dyes. Groups that enhance the
pharmacodynamic properties, include groups that improve oligomer uptake, enhance oligomer resistance to degradation, and/or strengthen sequence-specific hybridization with RNA. Groups that enhance the pharmacokinetic properties, include groups that improve oligomer uptake,
distribution, metabolism or excretion. Representative conjugate groups can be disclosed in International Patent Application PCT/US92/09196, filed Oct. 23, 1992 the entire disclosure of which is incorporated herein by reference. Conjugate moieties can include but are not limited to lipid moieties, such as a cholesterol moiety (Letsinger et al, Proc. Natl. Acad. Sci. USA, 1989, 86, 6553-6556), cholic acid (Manoharan et al, Bioorg. Med. Chem. Let., 1994, 4, 1053-1060), a thioether, e.g., hexyl-S-tritylthiol (Manoharan et al, Ann. N. Y. Acad. Sci., 1992, 660, 306-309; Manoharan et al, Bioorg. Med. Chem. Let., 1993, 3, 2765-2770), a thiocholesterol (Oberhauser et. al, Nucl. Acids Res., 1992, 20, 533-538), an aliphatic chain, e.g., dodecandiol or undecyl residues (Saison-Behmoaras et al, EMBO J., 1991, 10, 1111-1118; Kabanov et al, FEBS Lett., 1990, 259, 327-330; Svinarchuk et al, Biochimie, 1993, 75, 49-54), a phospholipid; e.g., di hexadecyl-rac-glycerol or triethylammonium l,2-di-0-hexadecyl-rac-glycero-3-H-phosphonate (Manoharan et al, Tetrahedron Lett., 1995, 36, 3651-3654; Shea et al, Nucl. Acids Res., 1990, 18, 3777-3783), a polyamine or a polyethylene glycol chain (Manoharan et al, Nucleosides & Nucleotides, 1995, 14, 969-973), or adamantane acetic acid (Manoharan et al, Tetrahedron Lett., 1995, 36, 3651-3654), a palmityl moiety (Mishra et al, Biochim. Biophys. Acta, 1995, 1264, 229-237), or an octadecylamine or hexylamino-carbonyl-oxy cholesterol moiety (Crooke et al, J. Pharmacol. Exp. Ther., 1996, 277, 923-937. Oligonucleotides can also be conjugated to active drug substances, for example, aspirin, warfarin, phenylbutazone, ibuprofen, suprofen, fenbufen, ketoprofen, (S)-(+)-pranoprofen, carprofen, dansylsarcosine, 2,3,5-triiodobenzoic acid, flufenamic acid, folinic acid, a benzothiadiazide, chlorothiazide, a diazepine, indomethicin, a barbiturate, a cephalosporin, a sulfa drug, an antidiabetic, an antibacterial or an antibiotic.
Oligonucleotide-drug conjugates and their preparation can be described in U.S. patent application Ser. No. 09/334,130 (filed Jun. 15, 1999) which is incorporated herein by reference in its entirety.
[00385] Representative United States patents that teach the preparation of such
oligonucleotide conjugates include, but are not limited to, U.S. Pat. Nos. : 4,828,979; 4,948,882; 5,218,105; 5,525,465; 5,541,313; 5,545,730; 5,552,538; 5,578,717, 5,580,731; 5,580,731;
5,591,584; 5,109,124; 5,118,802; 5,138,045; 5,414,077; 5,486,603; 5,512,439; 5,578,718;
5,608,046; 4,587,044; 4,605,735; 4,667,025; 4,762,779; 4,789,737; 4,824,941; 4,835,263;
4,876,335; 4,904,582; 4,958,013; 5,082,830; 5,112,963; 5,214,136; 5,082,830; 5,112,963;
5,214,136; 5,245,022; 5,254,469; 5,258,506; 5,262,536; 5,272,250; 5,292,873; 5,317,098;
5,371,241, 5,391,723; 5,416,203, 5,451,463; 5,510,475; 5,512,667; 5,514,785; 5,565,552;
5,567,810; 5,574,142; 5,585,481; 5,587,371 ; 5,595,726; 5,597,696; 5,599,923; 5,599,928 and 5,688,941, each of which is herein incorporated by reference.
[00386] It is not necessary for all positions in a given compound to be uniformly modified, and more than one of the aforementioned modifications can be incorporated in a single compound or even at a single nucleoside within an oligonucleotide. Antisense compounds, which can be chimeric compounds are also provided herein. "Chimeric" antisense compounds or "chimeras," can be antisense compounds, particularly oligonucleotides, which contain two or more chemically distinct regions, each made up of at least one monomer unit, i.e., a nucleotide in the case of an oligonucleotide compound. These oligonucleotides can contain at least one region wherein the oligonucleotide is modified so as to confer upon the oligonucleotide increased resistance to nuclease degradation, increased cellular uptake, and/or increased binding affinity for the target nucleic acid. An additional region of the oligonucleotide can serve as a substrate for enzymes capable of cleaving R A:DNA or R A:R A hybrids. By way of example, R ase H is a cellular endonuclease which cleaves the RNA strand of an R A:DNA duplex. Activation of RNase H, therefore, results in cleavage of the RNA target, thereby greatly enhancing the efficiency of oligonucleotide inhibition of gene expression. Consequently, comparable results can often be obtained with shorter oligonucleotides when chimeric oligonucleotides can be used, compared to phosphorothioate deoxyoligonucleotides hybridizing to the same target region. Cleavage of the RNA target can be routinely detected by gel electrophoresis and, if necessary, associated nucleic acid hybridization techniques known in the art.
[00387] Chimeric antisense compounds can be formed as composite structures of two or more oligonucleotides, modified oligonucleotides, oligonucleosides and/or oligonucleotide mimetics as described above. Such compounds have also been referred to in the art as hybrids or
"gapmers". Representative U.S. patents that teach the preparation of such hybrid structures include, but are not limited to, U.S. Pat. Nos. : 5,013,830; 5,149,797; 5,220,007; 5,256,775; 5,366,878; 5,403,711; 5,491,133; 5,565,350; 5,623,065; 5,652,355; 5,652,356; and 5,700,922, each of which is herein incorporated by reference in its entirety.
[00388] The antisense compounds provided herein can be conveniently and routinely made through the well-known technique of solid phase synthesis. Equipment for such synthesis is sold by several vendors including, for example, Applied Biosy stems (Foster City, Calif. ). Any other
refers to for such synthesis known in the art can additionally or alternatively be employed. It is well known to use similar techniques to prepare oligonucleotides such as the phosphorothioates and alkylated derivatives.
[00389] The antisense compounds can be synthesized in vitro and do not include antisense compositions of biological origin, or genetic vector constructs designed to direct the in vivo synthesis of antisense molecules.
[00390] The compounds can also be admixed, encapsulated, conjugated or otherwise associated with other molecules, molecule structures or mixtures of compounds, as for; example, liposomes, receptor targeted molecules, oral, rectal, topical or other formulations, for assisting in uptake, distribution and/or absorption. Representative United States patents that teach the preparation of such uptake, distribution and/or absorption assisting formulations include, but are not limited to, U.S. Pat. Nos. : 5,108,921; 5,354,844; 5,416,016; 5,459,127; 5,521,291;
5,543,158; 5,547,932; 5,583,020; 5,591,721 ; 4,426,330; 4,534,899; 5,013,556; 5,108,921;
5,213,804; 5,227,170; 5,264,221; 5,356,633; 5,395,619; 5,416,016; 5,417,978; 5,462,854;
5,469,854; 5,512,295; 5,527,528; 5,534,259; 5,543,152; 5,556,948; 5,580,575; and 5,595,756, each of which is herein incorporated by reference.
[00391] Antisense compounds provided herein encompass any pharmaceutically acceptable salts, esters, or salts of such esters, or any other compound which, upon administration to an animal including a human, is capable of providing (directly or indirectly) the biologically active metabolite or residue thereof. Accordingly, for example, the examples can also include prodrugs and pharmaceutically acceptable salts of the compounds, pharmaceutically acceptable salts of such prodrugs, and other bioequivalents.
[00392] The term "prodrug" refers to a therapeutic agent that is prepared in an inactive form that is converted to an active form (i.e., drug) within the body or cells thereof by the action of endogenous enzymes or other chemicals and/or conditions. In particular, prodrug versions of the oligonucleotides can be prepared as SATE [(S-acetyl-2-thioethyl) phosphate] derivatives according to the methods disclosed in WO 93/24510 to Gosselin et al., published Dec. 9, 1993 or in WO 94/26764 and U.S. Pat. No. 5,770,713 to Imbach et al.
[00393] The term "pharmaceutically acceptable salts" refers to physiologically and pharmaceutically acceptable salts of the compounds: i.e., salts that retain the desired biological activity of the parent compound and do not impart undesired toxicological effects thereto.
[00394] Pharmaceutically acceptable base addition salts can be formed with metals or amines, such as alkali and alkaline earth metals or organic amines. Examples of metals that can be used as cations can be sodium, potassium, magnesium, calcium, and the like. Examples of suitable amines can be Ν,Ν'-dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, dicyclohexylamine, ethylenediamine, N-methylglucamine, and procaine (see, for example, Berge et al., "Pharmaceutical Salts," J. of Pharma Sci., 1977, 66, 1-19). The base addition salts of said acidic compounds can be prepared by contacting the free acid form with a sufficient amount of the desired base to produce the salt in the conventional manner. The free acid form can be regenerated by contacting the salt form with an acid and isolating the free acid in the
conventional manner. The free acid forms differ from their respective salt forms somewhat in certain physical properties such as solubility in polar solvents, but otherwise the salts can be equivalent to their respective free acid for purposes of provided herein. As used herein, a "pharmaceutical addition salt" includes a pharmaceutically acceptable salt of an acid form of one of the components of the compositions. These include organic or inorganic acid salts of the amines. Exemplary acid salts can be the hydrochlorides, acetates, salicylates, nitrates and phosphates. Other suitable pharmaceutically acceptable salts can be well known to those skilled in the art and include basic salts of a variety of inorganic and organic acids, such as, for example, with inorganic acids, such as for example hydrochloric acid, hydrobromic acid, sulfuric acid or phosphoric acid; with organic carboxylic, sulfonic, sulfo or phospho acids or N-substituted sulfamic acids, for example acetic acid, propionic acid, glycolic acid, succinic acid, maleic acid, hydroxymaleic acid, methylmaleic acid, fumaric acid, malic acid, tartaric acid, lactic acid, oxalic acid, gluconic acid, glucaric acid, glucuronic acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, salicylic acid, 4-aminosalicylic acid, 2-phenoxybenzoic acid, 2-acetoxybenzoic acid, embonic acid, nicotinic acid or isonicotinic acid; and with amino acids, such as the 20 alpha-amino acids involved in the synthesis of proteins in nature, for example glutamic acid or aspartic acid, and also with phenylacetic acid, methanesulfonic acid, ethanesulfonic acid, 2- hydroxyethanesulfonic acid, ethane- 1 ,2-disulfonic acid, benzenesulfonic acid, 4- methylbenzenesulfonic acid, naphthalene-2-sulfonic acid, naphthalene- 1, 5 -disulfonic acid, 2- or 3-phosphoglycerate, glucose-6-phosphate, N-cyclohexylsulfamic acid (with the formation of cyclamates); or with other acid organic compounds, such as ascorbic acid. Pharmaceutically acceptable salts of compounds can also be prepared with a pharmaceutically acceptable cation.
Suitable pharmaceutically acceptable cations can be well known to those skilled in the art and include alkaline, alkaline earth, ammonium and quaternary ammonium cations. Carbonates or hydrogen carbonates can also be possible.
[00395] For oligonucleotides, examples of pharmaceutically acceptable salts include but are not limited to (a) salts formed with cations such as sodium, potassium, ammonium, magnesium, calcium, polyamines' such as spermine and spermidine, etc.; (b) acid addition salts formed with inorganic acids, for example hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, nitric acid and the like; (c) salts formed with organic acids such as, for example, acetic acid, oxalic acid, tartaric acid, succinic acid, maleic acid, fumaric acid, gluconic acid, citric acid, malic acid, ascorbic acid, benzoic acid, tannic acid, palmitic acid, alginic acid, polyglutamic acid, naphthalenesulfonic acid, methanesulfonic acid, p-toluenesulfonic acid, naphthalenedisulfonic acid, polygalacturonic acid, and the like; and (d) salts formed from elemental anions such as chlorine, bromine, and iodine.
[00396] The antisense compounds of provided herein, can be utilized for diagnostics, therapeutics, prophylaxis and as research reagents and kits. For therapeutics, an animal, such as a human, suspected of having a disease or disorder which can be treated by modulating the expression of ApoB is treated by administering antisense compounds, provided herein. The compounds can be utilized in pharmaceutical compositions by adding an effective amount of an antisense compound to a suitable pharmaceutically acceptable diluent or carrier.
[00397] The antisense compounds can be useful for research and diagnostics, because these compounds hybridize to nucleic acids encoding ApoB, enabling sandwich and other assays to easily be constructed to exploit this fact. Hybridization of the antisense oligonucleotides with a nucleic acid encoding ApoB can be detected by refers to known in the art. Such refers to can include conjugation of an enzyme to the oligonucleotide, radiolabelling of the oligonucleotide or any other suitable detection refers to. Kits using such detection refers to for detecting the level of ApoB in a sample can also be prepared.
1.3 Pharmaceutical compositions
[00398] Also provided are pharmaceutical compositions and formulations, which include an antisense compound. The pharmaceutical compositions provided herein can be administered in a number of ways depending upon whether local or systemic treatment is desired and upon the area to be treated. Administration can be topical (including ophthalmic and to mucous
membranes including vaginal and rectal delivery), pulmonary, e.g., by inhalation or insufflation of powders or aerosols, including by nebulizer; intratracheal, intranasal, epidermal and transdermal), oral or parenteral.
[00399] Parenteral administration includes intravenous, intra-arterial, subcutaneous, intraperitoneal or intramuscular injection or infusion; or intracranial, e.g., intratracheal or intraventricular, administration. Antisense compounds with at least one 2'-0-methoxyethyl modification can be believed to be particularly useful for oral administration.
[00400] Pharmaceutical compositions and formulations for topical administration can include transdermal patches, ointments, lotions, creams, gels, drops, suppositories, sprays, liquids and powders. Conventional pharmaceutical carriers, aqueous, powder or oily bases, thickeners and the like can be necessary or desirable. Coated condoms, gloves and the like can also be useful. Topical formulations include those in which the antisense compounds provided herein, can be in admixture with a topical delivery agent such as lipids, liposomes, fatty acids, fatty acid esters, steroids, chelating agents and surfactants. Lipids and liposomes include neutral (e.g.,
dioleoylphosphatidyl DOPE ethanolamine, dimyristoylphosphatidyl choline DMPC,
distearolyphosphatidyl choline) negative (e.g., dimyristoylphosphatidyl glycerol DMPG) and cationic (e.g., dioleoyltetramethylammopropyl DOTAP and dioleoylphosphatidyl ethanolamine DOTMA). Antisense compounds can be encapsulated within liposomes or can form complexes thereto, in particular to cationic liposomes. Alternatively, antisense compounds can be complexed to lipids, in particular to cationic lipids. Fatty acids and esters include but are not limited arachidonic acid, oleic acid, eicosanoic acid, lauric acid, caprylic acid, capric acid, myristic acid, palmitic acid, stearic acid, linoleic acid, linolenic acid, dicaprate, tricaprate, monoolein, dilaurin, glyceryl 1-monocaprate, l-dodecylazacycloheptan-2-one, an acylcarnitine, an acylcholine, or a C 1-10 alkyl ester (e.g., isopropylmyristate IPM), monoglyceride, diglyceride or pharmaceutically acceptable salt thereof. Topical formulations can be described in detail in U.S. patent application Ser. No. 09/315,298 filed on Can 20, 1999, which is incorporated herein by reference in its entirety.
[00401] Compositions and formulations for oral administration include powders or granules, microparticulates, nanoparticulates, suspensions or solutions in water or non-aqueous media, capsules, gel capsules, sachets, tablets or minitablets. Thickeners, flavoring agents, diluents, emulsifiers, dispersing aids or binders can be desirable. Oral formulations can be those in which
antisense compounds provided herein, can be administered in conjunction with one or more penetration enhancers surfactants and chelators. Surfactants include fatty acids and/or esters or salts thereof, bile acids and/or salts thereof. Bile acids/salts include chenodeoxycholic acid (CDCA) and ursodeoxychenodeoxycholic acid (UDCA), cholic acid, dehydrocholic acid, deoxycholic acid, glucholic acid, glycholic acid, glycodeoxycholic acid, taurocholic acid, taurodeoxycholic acid, sodium tauro-24,25-dihydro-fusidate, sodium glycodihydrofusidate. Fatty acids include arachidonic acid, undecanoic acid, oleic acid, lauric acid, caprylic acid, capric acid, myristic acid, palmitic acid, stearic acid, linoleic acid, linolenic acid, dicaprate, tricaprate, monoolein, dilaurin, glyceryl 1-monocaprate, l-dodecylazacycloheptan-2-one, an acylcarnitine, an acylcholine, or a monoglyceride, a diglyceride or a pharmaceutically acceptable salt thereof (e.g., sodium). Combinations of penetration enhancers, for example, fatty acids/salts in combination with bile acids/salts can also be used. An exemplary combination is the sodium salt of lauric acid, capric acid and UDCA. Further penetration enhancers include polyoxyethylene-9- lauryl ether, polyoxyethylene-20-cetyl ether. Antisense compounds provided herein, can be delivered orally in granular form including sprayed dried particles, or complexed to form micro or nanoparticles. Antisense compound complexing agents include poly-amino acids; polyimines; polyacrylates; polyalkylacrylates, polyoxethanes, polyalkylcyanoacrylates; cationized gelatins, albumins, starches, acrylates, polyethyleneglycols (PEG) and starches; polyalkylcyanoacrylates; DEAE-derivatized polyimines, pollulans, celluloses and starches. Complexing agents include chitosan, N-trimethylchitosan, poly-L-lysine, polyhistidine, polyornithine, polyspermines, protamine, polyvinylpyridine, polythiodiethylamino-methylethylene P(TDAE),
polyaminostyrene (e.g.,p-amino), poly(methylcyanoacrylate), poly(ethylcyanoacrylate), poly(butylcyanoacrylate), poly(isobutylcyanoacrylate), poly(isohexylcynaoacrylate), DEAE- methacrylate, DEAE-hexylacrylate, DEAE-acrylamide, DEAE-albumin and DEAE-dextran, polymethylacrylate, polyhexylacrylate, poly(D,L-lactic acid), poly(DL-lactic-co-glycolic acid (PLGA), alginate, and polyethyleneglycol (PEG). Oral formulations for antisense compounds and their preparation can be described in detail in U.S. applications Ser. No. 08/886,829 (filed Jul. 1 , 1997), U.S. Ser. No. 09/108,673 (filed Jul. 1 , 1998), U.S. Ser. No. 09/256,515 (filed Feb. 23, 1999), U.S. Ser. No. 09/082,624 (filed Can 21 , 1998) and U.S. Ser. No. 09/315,298 (filed Can 20, 1999) each of which is incorporated herein by reference in their entirety.
[00402] Compositions and formulations for parenteral, intrathecal or intraventricular administration can include sterile aqueous solutions which can also contain buffers, diluents and other suitable additives such as, but not limited to, penetration enhancers, carrier compounds and other pharmaceutically acceptable carriers or excipients.
[00403] Pharmaceutical compositions provided herein include, but are not limited to, solutions, emulsions, and liposome-containing formulations. These compositions can be generated from a variety of components that include, but are not limited to, preformed liquids, self-emulsifying solids and self-emulsifying semisolids.
[00404] The pharmaceutical formulations provided herein, which can conveniently be presented in unit dosage form, can be prepared according to conventional techniques well known in the pharmaceutical industry. Such techniques include the step of bringing into association the active ingredients with the pharmaceutical carrier(s) or excipient(s). The formulations can be prepared by uniformly and intimately bringing into association the active ingredients with liquid carriers or finely divided solid carriers or both, and then, if necessary, shaping the product.
[00405] The compositions provided herein can be formulated into any of many possible dosage forms such as, but not limited to, tablets, capsules, gel capsules, liquid syrups, soft gels, suppositories, and enemas. The compositions provided herein can also be formulated as suspensions in aqueous, non-aqueous or mixed media. Aqueous suspensions can further contain substances, which increase the viscosity of the suspension including, for example, sodium carboxymethylcellulose, sorbitol and/or dextran. The suspension can also contain stabilizers.
[00406] In one embodiment, the pharmaceutical compositions can be formulated and used as foams. Pharmaceutical foams include formulations such as, but not limited to, emulsions, microemulsions, creams, jellies and liposomes. While basically similar in nature these formulations vary in the components and the consistency of the final product. The preparation of such compositions and formulations is generally known to those skilled in the pharmaceutical and formulation arts and can be applied to the formulation of the compositions provided herein.
[00407] In certain embodiments, pharmaceutical compositions comprise one or more oligonucleotides and one or more excipients. In certain such embodiments, excipients are selected from water, salt solutions, alcohol, polyethylene glycols, gelatin, lactose, amylase, magnesium stearate, talc, silicic acid, viscous paraffin, hydroxymethylcellulosem and polyvinylpyrrolidone.
[00408] In certain embodiments, a pharmaceutical composition is prepared using known techniques, including, but not limited to mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping or tabletting processes.
[00409] In certain embodiments, a pharmaceutical composition is a liquid (e.g. , a suspension, elixir and/or solution). In certain of such embodiments, a liquid pharmaceutical composition is prepared using ingredients known in the art, including, but not limited to, water, glycols, oils, alcohols, flavoring agents, preservatives, and coloring agents.
[00410] In certain embodiments, a pharmaceutical composition is a solid (e.g., a powder, tablet, and/or capsule). In certain of such embodiments, a solid pharmaceutical composition comprising one or more oligonucleotides is prepared using ingredients known in the art, including, but not limited to, starches, sugars, diluents, granulating agents, lubricants, binders, and disintegrating agents.
[00411] In certain embodiments, a pharmaceutical composition is formulated as a depot preparation. Certain such depot preparations are typically longer acting than non-depot preparations. In certain embodiments, such preparations are administered by implantation (for example subcutaneously or intramuscularly) or by intramuscular injection. In certain
embodiments, depot preparations are prepared using suitable polymeric or hydrophobic materials (for example an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
[00412] In certain embodiments, a pharmaceutical composition comprises a delivery system. Examples of delivery systems include, but are not limited to, liposomes and emulsions. Certain delivery systems are useful for preparing certain pharmaceutical compositions including those comprising hydrophobic compounds. In certain embodiments, certain organic solvents such as dimethylsulfoxide are used.
[00413] In certain embodiments, a pharmaceutical composition comprises one or more tissue- specific delivery molecules designed to deliver the one or more pharmaceutical agents to specific tissues or cell types. For example, in certain embodiments, pharmaceutical compositions include liposomes coated with a tissue-specific antibody.
[00414] In certain embodiments, a pharmaceutical composition comprises a co-solvent system. Certain of such co-solvent systems comprise, for example, benzyl alcohol, a nonpolar surfactant, a water-miscible organic polymer, and an aqueous phase. In certain embodiments,
such co-solvent systems are used for hydrophobic compounds. A non-limiting example of such a co-solvent system is the VPD co-solvent system, which is a solution of absolute ethanol comprising 3% w/v benzyl alcohol, 8% w/v of the nonpolar surfactant Polysorbate 80™, and 65% w/v polyethylene glycol 300. The proportions of such co-solvent systems may be varied considerably without significantly altering their solubility and toxicity characteristics.
Furthermore, the identity of co-solvent components may be varied: for example, other surfactants may be used instead of Polysorbate 80™; the fraction size of polyethylene glycol may be varied; other biocompatible polymers may replace polyethylene glycol, e.g., polyvinyl pyrrolidone; and other sugars or polysaccharides may substitute for dextrose.
[00415] In certain embodiments, a pharmaceutical composition comprises a sustained-release system. A non-limiting example of such a sustained-release system is a semi-permeable matrix of solid hydrophobic polymers. In certain embodiments, sustained-release systems may, depending on their chemical nature, release pharmaceutical agents over a period of hours, days, weeks or months.
[00416] In certain embodiments, a pharmaceutical composition is prepared for oral administration. In certain of such embodiments, a pharmaceutical composition is formulated by combining one or more oligonucleotides with one or more pharmaceutically acceptable carriers. Certain of such carriers enable pharmaceutical compositions to be formulated as tablets, pills, dragees, capsules, liquids, gels, syrups, slurries, suspensions and the like, for oral ingestion by a subject. In certain embodiments, pharmaceutical compositions for oral use are obtained by mixing oligonucleotide and one or more solid excipient. Suitable excipients include, but are not limited to, fillers, such as sugars, including lactose, sucrose, mannitol, or sorbitol; cellulose preparations such as, for example, maize starch, wheat starch, rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose, hydroxypropylmethyl-cellulose, sodium
carboxymethylcellulose, and/or polyvinylpyrrolidone (PVP). In certain embodiments, such a mixture is optionally ground and auxiliaries are optionally added. In certain embodiments, pharmaceutical compositions are formed to obtain tablets or dragee cores. In certain
embodiments, disintegrating agents {e.g., cross-linked polyvinyl pyrrolidone, agar, or alginic acid or a salt thereof, such as sodium alginate) are added.
[00417] In certain embodiments, dragee cores are provided with coatings. In certain such embodiments, concentrated sugar solutions may be used, which may optionally contain gum
arabic, talc, polyvinyl pyrrolidone, carbopol gel, polyethylene glycol, and/or titanium dioxide, lacquer solutions, and suitable organic solvents or solvent mixtures. Dyestuffs or pigments may be added to tablets or dragee coatings.
[00418] In certain embodiments, pharmaceutical compositions for oral administration are push-fit capsules made of gelatin. Certain of such push- fit capsules comprise one or more pharmaceutical agents in admixture with one or more filler such as lactose, binders such as starches, and/or lubricants such as talc or magnesium stearate and, optionally, stabilizers. In certain embodiments, pharmaceutical compositions for oral administration are soft, sealed capsules made of gelatin and a plasticizer, such as glycerol or sorbitol. In certain soft capsules, one or more pharmaceutical agents are be dissolved or suspended in suitable liquids, such as fatty oils, liquid paraffin, or liquid polyethylene glycols. In addition, stabilizers may be added.
[00419] In certain embodiments, pharmaceutical compositions are prepared for buccal administration. Certain of such pharmaceutical compositions are tablets or lozenges formulated in conventional manner.
[00420] In certain embodiments, a pharmaceutical composition is prepared for administration by injection (e.g., intravenous, subcutaneous, intramuscular, etc.). In certain of such
embodiments, a pharmaceutical composition comprises a carrier and is formulated in aqueous solution, such as water or physiologically compatible buffers such as Hanks's solution, Ringer's solution, or physiological saline buffer. In certain embodiments, other ingredients are included (e.g., ingredients that aid in solubility or serve as preservatives). In certain embodiments, injectable suspensions are prepared using appropriate liquid carriers, suspending agents and the like. Certain pharmaceutical compositions for injection are presented in unit dosage form, e.g., in ampoules or in multi-dose containers. Certain pharmaceutical compositions for injection are suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents. Certain solvents suitable for use in pharmaceutical compositions for injection include, but are not limited to, lipophilic solvents and fatty oils, such as sesame oil, synthetic fatty acid esters, such as ethyl oleate or triglycerides, and liposomes. Aqueous injection suspensions may contain substances that increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol, or dextran. Optionally, such suspensions may also contain suitable stabilizers or agents that increase the solubility of the pharmaceutical agents to allow for the preparation of highly concentrated solutions.
[00421] In certain embodiments, a pharmaceutical composition is prepared for transmucosal administration. In certain of such embodiments penetrants appropriate to the barrier to be permeated are used in the formulation. Such penetrants are generally known in the art.
[00422] In certain embodiments, a pharmaceutical composition is prepared for administration by inhalation. Certain of such pharmaceutical compositions for inhalation are prepared in the form of an aerosol spray in a pressurized pack or a nebulizer. Certain of such pharmaceutical compositions comprise a propellant, e.g. , dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas. In certain embodiments using a pressurized aerosol, the dosage unit may be determined with a valve that delivers a metered amount. In certain embodiments, capsules and cartridges for use in an inhaler or insufflator may be formulated. Certain of such formulations comprise a powder mixture of a pharmaceutical agent provided herein and a suitable powder base such as lactose or starch.
[00423] In certain embodiments, a pharmaceutical composition is prepared for rectal administration, such as a suppositories or retention enema. Certain of such pharmaceutical compositions comprise known ingredients, such as cocoa butter and/or other glycerides.
[00424] In certain embodiments, a pharmaceutical composition is prepared for topical administration. Certain of such pharmaceutical compositions comprise bland moisturizing bases, such as ointments or creams. Exemplary suitable ointment bases include, but are not limited to, petrolatum, petrolatum plus volatile silicones, lanolin and water in oil emulsions such as
Eucerin™, available from Beiersdorf (Cincinnati, Ohio). Exemplary suitable cream bases include, but are not limited to, Nivea™ Cream, available from Beiersdorf (Cincinnati, Ohio), cold cream (USP), Purpose Cream™, available from Johnson & Johnson (New Brunswick, N. J.), hydrophilic ointment (USP) and Lubriderm™, available from Pfizer (Morris Plains, N.J.).
[00425] In certain embodiments, a pharmaceutical composition comprises an oligonucleotide in a therapeutically effective amount. In certain embodiments, the therapeutically effective amount is sufficient to prevent, alleviate or ameliorate symptoms of a disease or to prolong the survival of the subject being treated. Determination of a therapeutically effective amount is well within the capability of those skilled in the art.
[00426] In certain embodiments, one or more oligonucleotides is formulated as a prodrug. In certain embodiments, upon in vivo administration, a prodrug is chemically converted to the biologically, pharmaceutically or therapeutically more active form of the oligonucleotide. In
certain embodiments, prodrugs are useful because they are easier to administer than the corresponding active form. For example, in certain instances, a prodrug may be more
bioavailable (e.g. , through oral administration) than is the corresponding active form. In certain instances, a prodrug may have improved solubility compared to the corresponding active form. In certain embodiments, prodrugs are less water soluble than the corresponding active form. In certain instances, such prodrugs possess superior transmittal across cell membranes, where water solubility is detrimental to mobility. In certain embodiments, a prodrug is an ester. In certain such embodiments, the ester is metabolically hydrolyzed to carboxylic acid upon administration. In certain instances the carboxylic acid containing compound is the corresponding active form. In certain embodiments, a prodrug comprises a short peptide (polyaminoacid) bound to an acid group. In certain of such embodiments, the peptide is cleaved upon administration to form the corresponding active form.
[00427] In certain embodiments, a prodrug is produced by modifying a pharmaceutically active compound such that the active compound will be regenerated upon in vivo administration. The prodrug can be designed to alter the metabolic stability or the transport characteristics of a drug, to mask side effects or toxicity, to improve the flavor of a drug or to alter other
characteristics or properties of a drug. By virtue of knowledge of pharmacodynamic processes and drug metabolism in vivo, those of skill in this art, once a pharmaceutically active compound is known, can design prodrugs of the compound (see, e.g., Nogrady (1985) Medicinal Chemistry A Biochemical Approach, Oxford University Press, New York, pages 388-392).
[00428] In certain embodiments, pharmaceutical composition including one or more pharmaceutical agents are useful for treating a conditions or disorders in a mammalian, and particularly in a human, subject. Suitable administration routes include, but are not limited to, oral, rectal, transmucosal, intestinal, enteral, topical, suppository, through inhalation, intrathecal, intraventricular, intraperitoneal, intranasal, intraocular and parenteral (e.g. , intravenous, intramuscular, intramedullary, and subcutaneous). In certain embodiments, pharmaceutical intrathecals are administered to achieve local rather than systemic exposures. For example, pharmaceutical compositions may be injected directly in the area of desired effect (e.g., in the renal or cardiac area).
[00429] In certain embodiments, a pharmaceutical composition is administered in the form of a dosage unit (e.g., tablet, capsule, bolus, etc.). In certain embodiments, such pharmaceutical
compositions comprise an oligonucleotide in a dose selected from 25 mg, 30 mg, 35 mg, 40 mg, 45 mg, 50 mg, 55 mg, 60 mg, 65 mg, 70 mg, 75 mg, 80 mg, 85 mg, 90 mg, 95 mg, 100 mg, 105 mg, 110 mg, 115 mg, 120 mg, 125 mg, 130 mg, 135 mg, 140 mg, 145 mg, 150 mg, 155 mg, 160 mg, 165 mg, 170 mg, 175 mg, 180 mg, 185 mg, 190 mg, 195 mg, 200 mg, 205 mg, 210 mg, 215 mg, 220 mg, 225 mg, 230 mg, 235 mg, 240 mg, 245 mg, 250 mg, 255 mg, 260 mg, 265 mg, 270 mg, 270 mg, 280 mg, 285 mg, 290 mg, 295 mg, 300 mg, 305 mg, 310 mg, 315 mg, 320 mg, 325 mg, 330 mg, 335 mg, 340 mg, 345 mg, 350 mg, 355 mg, 360 mg, 365 mg, 370 mg, 375 mg, 380 mg, 385 mg, 390 mg, 395 mg, 400 mg, 405 mg, 410 mg, 415 mg, 420 mg, 425 mg, 430 mg, 435 mg, 440 mg, 445 mg, 450 mg, 455 mg, 460 mg, 465 mg, 470 mg, 475 mg, 480 mg, 485 mg, 490 mg, 495 mg, 500 mg, 505 mg, 510 mg, 515 mg, 520 mg, 525 mg, 530 mg, 535 mg, 540 mg, 545 mg, 550 mg, 555 mg, 560 mg, 565 mg, 570 mg, 575 mg, 580 mg, 585 mg, 590 mg, 595 mg, 600 mg, 605 mg, 610 mg, 615 mg, 620 mg, 625 mg, 630 mg, 635 mg, 640 mg, 645 mg, 650 mg, 655 mg, 660 mg, 665 mg, 670 mg, 675 mg, 680 mg, 685 mg, 690 mg, 695 mg, 700 mg, 705 mg, 710 mg, 715 mg, 720 mg, 725 mg, 730 mg, 735 mg, 740 mg, 745 mg, 750 mg, 755 mg, 760 mg, 765 mg, 770 mg, 775 mg, 780 mg, 785 mg, 790 mg, 795 mg, and 800 mg. In certain such embodiments, a pharmaceutical composition comprises a dose of oligonucleotide selected from 25 mg, 50 mg, 75 mg, 100 mg, 150 mg, 200 mg, 250 mg, 300 mg, 350 mg, 400 mg, 500 mg, 600 mg, 700 mg, and 800mg. In certain embodiments, a pharmaceutical composition is comprises a dose of oligonucleotide selected from 50 mg, 100 mg, 150 mg, 200 mg, 250 mg, 300 mg, and
400 mg. In a specifc embodiment, the dose is 200 mg. In an embodiment, the dose is 160 mg.
[00430] In a further aspect, a pharmaceutical agent is sterile lyophilized oligonucleotide that is reconstituted with a suitable diluent, e.g., sterile water for injection. The reconstituted product is administered as a subcutaneous injection or as an intravenous infusion after dilution into saline. The lyophilized drug product consists of the oligonucleotide which has been prepared in water for injection, adjusted to pH 7.0-9.0 with acid or base during preparation, and then lyophilized. The lyophilized oligonucleotide may be 25-800 mg of the oligonucleotide. It is understood that this encompasses 25, 50, 75, 100, 125, 150, 175, 200, 225, 250, 275, 300, 325, 350, 375, 425, 450, 475, 500, 525, 550, 575, 600, 625, 650, 675, 700, 725, 750, 775, and 800 mg of lyophilized oligonucleotide. The lyophilized drug product may be packaged in a 2 mL Type I, clear glass vial (ammonium sulfate-treated), stoppered with a bromobutyl rubber closure and sealed with an
aluminum FLIP-OFF® overseal. In one embodiment, the lyophilized pharmaceutical agent comprises ISIS 301012.
[00431] The compositions may additionally contain other adjunct components conventionally found in pharmaceutical compositions, at their art-established usage levels. Thus, for example, the compositions may contain additional, compatible, pharmaceutically-active materials such as, for example, antipruritics, astringents, local anesthetics or anti-inflammatory agents, or may contain additional materials useful in physically formulating various dosage forms of the compositions provided herein, such as dyes, flavoring agents, preservatives, antioxidants, opacifiers, thickening agents and stabilizers. However, such materials, when added, should not unduly interfere with the biological activities of the components of the compositions provided herein. The formulations can be sterilized and, if desired, mixed with auxiliary agents, e.g., lubricants, preservatives, stabilizers, wetting agents, emulsifiers, salts for influencing osmotic pressure, buffers, colorings, flavorings and/or aromatic substances and the like which do not deleteriously interact with the oligonucleotide(s) of the formulation.
1.3.1. Emulsions
[00432] The compositions provided herein can be prepared and formulated as emulsions. Emulsions can be typically heterogeneous systems of one liquid dispersed in another in the form of droplets usually exceeding 0.1 μιη in diameter (Idson, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 199; Rosoff, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 245; Block in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 2, p. 335; Higuchi et al., in Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa., 1985, p. 301). Emulsions can be often biphasic systems comprising of two immiscible liquid phases intimately mixed and dispersed with each other. In general, emulsions can be either water-in-oil (w/o) or of the oil-in-water (o/w) variety. When an aqueous phase is finely divided into and dispersed as minute droplets into a bulk oily phase the resulting composition is called water-in-oil (w/o) emulsion. Alternatively, when an oily phase is finely divided into and dispersed as minute droplets into a bulk aqueous phase the resulting
composition is called an oil-in-water (o/w) emulsion. Emulsions can contain additional components in addition to the dispersed phases and the active drug which can be present as a
solution in either the aqueous phase, oily phase or itself as a separate phase. Pharmaceutical excipients such as emulsifiers, stabilizers, dyes, and anti-oxidants can also be present in emulsions as needed. Pharmaceutical emulsions can also be multiple emulsions that can be comprised of more than two phases such as, for example, in the case of oil-in-water-in-oil (o/w/o) and water-in-oil-in-water (w/o/w) emulsions. Such complex formulations often provide certain advantages that simple binary emulsions do not. Multiple emulsions in which oil droplets of an o/w emulsion enclose small water droplets constitute a w/o/w emulsion. Likewise a system of oil droplets enclosed in globules of water stabilized in an oily continuous provides an o/w/o emulsion.
[00433] Emulsions can be characterized by little or no thermodynamic stability. Often, the dispersed or discontinuous phase of the emulsion is well dispersed into the external or continuous phase and maintained in this form through the means of emulsifiers or the viscosity of the formulation. Either of the phases of the emulsion can be a semisolid or a solid, as is the case of emulsion- style ointment bases and creams. Other means of stabilizing emulsions entail the use of emulsifiers that can be incorporated into either phase of the emulsion. Emulsifiers can broadly be classified into four categories: synthetic surfactants, naturally occurring emulsifiers, absorption bases, and finely dispersed solids (Idson, in Pharmaceutical Dosage Forms,
Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 199).
[00434] Synthetic surfactants, also known as surface active agents, have found wide applicability in the formulation of emulsions and have been reviewed in the literature (Rieger, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 285; Idson, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds. ), Marcel Dekker, Inc., New York, N.Y., 1988, volume 1, p. 199). Surfactants can be typically amphiphilic and comprise a hydrophilic and a hydrophobic portion. The ratio of the hydrophilic to the hydrophobic nature of the surfactant has been termed the hydrophile/lipophile balance (HLB) and is a valuable tool in categorizing and selecting surfactants in the preparation of formulations. Surfactants can be classified into different classes based on the nature of the hydrophilic group: nonionic, anionic, cationic and amphoteric (Rieger, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 285).
[00435] Naturally occurring emulsifiers used in emulsion formulations include lanolin, beeswax, phosphatides, lecithin and acacia. Absorption bases possess hydrophilic properties such that they can soak up water to form w/o emulsions yet retain their semisolid consistencies, such as anhydrous lanolin and hydrophilic petrolatum. Finely divided solids have also been used as good emulsifiers especially in combination with surfactants and in viscous preparations. These include polar inorganic solids, such as heavy metal hydroxides, non-swelling clays such as bentonite, attapulgite, hectorite, kaolin, montmorillonite, colloidal aluminum silicate and colloidal magnesium aluminum silicate, pigments and nonpolar solids such as carbon or glyceryl tristearate.
[00436] A large variety of non-emulsifying materials can also be included in emulsion formulations and contribute to the properties of emulsions. These include fats, oils, waxes, fatty acids, fatty alcohols, fatty esters, humectants, hydrophilic colloids, preservatives and
antioxidants (Block, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 335; Idson, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 199).
[00437] Hydrophilic colloids or hydrocolloids include naturally occurring gums and synthetic polymers such as polysaccharides (for example, acacia, agar, alginic acid, carrageenan, guar gum, karaya gum, and tragacanth), cellulose derivatives (for example, carboxymethylcellulose and carboxypropylcellulose), and synthetic polymers (for example, carbomers, cellulose ethers, and carboxyvinyl polymers). These disperse or swell in water to form colloidal solutions that stabilize emulsions by forming strong interfacial films around the dispersed-phase droplets and by increasing the viscosity of the external phase.
[00438] Since emulsions often contain a number of ingredients such as carbohydrates, proteins, sterols and phosphatides that can readily support the growth of microbes, these formulations often incorporate preservatives. Commonly used preservatives included in emulsion formulations include methyl paraben, propyl paraben, quaternary ammonium salts,
benzalkonium chloride, esters of p-hydroxybenzoic acid, and boric acid. Antioxidants can also be commonly added to emulsion formulations to prevent deterioration of the formulation.
Antioxidants used can be free radical scavengers such as tocopherols, alkyl gallates, butylated
hydroxyanisole, butylated hydroxytoluene, or reducing agents such as ascorbic acid and sodium metabisulfite, and antioxidant synergists such as citric acid, tartaric acid, and lecithin.
[00439] The applications of emulsion formulations via dermatological, oral and parenteral routes and methods for their manufacture have been reviewed in the literature (Idson, in
Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds. ), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 199). Emulsion formulations for oral delivery have been very widely used because of reasons of ease of formulation, efficacy from an absorption and bioavailability standpoint. (Rosoff, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds. ), 1988, Marcel Dekker, Inc., New York N.Y., volume 1, p. 245; Idson, in
Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 199). Mineral-oil base laxatives, oil-soluble vitamins and high fat nutritive preparations can be among the materials that have commonly been
administered orally as oil-in- water emulsions.
[00440] In one embodiment, the compositions of antisense compounds and nucleic acids can be formulated as microemulsions. A microemulsion can be defined as a system of water, oil and amphiphile which is a single optically isotropic and thermodynamically stable liquid solution (Rosoff, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 245). Typically microemulsions can be systems that can be prepared by first dispersing an oil in an aqueous surfactant solution and then adding a sufficient amount of a fourth component, generally an intermediate chain-length alcohol to form a transparent system. Therefore, microemulsions have also been described as thermodynamically stable, isotropically clear dispersions of two immiscible liquids that can be stabilized by interfacial films of surface-active molecules (Leung and Shah, in: Controlled Release of Drugs: Polymers and Aggregate Systems, Rosoff, M., Ed., 1989, VCH Publishers, New York, pages 185-215). Microemulsions commonly can be prepared via a combination of three to five components that include oil, water, surfactant, cosurfactant and electrolyte. Whether the microemulsion is of the water-in-oil (w/o) or an oil-in-water (o/w) type is dependent on the properties of the oil and surfactant used and on the structure and geometric packing of the polar heads and hydrocarbon tails of the surfactant molecules (Schott, in Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa., 1985, p. 271).
[00441] The phenomeno logical approach utilizing phase diagrams has been extensively studied and has yielded a comprehensive knowledge, to one skilled in the art, of how to formulate microemulsions (Rosoff, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 245; Block, in
Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 335). Compared to conventional emulsions, microemulsions offer the advantage of solubilizing water-insoluble drugs in a formulation of thermodynamically stable droplets that can be formed spontaneously.
[00442] Surfactants used in the preparation of microemulsions include, but are not limited to, ionic surfactants, non-ionic surfactants, Brij 96, polyoxyethylene oleyl ethers, polyglycerol fatty acid esters, tetraglycerol monolaurate (ML310), tetraglycerol monooleate (MO310),
hexaglycerol monooleate (PO310), hexaglycerol pentaoleate (PO500), decaglycerol monocaprate (MCA750), decaglycerol monooleate (MO750), decaglycerol sequioleate (SO750), decaglycerol decaoleate (DAO750), alone or in combination with cosurfactants. The cosurfactant, usually a short-chain alcohol such as ethanol, 1-propanol, and 1-butanol, serves to increase the interfacial fluidity by penetrating into the surfactant film and consequently creating a disordered film because of the void space generated among surfactant molecules. Microemulsions can, however, be prepared without the use of cosurfactants and alcohol-free self-emulsifying microemulsion systems can be known in the art. The aqueous phase can typically be, but is not limited to, water, an aqueous solution of the drug, glycerol, PEG300, PEG400, polyglycerols, propylene glycols, and derivatives of ethylene glycol. The oil phase can include, but is not limited to, materials such as Captex 300, Captex 355, Capmul MCM, fatty acid esters, medium chain (C8-C12) mono, di, and tri-glycerides, polyoxyethylated glyceryl fatty acid esters, fatty alcohols, polyglycolized glycerides, saturated polyglycolized C8-C10 glycerides, vegetable oils and silicone oil.
[00443] Microemulsions can be particularly of interest from the standpoint of drug
solubilization and the enhanced absorption of drugs. Lipid based microemulsions (both o/w and w/o) have been proposed to enhance the oral bioavailability of drugs, including peptides
(Constantinides et al., Pharmaceutical Research, 1994, 11, 1385-1390; Ritschel, Meth. Find. Exp. Clin. Pharmacol, 1993, 13, 205). Microemulsions afford advantages of improved drug solubilization, protection of drug from enzymatic hydrolysis, possible enhancement of drug absorption due to surfactant-induced alterations in membrane fluidity and permeability, ease of
preparation, ease of oral administration over solid dosage forms, improved clinical potency, and decreased toxicity (Constantinides et al., Pharmaceutical Research, 1994, 11, 1385; Ho et al., J. Pharm. Sci., 1996, 85, 138-143). Often microemulsions can form spontaneously when their components can be brought together at ambient temperature. This can be particularly
advantageous when formulating thermolabile drugs, peptides or antisense compounds.
Microemulsions have also been effective in the transdermal delivery of active components in both cosmetic and pharmaceutical applications. It is expected that the microemulsion
compositions and formulations provided herein will facilitate the increased systemic absorption of antisense compounds and nucleic acids from the gastrointestinal tract, as well as improve the local cellular uptake of antisense compounds and nucleic acids within the gastrointestinal tract, vagina, buccal cavity and other can beas of administration.
[00444] Microemulsions provided herein can also contain additional components and additives such as sorbitan monostearate (Grill 3), Labrasol, and penetration enhancers to improve the properties of the formulation and to enhance the absorption of the antisense compounds and nucleic acids provided herein. Penetration enhancers used in the microemulsions provided herein can be classified as belonging to one of five broad categories-surfactants, fatty acids, bile salts, chelating agents, and non-chelating non-surfactants (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p. 92). Each of these classes has been discussed above.
1.3.2. Liposomes
[00445] There are organized surfactant structures besides microemulsions that have been studied and used for the formulation of drugs. These include monolayers, micelles, bilayers and vesicles. Vesicles, such as liposomes, have attracted great interest because of their specificity and the duration of action they offer from the standpoint of drug delivery. As used provided herein, the term "liposome" refers to a vesicle composed of amphiphilic lipids arranged in a spherical bilayer or bilayers.
[00446] Liposomes can be unilamellar or multilamellar vesicles which have a membrane formed from a lipophilic material and an aqueous interior. The aqueous portion contains the composition to be delivered. Cationic liposomes possess the advantage of being able to fuse to the cell wall. Non-cationic liposomes, although not able to fuse as efficiently with the cell wall, can be taken up by macrophages in vivo.
[00447] In order to cross intact mammalian skin, lipid vesicles must pass through a series of fine pores, each with a diameter less than 50 nm, under the influence of a suitable transdermal gradient. Therefore, it is desirable to use a liposome which is highly deformable and able to pass through such fine pores.
[00448] Further advantages of liposomes include; liposomes obtained from natural phospholipids can be biocompatible and biodegradable; liposomes can incorporate a wide range of water and lipid soluble drugs; liposomes can protect encapsulated drugs in their internal compartments from metabolism and degradation (Rosoff, in Pharmaceutical Dosage Forms, Lieberman, Rieger and Banker (Eds.), 1988, Marcel Dekker, Inc., New York, N.Y., volume 1, p. 245). Important considerations in the preparation of liposome formulations can be the lipid surface charge, vesicle size and the aqueous volume of the liposomes.
[00449] Liposomes can be useful for the transfer and delivery of active ingredients to the site of action. Because the liposomal membrane is structurally similar to biological membranes, when liposomes can be applied to a tissue, the liposomes start to merge with the cellular membranes. As the merging of the liposome and cell progresses, the liposomal contents can be emptied into the cell where the active agent can act.
[00450] Liposomal formulations have been the focus of extensive investigation as the mode of delivery for many drugs. There is growing evidence that for topical administration, liposomes present several advantages over other formulations. Such advantages include reduced sided effects related to high systemic absorption of the administered drug, increased accumulation of the administered drug at the desired target, and the ability to administer a wide variety of drugs, both hydrophilic and hydrophobic, into the skin.
[00451] Several reports have detailed the ability of liposomes to deliver agents including high- molecular weight DNA into the skin. Compounds including analgesics, antibodies, hormones and high-molecular weight DNAs have been administered to the skin. The majority of applications resulted in the targeting of the upper epidermis.
[00452] Liposomes fall into two broad classes. Cationic liposomes can be positively charged liposomes which interact with the negatively charged DNA molecules to form a stable complex. The positively charged DNA/liposome complex binds to the negatively charged cell surface and is internalized in an endosome. Due to the acidic pH within the endosome, the liposomes can be
ruptured, releasing their contents into the cell cytoplasm (Wang et al., Biochem. Biophys. Res. Commun., 1987, 147, 980-985).
[00453] Liposomes which can be pH-sensitive or negatively-charged, entrap DNA rather than complex with it. Since both the DNA and the lipid can be similarly charged, repulsion rather than complex formation occurs. Nevertheless, some DNA is entrapped within the aqueous interior of these liposomes. pH-sensitive liposomes have been used to deliver DNA encoding the thymidine kinase gene to cell monolayers in culture. Expression of the exogenous gene was detected in the target cells (Zhou et al., Journal of Controlled Release, 1992, 19, 269-274).
[00454] One major type of liposomal composition includes phospholipids other than naturally-derived phosphatidylcholine. Neutral liposome compositions, for example, can be formed from dimyristoyl phosphatidylcholine (DMPC) or dipalmitoyl phosphatidylcholine (DPPC). Anionic liposome compositions generally can be formed from dimyristoyl
phosphatidylglycerol, while anionic-fusogenic liposomes can be formed primarily from dioleoyl phosphatidylethanolamine (DOPE). Another type of liposomal composition is formed from phosphatidylcholine (PC) such as, for example, soybean PC, and egg PC. Another type is formed from mixtures of phospholipid and/or phosphatidylcholine and/or cholesterol.
[00455] Several studies have assessed the topical delivery of liposomal drug formulations to the skin. Application of liposomes containing interferon to guinea pig skin resulted in a reduction of skin herpes sores while delivery of interferon via other refers to (e.g., as a solution or as an emulsion) were ineffective (Weiner et al., Journal of Drug Targeting, 1992, 2, 405-410). Further, an additional study tested the efficacy of interferon administered as part of a liposomal formulation to the administration of interferon using an aqueous system, and concluded that the liposomal formulation was superior to aqueous administration (du Plessis et al., Antiviral Research, 1992, 18, 259-265).
[00456] Non-ionic liposomal systems have also been examined to determine their utility in the delivery of drugs to the skin, in particular systems comprising non-ionic surfactant and cholesterol. Non-ionic liposomal, formulations comprising Novasome™ I (glyceryl
dilaurate/cholesterol/polyoxyethylene-10-stearyl ether) and Novasome™ II (glyceryl
distearate/cholesterol/polyoxyethylene-10-stearyl ether) were used to deliver cyclosporin- A into the dermis of mouse skin. Results indicated that such non-ionic liposomal systems were effective
in facilitating the deposition of cyclosporin-A into different layers of the skin (Hu et al. S. T. P. Pharma. Sci., 1994, 4, 6, 466).
[00457] Liposomes also include "sterically stabilized" liposomes, a term which, as used herein, refers to liposomes comprising one or more specialized lipids that, when incorporated into liposomes, result in enhanced circulation lifetimes relative to liposomes lacking such specialized lipids. Examples of sterically stabilized liposomes can be those in which part of the vesicle-forming lipid portion of the liposome (A) comprises one or more glycolipids, such as monosialoganglioside GM1, or (B) is derivatized with one or more hydrophilic polymers, such as a polyethylene glycol (PEG) moiety. While not wishing to be bound by any particular theory, it is thought in the art that, at least for sterically stabilized liposomes containing gangliosides, sphingomyelin, or PEG-derivatized lipids, the enhanced circulation half-life of these sterically stabilized liposomes derives from a reduced uptake into cells of the reticuloendothelial system (RES) (Allen et al, FEBS Letters, 1987, 223, 42; Wu et al, Cancer Research, 1993, 53, 3765).
[00458] Various liposomes comprising one or more glycolipids can be known in the art. Papahadjopoulos et al. (Ann. N. Y. Acad. Sci., 1987, 507, 64) reported the ability of
monosialoganglioside GM1, galactocerebroside sulfate and phosphatidylinositol to improve blood half-lives of liposomes. These findings were expounded upon by Gabizon et al. (Proc. Natl. Acad. Sci. U.S. A., 1988, 85, 6949). U.S. Pat. No. 4,837,028 and WO 88/04924, both to Allen et al., disclose liposomes comprising (1) sphingomyelin and (2) the ganglioside GM1 or a galactocerebroside sulfate ester. U.S. Pat. No. 5,543,152 (Webb et al. ) discloses liposomes comprising sphingomyelin. Liposomes comprising 1 ,2-sn-dimyristoylphosphatidylcholine can be disclosed in WO 97/13499 (Lim et al. ).
[00459] Many liposomes comprising lipids derivatized with one or more hydrophilic polymers, and methods of preparation thereof, can be known in the art. Sunamoto et al. (Bull. Chem. Soc. Jpn., 1980, 53, 2778) described liposomes comprising a nonionic detergent, 2C1215G, that contains a PEG moiety. Ilium et al. (FEBS Lett., 1984, 167, 79) noted that hydrophilic coating of polystyrene particles with polymeric glycols results in significantly enhanced blood half-lives. Synthetic phospholipids modified by the attachment of carboxylic groups of polyalkylene glycols {e.g., PEG) can be described by Sears ( U.S. Pat. Nos. 4,426,330 and 4,534,899). Klibanov et al. (FEBS Lett., 1990, 268, 235) described experiments
demonstrating that liposomes comprising phosphatidylethanolamme (PE) derivatized with PEG
or PEG stearate have significant increases in blood circulation half- lives. Blume et al. (Biochimica et Biophysica Acta, 1990, 1029, 91) extended such observations to other PEG- derivatized phospholipids,, e.g., DSPE-PEG, formed from the combination of
distearoylphosphatidylethanolamine (DSPE) and PEG. Liposomes having covalently bound PEG moieties on their external surface can be described in European Patent No. EP 0 445 131 Bl and WO 90/04384 to Fisher. Liposome compositions containing 1-20 mole percent of PE derivatized with PEG, and methods of use thereof, can be described by Woodle et al. ( U.S. Pat. Nos.
5,013,556 and 5,356,633) and Martin et al. ( U.S. Pat. No. 5,213,804 and European Patent No. EP 0 496 813 Bl). Liposomes comprising a number of other lipid-polymer conjugates can be disclosed in WO 91/05545 and U.S. Pat. No. 5,225,212 (both to Martin et al. ) and in WO 94/20073 (Zalipsky et al. ) Liposomes comprising PEG-modified ceramide lipids can be described in WO 96/10391 (Choi et al. ). U.S. Pat. No. 5,540,935 (Miyazaki et al. ) and U.S. Pat. No. 5,556,948 (Tagawa et al. ) describes PEG-containing liposomes that can be further derivatized with functional moieties on their surfaces.
[00460] A limited number of liposomes comprising nucleic acids can be known in the art. WO 96/40062 to Thierry et al. discloses methods for encapsulating high molecular weight nucleic acids in liposomes. U.S. Pat. No. 5,264,221 to Tagawa et al. discloses protein-bonded liposomes and asserts that the contents of such liposomes can include an antisense RNA. U.S. Pat. No. 5,665,710 to Rahman et al. describes certain methods of encapsulating oligodeoxynucleotides in liposomes. WO 97/04787 to Love et al. discloses liposomes comprising antisense antisense compounds targeted to the raf gene.
[00461] Transfersomes can be yet another type of liposomes, and can be highly deformable lipid aggregates which can be attractive candidates for drug delivery vehicles. Transfersomes can be described as lipid droplets which can be so highly, deformable that they can be easily able to penetrate through pores which can be smaller than the droplet. Transfersomes can be adaptable to the environment in which they can be used, e.g., they can be self-optimizing (adaptive to the shape of pores in the skin), self-repairing, frequently reach their targets without fragmenting, and often self-loading. To make transfersomes it is possible to add surface edge-activators, usually surfactants, to a standard liposomal composition. Transfersomes have been used to deliver serum albumin to the skin. The transfersome-mediated delivery of serum albumin has been shown to be as effective as subcutaneous injection of a solution containing serum albumin.
[00462] Surfactants find wide application in formulations such as emulsions (including microemulsions) and liposomes. The most common way of classifying and ranking the properties of the many different types of surfactants, both natural and synthetic, is by the use of the hydrophile/lipophile balance (HLB). The nature of the hydrophilic group (also known as the "head") provides the most useful refers to for categorizing the different surfactants used in formulations (Rieger, in Pharmaceutical Dosage Forms, Marcel Dekker, Inc., New York, N. Y., 1988, p. 285).
[00463] If the surfactant molecule is not ionized, it is classified as a nonionic surfactant. Nonionic surfactants find wide application in pharmaceutical and cosmetic products and can be usable over a wide range of pH values. In general their HLB values range from 2 to about 18 depending on their structure. Nonionic surfactants include nonionic esters such as ethylene glycol esters, propylene glycol esters, glyceryl esters, polyglyceryl esters, sorbitan esters, sucrose esters, and ethoxylated esters. Nonionic alkanolamides and ethers such as fatty alcohol ethoxylates, propoxylated alcohols, and ethoxylated/propoxylated block polymers can also be included in this class. The polyoxyethylene surfactants can be the most popular members of the nonionic surfactant class.
[00464] If the surfactant molecule carries a negative charge when it is dissolved or dispersed in water, the surfactant is classified as anionic. Anionic surfactants include carboxylates such as soaps, acyl lactylates, acyl amides of amino acids, esters of sulfuric acid such as alkyl sulfates and ethoxylated alkyl sulfates, sulfonates such as alkyl benzene sulfonates, acyl isethionates, acyl taurates and sulfosuccinates, and phosphates. The most important members of the anionic surfactant class can be the alkyl sulfates and the soaps.
[00465] If the surfactant molecule carries a positive charge when it is dissolved or dispersed in water, the surfactant is classified as cationic. Cationic surfactants include quaternary ammonium salts and ethoxylated amines. The quaternary ammonium salts can be the most used members of this class.
[00466] If the surfactant molecule has the ability to carry either a positive or negative charge, the surfactant is classified as amphoteric. Amphoteric surfactants include acrylic acid
derivatives, substituted alkylamides, N-alkylbetaines and phosphatides.
[00467] The use of surfactants in drug products, formulations and in emulsions has been reviewed (Rieger, in Pharmaceutical Dosage Forms, Marcel Dekker, Inc., New York, N. Y., 1988, p. 285).
1.3.3. Penetration Enhancers
[00468] In one embodiment, provided herein, various penetration enhancers to effect the efficient delivery of nucleic acids, particularly antisense compounds, to the skin of animals. Most drugs can be present in solution in both ionized and nonionized forms. However, usually only lipid soluble or lipophilic drugs readily cross cell membranes. It has been discovered that even non-lipophilic drugs can cross cell membranes if the membrane to be crossed is treated with a penetration enhancer. In addition to aiding the diffusion of non-lipophilic drugs across cell membranes, penetration enhancers also enhance the permeability of lipophilic drugs.
[00469] Penetration enhancers can be classified as belonging to one of five broad categories, i.e., surfactants, fatty acids, bile salts, chelating agents, and non-chelating non-surfactants (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p. 92). Each of the above mentioned classes of penetration enhancers can be described below in greater detail.
1.3.3.1 Surfactants
[00470] In connection with provided herein, surfactants (or "surface-active agents") can be chemical entities which, when dissolved in an aqueous solution, reduce the surface tension of the solution or the interfacial tension between the aqueous solution and another liquid, with the result that absorption of antisense compounds through the mucosa is enhanced. In addition to bile salts and fatty acids, these penetration enhancers include, for example, sodium lauryl sulfate, polyoxyethylene-9-lauryl ether and polyoxyethylene-20-cetyl ether) (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p. 92); and perfluorochemical emulsions, such as FC-43. Takahashi et al., J. Pharm. Pharmacol, 1988, 40, 252).
1.3.3.2 Fatty acids
[00471] Various fatty acids and their derivatives which act as penetration enhancers include, for example, oleic acid, lauric acid, capric acid (n-decanoic acid), myristic acid, palmitic acid, stearic acid, linoleic acid; linolenic acid, dicaprate, tricaprate, monoolein (1-monooleoyl-rac- glycerol), dilaurin, caprylic acid, arachidonic acid, glycerol 1-monocaprate, 1- dodecylazacycloheptan-2-one, acylcarnitines, acylchoines, CI -10 alkyl esters thereof (e.g.,
methyl, isopropyl and t-butyl), and mono- and di-glycerides thereof (i.e., oleate, laurate, caprate, myristate, palmitate, stearate, linoleate, etc.) (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p. 92; Muranishi, Critical Reviews in Therapeutic Drug Carrier Systems, 1990, 7, 1-33; El Hariri et al, J. Pharm. Pharmacol, 1992, 44, 651-654).
1.3.3.3 Bile salts
[00472] The physiological role of bile includes the facilitation of dispersion and absorption of lipids and fat-soluble vitamins (Brunton, Chapter 38 in: Goodman & Gilman's The
Pharmacological Basis of Therapeutics, 9th Ed., Hardman et al. Eds., McGraw-Hill, New York, 1996, pp. 934-935). Various natural bile salts, and their synthetic derivatives, act as penetration enhancers. Thus the term "bile salts" includes any of the naturally occurring components of bile as well as any of their synthetic derivatives. The bile salts include, for example, cholic acid (or its pharmaceutically acceptable sodium salt, sodium cholate), dehydrocholic acid (sodium dehydrocholate), deoxycholic acid (sodium deoxycholate), glucholic acid (sodium glucholate), glycholic acid (sodium glycocholate), glycodeoxycholic acid (sodium glycodeoxycholate), taurocholic acid (sodium taurocholate), taurodeoxycholic acid (sodium taurodeoxycholate), chenodeoxycholic acid (sodium chenodeoxycholate), ursodeoxycholic acid (UDCA), sodium tauro-24,25-dihydro-fusidate (STDHF), sodium glycodihydrofusidate and polyoxyethylene-9- lauryl ether (POE) (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, page 92; Swinyard, Chapter 39 In: Remington's Pharmaceutical Sciences, 18th Ed., Gennaro, ed., Mack Publishing Co., Easton, Pa., 1990, pages 782-783; Muranishi, Critical Reviews in
Therapeutic Drug Carrier Systems, 1990, 7, 1-33; Yamamoto et al., J. Pharm. Exp. Ther., 1992, 263, 25; Yamashita et al, J. Pharm. Sci., 1990, 79, 579-583).
1.3.3.4 Chelating Agents
[00473] Chelating agents, as used in connection with provided herein, can be defined as compounds that remove metallic ions from solution by forming complexes therewith, with the result that absorption of antisense compounds through the mucosa is enhanced. With regards to their use as penetration enhancers provided herein, chelating agents have the added advantage of also serving as DNase inhibitors, as most characterized DNA nucleases require a divalent metal ion for catalysis and can be thus inhibited by chelating agents (Jarrett, J. Chromatogr., 1993, 618, 315-339). Chelating agents include but are not limited to disodium ethylenediaminetetraacetate (EDTA), citric acid, salicylates (e.g., sodium salicylate, 5 -methoxy salicylate and homovanilate),
N-acyl derivatives of collagen, laureth-9 and N-amino acyl derivatives of beta-diketones (enamines) (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, page 92; Muranishi, Critical Reviews in Therapeutic Drug Carrier Systems, 1990, 7, 1-33; Buur et al., J. Control ReL, 1990, 14, 43-51).
1.3.3.5 Non-chelating non-surfactants
[00474] As used herein, non-chelating non-surfactant penetration enhancing compounds can be defined as compounds that demonstrate insignificant activity as chelating agents or as surfactants but that nonetheless enhance absorption of antisense compounds through the alimentary mucosa (Muranishi, Critical Reviews in Therapeutic Drug Carrier Systems, 1990, 7, 1-33). This class of penetration enhancers includes, for example, unsaturated cyclic ureas, 1- alkyl- and 1-alkenylazacycloalkanone derivatives (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, page 92); and non-steroidal anti-inflammatory agents such as diclofenac sodium, indomethacin and phenylbutazone (Yamashita et al., J. Pharm. Pharmacol., 1987, 39, 621-626).
[00475] Agents that enhance uptake of antisense compounds at the cellular level can also be added to the pharmaceutical and other compositions provided herein. For example, cationic lipids, such as lipofectin (Junichi et al, U.S. Pat. No. 5,705,188), cationic glycerol derivatives, and polycationic molecules, such as polylysine (Lollo et al., PCT Application WO 97/30731), can also be known to enhance the cellular uptake of antisense compounds.
[00476] Other agents can be utilized to enhance the penetration of the administered nucleic acids, including glycols such as ethylene glycol and propylene glycol, pyrrols such as 2-pyrrol, azones, and terpenes such as limonene and menthone.
1.3.3.6 Carriers
[00477] Certain compositions provided herein also incorporate carrier compounds in the formulation. As used herein, "carrier compound" or "carrier" can refer to a nucleic acid, or analog thereof, which is inert (i.e., does not possess biological activity per se) but is recognized as a nucleic acid by in vivo processes that reduce the bioavailability of a nucleic acid having biological activity by, for example, degrading the biologically active nucleic acid or promoting its removal from circulation. The co administration of a nucleic acid and a carrier compound, typically with an excess of the latter substance, can result in a substantial reduction of the amount of nucleic acid recovered in the liver, kidney or other extracirculatory reservoirs,
presumably due to competition between the carrier compound and the nucleic acid for a common receptor. For example, the recovery of a partially phosphorothioate antisense compound in hepatic tissue can be reduced when it is co administered with polyinosinic acid, dextran sulfate, polycytidic acid or 4-acetamido-4'isothiocyano-stilbene-2,2'-disulfonic acid (Miyao et al., Antisense Res. Dev., 1995, 5, 115-121; Takakura et al., Antisense & Nucl. Acid Drug Dev., 1996, 6, 177-183).
1.3.4. Excipients
[00478] In contrast to a carrier compound, a "pharmaceutical carrier" or "excipient" is a pharmaceutically acceptable solvent, suspending agent or any other pharmacologically inert vehicle for delivering one or more nucleic acids to an animal. The excipient can be liquid or solid and is selected, with the planned manner of administration in mind, so as to provide for the desired bulk, consistency, etc., when combined with a nucleic acid and the other components of a given pharmaceutical composition. Typical pharmaceutical carriers include, but are not limited to, binding agents (e.g., pregelatinized maize starch, polyvinylpyrrolidone or hydroxypropyl methylcellulose, etc.); fillers (e.g., lactose and other sugars, microcrystalline cellulose, pectin, gelatin, calcium sulfate, ethyl cellulose, polyacrylates or calcium hydrogen phosphate, etc.); lubricants (e.g., magnesium stearate, talc, silica, colloidal silicon dioxide, stearic acid, metallic stearates, hydrogenated vegetable oils, corn starch, polyethylene glycols, sodium benzoate, sodium acetate, etc.); disintegrants (e.g., starch, sodium starch glycolate, etc.); and wetting agents (e.g., sodium lauryl sulphate, etc.).
[00479] Pharmaceutically acceptable organic or inorganic excipient suitable for non- parenteral administrations which do not deleteriously react with nucleic acids can also be used to formulate the compositions provided herein. Suitable pharmaceutically acceptable carriers include, but are not limited to, water, salt solutions, alcohols, polyethylene glycols, gelatin, lactose, amylose, magnesium stearate, talc, silicic acid, viscous paraffin,
hydroxymethylcellulose, polyvinylpyrrolidone and the like.
[00480] Formulations for topical administration of nucleic acids can include sterile and non- sterile aqueous solutions, non-aqueous solutions in common solvents such as alcohols, or solutions of the nucleic acids in liquid or solid oil bases. The solutions can also contain buffers, diluents and other suitable additives. Pharmaceutically acceptable organic or inorganic excipients
suitable for non-parenteral administration which do not deleteriously react with nucleic acids can be used.
[00481] Suitable pharmaceutically acceptable excipients include, but are not limited to, water, salt solutions, alcohol, polyethylene glycols, gelatin, lactose, amylose, magnesium stearate, talc, silicic acid, viscous paraffin, hydroxymethylcellulose, polyvinylpyrrolidone and the like.
1.3.5. Pulsatile Delivery
[00482] The compounds provided herein can also be administered by pulsatile delivery.
"Pulsatile delivery" refers to a pharmaceutical formulations that delivers a first pulse of drug combined with a penetration enhancer and a second pulse of penetration enhancer to promote absorption of drug which is not absorbed upon release with the first pulse of penetration enhancer.
[00483] One embodiment provided herein is a delayed release oral formulation for enhanced intestinal drug absorption, comprising:
(a) a first population of carrier particles comprising said drug and a penetration enhancer, wherein said drug and said penetration enhancer can be released at a first location in the intestine; and
(b) a second population of carrier particles comprising a penetration enhancer and a delayed release coating or matrix, wherein the penetration enhancer is released at a second location in the intestine downstream from the first location, whereby absorption of the drug is enhanced when the drug reaches the second location.
[00484] Alternatively, the penetration enhancer in (a) and (b) is different.
[00485] This enhancement is obtained by encapsulating at least two populations of carrier particles. The first population of carrier particles comprises a biologically active substance and a penetration enhancer, and the second (and optionally additional) population of carrier particles comprises a penetration enhancer and a delayed release coating or matrix.
[00486] A "first pass effect" that applies to orally administered drugs is degradation due to the action of gastric acid and various digestive enzymes. One means of ameliorating first pass clearance effects is to increase the dose of administered drug, thereby compensating for proportion of drug lost to first pass clearance. Although this can be readily achieved with intravenous, administration by, for example, simply providing more of the drug to an animal, other factors influence the bioavailability of drugs administered via non-parenteral refers to. For
example, a drug can be enzymatically or chemically degraded in the alimentary canal or blood stream and/or can be impermeable or semipermeable to various mucosal membranes.
[00487] It is also contemplated that these pharmaceutical compositions can be capable of enhancing absorption of biologically active substances when administered via the rectal, vaginal, nasal or pulmonary routes. It is also contemplated that release of the biologically active substance can be achieved in any part of the gastrointestinal tract.
[00488] Liquid pharmaceutical compositions of antisense compound can be prepared by combining the antisense compound with a suitable vehicle, for example sterile pyrogen free water, or saline solution. Other therapeutic compounds can optionally be included.
[00489] Provided herein, also contemplates the use of solid particulate compositions. Such compositions can comprise particles of antisense compound that can be of respirable size. Such particles can be prepared by, for example, grinding dry antisense compound by conventional refers to, fore example with a mortar and pestle, and then passing the resulting powder composition through a 400 mesh screen to segregate large particles and agglomerates. A solid particulate composition comprised of an active antisense compound can optionally contain a dispersant which serves to facilitate the formation of an aerosol, for example lactose.
[00490] In accordance with provided herein, antisense compound compositions can be aerosolized. Aerosolization of liquid particles can be produced by any suitable refers to, such as with a nebulizer. See, for example, U.S. Pat. No. 4,501,729. Nebulizers can be commercially available devices which transform solutions or suspensions into a therapeutic aerosol mist either by means of acceleration of a compressed gas, typically air or oxygen, through a narrow venturi orifice or by means of ultrasonic agitation. Suitable nebulizers include those sold by Blairex® under the name PARI LC PLUS, PARI DURA-NEB 2000, PARI-BABY Size, PARI PRONEB Compressor with LC PLUS, PARI WALKHALER Compressor/Nebulizer System, PARI LC PLUS Reusable Nebulizer, and PARI LC Jet+ ©Nebulizer.
[00491] Exemplary formulations for use in nebulizers consist of an antisense compound in a liquid, such as sterile, pyragen free water, or saline solution, wherein the antisense compound comprises up to about 40% w/w of the formulation. Can, the antisense compound comprises less than 20%) w/w. If desired, further additives such as preservatives (for example, methyl hydroxybenzoate) antioxidants, and flavoring agents can be added to the composition.
[00492] Solid particles comprising an antisense compound can also be aerosolized using any solid particulate medicament aerosol generator known in the art. Such aerosol generators produce respirable particles, as described above, and further produce reproducible metered dose per unit volume of aerosol. Suitable solid particulate aerosol generators include insufflators and metered dose inhalers. Metered dose inhalers can be used in the art and can be useful provided herein.
[00493] Can, liquid or solid aerosols can be produced at a rate of from about 10 to 150 liters per minute, more can from about 30 to 150 liters per minute, and most can about 60 liters per minute.
[00494] Enhanced bioavailability of biologically active substances is also achieved via the oral administration of the compositions and methods provided herein. The term "bioavailability" refers to a measurement of what portion of an administered drug reaches the circulatory system when a non-parenteral mode of administration is used to introduce the drug into an animal.
[00495] Penetration enhancers include, but are not limited to, members of molecular classes such as surfactants, fatty acids, bile salts, chelating agents, and non-chelating non-surfactant molecules. (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p. 92).
[00496] Carriers can be inert molecules that can be included in the compositions provided herein to interfere with processes that lead to reduction in the levels of bioavailable drug.
1.3.6. Chewable oral formulation
[00497] In addition to the solid dosage forms disclosed throughout, provided herein can be chewable oral formulations, In some embodiments, the formulations will comprise (or consist essentially of) an effective amount of a pharmaceutical composition along with suitable excipients that allow the formulations to be chewed by the patient, in additional embodiments, the formulations can further comprise one or more taste -masking or sweetening agents, such as those described herein. In one embodiment, sucralose is used in the chewable formulations. Additional active agents, such as those described herein, can also optionally be added to the chewable formulations. The amount of a pharmaceutical composition , other optional active agents and sweetening agents (e.g., sucralose) in the chewable formulations of provided herein, can be readily determinable by those of ordinary skill in the art, and include those amounts and combinations described herein. For example, the chewable formulations of provided herein, comprise (or consist essentially of) a pharmaceutical composition and about 0. 05% to about 0.
15% sucralose. Such chewable formulations can be especially useful in patient populations where compliance is an issue, such as children, the elderly, and patients who can have difficulty swallowing or using spray/inhalable formulations.
[00498] The formulations can also contain colorants to improve the appearance of the chewable formulations, especially since an attractive coloration imparted by a colorant can improve patient compliance. The relative amounts of the colorants selected will vary depending upon the particular hue of the subject colorants and the resultant color desired.
[00499] Any standard pharmaceutically acceptable excipient can be used in the chewable tablet formulations which provides adequate compression such as diluents (e.g., mannitol, xylitol, maltitol, lactitol, sorbitol, lactose, sucrose, and compressible sugars such as DiPac® (dextrinized sucrose), available from Austin Products Inc. (Holmdel, N. J.), binders,
disintegrants, splitting or swelling agents (e.g., polyvinyl polypyrrolidone, croscarmellose sodium (e.g., Ac-Di-Sol available from FMC BioPolymer, Philadelphia, Pa.), starches and derivatives, cellulose and derivatives, microcrystalline celluloses, such as Avicel™ PH 101 or Avicel™ CE- 15 (a microcrystalline modified with guar gum), both available from FMC
BioPolymer, (Philadelphia, Pa. ), lubricating agents (e.g., magnesium stearate), and flow agents (e.g., colloidal silicon dioxide, such as Cab-O-Sil M5® available from Cabot Corporation, Kokomo, Ind.).
[00500] Suitable amounts of sweetener (e.g., sucralose) used in the chewable formulations, will be familiar to, and can be readily determined by, those skilled in the art. In certain embodiments, the sweetener is present in an amount from about 0. 05%> to about 5. 0%> (e.g., about 0. 05%, about 0. 1%, about 0. 125%, about 0. 15%, about 0. 2%, about 0. 3%, about 0. 4%, about 0. 5%, about 0. 6%, about 0. 7%, about 0. 8%, about 0. 9%, about 1%, about 1. 25% about 1. 5%), about 1. 75%), about 2%, about 2. 25%, about 2. 5%, about 2. 75%, about 3%, about 3. 25%, about 3. 5%, about 3. 75%, about 4%, about 4. 25%, about 4. 5%, about 4. 75% or about 5%). Those or ordinary skill in the art will appreciate that the amount of sweetener can vary depending on the strength of the particular sweetener used and the levels approved by the regulatory authorities for use in pharmaceutical products.
[00501] Suitable cyclodextrins for use in the chewable formulations of provided herein, include α, β, or γ cyclodextrins, or an alkylated or hydroxyalkylated derivatives thereof, such as heptakis (2,6-di-o-methyl)-P-cyclodextrin (DIMEB), randomly methylated β-cyclodextrin
(RAMEB), and hydroxypropyl β-cyclodextrin (HPpCD). A suitable cyclodextrin is β- cyclodextrin (available from Cerestar USA, Inc., Hammond, Ind. or from Roquette America, Inc., Keokuk. IA under the trade name Kleptose™). If desired, the complex of the active substance with cyclodextrin can be prepared in advance, for example, by malaxating or granulating a pharmaceutical composition and any additional active substance(s) and the cyclodextrin in the presence of water, or by preparing an aqueous solution containing a pharmaceutical composition and any additional active substance(s) and the cyclodextrin in the desired molar ratio. Alternatively, the pharmaceutical composition and any additional active substance(s) and the cyclodextrin can be simply mixed with other excipients and adjuvants.
[00502] A typical manufacturing process for making either a single layer or bi-layer chewable tablet generally involves blending of the desired ingredients to form a uniform distribution of the pharmaceutical composition (and any other active agent(s)), excipients (e.g., colorants and flavoring agents as well as others). If desired, an inclusion complex of a pharmaceutical composition and any other active agent(s) and cyclodextrin (e.g., β-cyclodextrin) can be formed prior to blending into the mixture by malaxating a pharmaceutical composition and any other active agent(s) and cyclodextrin in the presence of water in a planetary mixer for about 20 minutes. The mixture is then dried in a drying oven. After drying, the complex is mixed with any color/flavoring blend. The blend is then compressed into a single layer or bi-layer tablet using standard methods well-known to those skilled in the art (e.g., Kilian T-100 tablet press or Courtoy 292/43 rotary bi-layer press). The colorants and flavoring agents can be added to both layers to form a uniform presentation of the tablet. Methods for preparation of chewable tablets and various components for use in the tablets can be found throughout the detailed description section and the Examples of U.S. Patent Publication No. 2003/0215503, the disclosure of which is incorporated by reference herein for all purposes. Additional chewable/orally dissolving tablets, and methods for their manufacture, can be disclosed in U.S. Patent Publication No. 2004/0265372 and U.S. Patent No. 6,270,790, the disclosures of each of which can be incorporated by reference herein for all purposes
1.3.7. Other Components
[00503] The compositions provided herein can additionally contain other adjunct components conventionally found in pharmaceutical compositions, at their art-established usage levels. Thus, for example, the compositions can contain additional, compatible, pharmaceutically-active
materials such as, for example, antipruritics, astringents, local anesthetics or anti-inflammatory agents, or can contain additional materials useful in physically formulating various dosage forms of the compositions provided herein, such as dyes, flavoring agents, preservatives, antioxidants, opacifiers, thickening agents and stabilizers. However, such materials, when added, should not unduly interfere with the biological activities of the components of the compositions provided herein. The formulations can be sterilized and, if desired, mixed with auxiliary agents, e.g., lubricants, preservatives, stabilizers, wetting agents, emulsifiers, salts for influencing osmotic pressure, buffers, colorings, flavorings and/or aromatic substances and the like which do not deleteriously interact with the nucleic acid(s) of the formulation.
[00504] Aqueous suspensions can contain substances which increase the viscosity of the suspension including, for example, sodium carboxymethylcellulose, sorbitol and/or dextran. The suspension can also contain stabilizers.
[00505] Certain embodiments, provide pharmaceutical compositions containing (a) an antisense compound and (b) one or more other chemotherapeutic agents which function by a non-antisense mechanism. Examples of such chemotherapeutic agents include but are not limited to daunorubicin, daunomycin, dactinomycin, doxorubicin, epirubicin, idarubicin, esorubicin, bleomycin, mafosfamide, ifosfamide, cytosine arabinoside, bis-chloroethylnitrosurea, busulfan, mitomycin C, actinomycin D, mithramycin, prednisone, hydroxyprogesterone, testosterone, tamoxifen, dacarbazine, procarbazine, hexamethylmelamine, pentamethylmelamine,
mitoxantrone, amsacrine, chlorambucil, methylcyclohexylnitrosurea, nitrogen mustards, melphalan, cyclophosphamide, 6-mercaptopurine, 6-thioguanine, cytarabine, 5-azacytidine, hydroxyurea, deoxycoformycin, 4-hydroxyperoxycyclophosphoramide, 5-fluorouracil (5-FU), 5- fluorodeoxyuridine (5-FUdR), methotrexate (MTX), colchicine, taxol, vincristine, vinblastine, etoposide (VP- 16), trimetrexate, irinotecan, topotecan, gemcitabine, teniposide, cisplatin and diethylstilbestrol (DES). See, generally, The Merck Manual of Diagnosis and Therapy, 15th Ed. 1987, pp. 1206-1228, Berkow et al., eds., Rahway, N. J. When used with the compounds , such chemotherapeutic agents can be used ly (e.g., 5-FU and antisense compound), sequentially (e.g., 5-FU and antisense compound for a period of time followed by MTX and antisense compound), or in combination with one or more other such chemotherapeutic agents (e.g., 5-FU, MTX and antisense compound, or 5-FU, radiotherapy and antisense compound). Anti-inflammatory drugs, including but not limited to nonsteroidal anti -inflammatory drugs and corticosteroids, and
antiviral drugs, including but not limited to ribivirin, vidarabine, acyclovir and ganciclovir, can also be combined in compositions. See, generally, The Merck Manual of Diagnosis and Therapy, 15th Ed., Berkow et al, eds., 1987, Rahway, N.J., pages 2499-2506 and 46-49, respectively). Other non-antisense chemotherapeutic agents can also be used. Two or more combined compounds can be used together or sequentially.
[00506] In another related embodiment, compositions can contain an antisense compound, particularly antisense compounds, targeted to a first nucleic acid and one or more additional antisense compounds targeted to a second nucleic acid target. Numerous examples of antisense compounds can be known in the art. Two or more combined compounds can be used together or sequentially.
[00507] The formulation of therapeutic compositions and their subsequent administration is believed to be within the skill of those in the art. Dosing is dependent on severity and
responsiveness of the disease state to be treated, with the course of treatment lasting from several days to several months, or until a cure is effected or a diminution of the disease state is achieved. Optimal dosing schedules can be calculated from measurements of drug accumulation in the body of the patient. Persons of ordinary skill can easily determine optimum dosages, dosing methodologies and repetition rates. Optimum dosages can vary depending on the relative potency of antisense compounds, and can generally be estimated based on EC50s found to be effective in in vitro and in vivo animal models. In general, dosage is from 0. 01 μg to 100 g per kg of body weight, and can be given once or more daily, weekly, monthly or yearly, or even once every 2 to 20 years. Persons of ordinary skill in the art can easily estimate repetition rates for dosing based on measured residence times and concentrations of the drug in bodily fluids or tissues. Following successful treatment, it can be desirable to have the patient undergo maintenance therapy to prevent the recurrence of the disease state, wherein the antisense compound is administered in maintenance doses, ranging from 0. 01 μg to 100 g per kg of body weight, once or more daily, to once every 20 years.
1.4 Methods of Administration, Dosing, and Scheduling
[00508] In one embodiment, administration of an antisense compound is parenteral administration. Parenteral administration can be intravenous or subcutaneous administration. Accordingly, in another embodiment, administration of an antisense compound is intravenous or
subcutaneous administration. Administration can include multiple doses of an antisense compound.
[00509] In another embodiment, the antisense compound is administered to a subject by, but not including oral, intradermal, intramuscular, intraperitoneal, intravenous, topical,
subcutaneous, percutaneous, intranasal, and inhalation routes, and via scarification (scratching the top layers of skin, e.g., using a bifurcated needle. Specifically, subcutaneous or intravenous routes can be used.
[00510] For the administration with intranasally or by inhalation the preparation for use according to provided herein, can be conveniently delivered in the form of an aerosol spray.
[00511] In particular embodiments, the compositions can be administered to the subject via oral administration. Methods of oral administration can be accomplished via liquid or solid form, and particularly in solid form such as in tablet or capsule form, using approaches and
mechanisms described elsewhere herein and others that will be familiar to the ordinarily skilled artisan.
[00512] Suitable dosages {e.g., amounts, volumes, etc.) of the compositions will be apparent from the description herein, including the Examples below. Thus in one embodiment, provided herein are pharmaceutical compositions for the treatment of MACE, including the underlying causes, but not limited to hypercholesterolemia and hypertension.
[00513] In certain embodiments, the compositions can be administered to the patient in a single dosage comprising a therapeutically effective amount of each of an antisense compounds. In some embodiments, the compositions can be administered to the patient in a single does comprising a therapeutically effective amount of an antisense compound and, one or more pharmaceutical compositions as described herein, each in a therapeutically effective {i.e., MACE-treating or MACE-preventing amount).
[00514] In some embodiments, the compositions can be administered to the patient in a single, daily dosage form, once per day. In other embodiments, the compositions can be administered to the patient two or more {i.e., two, three, four or more) times per day, or as needed according to the particular treatment regiment designed by the patient's physician.
[00515] The amount of the compositions administered each time throughout the treatment period can be the same; alternatively, the amount administered each time during the treatment period can vary {e.g., the amount administered at a given time can be more or less than the
amount administered previously). For example, doses given during maintenance therapy can be lower than those administered during the acute phase of treatment. Appropriate dosing schedules depending on the specific circumstances will be apparent to persons of ordinary skill in the art.
[00516] In certain embodiments, provided herein MACE rates can be remarkably lower following treatment with the antisense compound described herein. In another embodiment, the antisense compound is administered in a dosage to achieve at least a 40% reduction, at least a 45% reduction, at least a 50% reduction, or at least a 55% reduction in MACE. In another embodiment, the antisense compound is administered in a dosage to achieve at least a 60% reduction in MACE. In more specific embodiments, the antisense compound is administered to in a dosage of at least a 65% reduction, at least a 70% reduction, at least a 75% reduction, at least a 75%) reduction, at least an 80%> reduction, at least an 85% reduction, 85% reduction, or at least a 90% reduction in MACE.
[00517] In some embodiments, the reduction in MACE correlates with a refers to reduction in LDL cholesterol. In a specific embodiment, the reduction in MACE correlates with a refers to reduction in LDL cholesterol of approximately -49 to -113 mg/dL.
[00518] In certain embodiments, provided herein administration of the antisense compound decreases total serum cholesterol, ApoB , serum LDL, serum VLDL, serum triglycerides, serum apolipoprotein(a) and/or fatty free acids in the subject. In another embodiment administration of the antisense compound decreases LDL cholesterol. In more specific embodiments,
administration of the antisense compound causes reductions in atherogenic lipoproteins in plasma.
[00519] In other embodiments, the antisense compound is administered to patients with a baseline LDL of approximately >100 mg/dl.
[00520] In another embodiment, provided herein subjects can be administered with at least 50 mg of the antisense compound, at least 100 mg of the antisense compound, at least 200 mg of the antisense compound or at least 300 mg of the antisense compound. In a more specific
embodiment, subjects can be administered with at least 50 mg per day, at least 100 mg per day, at least 200 mg per day, or at least 300 mg per day of the antisense compound. In yet another embodiment subjects receive subcutaneous antisense compound at least 50 mg per day, at least 100 mg per day, at least 200 mg per day, or at least 300 mg per day.
[00521] In certain embodiments, the antisense compound is administered at 200 mg/week. In some embodiments, the 200 mg/week is adminitered one time per week. In some embodiments, the subject is administered a single 200 mg/dose per week. In other embodiments, the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week. In some embodiments, the antisense compound is administered weekly for at least 12 months. In one embodiment, the antisense compound is administered subcutaneously (s.c).
[00522] In certain embodiments, provided herein subjects receive 200 mg of the antisense compound on days 1 , 4, 8, and 1 1 , followed by once-weekly injections to a total of
approximately 6 weeks. In another embodiment, the antisense compound described herein is administered as a single dose once weekly for 26 weeks.
[00523] In other embodiments, subjects receive a placebo for the first 6 months followed by treatment with the antisense compound for at least 12 months. In some embodiments, provided herein subjects receive blinded antisense compound for 6 months followed by at least 6 months of open labeled treatment. In some embodiments, provided herein subjects can be administered with the antisense compound for at least 12 months.
[00524] In other embodiments, provided herein substantial reductions in refers to LDL cholesterol can be seen by week 5, with near maximal effects observed at approximately 17 weeks.
[00525] In certain embodiments, the dosage of the active ingredient depends upon the mode of administration as upon the subject, and their age, weight, condition, and the subject
pharmacokinetic data.
[00526] It will be readily apparent to one of ordinary skill in the relevant arts that other suitable modifications and adaptations to the methods and applications described herein can be obvious and can be made without departing from the scope or any embodiment thereof.
[00527] In certain embodiments, pharmaceutical compositions are administered according to a dosing regimen. In certain such embodiments, the dosing regimen comprises an induction phase and a maintenance phase.
[00528] In certain embodiments, the induction phase includes one, two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, or more than twenty doses.
[00529] In certain embodiments, the induction phase lasts from one day to six months. In certain embodiments an induction phase lasts from one week to five months as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts from one week to five months as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts from two weeks to five months as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts from three weeks to four months as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts from five weeks to three months as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts five weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts six weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts seven weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts eight weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts nine weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts ten weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts eleven weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts twelve weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts thirteen weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an
induction phase lasts fourteen weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts fifteen weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts sixteen weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts seventeen weeks as measured from
administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts eighteen weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts nineteen weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts twenty weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts twenty-one weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts twenty-two weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts twenty-three weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts twenty-four weeks as measured from administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain embodiments an induction phase lasts twenty- five weeks as measured from
administration of the first dose of the induction phase to administration of the first dose of the maintenance phase. In certain such embodiments, the doses administered during the induction phase are lower than the doses administered during the maintenance phase.
[00530] In certain embodiments, the dose administered during the induction phase is lower than the dose administered during the maintenance phase to avoid undesired side effects. In certain embodiments, the undesired side effect is liver toxicity. In certain such embodiments, the undesired side effect is increased ALT. In certain such embodiments, the lower induction dose
provides time for lipid metabolism in the liver to compensate for the decreased production of ApoB. In certain such embodiments, mild increases in ALT reflect rapid lipid-lowering activity.
[00531] In certain embodiments where the induction phase includes more than one dose, the doses administered during the induction phase are all the same amount as one another. In certain embodiments, the doses administered during the induction phase are not all the same amount. In certain such embodiments, the doses increase over time. In certain embodiments, the doses decrease over time.
[00532] In certain embodiments, an induction dose is administered by parenteral
administration. In certain such embodiments, the parenteral administration is subcutaneous administration. In certain such embodiments, the parenteral administration is intravenous infusion.
[00533] In certain embodiments, the doses during the induction phase are selected from 25 mg, 30 mg, 35 mg, 40 mg, 45 mg, 50 mg, 55 mg, 60 mg, 65 mg, 70 mg, 75 mg, 80 mg, 85 mg, 90 mg, 95 mg, 100 mg, 105 mg, 110 mg, 115 mg, 120 mg, 125 mg, 130 mg, 135 mg, 140 mg,
145 mg, 150 mg, 155 mg, 160 mg, 165 mg, 170 mg, 175 mg, 180 mg, 185 mg, 190 mg, 195 mg,
200 mg, 205 mg, 210 mg, 215 mg, 220 mg, 225 mg, 230 mg, 235 mg, 240 mg, 245 mg, 250 mg,
255 mg, 260 mg, 265 mg, 270 mg, 270 mg, 280 mg, 285 mg, 290 mg, 295 mg, 300 mg, 305 mg,
310 mg, 315 mg, 320 mg, 325 mg, 330 mg, 335 mg, 340 mg, 345 mg, 350 mg, 355 mg, 360 mg,
365 mg, 370 mg, 375 mg, 380 mg, 385 mg, 390 mg, 395 mg, 400 mg, 405 mg, 410 mg, 415 mg,
420 mg, 425 mg, 430 mg, 435 mg, 440 mg, 445 mg, 450 mg, 455 mg, 460 mg, 465 mg, 470 mg,
475 mg, 480 mg, 485 mg, 490 mg, 495 mg, 500 mg, 505 mg, 510 mg, 515 mg, 520 mg, 525 mg,
530 mg, 535 mg, 540 mg, 545 mg, 550 mg, 555 mg, 560 mg, 565 mg, 570 mg, 575 mg, 580 mg,
585 mg, 590 mg, 595 mg, 600 mg, 605 mg, 610 mg, 615 mg, 620 mg, 625 mg, 630 mg, 635 mg,
640 mg, 645 mg, 650 mg, 655 mg, 660 mg, 665 mg, 670 mg, 675 mg, 680 mg, 685 mg, 690 mg,
695 mg, 700 mg, 705 mg, 710 mg, 715 mg, 720 mg, 725 mg, 730 mg, 735 mg, 740 mg, 745 mg,
750 mg, 755 mg, 760 mg, 765 mg, 770 mg, 775 mg, 780 mg, 785 mg, 790 mg, 795 mg, and 800 mg. In certain such embodiments, the doses during the induction phase are selected from 25 mg, 50 mg, 75 mg, 100 mg, 150 mg, 200 mg, 250 mg, 300 mg, 350 mg, 400 mg, 500 mg, 600 mg, 700 mg, and 800mg. In certain such embodiments, the doses during the induction phase are selected from 100 mg, 125 mg, 150 mg, 175 mg, 200 mg, 225 mg, 250 mg, 275 mg, 300 mg, 325 mg, 350 mg, 375 mg, and 400 mg. In certain such embodiments, the doses during the induction
phase are selected from 100 mg, 125 mg, 150 mg, 175 mg, 200 mg, 225 mg, and 250 mg. In certain embodiments, the dose administered during the induction phase is 100 mg. In certain embodiments, the dose administered during the induction phase is 125 mg. In certain
embodiments the dose administered during the induction phase is 150 mg. In certain
embodiments the dose administered during the induction phase is 175 mg. In certain
embodiments the dose administered during the induction phase is 200 mg. In certain
embodiments the dose administered during the induction phase is 225 mg. In certain
embodiments the dose administered during the induction phase is 250 mg. In certain
embodiments the dose administered during the induction phase is 300 mg. In certain
embodiments the dose administered during the induction phase is 325 mg. In certain
embodiments the dose administered during the induction phase is 350 mg. In certain
embodiments the dose administered during the induction phase is 375 mg. In certain
embodiments the dose administered during the induction phase is 400 mg.
[00534] In certain embodiments, where subcutaneous administration is desired, an induction dose may be administered in two or more subcutaneous injections. In certain such embodiments, when the desired induction dose requires a volume not easily accomodated by a single injection, two or more subcutaneous injections may be used to achieve the desired induction dose. In certain such embodiments, two or more subcutaneous injections may be used to administer the desired induction dose and minimize or eliminate an injection site reaction in a subject.
[00535] In certain embodiments, dose, dose frequency, and duration of the induction phase may be selected to achieve a desired effect. In certain embodiments, those variables are adjusted to result in a desired concentration of pharmaceutical agent in a subject. For example, in certain embodiments, dose and dose frequency are adjusted to provide plasma concentration of a pharmaceutical agent at an amount sufficient to achieve a desired effect. In certain of such embodiments the plasma concentration is maintained above the minimal effective concentration (MEC). In certain embodiments, pharmaceutical compositions are administered with a dosage regimen designed to maintain a concentration above the MEC for 10-90% of the time, between 30-90% of the time, or between 50-90% of the time.
[00536] In certain embodiments, doses, dose frequency, and duration of the induction phase may be selected to achieve a desired plasma trough concentration of a pharmaceutical composition. In certain such embodiments, the pharmaceutical composition is an
oligonucleotide. In certain such embodiments, the desired plasma trough concentration is from 5- 100 ng/mL. In certain such embodiments, the desired plasma trough concentration is from 5-50 ng/mL. In certain such embodiments, the desired plasma trough concentration is from 10-40 ng/mL. In certain such embodiments, the desired plasma trough concentration is from 15-35 ng/mL. In certain such embodiments, the desired plasma trough concentration is from 20-30 ng/mL.
[00537] In certain embodiments, dose, dose frequency, and duration of the induction phase may be selected to achieve a desired effect within five to thirteen weeks. In certain such embodiments, the dose is the same and the dose frequency is varied to achieve the desired effect within five to thirteen weeks. In certain such embodiments, the dose increases over time and the dose frequency remains constant. In certain such embodiments, doses and dose frequency are selected to achieve a desired effect within six to 13 weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within six weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within seven weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within eight weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within nine weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within ten weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within eleven weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within twelve weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within thirteen weeks. In certain such embodiments, one or more doses of the induction phase is greater than one or more doses of the maintenance phase. In certain such embodiments, each of the induction doses is greater than each of the maintenance doses.
[00538] In certain embodiments, doses, dose frequency, and duration of the induction phase may be selected to achieve a desired effect within 13 to 25 weeks. In certain such embodiments, the dose is the same and the dose frequency is varied to achieve the desired effect within 13 to 25 weeks. In certain such embodiments, the dose increases over time and the dose frequency remains constant. In certain such embodiments, doses and frequency are selected to achieve a desired effect within thirteen weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within fourteen weeks. In certain such embodiments, doses
and frequency are selected to achieve a desired effect within fifteen weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within sixteen weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within seventeen weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within eighteen weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within nineteen weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within twenty weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within twenty-one weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within twenty-two weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within twenty-three weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within twenty-four weeks. In certain such embodiments, doses and frequency are selected to achieve a desired effect within twenty- five weeks. In certain embodiments, one or more doses of the induction phase is less than one or more doses of the maintenance phase. In certain such embodiments, each dose of the induction phase is less than each dose of the maintenance phase.
[00539] In certain embodiments, it is desirable to achieve a desired effect as quickly as possible. In such embodiments, an induction phase with a high dose and/or high dose frequency may be desirable. Such embodiments may include administration to subjects with very high cholesterol concentrations.
[00540] In certain embodiments, it is desirable to mitigate an undesired side effect. In certain such embodiments, an induction phase with a low dose and/or low dose frequency and/or long duration may be desirable. For example, a long induction phase, with relatively low doses, may result in better tolerance of the pharmaceutical agent. Certain such embodiments, result in physiological changes that result in reduced overall side effects. In certain embodiments, such a dose regimen results in reduced liver toxicity when compared to higher initial doses and/or frequency. Such embodiments may include gradual increases of dose over time.
[00541] In certain embodiments in which a pharmaceutical composition is administered locally, the dosage regimen is selected to achieve a desired local concentration of a
pharmaceutical agent provided herein.
[00542] In certain embodiments, doses, dose frequency, and duration of the induction phase may be selected to achieve an acceptable safety profile. For example, in certain embodiments, such variables may be selected to mitigate toxicity of the pharmaceutical composition. In certain such embodiments, such variables are selected to mitigate liver toxicity. In certain such embodiments, such variables are selected to mitigate renal toxicity. In certain such embodiments, doses increase over time. In certain embodiments, one or more doses of the induction phase is lower than one or more doses of the maintenance phase. In certain such embodiments, a safety profile is not acceptable when ALT is 5-10 times the upper limit of normal. In certain such embodiments, a safety profile is not acceptable when ALT is 5-10 times the upper limit of normal, and bilirubin is elevated two or more times the upper limit of normal. In certain such embodiments, an acceptable safety profile comprises ALT elevations that are above three times the upper limit of normal, but do not exceed five times the upper limit of normal. In certain such embodiments, and acceptable safety profile comprises ALT elevations that are above three times the upper limit of normal, but do not exceed five times the upper limit of normal, and bilirubin elevations that do not exceed two times the upper limit of normal. In certain such embodiments, when administration of a pharmaceutical composition results in ALT elevations that are above three times the upper limit of normal, the dose and/or dose frequency is adjusted to mitigate the ALT elevation. In certain such embodiments, when administration of a pharmaceutical composition provided herein results in ALT elevations that are above three times the upper limit of normal, and bilirubin concentrations that are above two times the upper limit of normal, the dose and/or dose frequency is adjusted to mitigate the ALT elevation and bilirubin elevation. In certain such embodiments, the dose and/or dose frequency is adjusted to mitigate the bilirubin elevation alone.
[00543] In certain embodiments, the maintenance phase includes one, two three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, or more than twenty doses.
[00544] In certain embodiments, the maintenance phase lasts from one day to the lifetime of the subject. In certain embodiments the maintenance phase lasts from one week to twenty years as measured from administration of the last dose of the induction phase to administration of the last dose of the maintenance phase. In certain embodiments the maintenance phase lasts from two weeks to fifteen years as measured from administration of the last dose of the induction
phase to administration of the last dose of the maintenance phase. In certain embodiments the maintenance phase lasts three weeks to ten years as measured from administration of the last dose of the induction phase to administration of the last dose of the maintenance phase. In certain embodiments the maintenance phase lasts from four weeks to ten years as measured from administration of the last dose of the induction phase to administration of the last dose of the maintenance phase. In certain embodiments the maintenance phase lasts as long as the dose continues to be needed, effective, and tolerated.
[00545] In certain embodiments where the maintenance phase includes more than one dose, the doses administered during the maintenance phase are all the same as one another. In certain embodiments, the doses administered during the maintenance phase are not all the same. In certain such embodiments, the doses increase over time. In certain embodiments, the doses decrease over time.
[00546] In certain embodiments, a maintenance dose is administered by parenteral administration. In certain such embodiments, the parenteral administration is subcutaneous administration. In certain such embodiments, the parenteral administration is intravenous infusion.
[00547] In certain embodiments, the doses during the maintenance phase are selected from 25 mg, 30 mg, 35 mg, 40 mg, 45 mg, 50 mg, 55 mg, 60 mg, 65 mg, 70 mg, 75 mg, 80 mg, 85 mg, 90 mg, 95 mg, 100 mg, 105 mg, 110 mg, 115 mg, 120 mg, 125 mg, 130 mg, 135 mg, 140 mg,
145 mg, 150 mg, 155 mg, 160 mg, 165 mg, 170 mg, 175 mg, 180 mg, 185 mg, 190 mg, 195 mg,
200 mg, 205 mg, 210 mg, 215 mg, 220 mg, 225 mg, 230 mg, 235 mg, 240 mg, 245 mg, 250 mg,
255 mg, 260 mg, 265 mg, 270 mg, 270 mg, 280 mg, 285 mg, 290 mg, 295 mg, 300 mg, 305 mg,
310 mg, 315 mg, 320 mg, 325 mg, 330 mg, 335 mg, 340 mg, 345 mg, 350 mg, 355 mg, 360 mg,
365 mg, 370 mg, 375 mg, 380 mg, 385 mg, 390 mg, 395 mg, 400 mg, 405 mg, 410 mg, 415 mg,
420 mg, 425 mg, 430 mg, 435 mg, 440 mg, 445 mg, 450 mg, 455 mg, 460 mg, 465 mg, 470 mg,
475 mg, 480 mg, 485 mg, 490 mg, 495 mg, 500 mg, 505 mg, 510 mg, 515 mg, 520 mg, 525 mg,
530 mg, 535 mg, 540 mg, 545 mg, 550 mg, 555 mg, 560 mg, 565 mg, 570 mg, 575 mg, 580 mg,
585 mg, 590 mg, 595 mg, 600 mg, 605 mg, 610 mg, 615 mg, 620 mg, 625 mg, 630 mg, 635 mg,
640 mg, 645 mg, 650 mg, 655 mg, 660 mg, 665 mg, 670 mg, 675 mg, 680 mg, 685 mg, 690 mg,
695 mg, 700 mg, 705 mg, 710 mg, 715 mg, 720 mg, 725 mg, 730 mg, 735 mg, 740 mg, 745 mg,
750 mg, 755 mg, 760 mg, 765 mg, 770 mg, 775 mg, 780 mg, 785 mg, 790 mg, 795 mg, and 800
mg. In certain such embodiments, the doses during the maintenance phase are selected from 25 mg, 50 mg, 75 mg, 100 mg, 150 mg, 200 mg, 250 mg, 300 mg, 350 mg, 400 mg, 500 mg, 600 mg, 700 mg, and 800mg. In certain such embodiments, the doses during the maintenance phase are selected from 100 mg, 125 mg, 150 mg, 175 mg, 200 mg, 225 mg, 250 mg, 275 mg, 300 mg, 325 mg, 350 mg, 375 mg, and 400 mg. In certain such embodiments, the doses during the maintenance phase are selected from 100 mg, 125 mg, 150 mg, 175 mg, 200 mg, 225 mg, and 250 mg. In certain embodiments, the dose administered during the maintenance phase is 100 mg. In certain embodiments, the dose administered during the maintenance phase is 125 mg. In certain embodiments the dose administered during the maintenance phase is 150 mg. In certain embodiments the dose administered during the maintenance phase is 175 mg. In certain embodiments the dose administered during the maintenance phase is 200 mg. In certain embodiments the dose administered during the maintenance phase is 225 mg. In certain embodiments the dose administered during the maintenance phase is 250 mg. In certain embodiments the dose administered during the maintenance phase is 275 mg. In certain embodiments the dose administered during the maintenance phase is 300 mg.
[00548] In certain embodiments, where subcutaneous administration is desired, a maintenance dose may be administered in two or more subcutaneous injections. In certain such embodiments, when the desired maintenance dose requires a volume not easily accomodated by a single injection, two or more subcutaneous injections may be used to achieve the desired maintenance dose. In certain such embodiments, two or more subcutaneous injections may be used to administer the desired maintenance dose and minimize or eliminate an injection site reaction in a subject.
[00549] In certain embodiments, doses, dose frequency, and duration of the maintenance phase may be selected to achieve a desired effect. In certain embodiments, those variables are adjusted to result in a desired concentration of pharmaceutical agent in a subject. For example, in certain embodiments, dose and dose frequency are adjusted to provide plasma concentration of a pharmaceutical agent at an amount sufficient to achieve a desired effect. In certain of such embodiments the plasma concentration is maintained above the minimal effective concentration (MEC). In certain embodiments, pharmaceutical compositions are administered with a dosage regimen designed to maintain a concentration above the MEC for 10-90% of the time, between 30-90% of the time, or between 50-90% of the time.
[00550] In certain embodiments, doses, dose frequency, and duration of the maintenance phase may be selected to achieve a desired plasma trough concentration of a pharmaceutical composition. In certain such embodiments, the pharmaceutical composition is an
oligonucleotide. In certain such embodiments, the desired plasma trough concentration is from 5- 100 ng/mL. In certain such embodiments, the desired plasma trough concentration is from 5-50 ng/mL. In certain such embodiments, the desired plasma trough concentration is from 10-40 ng/mL. In certain such embodiments, the desired plasma trough concentration is from 15-35 ng/mL. In certain such embodiments, the desired plasma trough concentration is from 20-30 ng/mL.
[00551] In certain embodiments, doses, dose frequency, and duration of the maintenance phase may be selected to achieve a desired safety profile. For example, in certain embodiments, such variables may be selected to mitigate toxicity of the pharmaceutical composition. In certain such embodiments, such variables are selected to mitigate liver toxicity. In certain such embodiments, such variables are selected to mitigate renal toxicity. In certain such embodiments, doses increase over time.
[00552] In certain embodiments, doses, dose frequency, and duration of the maintenance phase may be adjusted from time to time to achieve a desired effect. In certain embodiments, subjects are monitored for effects (therapeutic and/or toxic effects) and doses, dose frequency, and/or duration of the maintenance phase may be adjusted based on the results of such monitoring.
[00553] It will be recognized by one of ordinary skill in the art that doses, dose frequency, and duration of the induction phase and for the maintenance phase may be manipulated
independently to achieve a desired effect. For example, in certain embodiments, provided are dosage regimens listed in Tables A-F, below. One of skill in the art will recognize that the variables in the table can be selected and combined independently. The table is included solely to illustrate how the variables may be combined and is does not limit the invention. Moreover, the present invention is not limited to the variables listed on the table.
[00554] In certain embodiments, the antisense compound is administered at 200 mg/week. In some embodiments, the 200 mg/week is adminitered one time per week. In some embodiments, the subject is administered a single 200 mg/dose per week. In other embodiments, the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week.
In some embodiments, the antisense compound is administered for at least 12 months. In one embodiment, the antisense compound is administered subcutaneously (s.c).
[00555] In certain embodiments, the antisense compound is administered at 200 mg/week during the induction phase. In some embodiments, the 200 mg/week is adminitered one time per week. In some embodiments, the subject is administered a single 200 mg/dose per week. In other embodiments, the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week. In one embodiment, the antisense compound is administered subcutaneously (s.c).
[00556] In certain embodiments, the antisense compound is administered at 200 mg/week during the maintenance phase. In some embodiments, the 200 mg/week is adminitered one time per week. In some embodiments, the subject is administered a single 200 mg/dose per week. In other embodiments, the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week. In one embodiment, the antisense compound is administered subcutaneously (s.c).
[00557] In certain embodiments, the antisense compound is administered at 200 mg/week during the induction phase and during the maintenance phase. In some embodiments, the 200 mg/week is adminitered one time per week. In some embodiments, the subject is administered a single 200 mg/dose per week. In other embodiments, the 200 mg/week is split into two or more doses (e.g., 2, 3, 4, 5, 6 or 7 doses) over the course of a week. In one embodiment, the antisense compound is administered subcutaneously (s.c).
[00558] In certain embodiments, a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein an induction dose of 200-400 mg is
administered once per week for at least 8 weeks, followed by a maintenance phase, wherein a maintenance dose of 100-300 mg is administered at intervals ranging from one per week to once per three months, for as long as needed to sustain the desired effect. In certain embodiments the induction dose is administered once per week for 8-20 weeks. In certain embodiments the induction dose is administered once per week for 10-15 weeks. In certain embodiments, the induction dose is administered once per week for at least 12 weeks. In certain embodiments the induction dose is administered once per week for at least 14 weeks. In certain embodiments the induction dose is administered once per week for at least 16 weeks. In certain such embodiments, the induction dose is 200 mg. In certain such embodiments, the induction dose is 300 mg. In
certain such embodiments, the induction dose is 400 mg. In certain such embodiments, the maintenance dose ranges from 200-300 mg. In certain such embodiments, the maintenance dose is 150 mg. In certain such embodiments, the maintenance dose is 200 mg. In certain such embodiments, the maintenance dose is 250 mg. In certain such embodiments, the maintenance dose is 300 mg. In certain such embodiments, the maintenance dose is administered once per week. In certain such embodiments, the maintenance dose is administered once per month. In certain such embodiments, the maintenance dose is administered once per three months. In certain such embodiments, the maintenance dose is administered for at least 6 months. In certain such embodiments, the maintenance dose is administered for at least one year. In certain such embodiments, the maintenance dose is administered for up to five years. In certain such embodiments the maintenance dose is administered for up to ten years. In certain such embodiments the maintenance dose is administered for as long as is necessary to sustain the desired effect. In certain such embodiments, the frequency of administration of the maintenance dose is adjusted to achieved desired efficacy and/or desired safety profile. In certain such embodiments, the frequency of the maintenance dose is adjusted to achieve a desired plasma trough concentration of oligonucleotide. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 15-40 ng/mL. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 20-30 ng/mL. In certain such embodiments, the desired effect is selected from reduced ApoB, reduced LDL-C, reduced VLDL-C, reduced IDL-C, reduced non-HDL-C, reduced serum triglycerides, reduced liver triglycerides, reduced Lp(a), reduced Ox-LDL-C, and reduced small dense LDL particles. In certain such embodiments, the subject has polygenic
hypercholesterolemia. In certain such embodiments, the subject has familial
hypercholesterolemia. In certain such embodiments, the subject has homozygous familial hypercholesterolemia. In certain such embodiments, the subject has heterozygous familial hypercholesterolemia. In certain such embodiments, the pharmaceutical composition is coadministered with a statin. In certain such embodiments, the subject is intolerant to statins. In certain such embodiments, the subject is not meeting LDL-C target on current therapy. Non- limiting examples of certain dosing regimens are illustrated in Table A.
Table A
Certain Dosing Regimens
Induction Phase Maintenance Phase
Doses Dose frequency Duration Doses Dose Frequency Duration
300 mg Once / week 8-20 weeks 150 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
300 mg Once / week 8-20 weeks 200 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
300 mg Once / week 8-20 weeks 250 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
300 mg Once / week 12-16 weeks 150 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
300 mg Once / week 12-16 weeks 200 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
300 mg Once / week 12-16 weeks 250 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years
Induction Phase Maintenance Phase
Doses Dose frequency Duration Doses Dose Frequency Duration
Once / three months Up to ten years
[00559] In certain embodiments, a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein a 200 mg dose is administered once per week for 13 weeks, followed by a maintenance phase, wherein a dose ranging from 80-200 mg is administered at intervals ranging from once per week to once per three months, for as long as needed to sustain the desired effect. In certain such embodiments, the maintenance dose ranges from 100-150 mg. In certain such embodiments, the maintenance dose is 100 mg. In certain such embodiments, the maintenance dose is 125 mg. In certain such embodiments, the maintenance dose is 140 mg. In certain such embodiments, the maintenance dose is 150 mg. In certain such embodiments, the maintenance dose is 175 mg. In certain such embodiments, the maintenance dose is 180 mg. In certain such embodiments, the maintenance dose is 200 mg. In certain such embodiments, the maintenance dose is administered once per week. In certain such
embodiments, the maintenance dose is administered once per month. In certain such
embodiments, the maintenance dose is administered once per three months. In certain such embodiments, the maintenance dose is administered for at least 6 months. In certain such embodiments, the maintenance dose is administered for at least one year. In certain such embodiments, the maintenance dose is administered for up to five years. In certain such embodiments the maintenance dose is administered for up to ten years. In certain such embodiments the maintenance dose is administered for as long as is necessary to sustain the desired effect. In certain such embodiments, the frequency of administration of the maintenance dose is adjusted to achieved desired efficacy and/or desired safety profile. In certain such embodiments, the frequency of the maintenance dose is adjusted to achieve a desired plasma trough concentration of oligonucleotide. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 15-40 ng/mL. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 20-30 ng/mL. In certain such embodiments, the desired effect is selected from reduced ApoB, reduced LDL-C, reduced VLDL-C, reduced IDL-C, reduced non-HDL-C, reduced serum triglycerides, reduced liver triglycerides, reduced Lp(a), reduced Ox-LDL-C, and reduced small dense LDL particles. In certain such embodiments, the subject has polygenic
hypercholesterolemia. In certain such embodiments, the subject has familial
hypercholesterolemia. In certain such embodiments, the pharmaceutical composition is coadministered with a statin. In certain such embodiments, the subject is intolerant to statins. In certain such embodiments, the subject is not meeting LDL-C target on current therapy. Non- limiting examples of certain dosing regimens are illustrated in Table B.
Table B
Certain Dosing Regimens

Doses Dose frequency Duration Doses Dose Frequency Duration
Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
200 mg Once / week 13 weeks 175 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
200 mg Once / week 13 weeks 180 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
200 mg Once / week 13 weeks 200 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
[00560] In certain embodiments, a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein a 300 mg dose is administered once per week for 13 weeks, followed by a maintenance phase, wherein a dose ranging from 100-250 mg is administered at intervals ranging from once per week to once per three months, for as long as needed to sustain the desired effect. In certain such embodiments, the maintenance dose is 100 mg. In certain such embodiments, the maintenance dose is 125 mg. In certain such embodiments, the maintenance dose is 150 mg. In certain such embodiments, the maintenance dose is 175 mg. In certain such embodiments, the maintenance dose is 200 mg. In certain such embodiments, the maintenance dose is 250 mg. In certain such embodiments, the maintenance dose is administered once per week. In certain such embodiments, the maintenance dose is administered once per
month. In certain such embodiments, the maintenance dose is administered once per three months. In certain such embodiments, the maintenance dose is administered for at least 6 months. In certain such embodiments, the maintenance dose is administered for at least one year. In certain such embodiments, the maintenance dose is administered for up to five years. In certain such embodiments the maintenance dose is administered for up to ten years. In certain such embodiments the maintenance dose is administered for as long as is necessary to sustain the desired effect. In certain such embodiments, the frequency of administration of the maintenance dose is adjusted to achieved desired efficacy and/or desired safety profile. In certain such embodiments, the frequency of the maintenance dose is adjusted to achieve a desired plasma trough concentration of oligonucleotide. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 15-40 ng/mL. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 20-30 ng/mL. In certain such embodiments, plasma trough concentration of the administered antisense oligonucleotide is 15-40 ng/mL. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 20-30 ng/mL. In certain such embodiments, the desired effect is selected from reduced ApoB, reduced LDL-C, reduced VLDL-C, reduced IDL-C, reduced non-HDL-C, reduced serum triglycerides, reduced liver triglycerides, reduced Lp(a), reduced Ox-LDL-C, and reduced small dense LDL particles. In certain such embodiments, the subject has polygenic hypercholesterolemia. In certain such embodiments, the subject has familial hypercholesterolemia. In certain such embodiments, the pharmaceutical composition is co-administered with a statin. In certain such embodiments, the subject is intolerant to statins. In certain such embodiments, the subject is not meeting LDL-C target on current therapy. Non-limiting examples of certain dosing regimens are illustrated in Table C.
Table C
Certain Dosing Regimens

Doses Dose frequency Duration Doses Dose Frequency Duration
Once / two months Up to five years Once / three months Up to ten years
300 mg Once / week 13 weeks 125 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
300 mg Once / week 13 weeks 150 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
300 mg Once / week 13 weeks 200 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
300 mg Once / week 13 weeks 250 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
[00561] In certain embodiments, a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein a 100 mg dose is administered once per week for 13 weeks, followed by a maintenance phase, wherein a dose ranging from 100-300 mg is administered at intervals ranging from once per week to once per three months, for as long as needed to sustain the desired effect. In certain such embodiments, the dose ranges from 150-250 mg. In certain such embodiments, the maintenance dose is 100 mg. In certain such embodiments, the maintenance dose is 125 mg. In certain such embodiments, the maintenance dose is 150 mg.
In certain such embodiments, the maintenance dose is 175 mg. In certain such embodiments, the maintenance dose is 200 mg. In certain such embodiments, the maintenance dose is 225 mg. In certain such embodiments, the maintenance dose is 250 mg. In certain such embodiments, the maintenance dose is 275 mg. In certain such embodiments, the maintenance dose is 300 mg. In certain such embodiments, the maintenance dose is administered once per week. In certain such embodiments, the maintenance dose is administered once per month. In certain such
embodiments, the maintenance dose is administered once per three months. In certain such embodiments, the maintenance dose is administered for at least 6 months. In certain such embodiments, the maintenance dose is administered for at least one year. In certain such embodiments, the maintenance dose is administered for up to five years. In certain such embodiments the maintenance dose is administered for up to ten years. In certain such embodiments the maintenance dose is administered for as long as is necessary to sustain the desired effect. In certain such embodiments, the frequency of administration of the maintenance dose is adjusted to achieved desired efficacy and/or desired safety profile. In certain such embodiments, the frequency of the maintenance dose is adjusted to achieve a desired plasma trough concentration of oligonucleotide. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 15-40 ng/mL. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 20-30 ng/mL. In certain such embodiments, the desired effect is selected from reduced ApoB, reduced LDL-C, reduced VLDL-C, reduced IDL-C, reduced non-HDL-C, reduced serum triglycerides, reduced liver triglycerides, reduced Lp(a), reduced Ox-LDL-C, and reduced small dense LDL particles. In certain such embodiments, the subject has polygenic
hypercholesterolemia. In certain such embodiments, the subject has familial
hypercholesterolemia. In certain such embodiments, the pharmaceutical composition is coadministered with a statin. In certain such embodiments, the subject is intolerant to statins. In certain such embodiments, the subject is not meeting LDL-C target on current therapy. Non- limiting examples of certain dosing regimens are illustrated in Table D.
Table D
Certain Dosing Regimens
 Induction Phase Maintenance Phase
Induction Phase Maintenance Phase
Doses Dose frequency Duration Doses Dose Frequency Duration
Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
100 mg Once / week 13 weeks 250 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
100 mg Once / week 13 weeks 275 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
100 mg Once / week 13 weeks 300 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
[00562] In certain embodiments, a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein a dose ranging from 100-200 mg is administered once per week for 13 weeks, followed by a maintenance phase, wherein a dose ranging from 100-300 mg is administered at intervals ranging from once per week to once per three months, for as long as needed to sustain the desired effect. In certain such embodiments, the induction dose is 100 mg. In certain such embodiments, the induction dose is 125 mg. In certain such embodiments, the induction dose is 150 mg. In certain such embodiments, the induction dose is 175 mg. In certain such embodiments, the induction dose is 200 mg. In certain such
embodiments, an during an induction phase four doses of 100 mg are followed by five doses of 150 mg which are followed by four doses of 200 mg. In certain such embodiments during an
induction phase four doses of 100 mg are followed by four doses of 150 mg which are followed by five doses of 200 mg. In certain such embodiments five doses of 100 mg are followed by four doses of 150 mg which are followed by four doses of 200 mg. In certain such embodiments, the maintenance dose is higher than the induction dose. In certain such embodiments, the maintenance dose is 100 mg. In certain such embodiments, the maintenance dose is 125 mg. In certain such embodiments, the maintenance dose is 150 mg. In certain such embodiments, the maintenance dose is 175 mg. In certain such embodiments, the maintenance dose is 200 mg. In certain such embodiments, the maintenance dose is 225 mg. In certain such embodiments, the maintenance dose is 250 mg. In certain such embodiments, the maintenance dose is 275 mg. In certain such embodiments, the maintenance dose is 300 mg. In certain such embodiments, the maintenance dose is administered once per week. In certain such embodiments, the maintenance dose is administered once per month. In certain such embodiments, the maintenance dose is administered once per three months. In certain such embodiments, the maintenance dose is administered for at least 6 months. In certain such embodiments, the maintenance dose is administered for at least one year. In certain such embodiments, the maintenance dose is administered for up to five years. In certain such embodiments the maintenance dose is administered for up to ten years. In certain such embodiments the maintenance dose is administered for as long as is necessary to sustain the desired effect. In certain such
embodiments, the amount or frequency of the induction dose is adjusted to achieve desired efficacy and/or desired safety profile. In certain such embodiments, the amount or frequency of the induction dose is adjusted to achieve a desired plasma trough concentration of antisense oligonucleotide. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 15-40 ng/mL. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 20-30 ng/mL. In certain such embodiments, the amount or frequency of administration of the maintenance dose is adjusted to achieved desired efficacy and/or desired safety profile. In certain such embodiments, the frequency of the maintenance dose is adjusted to achieve a desired plasma trough
concentration of oligonucleotide. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 15-40 ng/mL. In certain such embodiments, the plasma trough concentration of the administered antisense oligonucleotide is 20-30 ng/mL. In certain such embodiments, the desired effect is selected from reduced ApoB, reduced LDL-C,
reduced VLDL-C, reduced IDL-C, reduced non-HDL-C, reduced serum triglycerides, reduced liver triglycerides, reduced Lp(a), reduced Ox-LDL-C, and reduced small dense LDL particles. In certain such embodiments, the subject has polygenic hypercholesterolemia. In certain such embodiments, the subject has familial hypercholesterolemia. In certain such embodiments, the pharmaceutical composition is co-administered with a statin. In certain such embodiments, the subject is intolerant to statins. In certain such embodiments, the subject is not meeting LDL-C target on current therapy. Non-limiting examples of certain dosing regimens are illustrated in Table E.
Table E - Certain Dosing Regimens

embodiments, the duration of the induction phase is 16 weeks. In certain such embodiments, the duration of the induction phase is 17 weeks. In certain such embodiments, the duration of the induction phase is 18 weeks. In certain such embodiments, the duration of the induction phase is 19 weeks. In certain such embodiments, the duration of the induction phase is 20 weeks. In certain such embodiments, the maintenance dose is higher than the induction dose. In certain such embodiments, the maintenance dose ranges from 100-300 mg. In certain such
embodiments, the maintenance dose ranges from 100-200 mg. In certain such embodiments, the maintenance dose is 100 mg. In certain such embodiments, the maintenance dose is 125 mg. In certain such embodiments, the maintenance dose is 150 mg. In certain such embodiments, the maintenance dose is 175 mg. In certain such embodiments, the maintenance dose is 200 mg. In certain such embodiments, the maintenance dose is 225 mg. In certain such embodiments, the maintenance dose is 250 mg. In certain such embodiments, the maintenance dose is 275 mg. In certain such embodiments, the maintenance dose is 300 mg. In certain such embodiments, the maintenance dose is administered once per week. In certain such embodiments, the maintenance dose is administered once per month. In certain such embodiments, the maintenance dose is administered once per three months. In certain such embodiments, the maintenance dose is administered for at least 6 months. In certain such embodiments, the maintenance dose is administered for at least one year. In certain such embodiments, the maintenance dose is administered for up to five years. In certain such embodiments the maintenance dose is administered for up to ten years. In certain such embodiments the maintenance dose is administered for as long as is necessary to sustain the desired effect. In certain such
embodiments, the frequency of administration of the maintenance dose is adjusted to achieved desired efficacy and/or desired safety profile. In certain such embodiments, the desired effect is
selected from reduced ApoB, reduced LDL-C, reduced VLDL-C, reduced IDL-C, reduced non- HDL-C, reduced serum triglycerides, reduced liver triglycerides, reduced Lp(a), reduced Ox- LDL-C, and reduced small dense LDL particles. In certain such embodiments, the subject has polygenic hypercholesterolemia. In certain such embodiments, the subject has familial hypercholesterolemia. In certain such embodiments, the pharmaceutical composition is coadministered with a statin. In certain such embodiments, the subject is intolerant to statins. In certain such embodiments, the subject is not meeting LDL-C target on current therapy. Non- limiting examples of certain dosing regimens are illustrated in Table F.
Table F
Certain Dosing Regimens

Doses Dose frequency Duration Doses Dose Frequency Duration
Once / two months Up to five years Once / three months Up to ten years
100 mg Once / week 14-20 weeks 200 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
100 mg Once / week 14-20 weeks 225 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
100 mg Once / week 14-20 weeks 275 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
100 mg Once / week 14-20 weeks 300 mg Once / week At least 6 months
Once / two weeks At least one year Once / three weeks At least two years Once / month At least five years Once / two months Up to five years Once / three months Up to ten years
[00564] In a particular embodiment, a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein a dose ranging from 100-300 mg is administered once per week for 13 weeks, followed by a maintenance phase, wherein a dose ranging from 80-200 mg is administered once per week for as long as needed, effective, and/or tolerated. In certain of such embodiments, the pharmaceutical composition is administered subcutaneously during the induction phase and/or the maintenance phase. In certain of such embodiments, the subject is afflicted with familial hypercholesterolemia (either heterozygous or
homozygous), non-familial hypercholesterolemia, or polygenic hypercholesterolemia. In certain of such embodiments, the maintenance phase lasts from one day to the end of the subject's lifetime or any fraction thereof as discussed above. In certain of such embodiments, the induction dose is 100 mg, and the maintenance dose is 80 mg, 100 mg, 140 mg, 180 mg, or 200 mg. In certain of such embodiments, the induction dose is 200 mg, and the maintenance dose is 80 mg, 100 mg, 140 mg, 180 mg, or 200 mg. In certain of such embodiments, the induction dose is 300 mg, and the maintenance dose is 80 mg, 100 mg, 140 mg, 180 mg, or 200 mg.
[00565] In certain of such embodiments, the administration at the end of the induction phase achieves a reduction in plasma concentration of ApoB of from about -28% to -65%. In certain of such embodiments, the administration after 13 weeks of the maintenance phase achieves a reduction in plasma concentration of ApoB of from about -32% to -48%, from about -35% to about -52%o, from about -40%> to about -60%>, from about -43% to about -65%, or from about - 45% to about -67%. In certain of such embodiments, the administration at the end of the induction phase achieves a reduction in plasma concentration of LDL-Col from about -26% to - 60%. In certain of such embodiments, the administration after 13 weeks of the maintenance phase achieves a reduction in plasma concentration of LDL-Col from about -29% to -44%, from about -32%o to about -48%, from about -37% to about -55%, from about -40% to about -61%, or from about -42% to about -63%.
[00566] In certain of such embodiments, the administration at the end of the induction phase achieves a plasma trough concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 11 to 38 ng/mL. In certain of such embodiments, the administration after 13 weeks of the maintenance phase achieves a plasma trough concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 7 to 27 ng/mL, from about 8 to 31 ng/mL, from about 11 to 38 ng/mL, from about 13 to 46 ng/mL, or from about 14 to 50 ng/mL. In certain of such embodiments, the administration at the end of the induction phase achieves a liver concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 55 to 190 μg/G. In certain of such embodiments, the administration after 13 weeks of the maintenance phase achieves a liver concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 38 to 133 μ^β, from about 44 to 152 μ^ϋ, from about 55 to 190 μ^ϋ, from about 66 to 228 μ^ϋ, or from about 7 to 247 μ^ϋ.
[00567] In certain of such embodiments, the administration at the end of the induction phase achieves a reduction in plasma concentration of ApoB of from about -34% to -77%. In certain of such embodiments, the administration after 13 weeks of the maintenance phase achieves a reduction in plasma concentration of ApoB of from about -38% to -58%, from about -43% to about -65%, from about -47% to about -70%, or from about -49% to about -74%. In certain of such embodiments, the administration at the end of the induction phase achieves a reduction in plasma concentration of LDL-Col from about -31% to -73%. In certain of such embodiments, the administration after 13 weeks of the maintenance phase achieves a reduction in plasma concentration of LDL-Col from about -35% to -54%, from about -40% to about -61%, from about -44% to about -66%, or from about -46% to about -70%.
[00568] In certain of such embodiments, the administration at the end of the induction phase achieves a plasma trough concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 16 to 57 ng/mL. In certain of such embodiments, the administration after 13 weeks of the maintenance phase achieves a plasma trough concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 10 to 37 ng/mL, from about 13 to 46 ng/mL, from about 16 to 55 ng/mL, or from about 18 to 65 ng/mL. In certain of such embodiments, the administration at the end of the induction phase achieves a liver concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 82 to 285 μg/G. In certain of such embodiments, the administration after 13 weeks of the maintenance phase achieves a liver concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 52 to 181 μg/G, from about 66 to 228 μ^ϋ, from about 80 to 276 μ^ϋ, or from about 94 to 323 μg/G.
[00569] In another particular embodiment, a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein a dose ranging from 100-300 mg is administered once per week for 13 weeks, followed by a maintenance phase, wherein a dose ranging from 100-200 mg is administered once per week for as long as needed, effective, and/or tolerated, wherein the efficacy and/or the tolerability of the antisense oligonucleotide is monitored during the induction phase, the maintenance phase, or both, or any portion thereof. In certain of such embodiments, the pharmaceutical composition is administered subcutaneously during the induction phase and/or the maintenance phase. In certain of such embodiments, the subject is afflicted with familial hypercholesterolemia (either heterozygous or homozygous),
non- familial hypercholesterolemia, or polygenic hypercholesterolemia. In certain of such embodiments, the maintenance phase lasts from one day to the end of the subject's lifetime or any fraction thereof as discussed above.
[00570] In certain of such embodiments, the rate of reduction in the plasma concentration of ApoB is monitored during the induction and/or maintenance phases. In certain of such embodiments, the plasma concentration of ApoB is monitored during the induction and/or maintenance phases. In certain embodiments, if the rate of reduction in the plasma concentration of ApoB exceeds 30 mg/dL*day, the dose of pharmaceutical composition is altered, e.g., reduced. In certain embodiments, if the rate of reduction in the plasma concentration of ApoB exceeds 30 mg/dL* day, the frequency of administration of pharmaceutical composition is altered, e.g., reduced. In certain embodiments, if the plasma concentration of ApoB falls below about 50 mg/dL, the dose of pharmaceutical composition is altered, e.g., reduced. In certain embodiments, if the plasma concentration of ApoB falls below about 50 mg/dL, the frequency of administration of pharmaceutical composition is altered, e.g., reduced. In certain embodiments, if the plasma concentration of ApoB falls below about 60 mg/dL, the dose of pharmaceutical composition is altered, e.g. , reduced. In certain embodiments, if the plasma concentration of ApoB falls below about 60 mg/dL, the frequency of administration of pharmaceutical composition is altered, e.g. , reduced.
[00571] In certain embodiments, if the rate of reduction in the plasma concentration of ApoB exceeds 30 mg/dL* day and the plasma concentration of ApoB falls below about 50 mg/dL, the dose of pharmaceutical composition is altered, e.g., reduced. In certain embodiments, if the rate of reduction in the plasma concentration of ApoB exceeds 30 mg/dL*day and the plasma concentration of ApoB falls below about 50 mg/dL, the frequency of administration of pharmaceutical composition is altered, e.g., reduced. In certain embodiments, if the rate of reduction in the plasma concentration of ApoB exceeds 30 mg/dL* day and the plasma concentration of ApoB falls below about 60 mg/dL, the dose of pharmaceutical composition is altered, e.g., reduced. In certain embodiments, if the rate of reduction in the plasma
concentration of ApoB exceeds 30 mg/dL* day and the plasma concentration of ApoB falls below about 60 mg/dL, the frequency of administration of pharmaceutical composition is altered, e.g., reduced.
[00572] In another particular embodiment, a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein a dose ranging from 100 to 200 mg is administered once per week for 13 weeks, and a maintenance phase, wherein a dose ranging from 200 to 300 mg is administered once per week for as long as needed, effective, and/or tolerated, wherein the tolerability and/or efficacy of the pharmaceutical composition is assessed during or at the end of the induction phase, or any portion thereof. In certain of such embodiments, the dose of the maintenance phase is increased relative to the dose of the maintenance phase if the dose of the induction phase is well-tolerated and treatment goals are not met. In certain of such embodiments, the pharmaceutical composition is administered subcutaneously during the induction phase and/or the maintenance phase. In certain of such embodiments, the subject is afflicted with familial hypercholesterolemia (either heterozygous or homozygous), non-familial hypercholesterolemia, or polygenic hypercholesterolemia. In certain of such embodiments, the maintenance phase lasts from one day to the end of the subject's lifetime or any fraction thereof as discussed above. In certain of such embodiments, the induction dose is 100 mg, and the maintenance dose is 200 mg. In certain of such embodiments, the induction dose is 200 mg, and the maintenance dose is 300 mg. In certain embodiments, the treatment goals are assessed by monitoring plasma concentration of ApoB, LDL-C, VLDL-C, non-HDL-C, HDL-C, ApoAl, total cholesterol, triglycerides, and Lp(a). In certain
embodiments, tolerability is assessed by monitoring ALT activity, AST activity, and plasma bilirubin concentrations.
[00573] In certain of such embodiments, the administration at the end of the induction phase achieves a reduction in plasma concentration of ApoB of from about -17% to -40%. In certain of such embodiments, the administration after 26 weeks of the maintenance phase achieves a reduction in plasma concentration of ApoB of from about -42% to -63%. In certain of such embodiments, the administration at the end of the induction phase achieves a reduction in plasma concentration of LDL-Col from about -14% to -35%. In certain of such embodiments, the administration after 13 weeks of the maintenance phase achieves a reduction in plasma concentration of LDL-Col from about -39% to -60%.
[00574] In certain of such embodiments, the administration at the end of the induction phase achieves a plasma trough concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 5 to 19 ng/mL. In certain of such embodiments, the
administration after 26 weeks of the maintenance phase achieves a plasma trough concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 12 to 44 ng/mL. In certain of such embodiments, the administration at the end of the induction phase achieves a liver concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 27 to 95 μg/G. In certain of such embodiments, the administration after 26 weeks of the maintenance phase achieves a liver concentration of an oligonucleotide administered as part of the pharmaceutical composition of from about 63 to 220 μg/G.
[00575] In another particular embodiment, a method of administering a pharmaceutical composition to a subject comprises an induction phase, wherein a dose ranging from 100 to 200 mg is administered once per week for 13 weeks, and a maintenance phase, wherein a dose ranging from 200 to 300 mg is administered once every one or two weeks for as long as needed, effective, and/or tolerated, wherein the tolerability and/or efficacy of the pharmaceutical composition is assessed during or at the end of the induction phase, or any portion thereof. In certain of such embodiments, the frequency of administration of dose during the maintenance phase is reduced relative if the dose of the induction phase is not well-tolerated and/or treatment goals are met. In certain of such embodiments, the pharmaceutical composition is administered subcutaneously during the induction phase and/or the maintenance phase. In certain of such embodiments, the subject is afflicted with familial hypercholesterolemia (either heterozygous or homozygous), non-familial hypercholesterolemia, or polygenic hypercholesterolemia. In certain of such embodiments, the maintenance phase lasts from one day to the end of the subject's lifetime or any fraction thereof as discussed above. In certain of such embodiments, the induction dose is 100 mg, and the maintenance dose is 200 mg. In certain of such embodiments, the induction dose is 200 mg, and the maintenance dose is 300 mg. In certain embodiments, the treatment goals are assessed by monitoring plasma concentration of ApoB, LDL-C, VLDL-C, non-HDL-C, HDL-C, ApoAl, total cholesterol, triglycerides, and Lp(a). In certain
embodiments, tolerability is assessed by monitoring ALT levels, AST levels, plasma bilirubin concentrations or total bilirubin.
1.5 Combination Therapy
[00576] In certain embodiments, an antisense compound can be co-administered with one or more other pharmaceutical agents. In certain embodiments, such one or more other
pharmaceutical agents is designed to treat the same disease or condition as the one or more
antisense compound. In some embodiments, such one or more other pharmaceutical agents is designed to treat a different disease or condition as the one or more antisense compound. In other embodiments, such one or more other pharmaceutical agents is designed to treat an undesired effect of the one or more antisense compound. In certain embodiments, the one or more antisense compound is co-administered with another pharmaceutical agent to treat an undesired effect of that other pharmaceutical agent. In some embodiments, the one or more antisense compound, and one or more other pharmaceutical agents are administered at the same time. In other embodiments, the one or more antisense compound, and one or more other pharmaceutical agents are administered at different times. In certain embodiments, one or more pharmaceutical compositions described herein, and one or more other pharmaceutical agents can be prepared together in a single formulation. In some embodiments, one or more pharmaceutical compositions described herein, and one or more other pharmaceutical agents are prepared separately. For example, a composition can comprise a pharmaceutical agent for separate, sequential, or simultaneous administration with an antisense compound.
[00577] In certain embodiments, pharmaceutical agents that can be co-administered with a pharmaceutical composition described herein include lipid- lowering agents or LXR agonists. In some embodiments, pharmaceutical agents that can be co-administered with a pharmaceutical composition described herein, include, but are not limited to atorvastatin, simvastatin, rosuvastatin, and ezetimibe. In other embodiments, the lipid-lowering agent is administered prior to administration of a pharmaceutical composition described herein. In some embodiments, the lipid-lowering agent is administered following administration of a pharmaceutical composition described herein. In certain embodiments, the lipid-lowering agent is administered at the same time as a pharmaceutical composition described herein. In other embodiments, the dose of a coadministered lipid-lowering agent is the same as the dose that would be administered if the lipid- lowering agent was administered alone. In some embodiments, the dose of a co-administered lipid-lowering agent is lower than the dose that would be administered if the lipid-lowering agent was administered alone. In certain embodiments, the dose of a co-administered lipid-lowering agent is greater than the dose that would be administered if the lipid-lowering agent was administered alone.
[00578] In certain embodiments, a co-administered lipid-lowering agent is a HMG-CoA reductase inhibitor. In some embodiments, the HMG-CoA reductase inhibitor is a statin. In some
embodiments, the statin is selected from, for example, atorvastatin, simvastatin, pravastatin, fluvastatin, and rosuvastatin.
[00579] In certain embodiments, a co-administered lipid-lowering agent is a cholesterol absorption inhibitor. In some embodiments, cholesterol absorption inhibitor is ezetimibe.
[00580] In certain embodiments, a co-administered lipid-lowering agent is a co-formulated HMG-CoA reductase inhibitor and cholesterol absorption inhibitor. In some embodiments, the co-formulated lipid-lowering agent is ezetimibe/simvastatin.
[00581] In certain embodiments, a co-administered lipid-lowering agent is a microsomal triglyceride transfer protein inhibitor (MTP inhibitor).
[00582] In certain embodiments, a co-administered lipid-lowering agent is an oligonucleotide selected from an oligonucleotide targeted to PCSK9, an oligonucleotide targeted to ACAT-2, an oligonucleotide targeted to endothelial lipase, and an oligonucleotide targeted to CETP.
[00583] In certain embodiments, a co-administered lipid-lowering agent is an oligonucleotide targeted to ApoB.
[00584] In certain embodiments, a co-administered pharmaceutical agent is a bile acid sequestrant. In some embodiments, the bile acid sequestrant is selected from, for example, cholestyramine, colestipol, and colesevelam.
[00585] In some embodiments, a co-administered pharmaceutical agent is a nicotinic acid. In some embodiments, the nicotinic acid is selected from immediate release nicotinic acid, extended release nicotinic acid, and sustained release nicotinic acid.
[00586] In other embodiments, a co-administered pharmaceutical agent is a fibric acid. In some embodiments, a fibric acid is selected from gemfibrozil, fenofibrate, clofibrate, bezafibrate, and ciprofibrate.
[00587] In certain embodiments, a co-administered pharmaceutical agent is an
antihypertensive agent. In some embodiments, the antihypertensive agent is selected from, for example, angiotensin-converting enzyme (ACE) inhibitors, beta and alpha adrenergic blockers, calcium-channel blockers, renin inhibitors, aldosterone receptor antagonists, and angiotensin- receptor blockers. In some embodiments, the antihypertensive agent is administered at the same time as a pharmaceutical composition described herein. In some embodiments, the dose of a coadministered antihypertensive agent is the same as the dose that would be administered if the antihypertensive agent was administered alone. In other embodiments, the dose of a co-
administered antihypertensive agent is lower than the dose that would be administered if the antihypertensive agent was administered alone. In certain embodiments, the dose of a coadministered antihypertensive agent is greater than the dose that would be administered if the antihypertensive agent was administered alone.
[00588] In certain embodiments, the angiotensin-converting enzyme inhibitor is selected from, for example, captopril, enalapril, fosinopril, lisinopril, perindopril, quinapril, and ramipril.
[00589] In some embodiments, the beta adrenergic blocker is selected from, for example, atenolol, metoprolol, propranolol, timolol, and oxprenolol.
[00590] In other embodiments, the alpha adrengergic blocker is selected from, for example, doxazosin, indoramin, prozosin, terazosin, tolazoline, and phentolamine.
[00591] In certain embodiments, the calcium-channel blocker is selected from, for example, dihydropyridines. In some embodiments, the dihydropyridine is selected from, for example, amlodipine, cinidipine, felodipine, isradipine, mad nimodipine.
[00592] In some embodiments, the calcium-channel blocker is selected from, for example, non-dihydropyridines. In some embodiments, the non-dihydropyridine is selected from, for example, diltiazem, or verapamil.
[00593] In some embodiments, the renin inhibitor is selected from for example, Aliskiren.
[00594] In some embodiments, the aldosterone receptor antagonist is selected from for example, elerenone or spironolactone.
[00595] In some embodiments, the angiotensin-receptor blocker is selected from for example, azilsartan, candesartan eprosartan, irbesartan, losartan, olmesartan, telmisartan, and valsartan.
[00596] In certain embodiments, a co-administered pharmaceutical agent is a thrombolytic agent. In some embodiments, the thrombolytic agent is administered at the same time as a pharmaceutical composition described herein. In some embodiments, the dose of a coadministered thrombolytic agent is the same as the dose that would be administered if the thrombolytic agent was administered alone. In some embodiments, the dose of a co-administered thrombolytic agent is lower than the dose that would be administered if the thrombolytic agent was administered alone. In some embodiments, the dose of a co-administered thrombolytic agent is greater than the dose that would be administered if the thrombolytic agent was administered alone.
[00597] In some embodiments, the thrombolytic agent is selected from, for example, eminase, retavase, streptase, t-PA, TNKase, abbokinase, and kinlytic.
[00598] In certain embodiments, a co-administered pharmaceutical agent is an antiplatelet agent. In some embodiments, the antiplatelet agent is administered at the same time as a pharmaceutical composition described herein. In some embodiments, the dose of a coadministered antiplatelet agent is the same as the dose that would be administered if the antiplatelet agent was administered alone. In some embodiments, the dose of a co-administered antiplatelet agent is lower than the dose that would be administered if the antiplatelet agent was administered alone. In some embodiments, the dose of a co-administered antiplatelet agent is greater than the dose that would be administered if the antiplatelet agent was administered alone.
[00599] In some embodiments, the antiplatelet agent is selected from, for example, cyclooxygenase inhibitors, adenosine diphosphate (ADP) receptor inhibitors, phosphodiesterase inhibitors, glycoprotein IIB/IIIA inhibitors, adenosine reuptake inhibitors, and thromboxane inhibitors.
[00600] In some embodiments, the cyclooxygenase inhibitor is selected from, for example aspirin, or triflusal. In some embodiments, the adenosine diphosphate (ADP) receptor inhibitor is selected from, for example, clopidogrel, prasugrel, ticagrelor, ticlopidine, phosphodiesterase inhibitors, and cilostazol.
[00601] In some embodiments, the Glycoprotein IIB/IIIA inhibitor is selected from, for example, abciximab, eptifibatide, and tirofiban.
[00602] In some embodiments, the adenosine reuptake inhibitor is selected from, for example, dipyridamole.
[00603] In some embodiments, the thromboxane inhibitor is selected from, for example, thromboxane synthase inhibitors and thromboxane receptor antagonists.
[00604] Further examples of pharmaceutical agents that can be co-administered with a pharmaceutical composition described herein include, but are not limited to, antiarrhythmic agents; azetidinone-based cholesterol absorption inhibitors; niacin; niacin derivatives; PPAR agonists; PPAR antagonists; antiplatelet drugs; corticosteroids, including but not limited to prednisone; LXR agonists; immunoglobulins, including, but not limited to intravenous immunoglobulin (IVIg); analgesics (e.g., acetaminophen); anti-inflammatory agents, including, but not limited to non-steroidal anti-inflammatory drugs (e.g., ibuprofen, COX-1 inhibitors, and
COX-2, inhibitors); salicylates; antibiotics; antivirals; antifungal agents; antidiabetic agents (e.g., biguanides, glucosidase inhibitors, insulins, sulfonylureas, and thiazolidenediones); adrenergic modifiers; diuretics; hormones (e.g., anabolic steroids, androgen, estrogen, calcitonin, progestin, somatostan, and thyroid hormones); immunomodulators; muscle relaxants; antihistamines;
osteoporosis agents (e.g., biphosphonates, calcitonin, and estrogens); prostaglandins,
antineoplastic agents; psychotherapeutic agents; sedatives; poison oak or poison sumac products; antibodies; and vaccines.
[00605] In certain embodiments, the pharmaceutical compositions described herein are administered in conjunction with a lipid-lowering therapy. In some embodiments, a lipid- lowering therapy is therapeutic lifestyle change. In some embodiments, a lipid-lowering therapy is LDL apheresis.
[00606] In certain embodiments, the pharmaceutical compositions described herein and one or more other pharmaceutical agents as described herein provides use in the manufacture of a medicament for the treatment, prevention, or management of a disease or conditions described herein.
1.6 Targeted delivery
[00607] In another embodiment, the antisense compound is targeted to a specific tissue, organ or location in the body. Exemplary targets include liver, lung, kidney, heart, and atherosclerotic plaques within a blood vessel. Methods of targeting compounds can be well known in the art.
[00608] In one embodiment, the compound is targeted by direct or local administration. For example, when targeting a blood vessel, the compound is administered directly to the relevant portion of the vessel from inside the lumen of the vessel, e.g., single balloon or double balloon catheter, or through the adventitia with material aiding slow release of the compound, e.g., a pluronic gel system as described by Simons et al., Nature 359: 67-70 (1992). Other slow release techniques for local delivery of the compound to a vessel include coating a stent with the compound. Methods of delivery of antisense compounds to a blood vessel can be disclosed in U.S. Pat. No. 6,159,946, which is incorporated by reference in its entirety.
[00609] When targeting a particular tissue or organ, the compound can be administered in or around that tissue or organ. For example, U.S. Pat. No. 6,547,787, incorporated herein by reference in its entirety, discloses methods and devices for targeting therapeutic agents to the heart. In one embodiment, administration occurs by direct injection or by injection into a blood
vessel associated with the tissue or organ. For example, when targeting the liver, the compound can be administered by injection or infusion through the portal vein.
[00610] In another embodiment, methods of targeting a compound are provided which include associating the compound with an agent that directs uptake of the compound by one or more cell types. Exemplary agents include lipids and lipid-based structures such as liposomes generally in combination with an organ- or tissue-specific targeting moiety such as, for example, an antibody, a cell surface receptor, a ligand for a cell surface receptor, a polysaccharide, a drug, a hormone, a hapten, a special lipid and a nucleic acid as described in U.S. Pat. No. 6,495,532, the disclosure of which is incorporated herein by reference in its entirety. U.S. Pat. No. 5,399,331 , the disclosure of which is incorporated herein by reference in its entirety, describes the coupling of proteins to liposomes through use of a cross linking agent having at least one maleimido group and an amine reactive function; U.S. Pat. Nos. 4,885,172, 5,059,421 and 5,171 ,578, the disclosures of which can be incorporated herein by reference in their entirety, describe linking proteins to liposomes through use of the glycoprotein streptavidin and coating targeting liposomes with polysaccharides. Other lipid based targeting agents include, for example, micelle and crystalline products as described in U.S. Pat. No. 6,217,886, the disclosure of which is incorporated herein by reference in its entirety.
[00611] In another embodiment, targeting agents include porous polymeric microspheres, which can be derived from copolymeric, and homopolymeric polyesters containing hydrolyzable ester linkages, which can be biodegradable, as described in U.S. Pat. No. 4,818,542, the disclosure of which is incorporated herein by reference in its entirety. Typical polyesters include polyglycolic (PGA) and polylactic (PLA) acids, and copolymers of glycolide and L(-lactide) (PGL), which can be particularly suited for the methods and compositions provided herein in that they exhibit low human toxicity and can be biodegradable. The particular polyester or other polymer, oligomer, or copolymer utilized as the microspheric polymer matrix is not critical and a variety of polymers can be utilized depending on desired porosity, consistency, and shape and size distribution. Other biodegradable or bioerodable polymers or copolymers include, for example, gelatin, agar, starch, arabinogalactan, albumin, collagen, natural and synthetic materials or polymers, such as, poly(8-caprolactone), poly(8-caprolactone-CO-lactic acid), poly(8- caprolactone-CO-glycolic acid), poly(P-hydroxy butyric acid), polyethylene oxide, polyethylene, poly(alkyl-2-cyanoacrylate), (e.g., methyl, ethyl, butyl), hydrogels such as poly(hydroxyethyl
methacrylate), polyamides (e.g., polyacrylamide), poly(amino acids) (i.e., L-leucine, L-aspartic acid, β-methyl-L-aspartate, β-benzyl-L-aspartate, glutamic acid), poly(2-hydroxyethyl DL- aspartamide), poly(ester urea), poly(L-phenylalanine/ethylene glycol/1, 6-diisocyanatohexane) and poly(methyl methacrylate). The exemplary natural and synthetic polymers suitable for targeted delivery can be either readily available commercially or can be obtainable by
condensation polymerization reactions from the suitable monomers or, comonomers or oligomers.
[00612] In still another embodiment, U.S. Pat. No. 6,562,864, the disclosure of which is incorporated herein by reference in its entirety, describes catechins, including epi and other carbo-cationic isomers and derivatives thereof, which as monomers, dimers and higher multimers can form complexes with nucleophilic and cationic bioactive agents for use as delivery agents. Catechin multimers have a strong affinity for polar proteins, such as those residing in the vascular endothelium, and on cell/organelle membranes and can be particularly useful for targeted delivery of bioactive agents to select sites in vivo. In treatment of vascular diseases and disorders, such as atherosclerosis and coronary artery disease, delivery agents include substituted catechin multimers, including amidated catechin multimers which can be formed from reaction between catechin and nitrogen containing moities such as ammonia.
[00613] Other targeting strategies of the antisense compound include ADEPT (antibody- directed enzyme prodrug therapy), GDEPT (gene-directed EPT) and VDEPT (virus-directed EPT) as described in U.S. Pat. No. 6,433,012, the disclosure of which is incorporated herein by reference in its entirety.
[00614] In another embodiment medical devices and kits can be used for targeted delivery, wherein the device is, for example, a syringe, stent, or catheter. Kits include a device for administering a compound and a container comprising an antisense compound provided herein. In another embodiment, the compound is preloaded into the device. In other embodiments, the kit provides instructions for methods of administering the compound and dosages. U.S. patents describing medical devices and kits for delivering an antisense compound include U.S. Pat. Nos. 6,368,356; 6,344,035; 6,344,028; 6,287,285; 6,200,304; 5,824,049; 5,749,915; 5,674,242;
5,670,161; 5,609,629; 5,593,974; and 5,470,307 (all incorporated herein by reference in their entirety).
1.7 Certain Indications
[00615] In certain embodiments, provided herein are methods of treating a subject comprising administering one or more pharmaceutical agents provided herein. In certain embodiments, such subject has hypercholesterolemia, hyperlipidemia, non-familial hypercholesterolemia, familial hypercholesterolemia, heterozygous familial hypercholesterolemia, homozygous familial hypercholesterolemia, coronary heart disease, atherosclerosis, mixed dyslipidemia, diabetic dyslipidemia. In certain embodiments, such subject has been identified as having one or more CHD risk equivalents. In certain embodiments, such subject has been identified has having major risk factors for coronary heart disease. In certain embodiments, such subject has been identified as having one or more CHD risk factors. In certain embodiments, such subject has been identified being at risk for atherosclerosis. In certain embodiments, such subject has been identified as having a history of coronary heart disease. In certain embodiments, such subject has been identified as having a family history of early onset coronary heart disease.
[00616] In certain embodiments, the subject has been identified as having elevated
cholesterol. In certain embodiments, the subject has been identified as in need of lipid lowering therapy. In certain such embodiments, the subject has been identified as in need of lipid- lowering therapy according to the guidelines established by the Adult Treatment Panel III of the National Cholesterol Education Program (NCEP) in 2001 and modified by the Coordinating Committee of the NCEP in 2004 (Grundy et al, Circulation, 2004, 110, 227-239). In certain such
embodiments, the subject in need of lipid-lowering therapy has LDL-C above 190 mg/dL. In certain such embodiments, the subject in need of lipid-lowering therapy has LDL-C above 160 mg/dL. In certain such embodiments the subject in need of lipid-lowering therapy has LDL-C above 130 mg/dL. In certain such embodiments, the subject in need of lipid-lowering therapy should maintain LDL-C below 160 mg/dL. In certain such embodiments, the subject in need of lipid-lowering therapy should maintain LDL-C below 130 mg/dL. In certain such embodiments, the subject in need of lipid-lowering therapy should maintain LDL-C below 100 mg/dL. In certain such embodiments the subject should maintain LDL-C below 70 mg/dL.
[00617] In certain embodiments, provided are methods for reducing ApoB concentration in a subject. In certain embodiments, provided is method for reducing ApoB-containing lipoprotein concentration in a subject. In certain embodiments, provided are methods for reducing LDL-C concentration in a subject. In certain embodiments, provided are methods for reducing VLDL-C concentration in a subject. In certain embodiments, provided are methods for reducing IDL-C
concentration in a subject. In certain embodiments, provided are methods for reducing non-HDL- C concentration in a subject. In certain embodiments, provided are methods for reducing Lp(a) concentration in a subject. In certain embodiments, provided are methods for reducing serum triglyceride concentration in a subject. In certain embodiments provided are methods for reducing Ox-LDL-C concentration in a subject. In certain such embodiments, the reduction in ApoB, LDL-C, VLDL-C, IDL-C, total cholesterol, non-HDL-C, Lp(a), triglyerides, or Ox-LDL- C is, independently, selected from at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, and at least 100%. In certain such embodiments, the reduction in ApoB, LDL-C, VLDL-C, IDL-C, total cholesterol, non-HDL-C, Lp(a), triglyerides, or Ox-LDL-C is, independently, selected from at least 20%, at least 30%, at least 40%, at least 50%, at least 60%, and at least 70%. In certain such embodiments, the reduction in ApoB, LDL-C, VLDL-C, IDL-C, total cholesterol, non-HDL-C, Lp(a), triglyerides, or Ox-LDL-C is, independently, selected from at least 40%>, at least 50%>, at least 60%), and at least 70%>.
[00618] In certain embodiments, provided is method for raising HDL-C concentration in a subject.
[00619] In certain embodiments, the methods provided herein do not lower HDL-C. In certain embodiments, the methods provided herein do not result in accumulation of lipids in the liver.
[00620] In certain embodiments, provided are methods for lowering ApoB concentration in a subject while reducing side effects associated with treatment. In certain such embodiments, a side effect is liver toxicity. In certain such embodiments, a side effect is abnormal liver function. In certain such embodiments, a side effect is liver inflammation or other adverse event that occurs in the liver. In certain such embodiments, a side effect is elevated alanine
aminotransferase (ALT). In certain such embodiments, a side effect is elevated aspartate aminotransferase (AST). For example, certain dosing regimens result in effective lowering of ApoB concentration with less liver toxicity than has been observed from studies employing different dosing regimens. In certain embodiments, dosing regimens result in effective lowering of ApoB with less elevation in ALT. In certain such embodiments, the amount of an induction dose administered is lower than the amount of a maintenance dose administered.
[00621] In certain embodiments, provided are methods for lowering ApoB concentration in a subject who is not reaching target LDL-C levels as a result of lipid-lowering therapy. In certain such embodiments, ISIS 301012 is the only lipid-lowering agent administered to the subject. In certain such embodiments, the subject has not complied with recommended lipid-lowering therapy. In certain such embodiments, a pharmaceutical composition provided herein is coadministered with an additional different lipid-lowering therapy. In certain such embodiments, an additional lipid-lowering therapy is LDL-apheresis. In certain such embodiments, an additional lipid-lowering therapy is a statin. In certain such embodiments, an additional lipid- lowering therapy is ezetimibe.
[00622] In certain embodiments, provided are methods for lowering ApoB concentration in a statin-intolerant subject. In certain such embodiments, the subject has creatine kinase
concentration increases as a result of statin administration. In certain such embodiments, the subject has liver function abnormalities as a result of statin administration. In certain such embodiments the subject has muscle aches as a result of statin administration. In certain such embodiments the subject has central nervous system side effects as a result of statin
administration. In certain embodiments, the subject has not complied with recommended statin administration.
[00623] In certain embodiments, provided are methods for lowering liver triglycerides in a subject. In certain such embodiments, the subject has elevated liver triglycerides. In certain such embodiments, the subject has steatohepatitis. In certain such embodiments, the subject has steatosis. In certain such embodiments, liver triglyceride levels are measured by magnetic resonance imaging.
[00624] In certain embodiments, provided are methods for reducing coronary heart disease risk in a subject. In certain embodiments provided are methods for slowing the progression of atherosclerosis in a subject. In certain such embodiments provided are methods for stopping the progression of atherosclerosis in a subject. In certain such embodiments provided are methods for reducing the size and/or prevalence of atherosclerotic plaques in a subject. In certain embodiments the methods provided reduce a subject's risk of developing atherosclerosis.
[00625] In certain embodiments the methods provided improve the cardiovascular outcome in a subject. In certain such embodiments improved cardiovascular outcome is the reduction of the risk of developing coronary heart disease. In certain such embodiments, improved cardiovascular
outcome is a reduction in the occurance of one or more major cardiovascular events, which include, but are not limited to, myocardial infarction, reinfarction, stroke, cardiogenic shock, pulmonary edema, cardiac arrest, and atrial dysrhythmia. In certain of the methods of preventing a MACE provided herein, the MACE is death. In certain such embodiments, the improved cardiovascular outcome is evidenced by improved carotid intimal media thickness. In certain such embodiments, improved carotid intimal media thickness is a decrease in thickness. In certain such embodiments, improved carotid intimal media thickness is a prevention an increase of intimal media thickness.
1.8 Kits
[00626] Also provided is a pharmaceutical pack or kit comprising one or more containers filled with one or more of the ingredients of the pharmaceutical compositions provided herein, such as an antisense compound provided herein. Optionally associated with such container(s) can be a notice in the form prescribed by a governmental agency regulating the manufacture, use or sale of pharmaceuticals or biological products, which notice reflects approval by the agency of manufacture, use or sale for human administration.
[00627] Also provided herein can be kits that can be used in the above methods. In one embodiment, a kit is provided for treating, preventing, or managing MACEs. In a specific embodiment, a kit comprises one of more antisense compounds provided herein. In a more specific embodiment, the kits provided herein in contain a therapeutically effective amount of an antisense compound provided herein. In another embodiment, a kit comprises an antisense compound and one or more other pharmaceutical agents provided herein. In another
embodiment, the kits provided herein include a therapeutically effective amount of an antisense compound, and a therapeutically effective amount of one or more pharmaceutical agents, along with instructional material which describes administering the composition comprising the inhibitor to a patient in need thereof. This should be construed to include other embodiments, of kits that can be known to those skilled in the art, such as a kit comprising a (can sterile) solvent suitable for dissolving or suspending the composition prior to administering the compound to a subject.
[00628] In another embodiment, provided herein, is a pharmaceutical pack or kit comprising one or more containers filled with one or more of the agents of the pharmaceutical composition provided herein. In specific embodiments, the kit comprises a pharmaceutical composition
including a therapeutically effective amount of an antisense compound, and a therapeutically effective amount of one or more pharmaceutical agents. For example, the kit can include four (4) containers each having one pharmaceutical agent; three (3) containers, one container including two agents and the other two containers each including one agent; two (2) containers, one container including one agent and the other container including the three remaining agents, or two (2) containers, with each container including two agents; or one container including a mixture of all agents.
[00629] In a specific embodiment, the kit is provided including two containers, as dual entities (each container having two agents) or triple entities, (with one container having three agents and the remaining agent in the other container) for treating a subject in need of treatment of MACE. In some embodiments, the subject is a human.
[00630] It is understood that modifications which do not substantially affect the activity of the various embodiments, described herein can also be included within the definition provided herein. Accordingly, the following examples can be intended to illustrate but not limit provided herein.
2. EXAMPLES
2.1 Example 1 - Experimental protocols
2.1.1. Nucleoside Phosphoramidites for Oligonucleotide Synthesis Deoxy and 2'-alkoxy amidites
[00631 ] 2 ' -Deoxy and 2 ' -methoxy beta-cyanoethyldiisopropyl phosphoramidites were purchased from commercial sources (e.g.,Chemgenes, Needham MA or Glen Research, Inc. Sterling VA). Other 2'-0-alkoxy substituted nucleoside amidites can be prepared as described in U.S. Patent 5,506,351, herein incorporated by reference. For oligonucleotides synthesized using 2'-alkoxy amidites, the standard cycle for unmodified oligonucleotides was utilized, except the wait step after pulse delivery of tetrazole and base was increased to 360 seconds.
[00632] Oligonucleotides containing 5-methyl-2'-deoxycytidine (5-Me-C) nucleotides were synthesized according to published methods [Sanghvi, et. al, Nucleic Acids Research, 1993, 21, 3197-3203] using commercially available phosphoramidites (Glen Research, Sterling VA or ChemGenes, Needham MA).
2.1.2. 2'-Fluoro amidites and 2'-Fluorodeoxyadenosine amidites
[00633] 2'-fluoro oligonucleotides were synthesized as described previously [Kawasaki, et. al, J. Med. Chem., 1993, 36, 831-841] and United States patent 5,670,633, herein incorporated by reference. Briefly, the protected nucleoside N6-benzoyl-2'-deoxy-2'-fluoroadenosine was synthesized utilizing commercially available 9-beta-D-arabinofuranosyladenine as starting material and by modifying literature procedures whereby the 2'-alpha-fluoro atom is introduced by a SN2-displacement of a 2'-beta-trityl group. Thus N6-benzoyl-9-beta-D- arabinofuranosyladenine was selectively protected in moderate yield as the 3 ',5'- ditetrahydropyranyl (THP) intermediate. Deprotection of the THP and N6-benzoyl groups was accomplished using standard methodologies and standard methods were used to obtain the 5'- dimethoxytrityl-(DMT) and 5'-DMT-3'-phosphoramidite intermediates.
2.1.3. 2'-Fluorodeoxyguanosine
[00634] The synthesis of 2'-deoxy-2'-fluoroguanosine was accomplished using
tetraisopropyldisiloxanyl (TPDS) protected 9-beta-D-arabinofuranosylguanine as starting material, and conversion to the intermediate diisobutyrylarabinofuranosylguanosine.
Deprotection of the TPDS group was followed by protection of the hydroxyl group with THP to give diisobutyryl di-THP protected arabinofuranosylguanine. Selective O-deacylation and triflation was followed by treatment of the crude product with fluoride, then deprotection of the THP groups.
[00635] Standard methodologies were used to obtain the 5' -DMT- and 5 '-DMT-3 '- phosphoramidites .
2.1.4. 2'-Fluorouridine
[00636] Synthesis of 2'-deoxy-2'-fluorouridine was accomplished by the modification of a literature procedure in which 2,2'-anhydro-l-beta-D-arabinofuranosyluracil was treated with 70% hydrogen fluoride-pyridine. Standard procedures were used to obtain the 5'-DMT and 5'- DMT-3 'phosphoramidites.
2.1.5. 2'-Fluorodeoxycytidine
[00637] 2 ' -deoxy-2 ' -fluorocytidine was synthesized via amination of 2 ' -deoxy-2 ' - fluorouridine, followed by selective protection to give N4-benzoyl-2'-deoxy-2'-fluorocytidine. Standard procedures were used to obtain the 5'-DMT and 5 '-DMT-3 'phosphoramidites.
2.1.6. 2'-0-(2-Methoxyethyl) modified amidites
[00638] 2'-0-Methoxyethyl-substituted nucleoside amidites can be prepared as follows, or alternatively, as per the methods of Martin, P., Helvetica Chimica Acta, 1995, 78, 486-504.
2.1.7. 2,2 '- Anhydro [ l-(beta-D-ar abinofuranosyl)-5-methyluridine]
[00639] 5-Methyluridine (ribosylthymine, commercially available through Yamasa, Choshi, Japan) (72. 0 g, 0. 279 M), diphenylcarbonate (90. 0 g, 0. 420 M) and sodium bicarbonate (2. 0 g, 0. 024 M) were added to DMF (300 mL). The mixture was heated to reflux, with stirring, allowing the evolved carbon dioxide gas to be released in a controlled manner. After 1 hour, the slightly darkened solution was concentrated under reduced pressure. The resulting syrup was poured into diethylether (2. 5 L), with stirring. The product formed a gum. The ether was decanted and the residue was dissolved in a minimum amount of methanol (ca. 400 mL). The solution was poured into fresh ether (2. 5 L) to yield a stiff gum. The ether was decanted and the gum was dried in a vacuum oven (60°C at 1 mm Hg for 24 h) to give a solid that was crushed to a light tan powder (57 g, 85% crude yield). The NMR spectrum was consistent with the structure, contaminated with phenol as its sodium salt (ca. 5%). The material was used as is for further reactions (or it can be purified further by column chromatography using a gradient of methanol in ethyl acetate (10-25%) to give a white solid, mp 222-4°C).
2.1.8. 2 '-O-Methoxyethyl-5-methyluridine
[00640] 2,2'-Anhydro-5-methyluridine (195 g, 0. 81 M), tris(2-methoxyethyl)borate (231 g, 0. 98 M) and 2-methoxyethanol (1. 2 L) were added to a 2 L stainless steel pressure vessel and placed in a pre-heated oil bath at 160°C. After heating for 48 hours at 155-160°C, the vessel was opened and the solution evaporated to dryness and triturated with MeOH (200 mL). The residue was suspended in hot acetone (1 L). The insoluble salts were filtered, washed with acetone (150 mL) and the filtrate evaporated. The residue (280 g) was dissolved in CH3CN (600 mL) and evaporated. A silica gel column (3 kg) was packed in CH2Cl2/acetone/MeOH (20:5 :3) containing 0. 5% Et3NH. The residue was dissolved in CH2C12 (250 mL) and adsorbed onto silica (150 g) prior to loading onto the column. The product was eluted with the packing solvent to give 160 g (63%) of product. Additional material was obtained by reworking impure fractions.
2.1.9. 2'-0-Methoxyethyl-5'-0-dimethoxytrityl-5-methyluridine
[00641] 2'-0-Methoxyethyl-5-methyluridine (160 g, 0. 506 M) was co-evaporated with pyridine (250 mL) and the dried residue dissolved in pyridine (1. 3 L). A first aliquot of
dimethoxytrityl chloride (94. 3 g, 0. 278 M) was added and the mixture stirred at room temperature for one hour. A second aliquot of dimethoxytrityl chloride (94. 3 g, 0. 278 M) was added and the reaction stirred for an additional one hour. Methanol (170 mL) was then added to stop the reaction. HPLC showed the presence of approximately 70% product. The solvent was evaporated and triturated with CH3CN (200 mL). The residue was dissolved in CHC13 (1. 5 L) and extracted with 2x500 mL of saturated NaHC03 and 2x500 mL of saturated NaCl. The organic phase was dried over Na2S04, filtered and evaporated. 275 g of residue was obtained. The residue was purified on a 3. 5 kg silica gel column, packed and eluted with
EtOAc/hexane/acetone (5:5: 1) containing 0. 5% Et3NH. The pure fractions were evaporated to give 164 g of product. Approximately 20 g additional was obtained from the impure fractions to give a total yield of 183 g (57%).
2.1.10. 3 '-0- Acetyl-2 '-O-methoxyethyl-5 '-O-dimethoxytrityl-5-methyluridine
[00642] 2'-0-Methoxyethyl-5'-0-dimethoxytrityl-5-methyluridine (106 g, 0. 167 M), DMF/pyridine (750 mL of a 3: 1 mixture prepared from 562 mL of DMF and 188 mL of pyridine) and acetic anhydride (24. 38 mL, 0. 258 M) were combined and stirred at room temperature for 24 hours. The reaction was monitored by TLC by first quenching the TLC sample with the addition of MeOH. Upon completion of the reaction, as judged by TLC, MeOH (50 mL) was added and the mixture evaporated at 35°C. The residue was dissolved in CHC13 (800 mL) and extracted with 2x200 mL of saturated sodium bicarbonate and 2x200 mL of saturated NaCl. The water layers were back extracted with 200 mL of CHC13. The combined organics were dried with sodium sulfate and evaporated to give 122 g of residue (approx. 90%> product). The residue was purified on a 3. 5 kg silica gel column and eluted using
EtOAc/hexane(4: l). Pure product fractions were evaporated to yield 96 g (84%). An additional 1. 5 g was recovered from later fractions.
2.1.11. 3 '-0- Acetyl-2 '-O-methoxyethyl-5 '-0-dimethoxytrityl-5-methyl-4- triazoleuridine
[00643] A first solution was prepared by dissolving 3 '-O-acetyl-2' -O-methoxyethyl-5 '-0- dimethoxytrityl-5-methyluridine (96 g, 0. 144 M) in CH3CN (700 mL) and set aside.
Triethylamine (189 mL, 1. 44 M) was added to a solution of triazole (90 g, 1. 3 M) in CH3CN (1 L), cooled to -5°C and stirred for 0. 5 h using an overhead stirrer. POCl3 was added dropwise, over a 30 minute period, to the stirred solution maintained at 0-10°C, and the resulting mixture
stirred for an additional 2 hours. The first solution was added dropwise, over a 45 minute period, to the latter solution. The resulting reaction mixture was stored overnight in a cold room. Salts were filtered from the reaction mixture and the solution was evaporated. The residue was dissolved in EtOAc (1 L) and the insoluble solids were removed by filtration. The filtrate was washed with 1x300 mL of NaHC03 and 2x300 mL of saturated NaCl, dried over sodium sulfate and evaporated. The residue was triturated with EtOAc to give the title compound.
2.1.12. 2'-0-Methoxyethyl-5'-0-dimethoxytrityl-5-methylcytidine
[00644] A solution of 3 '-0-acetyl-2'-0-methoxyethyl-5 '-0-dimethoxytrityl-5-methyl-4- triazoleuridine (103 g, 0. 141 M) in dioxane (500 mL) and NH4OH (30 mL) was stirred at room temperature for 2 hours. The dioxane solution was evaporated and the residue azeotroped with MeOH (2x200 mL). The residue was dissolved in MeOH (300 mL) and transferred to a 2 liter stainless steel pressure vessel. MeOH (400 mL) saturated with NH3 gas was added and the vessel heated to 100°C for 2 hours (TLC showed complete conversion). The vessel contents were evaporated to dryness and the residue was dissolved in EtOAc (500 mL) and washed once with saturated NaCl (200 mL). The organics were dried over sodium sulfate and the solvent was evaporated to give 85 g (95%) of the title compound.
2.1.13. N4-Benzoyl-2'-0-methoxyethyl-5'-0-dimethoxytrityl-5-methyl- cytidine
[00645] 2'-0-Methoxyethyl-5'-0-dimethoxytrityl-5-methylcytidine (85 g, 0. 134 M) was dissolved in DMF (800 mL) and benzoic anhydride (37. 2 g, 0. 165 M) was added with stirring. After stirring for 3 hours, TLC showed the reaction to be approximately 95% complete. The solvent was evaporated and the residue azeotroped with MeOH (200 mL). The residue was dissolved in CHC13 (700 mL) and extracted with saturated NaHC03 (2x300 mL) and saturated NaCl (2x300 mL), dried over MgS04 and evaporated to give a residue (96 g). The residue was chromatographed on a 1. 5 kg silica column using EtOAc/hexane (1 : 1) containing 0. 5% Et3NH as the eluting solvent. The pure product fractions were evaporated to give 90 g (90%>) of the title compound.
2.1.14. N4-Benzoyl-2'-0-methoxyethyl-5'-0-dimethoxytrityl-5-methyl- cytidine-3 '-amidite
[00646] N4-Benzoyl-2'-0-methoxyethyl-5'-0-dimethoxytrityl-5-methylcytidine (74 g, 0. 10 M) was dissolved in CH2CI2 (1 L). Tetrazole diisopropylamine (7. 1 g) and 2-cyanoethoxy-tetra-
(isopropyl)phosphite (40. 5 mL, 0. 123 M) were added with stirring, under a nitrogen atmosphere. The resulting mixture was stirred for 20 hours at room temperature (TLC showed the reaction to be 95% complete). The reaction mixture was extracted with saturated NaHC03 (1x300 mL) and saturated NaCl (3x300 mL). The aqueous washes were back-extracted with CH2CI2 (300 mL), and the extracts were combined, dried over MgSC^ and concentrated. The residue obtained was chromatographed on a 1. 5 kg silica column using EtOAc/hexane (3: 1) as the eluting solvent. The pure fractions were combined to give 90. 6 g (87%) of the title compound.
2.1.15. 2'-0-(Aminooxyethyl) nucleoside amidites and 2'-0-(dimethylamino- oxyethyl) nucleoside amidites (or 2'-(Dimethylaminooxyethoxy) nucleoside amidites)
[00647] 2'-(Dimethylaminooxyethoxy) nucleoside amidites [also known in the art as 2'-0- (dimethylaminooxyethyl) nucleoside amidites] can be prepared as described in the following paragraphs. Adenosine, cytidine and guanosine nucleoside amidites can be prepared similarly to the thymidine (5-methyluridine) except the exocyclic amines can be protected with a benzoyl moiety in the case of adenosine and cytidine and with isobutyryl in the case of guanosine.
2.1.16. 5 '-0-tert-Butyldiphenylsilyl-02-2 '-anhydro-5-methyluridine
[00648] 02-2'-anhydro-5-methyluridine (Pro. Bio. Sint., Vcanbese, Italy, 100. Og, 0. 416 mmol), dimethylaminopyridine (0. 66g, 0. 013eq, 0. 0054mmol) were dissolved in dry pyridine (500 ml) at ambient temperature under an argon atmosphere and with mechanical stirring, tert- Butyldiphenylchlorosilane (125. 8g, 119. OmL, 1. leq, 0. 458mmol) was added in one portion. The reaction was stirred for 16 h at ambient temperature. TLC (Rf 0. 22, ethyl acetate) indicated a complete reaction. The solution was concentrated under reduced pressure to a thick oil. This was partitioned between dichloromethane (1 L) and saturated sodium bicarbonate (2x1 L) and brine (1 L). The organic layer was dried over sodium sulfate and concentrated under reduced pressure to a thick oil. The oil was dissolved in a 1 : 1 mixture of ethyl acetate and ethyl ether (600mL) and the solution was cooled to -10° C. The resulting crystalline product was collected by filtration, washed with ethyl ether (3x200 mL) and dried (40°C, 1mm Hg, 24 h) to 149g (74. 8%>) of white solid. TLC and NMR were consistent with pure product.
2.1.17. 5 '-O-tert-Butyldiphenylsilyl-2 '-0-(2-hydroxyethyl)-5-methyluridine
[00649] In a 2 L stainless steel, unstirred pressure reactor was added borane in tetrahydrofuran (1. 0 M, 2. 0 eq, 622 mL). In the fume hood and with manual stirring, ethylene glycol (350 mL, excess) was added cautiously at first until the evolution of hydrogen gas subsided. 5'-0-tert- Butyldiphenylsilyl-02-2'-anhydro-5-methyluridine (149 g, 0. 311 mol) and sodium bicarbonate (0. 074 g, 0. 003 eq) were added with manual stirring. The reactor was sealed and heated in an oil bath until an internal temperature of 160 °C was reached and then maintained for 16 h (pressure < 100 psig). The reaction vessel was cooled to ambient and opened. TLC (Rf 0. 67 for desired product and Rf 0. 82 for ara-T side product, ethyl acetate) indicated about 70% conversion to the product. In order to avoid additional side product formation, the reaction was stopped, concentrated under reduced pressure (10 to 1mm Hg) in a warm water bath (40-100°C) with the more extreme conditions used to remove the ethylene glycol. Alternatively, once the low boiling solvent is gone, the remaining solution can be partitioned between ethyl acetate and water; the product will be in the organic phase. The residue was purified by column
chromatography (2kg silica gel, ethyl acetate-hexanes gradient 1 : 1 to 4: 1). The appropriate fractions were combined, stripped and dried to product as a white crisp foam (84g, 50%), contaminated starting material (17. 4g) and pure reusable starting material 20g. The yield based on starting material less pure recovered starting material was 58%. TLC and NMR were consistent with 99% pure product.
2.1.18. 2 '-0-( [2-phthalimidoxy)ethyl] -5 '-f-butyldiphenylsilyl-5-methyluridine
[00650] 5'-0-tert-Butyldiphenylsilyl-2'-0-(2-hydroxyethyl)-5-methyluridine (20g, 36.
98mmol) was mixed with triphenylphosphine (11. 63g, 44. 36mmol) and N-hydroxyphthalimide (7. 24g, 44. 36mmol). It was then dried over P205 under high vacuum for two days at 40°C. The reaction mixture was flushed with argon and dry THF (369. 8mL, Aldrich, sure seal bottle) was added to get a clear solutioN.D.iethyl-azodicarboxylate (6. 98mL, 44. 36mmol) was added dropwise to the reaction mixture. The rate of addition is maintained such that resulting deep red coloration is just discharged before adding the next drop. After the addition was complete, the reaction was stirred for 4 hrs. By that time TLC showed the completion of the reaction
(ethylacetate:hexane, 60:40). The solvent was evaporated in vacuum. Residue obtained was placed on a flash column and eluted with ethyl acetate :hexane (60:40), to get 2'-0-([2- phthalimidoxy)ethyl]-5 '-t-butyldiphenylsilyl-5-methyluridine as white foam (21. 819 g, 86%).
2.1.19. 5'-0-tei"i-butyldiphenylsilyl-2'-0-[(2-formadoximinooxy)ethyl]-5- methyluridine
[00651] 2'-0-([2-phthalimidoxy)ethyl]-5'-^utyldiphenylsilyl-5-methyluridine (3. lg, 4. 5mmol) was dissolved in dry CH2CI2 (4. 5mL) and methylhydrazine (300mL, 4. 64mmol) was added drop wise at -10°C to O°C. After 1 h the mixture was filtered, the filtrate was washed with ice cold CH2C12 and the combined organic phase was washed with water, brine and dried over anhydrous Na2S04. The solution was concentrated to get 2'-0-(aminooxyethyl) thymidine, which was then dissolved in MeOH (67. 5mL). To this formaldehyde (20% aqueous solution, w/w, 1. 1 eq. ) was added and the resulting mixture was stirred for 1 h. Solvent was removed under vacuum; residue chromatographed to get 5'-0-tert-butyldiphenylsilyl-2'-0-[(2- formadoximinooxy) ethyl]-5-methyluridine as white foam (1. 95 g, 78%).
2.1.20. 5'-0-tei-i-Butyldiphenylsilyl-2'-0-[N,N-dimethylaminooxyethyl]-5- methyluridine
[00652] 5 '-0-tert-butyldiphenylsilyl-2'-0-[(2-formadoximinooxy)ethyl]-5-methyluridine (1. 77g, 3. 12mmol) was dissolved in a solution of 1M pyridinium p-toluenesulfonate (PPTS) in dry MeOH (30. 6mL). Sodium cyanoborohydride (0. 39g, 6. 13mmol) was added to this solution at 10°C under inert atmosphere. The reaction mixture was stirred for 10 minutes at 10°C. After that the reaction vessel was removed from the ice bath and stirred at room temperature for 2 h, the reaction monitored by TLC (5%> MeOH in CH2C12). Aqueous NaHC03 solution (5%>, lOmL) was added and extracted with ethyl acetate (2x20mL). Ethyl acetate phase was dried over anhydrous Na2S04, evaporated to dryness. Residue was dissolved in a solution of 1M PPTS in MeOH (30. 6mL). Formaldehyde (20%> w/w, 30mL, 3. 37mmol) was added and the reaction mixture was stirred at room temperature for 10 minutes. Reaction mixture cooled to 10°C in an ice bath, sodium cyanoborohydride (0. 39g, 6. 13mmol) was added and reaction mixture stirred at 10°C for 10 minutes. After 10 minutes, the reaction mixture was removed from the ice bath and stirred at room temperature for 2 hrs. To the reaction mixture 5% NaHC03 (25mL) solution was added and extracted with ethyl acetate (2x25mL). Ethyl acetate layer was dried over anhydrous Na2S04 and evaporated to dryness . The residue obtained was purified by flash column chromatography and eluted with 5% MeOH in CH2C12 to get 5'-0-tert-butyldiphenylsilyl-2'-0-[N,N- dimethylaminooxyethyl]-5-methyluridine as a white foam (14. 6g, 80%).
2.1.21. 2 '-0-(dimethylaminooxyethyl)-5-methyluridine
[00653] Triethylamine trihydrofluoride (3. 91mL, 24. Ommol) was dissolved in dry THF and triethylamine (1. 67mL, 12mmol, dry, kept over KOH). This mixture of triethylamine -2HF was then added to 5'-0-tert-butyldiphenylsilyl-2'-0- N,N-dimethylaminooxyethyl]-5-methyluridine (1. 40g, 2. 4mmol) and stirred at room temperature for 24 hrs. Reaction was monitored by TLC (5% MeOH in CH2CI2). Solvent was removed under vacuum and the residue placed on a flash column and eluted with 10% MeOH in CH2CI2 to get 2'-0-(dimethylaminooxyethyl)-5- methyluridine (766mg, 92. 5%).
2.1.22. 5'-0-DMT-2'-0-(dimethylaminooxyethyl)-5-methyluridine
[00654] 2'-0-(dimethylaminooxyethyl)-5-methyluridine (750mg, 2. 17mmol) was dried over P2O5 under high vacuum overnight at 40°C. It was then co-evaporated with anhydrous pyridine (20mL). The residue obtained was dissolved in pyridine (1 lmL) under argon atmosphere. 4- dimethylaminopyridine (26. 5mg, 2. 60mmol), 4,4'-dimethoxytrityl chloride (880mg, 2.
60mmol) was added to the mixture and the reaction mixture was stirred at room temperature until all of the starting material disappecan bed. Pyridine was removed under vacuum and the residue chromatographed and eluted with 10% MeOH in CH2C12 (containing a few drops of pyridine) to get 5'-0-DMT-2'-0-(dimethylamino-oxyethyl)-5-methyluridine (1. 13g, 80%).
2.1.23. 5'-0-DMT-2'-0-(2-N,N-dimethylaminooxyethyl)-5-methyluridine-3'- [(2-cyanoethyl)-N,N-diisopropylphosphoramidite]
[00655] 5 '-0-DMT-2'-0-(dimethylaminooxyethyl)-5-methyluridine (1. 08g, 1. 67mmol) was co-evaporated with toluene (20mL). To the residue Ν,Ν-diisopropylamine tetrazonide (0. 29g, 1. 67mmol) was added and dried over P205 under high vacuum overnight at 40°C. Then the reaction mixture was dissolved in anhydrous acetonitrile (8. 4mL) and 2-cyanoethyl-N,N,N1,N1- tetraisopropylphosphoramidite (2. 12mL, 6. 08mmol) was added. The reaction mixture was stirred at ambient temperature for 4 hrs under inert atmosphere. The progress of the reaction was monitored by TLC (hexane: ethyl acetate 1 :1). The solvent was evaporated, then the residue was dissolved in ethyl acetate (70mL) and washed with 5% aqueous NaHC03 (40mL). Ethyl acetate layer was dried over anhydrous Na2S04 and concentrated. Residue obtained was
chromatographed (ethyl acetate as eluent) to get 5'-0-DMT-2'-0-(2-N,N- dimethylaminooxyethyl)-5-methyluridine-3'-[(2-cyanoethyl)-N,N-diisopropylphosphoramidite] as a foam (1. 04g, 74. 9%).
2.1.24. 2'-(Aminooxyethoxy) nucleoside amidites
[00656] 2'-(Aminooxyethoxy) nucleoside amidites [also known in the art as 2'-0- (aminooxyethyl) nucleoside amidites] can be prepared as described in the following paragraphs. Adenosine, cytidine and thymidine nucleoside amidites can be prepared similarly.
2.1.25. N2-isobutyryl-6-0-diphenylcarbamoyl-2 '-0-(2-ethylacetyl)-5 '-0- (4,4'-dimethoxytrityl)guanosine-3'-[(2-cyanoethyl)-N,N- diisopropylphosphoramidite]
[00657] The 2'-0-aminooxyethyl guanosine analog can be obtained by selective 2'-0- alkylation of diaminopurine riboside. Multigram quantities of diaminopurine riboside can be purchased from Schering AG (Berlin) to provide 2'-0-(2-ethylacetyl) diaminopurine riboside along with a minor amount of the 3'-0-isomer. 2'-0-(2-ethylacetyl) diaminopurine riboside can be resolved and converted to 2'-0-(2-ethylacetyl)guanosine by treatment with adenosine deaminase. (McGee, D. P. C, Cook, P. D., Guinosso, C. J., WO 94/02501 Al 940203. ) Standard protection procedures should afford 2'-0-(2-ethylacetyl)-5'-0-(4,4'- dimethoxytrityl)guanosine and 2-N-isobutyryl-6-0-diphenylcarbamoyl-2'-0-(2-ethylacetyl)-5 '- 0-(4,4'-dimethoxytrityl)guanosine which can be reduced to provide 2-N-isobutyryl-6-0- diphenylcarbamoyl-2'-0-(2-hydroxyethyl)-5'-0-(4,4'-dimethoxytrityl)guanosine. As before the hydroxyl group can be displaced by N-hydroxyphthalimide via a Mitsunobu reaction, and the protected nucleoside can phosphitylated as usual to yield 2-N-isobutyryl-6-0- diphenylcarbamoyl-2'-0-([2-phthalmidoxy]ethyl)-5'-0-(4,4'-dimethoxytrityl)guanosine-3'-[(2- cyanoethyl)-N,N-diisopropylphosphoramidite].
2.1.26. 2'-dimethylaminoethoxyethoxy (2'-DMAEOE) nucleoside amidites
[00658] 2'-dimethylaminoethoxyethoxy nucleoside amidites (also known in the art as 2'-0- dimethylaminoethoxyethyl, i.e., 2'-0-CH2-0-CH2-N(CH2)2, or 2'-DMAEOE nucleoside amidites) can be prepared as follows. Other nucleoside amidites can be prepared similarly.
2.1.27. 2 '-0- [2(2-N,N-dimethylaminoethoxy)ethyl] -5-methyl uridine
[00659] 2[2-(Dimethylamino)ethoxy]ethanol (Aldrich, 6. 66 g, 50 mmol) is slowly added to a solution of borane in tetrahydrofuran (1 M, 10 mL, 10 mmol) with stirring in a 100 mL bomb. Hydrogen gas evolves as the solid dissolves. 02-,2'-anhydro-5-methyluridine (1. 2 g, 5 mmol), and sodium bicarbonate (2. 5 mg) can be added and the bomb is sealed, placed in an oil bath and heated to 155°C for 26 hours. The bomb is cooled to room temperature and opened. The crude solution is concentrated and the residue partitioned between water (200 mL) and hexanes (200
mL). The excess phenol is extracted into the hexane layer. The aqueous layer is extracted with ethyl acetate (3x200 mL) and the combined organic layers can be washed once with water, dried over anhydrous sodium sulfate and concentrated. The residue is columned on silica gel using methanol/methylene chloride 1 :20 (which has 2% triethylamine) as the eluent. As the column fractions can be concentrated a colorless solid forms which is collected to give the title compound as a white solid.
2.1.28. 5 '-O-dimethoxytrityl-2 '-0- [2(2-N,N-dimethylaminoethoxy)ethyl)] -5- methyl uridine
[00660] To 0.5 g (1.3 mmol) of 2'-0-[2(2-N,N-dimethylaminoethoxy)ethyl)]-5-methyl uridine in anhydrous pyridine (8 mL), triethylamine (0. 36 mL) and dimethoxytrityl chloride (DMT-C1, 0. 87 g, 2 eq. ) can be added and stirred for 1 hour. The reaction mixture is poured into water (200 mL) and extracted with CH2CI2 (2x200 mL). The combined CH2CI2 layers can be washed with saturated NaHC03 solution, followed by saturated NaCl solution and dried over anhydrous sodium sulfate. Evaporation of the solvent followed by silica gel chromatography using
MeOH:CH2Cl2:Et3N (20: 1, v/v, with 1% triethylamine) gives the title compound.
2.1.29. 5 '-O-Dimethox trityl-2 '-0- [2(2-N,N-dimethylaminoethoxy)ethyl)] -5- methyl uridine-3'-0-(cyanoethyl-N,N-diisopropyl)phosphoramidite
[00661] Diisopropylaminotetrazolide (0. 6 g) and 2-cyanoethoxy-N,N-diisopropyl
phosphoramidite (1. 1 mL, 2 eq. ) can be added to a solution of 5' -O-dimethoxytrityl-2 '-0-[2(2- N,N-dimethylaminoethoxy)ethyl)]-5-methyluridine (2. 17 g, 3 mmol) dissolved in CH2CI2 (20 mL) under an atmosphere of argon. The reaction mixture is stirred overnight and the solvent evaporated. The resulting residue is purified by silica gel flash column chromatography with ethyl acetate as the eluent to give the title compound.
2.2 Example 2
2.2.1. Oligonucleotide synthesis
[00662] Unsubstituted and substituted phosphodiester (P=0) oligonucleotides can be synthesized on an automated DNA synthesizer (Applied Biosystems model 380B) using standard phosphoramidite chemistry with oxidation by iodine.
[00663] Phosphorothioates (P=S) can be synthesized as for the phosphodiester
oligonucleotides except the standard oxidation bottle was replaced by 0. 2 M solution of 3H-1,2-
benzodithiole-3-one 1,1 -dioxide in acetonitrile for the stepwise thiation of the phosphite linkages. The thiation wait step was increased to 68 sec and was followed by the capping step. After cleavage from the CPG column and deblocking in concentrated ammonium hydroxide at 55°C (18 h), the oligonucleotides were purified by precipitating twice with 2. 5 volumes of ethanol from a 0. 5 M NaCl solution. Phosphinate oligonucleotides can be prepared as described in U.S. Patent 5,508,270, herein incorporated by reference.
[00664] Alkyl phosphonate oligonucleotides can be prepared as described in U.S. Patent 4,469,863, herein incorporated by reference.
[00665] 3 '-Deoxy-3 '-methylene phosphonate oligonucleotides can be prepared as described in
U.S. Patents 5,610,289 or 5,625,050, herein incorporated by reference.
[00666] Phosphoramidite oligonucleotides can be prepared as described in U.S. Patent,
5,256,775 or U.S. Patent 5,366,878, herein incorporated by reference.
[00667] Alkylphosphonothioate oligonucleotides can be prepared as described in published
PCT applications PCT/US94/00902 and PCT/US 93/06976 (published as WO 94/17093 and WO
94/02499, respectively), herein incorporated by reference.
[00668] 3 '-Deoxy-3 '-amino phosphoramidate oligonucleotides can be prepared as described in U.S. Patent 5,476,925, herein incorporated by reference.
[00669] Phosphotriester oligonucleotides can be prepared as described in U.S. Patent 5,023,243, herein incorporated by reference.
[00670] Borano phosphate oligonucleotides can be prepared as described in U.S. Patents 5,130,302 and 5,177,198, both herein incorporated by reference.
2.3 Example 3 - Oligonucleoside Synthesis
[00671] Methylenemethylimino linked oligonucleosides, also identified as MMI linked oligonucleosides, methylenedimethylhydrazo linked oligonucleosides, also identified as MDH linked oligonucleosides, and methylenecarbonylammo linked oligonucleosides, also identified as amide-3 linked oligonucleosides, and methyleneaminocarbonyl linked oligonucleosides, also identified as amide-4 linked oligonucleosides, as well as mixed backbone compounds having, for instance, alternating MMI and P=0 or P=S linkages can be prepared as described in U.S. Patents 5,378,825, 5,386,023, 5,489,677, 5,602,240 and 5,610,289, all of which can be herein
incorporated by reference.
[00672] Formacetal and thioformacetal linked oligonucleosides can be prepared as described in U.S. Patents 5,264,562 and 5,264,564, herein incorporated by reference.
[00673] Ethylene oxide linked oligonucleosides can be prepared as described in U.S. Patent 5,223,618, herein incorporated by reference.
2.4 Example 4 - DNA Synthesis
[00674] Peptide nucleic acids (PNAs) can be prepared in accordance with any of the various procedures referred to in Peptide Nucleic Acids (PNA): Synthesis, Properties and Potential Applications, Bioorganic & Medicinal Chemistry, 1996, 4, 5-23. They can also be prepared in accordance with U.S. Patents 5,539,082, 5,700,922, and 5,719,262, herein incorporated by reference.
2.5 Example 5 - Synthesis of Chimeric Oligonucleotides
[00675] Chimeric oligonucleotides, oligonucleosides or mixed
oligonucleotides/oligonucleosides can be of several different types. These include a first type wherein the "gap" segment of linked nucleosides is positioned between 5' and 3' "wing" segments of linked nucleosides and a second "open end" type wherein the "gap" segment is located at either the 3 ' or the 5 ' terminus of the oligomeric compound. Oligonucleotides of the first type can also be known in the art as "gapmers" or gapped oligonucleotides.
Oligonucleotides of the second type can also be known in the art as "hemimers" or "wingmers".
2.5.1. [2'-0-Me]-[2'-deoxy]-[2'-0-Me] Chimeric Phosphorothioate
Oligonucleotides
[00676] Chimeric oligonucleotides having 2'-0-alkyl phosphorothioate and 2'-deoxy phosphorothioate oligonucleotide segments can be synthesized using an Applied Biosystems automated DNA synthesizer Model 380B, as above. Oligonucleotides can be synthesized using the automated synthesizer and 2'-deoxy-5'-dimethoxytrityl-3'-0-phosphoramidite for the DNA portion and 5'-dimethoxytrityl-2'-0-methyl-3'-0-phosphoramidite for 5' and 3' wings. The standard synthesis cycle is modified by increasing the wait step after the delivery of tetrazole and base to 600 s repeated four times for RNA and twice for 2'-0-methyl. The fully protected oligonucleotide is cleaved from the support and the phosphate group is deprotected in 3 : 1 ammonia/ethanol at room temperature overnight then lyophilized to dryness. Treatment in methanolic ammonia for 24 hrs at room temperature is then done to deprotect all bases and sample was again lyophilized to dryness. The pellet is resuspended in 1M TBAF in THF for 24
hrs at room temperature to deprotect the 2' positions. The reaction is then quenched with 1M TEAA and the sample is then reduced to 1/2 volume by rotovac before being desalted on a G25 size exclusion column. The oligo recovered is then analyzed spectrophotometrically for yield and for purity by capillary electrophoresis and by mass spectrometry.
2.5.2. 2'-0-(2-Methoxyethyl)]~[2'-deoxy]~[2'-0-(Methoxyethyl)] Chimeric Phosphorothioate Oligonucleotides
[00677] [2'-0-(2-methoxyethyl)]--[2'-deoxy]~[-2'-0-(methoxyethyl)] chimeric
phosphorothioate oligonucleotides were prepared as per the procedure above for the 2'-0-methyl chimeric oligonucleotide, with the substitution of 2'-0-(methoxyethyl) amidites for the 2'-0- methyl amidites.
2.5.3. [2'-0-(2-Methoxyethyl)Phosphodiester]--[2'-deoxy Phosphoro- thioate]~[2'-0-(2-Methoxyethyl) Phosphodiester] Chimeric Oligonucleotides
[00678] [2'-0-(2-methoxyethyl phosphodiester]-- [2 '-deoxy phosphorothioate]-- [2 '-0- (methoxyethyl) phosphodiester] chimeric oligonucleotides can be prepared as per the above procedure for the 2'-0-methyl chimeric oligonucleotide with the substitution of 2'-0- (methoxy ethyl) amidites for the 2'-0-methyl amidites, oxidization with iodine to generate the phosphodiester internucleotide linkages within the wing portions of the chimeric structures and sulfurization utilizing 3,H-1,2 benzodithiole-3-one 1,1 dioxide (Beaucage Reagent) to generate the phosphorothioate internucleotide linkages for the center gap.
[00679] Other chimeric oligonucleotides, chimeric oligonucleosides and mixed chimeric oligonucleotides/oligonucleosides can be synthesized according to United States patent
5,623,065, herein incorporated by reference.
2.6 Example 6 - Oligonucleotide Isolation
[00680] After cleavage from the controlled pore glass column (Applied Biosystems) and deblocking in concentrated ammonium hydroxide at 55°C for 18 hours, the oligonucleotides or oligonucleosides can be purified by precipitation twice out of 0. 5 M NaCl with 2. 5 volumes ethanol. Synthesized oligonucleotides were analyzed by polyacrylamide gel electrophoresis on denaturing gels and judged to be at least 85% full length material. The relative amounts of phosphorothioate and phosphodiester linkages obtained in synthesis were periodically checked by 31P nuclear magnetic resonance spectroscopy, and for some studies oligonucleotides were
purified by HPLC, as described by Chiang et al., J. Biol. Chem. 1991, 266, 18162-18171. Results obtained with HPLC-purified material were similar to those obtained with non-HPLC purified material.
2.7 Example 7 - Oligonucleotide Synthesis - 96 Well Plate Format
[00681] Oligonucleotides were synthesized via solid phase P (III) phosphoramidite chemistry on an automated synthesizer capable of assembling 96 sequences simultaneously in a standard 96 well format. Phosphodiester internucleotide linkages were afforded by oxidation with aqueous iodine. Phosphorothioate internucleotide linkages were generated by sulfurization utilizing 3,H- 1,2 benzodithiole-3-one 1,1 dioxide (Beaucage Reagent) in anhydrous acetonitrile. Standard base-protected beta-cyanoethyldiisopropyl phosphoramidites were purchased from commercial vendors (e.g.,PE-Applied Biosystems, Foster City, CA, or Pharmacia, Piscataway, NJ). Nonstandard nucleosides can be synthesized as per known literature or patented methods. They can be utilized as base protected beta-cyanoethyldiisopropyl phosphoramidites.
[00682] Oligonucleotides were cleaved from support and deprotected with concentrated NH4OH at elevated temperature (55-60°C) for 12-16 hours and the released product then dried in vacuo. The dried product was then re-suspended in sterile water to afford a master plate from which all analytical and test plate samples can be then diluted utilizing robotic pipettors.
2.8 Example 8 - Oligonucleotide Analysis - 96 Well Plate Format
[00683] The concentration of oligonucleotide in each well was assessed by dilution of samples and UV absorption spectroscopy. The full-length integrity of the subject products was evaluated by capillary electrophoresis (CE) in either the 96 well format (BECKMAN P/ACE® MDQ) or, for ly prepared samples, on a commercial CE apparatus {e.g., BECKMAN P/ACE® 5000, ABI 270). Base and backbone composition was confirmed by mass analysis of the compounds utilizing electrospray-mass spectroscopy. All assay test plates were diluted from the master plate using single and multi-channel robotic pipettors. Plates were judged to be acceptable if at least 85% of the compounds on the plate were at least 85% full length.
2.9 Example 9
2.9.1. A. Cell culture and oligonucleotide treatment
[00684] The effect of antisense compounds on target nucleic acid expression can be tested in any of a variety of cell types provided that the target nucleic acid is present at measurable levels.
This can be routinely determined using, for example, PCR or Northern blot analysis. The following 7 cell types can be provided for illustrative purposes, but other cell types can be routinely used, provided that the target is expressed in the cell type chosen. This can be readily determined by methods routine in the art, for example Northern blot analysis, Ribonuclease protection assays, or RT PCR.
2.9.1.1 T-24 cells
[00685] The human transitional cell bladder carcinoma cell line T-24 was obtained from the American Type Culture Collection (ATCC) (Manassas, VA). T-24 cells were routinely cultured in complete McCoy's 5 A basal media (Gibco/Life Technologies, Gaithersburg, MD)
supplemented with 10% fetal calf serum (Gibco/Life Technologies, Gaithersburg, MD), penicillin 100 units per mL, and streptomycin 100 micrograms per mL (Gibco/Life
Technologies, Gaithersburg, MD). Cells were routinely passaged by trypsinization and dilution when they reached 90% confluence. Cells were seeded into 96-well plates (Falcon-Primaria #3872) at a density of 7000 cells/well for use in RT-PCR analysis.
[00686] For Northern blotting or other analysis, cells can be seeded onto 100 mm or other standard tissue culture plates and treated similarly, using appropriate volumes of medium and oligonucleotide.
2.9.1.2 A549 cells
[00687] The human lung carcinoma cell line A549 was obtained from the American Type Culture Collection (ATCC) (Manassas, VA). A549 cells were routinely cultured in DMEM basal media (Gibco/Life Technologies, Gaithersburg, MD) supplemented with 10%> fetal calf serum (Gibco/Life Technologies, Gaithersburg, MD), penicillin 100 units per mL, and streptomycin 100 micrograms per mL (Gibco/Life Technologies, Gaithersburg, MD). Cells were routinely passaged by trypsinization and dilution when they reached 90%> confluence.
2.9.1.3 NHDF cells
[00688] Human neonatal dermal fibroblast (NHDF) were obtained from the Clonetics Corporation (Walkersville MD). NHDFs were routinely maintained in Fibroblast Growth Medium (Clonetics Corporation, Walkersville MD) supplemented as recommended by the supplier. Cells were maintained for up to 10 passages as recommended by the supplier.
2.9.1.4 HEK cells
[00689] Human embryonic keratinocytes (HEK) were obtained from the Clonetics Corporation (Walkersville MD). HEKs were routinely maintained in Keratinocyte Growth Medium (Clonetics Corporation, Walkersville MD) formulated as recommended by the supplier. Cells were routinely maintained for up to 10 passages as recommended by the supplier.
2.9.1.5 HepG2 cells
[00690] The human hepatoblastoma cell line HepG2 was obtained from the American Type Culture Collection (Manassas, VA). HepG2 cells were routinely cultured in Eagle's MEM supplemented with 10% fetal calf serum, non-essential amino acids, and 1 mM sodium pyruvate (Gibco/Life Technologies, Gaithersburg, MD). Cells were routinely passaged by trypsinization and dilution when they reached 90%> confluence. Cells were seeded into 96-well plates (Falcon- Primaria #3872) at a density of 7000 cells/well for use in RT-PCR analysis.
[00691] For Northern blotting or other analyses, cells can be seeded onto 100 mm or other standard tissue culture plates and treated similarly, using appropriate volumes of medium and oligonucleotide.
2.9.1.6 AML12 cells
[00692] The AML12 (alpha mouse liver 12) cell line was established from hepatocytes from a mouse (CD1 strain, line MT42) transgenic for human TGF alpha. Cells can be cultured in a 1 :1 mixture of Dulbecco's modified Eagle's medium and Ham's F12 medium with 0. 005 mg/ml insulin, 0. 005 mg/ml transferrin, 5 ng/ml selenium, and 40 ng/ml dexamethasone, and 90%>; 10%) fetal bovine serum. For subculturing, spent medium is removed and fresh media of 0. 25% trypsin, 0. 03 % EDTA solution is added. Fresh trypsin solution (1 to 2 ml) is added and the culture is left to sit at room temperature until the cells detach.
[00693] Cells were routinely passaged by trypsinization and dilution when they reached 90%> confluence. Cells were seeded into 96-well plates (Falcon-Primaria #3872) at a density of 7000 cells/well for use in RT-PCR analysis.
[00694] For Northern blotting or other analyses, cells can be seeded onto 100 mm or other standard tissue culture plates and treated similarly, using appropriate volumes of medium and oligonucleotide.
2.9.1.7 Primary mouse hepatocytes
[00695] Primary mouse hepatocytes were prepared from CD-I mice purchased from Charles River Labs (Wilmington, MA) and were routinely cultured in Hepatoyte Attachment Media (Gibco) supplemented with 10% Fetal Bovine Serum (Gibco/Life Technologies, Gaithersburg, MD), 250nM dexamethasone (Sigma), and ΙΟηΜ bovine insulin (Sigma). Cells were seeded into 96-well plates (Falcon-Primaria #3872) at a density of 10000 cells/well for use in RT-PCR analysis.
[00696] For Northern blotting or other analyses, cells can be plated onto 100 mm or other standard tissue culture plates coated with rat tail collagen (200ug/mL) (Becton Dickinson) and treated similarly using appropriate volumes of medium and oligonucleotide.
2.9.1.8 Hep3B cells
[00697] The human hepatocellular carcinoma cell line Hep3B was obtained from the
American Type Culture Collection (Manassas, VA). Hep3B cells were routinely cultured in Dulbeccos's MEM high glucose supplemented with 10%> fetal calf serum, L-glutamine and pyridoxine hydrochloride (Gibco/Life Technologies, Gaithersburg, MD). Cells were routinely passaged by trypsinization and dilution when they reached 90%> confluence. Cells were seeded into 24-well plates (Falcon-Primaria #3846) at a density of 50,000 cells/well for use in RT-PCR analysis.
[00698] For Northern blotting or other analyses, cells can be seeded onto 100 mm or other standard tissue culture plates and treated similarly, using appropriate volumes of medium and oligonucleotide.
2.9.1.9 Rabbit primary hepatocytes
[00699] Primary rabbit hepatocytes were purchased from Invitro Technologies (Gaithersburg, MD) and maintained in Dulbecco's modified Eagle's medium (Gibco). When purchased, the cells had been seeded into 96-well plates for use in RT-PCR analysis and were confluent.
[00700] For Northern blotting or other analyses, cells can be seeded onto 100 mm or other standard tissue culture plates and treated similarly using appropriate volumes of medium and oligonucleotide.
2.9.1.10 HeLa cells
[00701] The human epitheloid carcinoma cell line HeLa was obtained from the American Tissue Type Culture Collection (Manassas, VA). HeLa cells were routinely cultured in DMEM,
high glucose (Invitrogen Corporation, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen Corporation, Carlsbad, CA). Cells were seeded into 24-well plates (Falcon-Primaria #3846) at a density of 50,000 cells/well for use in RT-PCR analysis. Cells were routinely passaged by trypsinization and dilution when they reached 90%> confluence. Cells 96-well plates (Falcon-Primaria #3872) at a density of 5,000 cells/well for use in RT-PCR analysis. For Northern blotting or other analyses, cells can be seeded onto 100 mm or other standard tissue culture plates and treated similarly, using appropriate volumes of medium and oligonucleotide.
2.9.1.11 Human mammary epithelial cells
[00702] Normal human mammary epithelial cells (HMECs) were obtained from the American Type Culture Collection (Manassas VA). HMECs were routinely cultured in DMEM low glucose (Gibco/Life Technologies, Gaithersburg, MD) supplemented with 10%> fetal calf serum (Gibco/Life Technologies, Gaithersburg, MD). Cells were routinely passaged by trypsinization and dilution when they reached 90%> confluence. Cells were seeded into 96-well plates (Falcon- Primaria #353872, BD Biosciences, Bedford, MA) at a density of 7000 cells/well for use in RT- PCR analysis. For Northern blotting or other analyses, cells can be seeded onto 100 mm or other standard tissue culture plates and treated similarly, using appropriate volumes of medium and oligonucleotide.
2.9.1.12 Treatment with antisense compounds:
[00703] When cells reached 80%> confluency, they were treated with oligonucleotide. For cells grown in 96-well plates, wells were washed once with 200 ΟΡΤΙ-ΜΕΜ™-1 reduced-serum medium (Gibco BRL) and then treated with 130 μΕ of ΟΡΤΙ-ΜΕΜ™-1 containing 3. 75 μg/mL LIPOFECTIN™ (Gibco BRL) and the desired concentration of oligonucleotide. After 4-7 hours of treatment, the medium was replaced with fresh medium. Cells were harvested 16-24 hours after oligonucleotide treatment.
[00704] The concentration of oligonucleotide used varies from cell line to cell line. To determine the optimal oligonucleotide concentration for a particular cell line, the cells can be treated with a positive control oligonucleotide at a range of concentrations. For human cells the positive control oligonucleotide is ISIS 13920, TCCGTCATCGCTCCTCAGGG, SEQ ID NO: 1, a 2'-0-methoxyethyl gapmer (2 '-O-methoxy ethyls shown in bold) with a phosphorothioate backbone which is targeted to human H-ras. For mouse or rat cells the positive control
oligonucleotide is ISIS 15770, ATGCATTCTGCCCCCAAGGA, SEQ ID NO: 2, a 2'-0- methoxy ethyl gapmer (2 '-O-methoxy ethyls shown in bold) with a phosphorothioate backbone which is targeted to both mouse and rat c-raf. The concentration of positive control
oligonucleotide that results in 80% inhibition of c-Ha-ras (for ISIS 13920) or c-raf (for ISIS 15770) mRNA is then utilized as the screening concentration for new oligonucleotides in subsequent experiments for that cell line. If 80% inhibition is not achieved, the lowest concentration of positive control oligonucleotide that results in 60% inhibition of H-ras or c-raf mRNA is then utilized as the oligonucleotide screening concentration in subsequent experiments for that cell line. If 60% inhibition is not achieved, that particular cell line is deemed as unsuitable for oligonucleotide transfection experiments. The concentrations of antisense oligonucleotides used herein can be from 5 nM to 300 nM.
2.9.1.13 B. Cell culture and oligonucleotide treatment
[00705] The effect of antisense compounds on target nucleic acid expression can be tested in any of a variety of cell types provided that the target nucleic acid is present at measurable levels. This can be routinely determined using, for example, PCR or Northern blot analysis. The following cell types can be provided for illustrative purposes, but other cell types can be routinely used, provided that the target is expressed in the cell type chosen. This can be readily determined by methods routine in the art, for example Northern blot analysis, Ribonuclease protection assays, or real-time PCR.
2.9.1.14 HepG2 cells
[00706] The human hepatoblastoma cell line HepG2 was obtained from the American Type Culture Collection (Manassas, VA). HepG2 cells were routinely cultured in Eagle's MEM supplemented with 10% fetal bovine serum, non-essential amino acids, and 1 mM sodium pyruvate (Invitrogen Life Technologies, Carlsbad, CA). Cells were routinely passaged by trypsinization and dilution when they reached 90%> confluence. Cells were seeded into 96-well plates (Falcon-Primaria #3872, BD Biosciences, Bedford, MA) at a density of approximately 7000 cells/well for use in antisense oligonucleotide transfection experiments. For Northern blotting or other analyses, cells can be seeded onto 100 mm or other standard tissue culture plates and treated similarly, using appropriate volumes of medium and oligonucleotide.
2.9.1.15 AML12 cells
[00707] The AML12 (alpha mouse liver 12) cell line was established from hepatocytes from a mouse (CD1 strain, line MT42) transgenic for human TGF alpha. Cells can be cultured in a 1 : 1 mixture of Dulbecco's modified Eagle's medium and Ham's F12 medium with 0. 005 mg/ml insulin, 0. 005 mg/ml transferrin, 5 ng/ml selenium, and 40 ng/ml dexamethasone, and 90%: 10% fetal bovine serum (medium and additives from Invitrogen Life Technologies, Carlsbad CA and Sigma-Aldrich, St. Louis, MO). For subculturing, spent medium is removed and fresh media of 0. 25% trypsin, 0. 03 % EDTA solution is added. Fresh trypsin solution (1 to 2 ml) is added and the culture is left to sit at room temperature until the cells detach. Cells were routinely passaged by trypsinization and dilution when they reached approximately 90% confluence. Cells were seeded into 96-well plates (Falcon-Primaria #3872, BD Biosciences, Bedford, MA) at a density of approximately 7000 cells/well for use in antisense oligonucleotide transfection experiments. For Northern blotting or other analyses, cells can be seeded onto 100 mm or other standard tissue culture plates and treated similarly, using appropriate volumes of medium and oligonucleotide.
2.9.1.16 Primary mouse hepatocytes
[00708] Primary mouse hepatocytes were prepared from CD-I mice purchased from Charles River Labs (Wilmington, MA) and were routinely cultured in Hepatoyte Attachment Media (Invitrogen Life Technologies, Carlsbad, CA) supplemented with 10%> Fetal Bovine Serum (Invitrogen Life Technologies, Carlsbad, CA), 250nM dexamethasone (Sigma), and ΙΟηΜ bovine insulin (both from Sigma-Aldrich, St. Louis, MO). Cells were seeded into 96-well plates (Falcon-Primaria #3872, BD Biosciences, Bedford, MA) at a density of approximately 10,000 cells/well for use in antisense oligonucleotide transfection experiments. For Northern blotting or other analyses, cells can be plated onto 100 mm or other standard tissue culture plates coated with rat tail collagen (200ug/mL) (BD Biosciences, Bedford, MA) and treated similarly using appropriate volumes of medium and oligonucleotide.
2.9.1.17 Hep3B cells
[00709] The human hepatocellular carcinoma cell line Hep3B was obtained from the
American Type Culture Collection (Manassas, VA). Hep3B cells were routinely cultured in Dulbeccos's MEM high glucose supplemented with 10%> fetal bovine serum, L-glutamine and pyridoxine hydrochloride (Invitrogen Life Technologies, Carlsbad, CA). Cells were routinely passaged by trypsinization and dilution when they reached approximately 90% confluence. Cells were seeded into 24-well plates (Falcon-Primaria #3846, BD Biosciences, Bedford, MA) at a
density of approximately 50,000 cells/well for use in antisense oligonucleotide transfection experiments. For Northern blotting or other analyses, cells can be seeded onto 100 mm or other standard tissue culture plates and treated similarly, using appropriate volumes of medium and oligonucleotide.
2.9.1.18 HeLa cells
[00710] The human epitheloid carcinoma ceil line HeLa was obtained from the American Tissue Type Culture Collection (Manassas, VA). HeLa cells were routinely cultured in DMEM, high glucose (Invitrogen Corporation, Carlsbad, CA) supplemented with 10% fetal bovine senim (Invitrogen Corporation, Carlsbad, CA). Cells were routinely passaged by trypsinization and dilution when they reached approximately 90% confluence. Cells were seeded onto 96-well plates (Falcon-Primaria #3872, BD Biosciences, Bedford, MA) at a density of approximately 5,000 cells/well for use in antisense oligonucleotide transfection experiments. Alternatively, cells were seeded into 24-well plates (Falcon-Primaria #3846, BD Biosciences, Bedford, MA) at a density of approximately 50,000 cells/well for use in RT-PCR analysis. For Northern blotting or other analyses, cells can be seeded onto 100 mm or other standard tissue culture plates and treated similarly, using appropriate volumes of medium and oligonucleotide.
2.9.1.19 Human mammary epithelial cells
[00711] Normal human mammary epithelial cells (HMECs) were obtained from the American Type Culture Collection (Manassas VA). HMECs were routinely cultured in DMEM low glucose supplemented with 10% fetal bovine serum (Invitrogen Corporation, Carlsbad, CA). Cells were routinely passaged by trypsinization and dilution when they reached approximately 90% confluence. Cells were seeded into 96-well plates (Falcon-Primaria #353872, BD
Biosciences, Bedford, MA) at a density of approximately 7000 cells/well for use in antisense oligonucleotide transfection experiments. For Northern blotting or other analyses, cells can be seeded onto 100 mm or other standard tissue culture plates and treated similarly, using appropriate volumes of medium and oligonucleotide.
2.9.1.20 Treatment with antisense compounds
[00712] When cells reached 65-75% confluency, they were treated with oligonucleotide. Oligonucleotide was mixed with LIPOFECTF ® Invitrogen Life Technologies, Carlsbad, CA) in OPTI-MEM® 1 reduced serum medium (Invitrogen Life Technologies, Carlsbad, CA) to
achieve the desired concentration of oligonucleotide and a LIPOFECTIN® concentration of 2. 5 or 3 μg/mL per 100 nM oligonucleotide. This transfection mixture was incubated at room temperature for approximately 0. 5 hours. For cells grown in 96-well plates, wells were washed once with 100 OPTI-MEM® 1 and then treated with 130 of the transfection mixture. Cells grown in 24-well plates or other standard tissue culture plates can be treated similarly, using appropriate volumes of medium and oligonucleotide. Cells can be treated and data can be obtained in duplicate or triplicate. After approximately 4-7 hours of treatment at 37°C, the medium containing the transfection mixture was replaced with fresh culture medium. Cells were harvested 16-24 hours after oligonucleotide treatment.
[00713] The concentration of oligonucleotide used varies from cell line to cell line. To determine the optimal oligonucleotide concentration for a particular cell line, the cells can be treated with a positive control oligonucleotide at a range of concentrations. For human cells the positive control oligonucleotide is ISIS 13920 (TCCGTCATCGCTCCTCAGGG, SEQ ID NO: 1 ; targeted to human H-ras), a chimeric oligonucleotide having a 9 nucleotide gap segment composed of 2'-deoxynucleotides, which is flanked on the 5' side and 3' sides by 3 nucleotide and 8 nucleotide wing segments, respectively. The wings can be composed of 2'-0- methoxyethyl nucleotides. For mouse or rat cells the positive control oligonucleotide is ISIS 15770 (ATGCATTCTGCCCCCAAGGA, SEQ ID NO: 2; targeted to rodent c-raf), a chimeric oligonucleotide having a 10 nucleotide gap segment composed of 2'-deoxynucleotides, which is flanked on the 5' side and 3' sides by 5 nucleotide wing segments. The wings can be composed of 2'-0-methoxyethyl nucleotides. Both compounds have phosphorothioate internucleoside (backbone) linkages and cytidines in the wing segments can be 5-methylcytidines. The concentration of positive control oligonucleotide that results in 80% inhibition of H-ras (for ISIS 13920) or c-raf (for ISIS 15770) mRNA is then utilized as the screening concentration for new oligonucleotides in subsequent experiments for that cell line. If 80% inhibition is not achieved, the lowest concentration of positive control oligonucleotide that results in 60% inhibition of H- ras or c-raf mRNA is then utilized as the oligonucleotide screening concentration in subsequent experiments for that cell line. If 60% inhibition is not achieved, that particular cell line is deemed as unsuitable for oligonucleotide transfection experiments. The concentrations of antisense oligonucleotides used herein can be from 5 nM to 300 nM.
2.10 Example 10 - Analysis of oligonucleotide inhibition of ApoB expression
[00714] Antisense modulation of ApoB expression can be assayed in a variety of ways known in the art. For example, ApoB mRNA levels can be quantitated by, e.g., Northern blot analysis, competitive polymerase chain reaction (PCR), or real-time PCR (RT-PCR). Real-time quantitative PCR is presently. RNA analysis can be performed on total cellular RNA or poly(A)+ mRNA. Methods of RNA isolation can be taught in, for example, Ausubel, F. M. et al. , Current Protocols in Molecular Biology, Volume 1 , pp . 4. 1. 1-4. 2. 9 and 4. 5. 1-4. 5. 3, John Wiley & Sons, Inc., 1993. Northern blot analysis is routine in the art and is taught in, for example, Ausubel, F.M. et al., Current Protocols in Molecular Biology, Volume 1, pp. 4. 2. 1-4. 2. 9, John Wiley & Sons, Inc., 1996. Real-time quantitative (PCR) can be conveniently accomplished using the commercially available ABI PRISM® 7700 Sequence Detection System, available from PE- Applied Biosystems, Foster City, CA and used according to manufacturer's instructions.
[00715] Protein levels of ApoB can be quantitated in a variety of ways well known in the art, such as immunoprecipitation, Western blot analysis (immunoblotting), ELISA or fluorescence- activated cell sorting (FACS). Antibodies directed to ApoB can be identified and obtained from a variety of sources, such as the MSRS catalog of antibodies (Aerie Corporation, Birmingham, MI), or can be prepared via conventional antibody generation methods. Methods for preparation of polyclonal antisera can be taught in, for example, Ausubel, F. M. et al., Current Protocols in Molecular Biology, Volume 2, pp. 11. 12. 1-11. 12. 9, John Wiley & Sons, Inc., 1997.
Preparation of monoclonal antibodies is taught in, for example, Ausubel, F. M. et al., Current Protocols in Molecular Biology, Volume 2, pp. 11. 4. 1-11. 11. 5, John Wiley & Sons, Inc., 1997.
[00716] Immunoprecipitation methods can be standard in the art and can be found at, for example, Ausubel, F. M. et al. , Current Protocols in Molecular Biology, Volume 2, pp. 10. 16. 1-10. 16. 11, John Wiley & Sons, Inc., 1998. Western blot (immunoblot) analysis is standard in the art and can be found at, for example, Ausubel, F. M. et al., Current Protocols in Molecular Biology, Volume 2, pp. 10. 8. 1-10. 8. 21, John Wiley & Sons, Inc., 1997. Enzyme-linked immunosorbent assays (ELISA) can be standard in the art and can be found at, for example, Ausubel, F. M. et al., Current Protocols in Molecular Biology, Volume 2, pp. 11. 2. 1-11. 2. 22, John Wiley & Sons, Inc., 1991.
2.11 Example 11 - Poly(A)+ mRNA isolation
[00717] Poly(A)+ mR A was isolated according to Miura et al., Clin. Chem. , 1996, 42, 1758-1764. Other methods for poly(A)+ mRNA isolation can be taught in, for example, Ausubel, F. M. et al., Current Protocols in Molecular Biology, Volume 1, pp. 4. 5. 1-4. 5. 3, John Wiley & Sons, Inc., 1993. Briefly, for cells grown on 96-well plates, growth medium was removed from the cells and each well was washed with 200 μΐ, cold PBS. 60 μΐ, lysis buffer (10 mM Tris-HCl, pH 7. 6, 1 mM EDTA, 0. 5 M NaCl, 0. 5% NP-40, 20 mM vanadyl-ribonucleoside complex) was added to each well, the plate was gently agitated and then incubated at room temperature for five minutes. 55 μΐ, of lysate was transferred to Oligo d(T) coated 96-well plates (AGCT Inc., Irvine CA). Plates were incubated for 60 minutes at room temperature, washed 3 times with 200 μΐ^ of wash buffer (10 mM Tris-HCl pH 7. 6, 1 mM EDTA, 0. 3 M NaCl). After the final wash, the plate was blotted on paper towels to remove excess wash buffer and then air-dried for 5 minutes. 60 μΐ, of elution buffer (5 mM Tris-HCl pH 7. 6), preheated to 70°C was added to each well, the plate was incubated on a 90°C hot plate for 5 minutes, and the eluate was then transferred to a fresh 96-well plate.
[00718] Cells grown on 100 mm or other standard plates can be treated similarly, using appropriate volumes of all solutions.
2.12 Example 12 - Total RNA Isolation
[00719] Total RNA was isolated using an RNEASY® 96 kit and buffers purchased from Qiagen Inc. (Valencia CA) following the manufacturer's recommended procedures. Briefly, for cells grown on 96-well plates, growth medium was removed from the cells and each well was washed with 200 μΐ, cold PBS. 100 Buffer RLT was added to each well and the plate vigorously agitated for 20 seconds. 100 μΐ, of 70% ethanol was then added to each well and the contents mixed by pipetting three times up and down. The samples were then transferred to the RNEASY® 96 well plate attached to a QIAvac® manifold fitted with a waste collection tray and attached to a vacuum source. Vacuum was applied for 15 seconds. 1 mL of Buffer RW1 was added to each well of the RNEASY® 96 plate and the vacuum again applied for 15 seconds. 1 mL of Buffer RPE was then added to each well of the RNEASY® 96 plate and the vacuum applied for a period of 15 seconds. The Buffer RPE wash was then repeated and the vacuum was applied for an additional 10 minutes. The plate was then removed from the QIAvac® manifold and blotted dry on paper towels. The plate was then re-attached to the QIAvac® manifold fitted
with a collection tube rack containing 1. 2 mL collection tubes. RNA was then eluted by pipetting 60 water into each well, incubating 1 minute, and then applying the vacuum for 30 seconds. The elution step was repeated with an additional 60 μΐ, water.
[00720] The repetitive pipetting and elution steps can be automated using a QIAGEN Bio- Robot 9604 (Qiagen, Inc., Valencia CA). Essentially, after lysing of the cells on the culture plate, the plate is transferred to the robot deck where the pipetting, DNase treatment and elution steps can be carried out.
2.13 Example 13
2.13.1. Real-time Quantitative PCR Analysis of ApoB mRNA Levels
[00721] Quantitation of ApoB mRNA levels was determined by real-time quantitative PCR using the ABI PRISM® 7700 Sequence Detection System (PE-Applied Biosystems, Foster City, CA) according to manufacturer's instructions. This is a closed-tube, non-gel-based, fluorescence detection system which allows high-throughput quantitation of polymerase chain reaction (PCR) products in real-time. As opposed to standard PCR, in which amplification products can be quantitated after the PCR is completed, products in real-time quantitative PCR can be quantitated as they accumulate. This is accomplished by including in the PCR reaction an oligonucleotide probe that anneals specifically between the forward and reverse PCR primers, and contains two fluorescent dyes. A reporter dye (e.g., JOE™, FAM™, or VIC™, obtained from either Operon Technologies Inc., Alameda, CA or PE-Applied Biosystems, Foster City, CA) is attached to the 5 ' end of the probe and a quencher dye (e.g., TAMRA™, obtained from either Operon
Technologies Inc., Alameda, CA or PE-Applied Biosystems, Foster City, CA) is attached to the 3 ' end of the probe. When the probe and dyes can be intact, reporter dye emission is quenched by the proximity of the 3 ' quencher dye. During amplification, annealing of the probe to the target sequence creates a substrate that can be cleaved by the 5 '-exonuclease activity of Taq
polymerase. During the extension phase of the PCR amplification cycle, cleavage of the probe by Taq polymerase releases the reporter dye from the remainder of the probe (and hence from the quencher moiety) and a sequence-specific fluorescent signal is generated. With each cycle, additional reporter dye molecules can be cleaved from their respective probes, and the fluorescence intensity is monitored at regular intervals by laser optics built into the ABI
PRISM® 7700 Sequence Detection System. In each assay, a series of parallel reactions containing serial dilutions of mRNA from untreated control samples generates a standard curve
that is used to quantitate the percent inhibition after antisense oligonucleotide treatment of test samples.
[00722] Prior to quantitative PCR analysis, primer-probe sets specific to the target gene being measured can be evaluated for their ability to be "multiplexed" with a GAPDH amplification reaction. In multiplexing, both the target gene and the internal standard gene GAPDH can be amplified concurrently in a single sample. In this analysis, mRNA isolated from untreated cells is serially diluted. Each dilution is amplified in the presence of primer-probe sets specific for GAPDH only, target gene only ("single-plexing"), or both (multiplexing). Following PCR amplification, standard curves of GAPDH and target mRNA signal as a function of dilution can be generated from both the single-plexed and multiplexed samples. If both the slope and correlation coefficient of the GAPDH and target signals generated from the multiplexed samples fall within 10% of their corresponding values generated from the single-plexed samples, the primer-probe set specific for that target is deemed multiplexable. Other methods of PCR can also be known in the art.
[00723] PCR reagents were obtained from PE- Applied Biosystems, Foster City, CA. RT-PCR reactions were carried out by adding 25 μΕ PCR cocktail (lx TAQMAN™ buffer A, 5. 5 mM MgCl2, 300 μΜ each of dATP, dCTP and dGTP, 600 μΜ of dUTP, 100 nM each of forward primer, reverse primer, and probe, 20 Units RNAse inhibitor, 1. 25 Units AMPLITAQ GOLD™, and 12. 5 Units MuLV reverse transcriptase) to 96 well plates containing 25 μΐ^ total RNA solution. The RT reaction was carried out by incubation for 30 minutes at 48°C. Following a 10 minute incubation at 95 °C to activate the AMPLITAQ GOLD™, 40 cycles of a two-step PCR protocol were carried out: 95°C for 15 seconds (denaturation) followed by 60°C for 1. 5 minutes (annealing/ extension) .
[00724] Gene target quantities obtained by real time RT-PCR can be normalized using either the expression level of GAPDH, a gene whose expression is constant, or by quantifying total RNA using RiboGreen™ (Molecular Probes, Inc. Eugene, OR). GAPDH expression is quantified by real time RT-PCR, by being run simultaneously with the target, multiplexing, or separately. Total RNA is quantified using RiboGreen™ RNA quantification reagent from Molecular Probes. Methods of RNA quantification by RiboGreen™ can be taught in Jones, L. J., et al, Analytical Biochemistry, 1998, 265, 368-374.
[00725] In this assay, 175 μΐ, of RiboGreen working reagent (RiboGreen reagent diluted 1 :2865 in lOmM Tris-HCl, 1 mM EDTA, pH 7. 5) is pipetted into a 96-well plate containing 25uL purified, cellular RNA. The plate is read in a CytoFluor 4000 (PE Applied Biosystems) with excitation at 480nm and emission at 520nm.
[00726] Probes and primers to human ApoB were designed to hybridize to a human ApoB sequence, using published sequence information (GenBank accession number NM 000384. 1, incorporated herein as SEQ ID NO: 3). For human ApoB the PCR primers were:
[00727] forward primer: TGCTAAAGGCACATATGGCCT (SEQ ID NO: 4)
[00728] reverse primer: CTCAGGTTGGACTCTCCATTGAG (SEQ ID NO: 5) and the PCR probe was: FAM-CTTGTCAGAGGGATCCTAACACTGGCCG-TAMRA
[00729] (SEQ ID NO: 6) where FAM (PE-Applied Biosystems, Foster City, CA) is the fluorescent reporter dye) and TAMRA (PE-Applied Biosystems, Foster City, CA) is the quencher dye.
[00730] For human GAPDH the PCR primers were:
[00731] forward primer: GAAGGTGAAGGTCGGAGTC (SEQ ID NO: 7)
[00732] reverse primer: GAAGATGGTGATGGGATTTC (SEQ ID NO: 8) and the PCR probe was: 5' JOE-CAAGCTTCCCGTTCTCAGCC-TAMRA 3' (SEQ ID NO: 9) where JOE (PE-Applied Biosystems, Foster City, CA) is the fluorescent reporter dye) and TAMRA (PE- Applied Biosystems, Foster City, CA) is the quencher dye.
[00733] Probes and primers to mouse ApoB were designed to hybridize to a mouse ApoB sequence, using published sequence information (GenBank accession number M35186, incorporated herein as SEQ ID NO: 10). For mouse ApoB the PCR primers were:
[00734] forward primer: CGTGGGCTCCAGCATTCTA (SEQ ID NO: 11)
[00735] reverse primer: AGTCATTTCTGCCTTTGCGTC (SEQ ID NO: 12) and the PCR probe was: FAM-CCAATGGTCGGGCACTGCTCAA-TAMRA (SEQ ID NO: 13) where FAM (PE-Applied Biosystems, Foster City, CA) is the fluorescent reporter dye) and TAMRA (PE- Applied Biosystems, Foster City, CA) is the quencher dye. For mouse GAPDH the PCR primers were:
[00736] forward primer: GGCAAATTCAACGGCACAGT (SEQ ID NO: 14)
[00737] reverse primer: GGGTCTCGCTCCTGGAAGAT (SEQ ID NO: 15) and the PCR probe was: 5' JOE-AAGGCCGAGAATGGGAAGCTTGTCATC-TAMRA 3 ' (SEQ ID NO: 16)
where JOE (PE-Applied Biosystems, Foster City, CA) is the fluorescent reporter dye) and TAMRA (PE-Applied Biosystems, Foster City, CA) is the quencher dye.
2.13.2. Real-time Quantitative PCR Analysis of ApoB mRNA Levels
[00738] Quantitation of ApoB mRNA levels was determined by real-time quantitative PCR using the ABI PRISM® 7700 Sequence Detection System (PE-Applied Biosystems, Foster City, CA) according to manufacturer's instructions. This is a closed-tube, non-gel-based, fluorescence detection system which allows high-throughput quantitation of polymerase chain reaction (PCR) products in real-time. As opposed to standard PCR, in which amplification products can be quantitated after the PCR is completed, products in real-time quantitative PCR can be quantitated as they accumulate. This is accomplished by including in the PCR reaction an oligonucleotide probe that anneals specifically between the forward and reverse PCR primers, and contains two fluorescent dyes. A reporter dye (e.g., JOE™, FAM™, or VIC™, obtained from either Operon Technologies Inc., Alameda, CA or PE-Applied Biosystems, Foster City, CA) is attached to the 5 ' end of the probe and a quencher dye (e.g., TAMRA™, obtained from either Operon
Technologies Inc., Alameda, CA or PE-Applied Biosystems, Foster City, CA) is attached to the 3 ' end of the probe. When the probe and dyes can be intact, reporter dye emission is quenched by the proximity of the 3 ' quencher dye. During amplification, annealing of the probe to the target sequence creates a substrate that can be cleaved by the 5 '-exonuclease activity of Taq
polymerase. During the extension phase of the PCR amplification cycle, cleavage of the probe by Taq polymerase releases the reporter dye from the remainder of the probe (and hence from the quencher moiety) and a sequence-specific fluorescent signal is generated. With each cycle, additional reporter dye molecules can be cleaved from their respective probes, and the fluorescence intensity is monitored at regular intervals by laser optics built into the ABI
PRISM® 7700 Sequence Detection System. In each assay, a series of parallel reactions containing serial dilutions of mRNA from untreated control samples generates a standard curve that is used to quantitate the percent inhibition after antisense oligonucleotide treatment of test samples.
[00739] Prior to quantitative PCR analysis, primer-probe sets specific to the target gene being measured can be evaluated for their ability to be "multiplexed" with a GAPDH amplification reaction. In multiplexing, both the target gene and the internal standard gene GAPDH can be amplified concurrently in a single sample. In this analysis, mRNA isolated from untreated cells is
serially diluted. Each dilution is amplified in the presence of primer-probe sets specific for GAPDH only, target gene only ("single-plexing"), or both (multiplexing). Following PCR amplification, standard curves of GAPDH and target mRNA signal as a function of dilution can be generated from both the single-plexed and multiplexed samples. If both the slope and correlation coefficient of the GAPDH and target signals generated from the multiplexed samples fall within 10% of their corresponding values generated from the single-plexed samples, the primer-probe set specific for that target is deemed multiplexable. Other methods of PCR can also be known in the art.
[00740] After isolation the RNA is subjected to sequential reverse transcriptase (RT) reaction and real-time PCR, both of which can be performed in the same well. RT and PCR reagents were obtained from Invitrogen Life Technologies (Carlsbad, CA). RT, real-time PCR was carried out in the same by adding 20 μΐ. PCR cocktail (2. 5x PCR buffer minus MgCl2, 6. 6 mM MgCl2, 375 μΜ each of dATP, dCTP, dCTP and dGTP, 375 nM each of forward primer and reverse primer, 125 nM of probe, 4 Units RNAse inhibitor, 1. 25 Units PLATINUM® Taq, 5 Units MuLV reverse transcriptase, and 2. 5x ROX dye) to 96-well plates containing 30 total RNA solution (20-200 ng). The RT reaction was carried out by incubation for 30 minutes at 48°C. Following a 10 minute incubation at 95 °C to activate the PLATINUM® Taq, 40 cycles of a two-step PCR protocol were carried out: 95°C for 15 seconds (denaturation) followed by 60°C for 1. 5 minutes (annealing/ extension) .
[00741] Gene target quantities obtained by real time PCR can be normalized using either the expression level of GAPDH, a gene whose expression is constant, or by quantifying total RNA using RIBOGREEN® (Molecular Probes, Inc. Eugene, OR). GAPDH expression is quantified by real time PCR, by being run simultaneously with the target, multiplexing, or separately. Total RNA is quantified using RIBOGREEN® RNA quantification reagent from Molecular Probes. Methods of RNA quantification by RIBOGREEN® can be taught in Jones, L. J., et al, Analytical Biochemistry, 1998, 265, 368-374.
[00742] In this assay, 175 μΐ of RIBOGREEN® working reagent (RIBOGREEN® reagent diluted 1 :2865 in lOmM Tris-HCl, 1 mM EDTA, pH 7. 5) is pipetted into a 96-well plate containing 25uL purified, cellular RNA. The plate is read in a CytoFluor 4000 (PE Applied Biosystems) with excitation at 480nm and emission at 520nm.
[00743] Probes and primers to human ApoB were designed to hybridize to a human ApoB sequence, using published sequence information (GENBANK® accession number NM_000384. 1, incorporated herein as SEQ ID NO: 3). For human ApoB the PCR primers can be:
[00744] forward primer: TGCTAAAGGCACATATGGCCT (SEQ ID NO: 4)
[00745] reverse primer: CTCAGGTTGGACTCTCCATTGAG (SEQ ID NO: 5) and the PCR probe is: FAM-CTTGTCAGAGGGATCCTAACACTGGCCG-TAMRA (SEQ ID NO: 6) where FAM™ (PE -Applied Biosystems, Foster City, CA) is the fluorescent reporter dye) and
TAMRA™ (PE-Applied Biosystems, Foster City, CA) is the quencher dye.
[00746] For human GAPDH the PCR primers can be:
[00747] forward primer: GAAGGTGAAGGTCGGAGTC (SEQ ID NO: 7)
[00748] reverse primer: GAAGATGGTGATGGGATTTC (SEQ ID NO: 8) and the PCR probe is: 5' JOE-CAAGCTTCCCGTTCTCAGCC-TAMRA 3' (SEQ ID NO: 9) where JOE™ (PE-Applied Biosystems, Foster City, CA) is the fluorescent reporter dye) and TAMRA™ (PE- Applied Biosystems, Foster City, CA) is the quencher dye.
[00749] Probes and primers to mouse ApoB were designed to hybridize to a mouse ApoB sequence, using published sequence information (GENBANK® accession number M35186, incorporated herein as SEQ ID NO: 10). For mouse ApoB the PCR primers can be:
[00750] forward primer: CGTGGGCTCCAGCATTCTA (SEQ ID NO: 11)
[00751] reverse primer: AGTCATTTCTGCCTTTGCGTC (SEQ ID NO: 12) and the PCR probe is: FAM-CCAATGGTCGGGCACTGCTCAA-TAMRA (SEQ ID NO: 13) where FAM™ (PE-Applied Biosystems, Foster City, CA) is the fluorescent reporter dye) and TAMRA™ (PE- Applied Biosystems, Foster City, CA) is the quencher dye. For mouse GAPDH the PCR primers can be:
[00752] forward primer: GGCAAATTCAACGGCACAGT (SEQ ID NO: 14)
[00753] reverse primer: GGGTCTCGCTCCTGGAAGAT (SEQ ID NO: 15) and the PCR probe is: 5' JOE-AAGGCCGAGAATGGGAAGCTTGTCATC-TAMRA 3' (SEQ ID NO: 16) where JOE™ (PE-Applied Biosystems, Foster City, CA) is the fluorescent reporter dye) and TAMRA™ (PE-Applied Biosystems, Foster City, CA) is the quencher dye.
2.14 Example 14 - Northern blot analysis of ApoB mRNA levels
[00754] Eighteen hours after antisense treatment, cell monolayers were washed twice with cold PBS and lysed in 1 mL RNAZOL® (TEL-TEST "B" Inc., Friendswood, TX). Total RNA
was prepared following manufacturer's recommended protocols. Twenty micrograms of total RNA was fractionated by electrophoresis through 1. 2% agarose gels containing 1. 1% formaldehyde using a MOPS buffer system (AMRESCO, Inc. Solon, OH). RNA was transferred from the gel to HYBOND®-N+ nylon membranes (Amersham Pharmacia Biotech, Piscataway, NJ) by overnight capillary transfer using a Northern/Southern Transfer buffer system (TEL- TEST "B" Inc., Friendswood, TX). RNA transfer was confirmed by UV visualization.
Membranes were fixed by UV cross-linking using a STRATALINKER® UV Crosslinker 2400 (Stratagene, Inc, La Jolla, CA) and then robed using QUICKHYB® hybridization solution (Stratagene, La Jolla, CA) using manufacturer's recommendations for stringent conditions.
[00755] To detect human ApoB, a human ApoB specific probe was prepared by PCR using the forward primer TGCTAAAGGCACATATGGCCT (SEQ ID NO: 4) and the reverse primer CTCAGGTTGGACTCTCCATTGAG (SEQ ID NO: 5). To normalize for variations in loading and transfer efficiency membranes were stripped and probed for human glyceraldehyde-3 - phosphate dehydrogenase (GAPDH) RNA (Clontech, Palo Alto, CA).
[00756] To detect mouse ApoB, a human ApoB specific probe was prepared by PCR using the forward primer CGTGGGCTCCAGCATTCTA (SEQ ID NO: 11) and the reverse primer
AGTCATTTCTGCCTTTGCGTC (SEQ ID NO: 12). To normalize for variations in loading and transfer efficiency membranes were stripped and probed for mouse glyceraldehyde-3 -phosphate dehydrogenase (GAPDH) RNA (Clontech, Palo Alto, CA).
[00757] Hybridized membranes were visualized and quantitated using a
PHOSPHORIMAGER® and IMAGEQUANT® Softwcan be V3. 3 (Molecular Dynamics,
Sunnyvale, CA). Data was normalized to GAPDH levels in untreated controls.
2.15 Example 15 - Antisense inhibition of human ApoB expression by chimeric phosphorothioate oligonucleotides having 2'-MOE wings and a deoxy gap
[00758] In accordance with provided herein, a series of oligonucleotides was designed to target different regions of the human ApoB RNA, using published sequence (GenBank accession number NM 000384. 1, incorporated herein as SEQ ID NO: 3). The oligonucleotides can be shown in Table 1. "Target site" indicates the first (5 '-most) nucleotide number on the particular target sequence to which the oligonucleotide binds. All compounds in Table 1 can be chimeric oligonucleotides ("gapmers") 20 nucleotides in length, composed of a central "gap" region consisting of ten 2'-deoxynucleotides, which is flanked on both sides (5' and 3' directions) by
five-nucleotide "wings". The wings can be composed of 2'-methoxyethyl (2'-MOE) nucleotides. The internucleoside (backbone) linkages can be phosphorothioate (P=S) throughout the oligonucleotide. All cytidine residues can be 5-methylcytidines. The compounds were analyzed for their effect on human ApoB mRNA levels in HepG2 cells by quantitative real-time PCR as described in other examples hereiN.D.ata can be averages from two experiments in which HepG2 cells were treated with 150 nM of the compounds in Table 1. If present, "N.D." indicates "no data".
Table 1
Inhibition of human ApoB mRNA levels by chimeric phosphorothioate oligonucleotides having
2'-MOE wings and a deoxy gap

SEQ ID NO SITE NO
147813 Coding 3 5891 ACAGCTGCCCAGTATGTTCT N.D. 50
147814 Coding 3 7087 CCCAATAAGATT A AACAA 34 51
147815 Coding 3 7731 TGGCCTACCAGAGACAGGTA 45 52
147816 Coding 3 7841 TCATACGTTTAGCCCAATCT 100 53
147817 Coding 3 7901 GCATGGTCCCAAGGATGGTC 0 54
147818 Coding 3 8491 AGTGATGGAAGCTGCGATAC 30 55
147819 Coding 3 9181 ATGAGCATCATGCCTCCCAG N.D. 56
147820 Coding 3 9931 GAACACATAGCCGAATGCCG 100 57
147821 Coding 3 10263 GTGGTGCCCTCTAATTTGTA N.D. 58
147822 Coding 3 10631 CCCGAGAAAGAACCGAACCC N.D. 59
147823 Coding 3 10712 TGCCCTGCAGCTTCACTGAA 19 60
147824 Coding 3 11170 GAAATCCCATAAGCTCTTGT N.D. 61
147825 Coding 3 12301 AGAAGCTGCCTCTTCTTCCC 72 62
147826 Coding 3 12401 TCAGGGTGAGCCCTGTGTGT 80 63
147827 Coding 3 12471 CTAATGGCCCCTTGATAAAC 13 64
147828 Coding 3 12621 ACGTTATCCTTGAGTCCCTG 12 65
147829 Coding 3 12741 TATATCCCAGGTTTCCCCGG 64 66
147830 Coding 3 12801 ACCTGGGACAGTACCGTCCC N.D. 67
147831 3'UTR 3 13921 CTGCCTACTGCAAGGCTGGC 0 68
147832 3'UTR 3 13991 AGAGACCTTCCGAGCCCTGG N.D. 69
147833 3'UTR 3 14101 A GA ACACAA AAAGAC C 25 70
[00759] As shown in Table l, SEQ ID NOs 17, 18, 19, 21, 23, 25, 27, 31, 38, 43, 46, 51, 52, 53, 55, 57, 62, 63 and 66 demonstrated at least 30% inhibition of human ApoB expression in this assay and can be therefore . The target sites to which these sequences can be complementary can be herein referred to as "active sites" and can be therefore sites for targeting by compounds of provided herein,. As ApoB exists in two forms in mammals (ApoB -48 and ApoB -100) which can be colinear at the amino terminus, antisense oligonucleotides targeting nucleotides 1-6530 hybridize to both forms, while those targeting nucleotides 6531-14121 can be specific to the long form of ApoB.
2.16 Example 16 - Antisense inhibition of human ApoB expression by chimeric phosphorothioate oligonucleotides having 2'-MOE wings and a deoxy gap-Dose Response Study
[00760] In accordance with provided herein, a subset of the antisense oligonuclotides in Example 15 were further investigated in dose-response studies. Treatment doses were 50, 150 and 250 nM. The compounds were analyzed for their effect on human ApoB mR A levels in HepG2 cells by quantitative real-time PCR as described in other examples hereiN.D.ata can be averages from two experiments and can be shown in Table 2.
Table 2
[00761] Inhibition of human ApoB mRNA levels by chimeric phosphorothioate
oligonucleotides having 2'-MOE wings and a deoxy gap.

2.17 Example 17 - Antisense inhibition of mouse ApoB expression by chimeric phosphorothioate oligonucleotides having 2'-MOE wings and a deoxy gap
[00762] In accordance with provided herein, a series of oligonucleotides was designed to target different regions of the mouse ApoB RNA, using published sequence (GenBank accession number M35186, incorporated herein as SEQ ID NO: 10). The oligonucleotides can be shown in Table 3. "Target site" indicates the first (5 '-most) nucleotide number on the particular target sequence to which the oligonucleotide binds. All compounds in Table 3 can be chimeric oligonucleotides ("gapmers") 20 nucleotides in length, composed of a central "gap" region consisting of ten 2'-deoxynucleotides, which is flanked on both sides (5' and 3' directions) by five-nucleotide "wings". The wings can be composed of 2'-methoxyethyl (2'-MOE) nucleotides. The internucleoside (backbone) linkages can be phosphorothioate (P=S) throughout the oligonucleotide. All cytidine residues can be 5-methylcytidines. The compounds were analyzed for their effect on mouse ApoB mRNA levels in primary mouse hepatocytes by quantitative realtime PCR as described in other examples herein. Primary mouse hepatocytes were treated with 150 nM of the compounds in Table 3. Data can be averages from two experiments. If present, "N.D." indicates "no data".
Table 3
Inhibition of mouse ApoB mRNA levels by chimeric phosphorothioate oligonucleotides having
2'-MOE wings and a deoxy gap

N SEQ ID NO SITE NO
147480 Coding 10 291 TCTCACCCTCATGCTCCATT 54 76
147481 Coding 10 421 TGACTGTCAAGGGTGAGCTG 24 77
147482 Coding 10 461 GTCCAGCCTAGGAACACTCA 59 78
147483 Coding 10 531 ATGTCAATGCCACATGTCCA N.D. 79
147484 Coding 10 581 TTCATCCGAGAAGTTGGGAC 49 80
147485 Coding 10 601 ATTTGGGACGAATGTATGCC 64 81
147486 Coding 10 711 AGTTGAGGAAGCCAGATTCA N.D. 82
147487 Coding 10 964 TTCCCAGTCAGCTTTAGTGG 73 83
147488 Coding 10 1023 AGCTTGCTTGTTGGGCACGG 72 84
147489 Coding 10 1111 CCTATACTGGCTTCTATGTT 5 85
147490 Coding 10 1191 TGAACTCCGTGTAAGGCAAG N.D. 86
147491 Coding 10 1216 GAGAAATCCTTCAGTAAGGG 71 87
147492 Coding 10 1323 CAATGGAATGCTTGTCACTG 68 88
147493 Coding 10 1441 GCTTCATTATAGGAGGTGGT 41 89
147494 Coding 10 1531 ACAACTGGGATAGTGTAGCC 84 90
147495 Coding 10 1631 GTTAGGACCAGGGATTGTGA 0 91
147496 Coding 10 1691 ACCATGGAAAACTGGCAACT 19 92
147497 Coding 10 1721 TGGGAGGAAAAACTTGAATA N.D. 93
147498 Coding 10 1861 TGGGCAACGATATCTGATTG 0 94
147499 Coding 10 1901 CTGCAGGGCGTCAGTGACAA 29 95
147500 Coding 10 1932 GCATCAGACGTGATGTTCCC N.D. 96
147501 Coding 10 2021 CTTGGTTAAACTAATGGTGC 18 97
147502 Coding 10 2071 ATGGGAGCATGGAGGTTGGC 16 98
147503 Coding 10 2141 AATGGATGATGAAACAGTGG 26 99
147504 Coding 10 2201 ATCAATGCCTCCTGTTGCAG N.D. 100
147505 Coding 10 2231 GGAAGTGAGACTTTCTAAGC 76 101
147506 Coding 10 2281 AGGAAGGAACTCTTGATATT 58 102
147507 Coding 10 2321 ATTGGCTTCATTGGCAACAC 81 103
147759 Coding 10 1 AGGTGAGGAAGTTGGAATTC 19 104
147760 Coding 10 121 TTGTTCCCTGAAGTTGTTAC N.D. 105
147761 Coding 10 251 GTTCATGGATTCCTTCAGGA 45 106
147762 Coding 10 281 ATGCTCCATTCTCACATGCT 46 107
147763 Coding 10 338 TGCGACTGTGTCTGATTTCC 34 108
147764 Coding 10 541 GTCCCTGAAGATGTCAATGC 97 109
147765 Coding 10 561 AGGCCCAGTTCCATGACCCT 59 110
147766 Coding 10 761 GGAGCCCACGTGCTGAGATT 59 111
147767 Coding 10 801 CGTCCTTGAGCAGTGCCCGA 5 112
147768 Coding 10 1224 CCCATATGGAGAAATCCTTC 24 113
147769 Coding 10 1581 CATGCCTGGAAGCCAGTGTC 89 114
147770 Coding 10 1741 GTGTTGAATCCCTTGAAATC 67 115
147771 Coding 10 1781 GGTAAAGTTGCCCATGGCTG 68 116
147772 Coding 10 1841 GTTATAAAGTCCAGCATTGG 78 117
147773 Coding 10 1931 CATCAGACGTGATGTTCCCT 85 118
147774 Coding 10 1956 TGGCTAGTTTCAATCCCCTT 84 119
147775 Coding 10 2002 CTGTCATGACTGCCCTTTAC 52 120
147776 Coding 10 2091 GCTTGAAGTTCATTGAGAAT 92 121
147777 Coding 10 2291 TTCCTGAGAAAGGAAGGAAC N.D. 122
147778 Coding 10 2331 TCAGATATACATTGGCTTCA 14 123
[00763] As shown in Table 3, SEQ ID Nos 71, 74, 76, 78, 81, 83, 84, 87, 88, 90, 101, 102, 103, 109, 111, 111, 114, 115, 116, 117, 118, 119, 120 and 121 demonstrated at least 50% inhibition of mouse ApoB expression in this assay and can be therefore . The target sites to which these sequences can be complementary can be herein referred to as "active sites" and can be therefore sites for targeting by compounds of provided herein,.
Example 18 - Antisense inhibition mouse ApoB expression by chimeric phosphorothioate oligonucleotides having 2'-MOE wings and a deoxy gap- Dose Response Study
[00764] In accordance with provided herein, a subset of the antisense oligonuclotides in Example 17 were further investigated in dose-response studies. Treatment doses were 50, 150 and 300 nM. The compounds were analyzed for their effect on mouse ApoB mRNA levels in primary hepatocytes cells by quantitative real-time PCR as described in other examples hereiN.D.ata can be averages from two experiments and can be shown in Table 4.
Table 4
Inhibition of mouse ApoB mRNA levels by chimeric phosphorothioate oligonucleotides having
2'-MOE wings and a deoxy gap

2.18 Example 19 - Western blot analysis of ApoB protein levels
[00765] Western blot analysis (immunoblot analysis) was carried out using standard methods. Cells were harvested 16-20 h after oligonucleotide treatment, washed once with PBS, suspended in Laemmli buffer (100 ul/well), boiled for 5 minutes and loaded on a 16% SDS-PAGE gel. Gels were run for 1. 5 hours at 150 V, and transferred to membrane for western blotting. Appropriate primary antibody directed to ApoB was used, with a radiolabeled or fluorescently labeled secondary antibody directed against the primary antibody species. Bands were visualized using a PHOSPHORIMAGER™ (Molecular Dynamics, Sunnyvale CA) or the ECL+ chemiluminescent detection system (Amersham Biosciences, Piscataway, NJ).
2.19 Example 20 - Effects of antisense inhibition of ApoB (ISIS 147764) in
C57BL/6 mice: Lean animals vs. High Fat Fed animals.
[00766] C57BL/6 mice, a strain reported to be susceptible to hyperlipidemia-induced atherosclerotic plaque formation were used in the following studies to evaluate antisense oligonucleotides as potential lipid lowering compounds in lean versus high fat fed mice.
[00767] Male C57BL/6 mice were divided into two matched groups; (1) wild-type control animals (lean animals) and (2) animals receiving a high fat diet (60% kcal fat). Control animals received saline treatment and were maintained on a normal rodent diet. After overnight fasting, mice from each group were dosed intraperitoneally every three days with saline or 50 mg/kg ISIS 147764 (SEQ ID No: 109) for six weeks. At study termination and forty eight hours after the final injections, animals were sacrificed and evaluated for target mRNA levels in liver, cholesterol and triglyceride levels, liver enzyme levels and serum glucose levels.
[00768] The results of the comparative studies can be shown in Table 5.
Table 5
Effects of ISIS 147764 treatment on ApoB mRNA, cholesterol, lipid, triglyceride, liver enzyme and glucose levels in lean and high fat mice.

[00769] It is evident from these data that treatment with ISIS 147764 lowered cholesterol as well as LDL and HDL lipoproteins and serum glucose in both lean and high fat mice and that the effects demonstrated can be, in fact, due to the inhibition of ApoB expression as supported by the decrease in mRNA levels. No significant changes in liver enzyme levels were observed, indicating that the antisense oligonucleotide was not toxic to either treatment group.
2.20 Example 21 - Effects of antisense inhibition of ApoB (ISIS 147764) on High Fat Fed Mice; 6 Week Timecourse Study
[00770] In accordance with provided herein, a 6-week timecourse study was performed to further investigate the effects of ISIS 147764 on lipid and glucose metabolism in high fat fed mice.
[00771] Male C57BL/6 mice (n=8) receiving a high fat diet (60% kcal fat) were evaluated over the course of 6 weeks for the effects of treatment with the antisense oligonucleotide, ISIS 147764. Control animals received saline treatment (50 mg/kg). A subset of animals received a daily oral dose (20 mg/kg) atorvastatin calcium (Lipitor®, Pfizer Inc. ). All mice, except atorvastatin-treated animals, were dosed intraperitoneally every three days (twice a week), after fasting overnight, with 5, 25, 50 mg/kg ISIS 147764 (SEQ ID No: 109) or saline (50 mg/kg) for six weeks. Serum cholesterol and lipoproteins were analyzed at 0, 2 and 6 week interim timepoints. At study termination, animals were sacrificed 48 hours after the final injections and evaluated for levels of target mRNA levels in liver, cholesterol, lipoprotein, triglyceride, liver enzyme (AST and ALT) and serum glucose levels as well as body, liver, spleen and fat pad weights.
2.21 Example 22 - Effects of antisense inhibition of ApoB (ISIS 147764) in high fat fed mice- mRNA expression in liver
[00772] Male C57BL/6 mice (n=8) receiving a high fat diet (60% kcal fat) were evaluated over the course of 6 weeks for the effects of ISIS 147764 on mRNA expression. Control animals received saline treatment (50 mg/kg). Mice were dosed intraperitoneally every three days (twice a week), after fasting overnight, with 5, 25, 50 mg/kg ISIS 147764 (SEQ ID No: 109) or saline (50 mg/kg) for six weeks. At study termination, animals were sacrificed 48 hours after the final injections and evaluated for levels of target mRNA levels in liver. ISIS 147764 showed a dose- response effect, reducing mRNA levels by 15, 75 and 88%> at doses of 5, 25 and 50 mg/kg, respectively.
[00773] Liver protein samples collected at the end of the treatment period were subjected to immunoblot analysis using an antibody directed to mouse ApoB protein (Gladstone Institute, San Francisco, CA). These data demonstrate that treatment with ISIS 147764 decreases ApoB protein expression in liver in a dose-dependent manner, in addition to reducing mRNA levels.
2.22 Example 23 - Effects of antisense inhibition of ApoB (ISIS 147764) on serum cholesterol and triglyceride levels
[00774] Male C57BL/6 mice (n=8) receiving a high fat diet (60%> kcal fat) were evaluated over the course of 6 weeks for the effects of ISIS 147764 on serum cholesterol and triglyceride levels. Control animals received saline treatment (50 mg/kg). Mice were dosed intraperitoneally
every three days (twice a week), after fasting overnight, with 5, 25, 50 mg/kg ISIS 147764 (SEQ ID No: 109) or saline (50 mg/kg) for six weeks.
[00775] Serum cholesterol levels were measured at 0, 2 and 6 weeks and this data is shown in Table 6. Values in the table can be expressed as percent inhibition and can be normalized to the saline control.
[00776] In addition to serum cholesterol, at study termination, animals were sacrificed 48 hours after the final injections and evaluated for triglyceride levels.
[00777] Mice treated with ISIS 147764 showed a reduction in both serum cholesterol (240 mg/dL for control animals and 225, 125 and 110 mg/dL for doses of 5, 25, and 50 mg/kg, respectively) and triglycerides (115 mg/dL for control animals and 125, 150 and 85 mg/dL for doses of 5, 25, and 50 mg/kg, respectively) to normal levels by study end. These data were also compared to the effects of atorvastatin calcium at an oral dose of 20 mg/kg which showed only a minimal decrease in serum cholesterol of 20 percent at study termination.
Table 6
Percent Inhibition of mouse ApoB cholesterol levels by ISIS 147764

2.23 Example 24 - Effects of antisense inhibition of ApoB (ISIS 147764) on lipoprotein levels
[00778] Male C57BL/6 mice (n=8) receiving a high fat diet (60% kcal fat) were evaluated over the course of 6 weeks for the effects of ISIS 147764 on lipoprotein (VLDL, LDL and HDL) levels. Control animals received saline treatment (50 mg/kg). Mice were dosed intraperitoneally every three days (twice a week), after fasting overnight, with 5, 25, 50 mg/kg ISIS 147764 (SEQ ID No: 109) or saline (50 mg/kg) for six weeks.
[00779] Lipoprotein levels were measured at 0, 2 and 6 weeks and this data is shown in Table 7. Values in the table can be expressed as percent inhibition and can be normalized to the saline control. Negative values indicate an observed increase in lipoprotein levels.
[00780] These data were also compared to the effects of atorvastatin calcium at a daily oral dose of 20 mg/kg at 0, 2 and 6 weeks.
[00781] These data demonstrate that at a dose of 50 mg/kg, ISIS 147764 is capable of lowering all categories of serum lipoproteins investigated to a greater extent than atorvastatin.
Table 7
Percent Inhibition of mouse ApoB lipoprotein levels by ISIS 147764 as compared to atorvastatin

2.24 Example 25 - Effects of antisense inhibition of ApoB (ISIS 147764) on serum AST and ALT levels
[00782] Male C57BL/6 mice (n=8) receiving a high fat diet (60% kcal fat) were evaluated over the course of 6 weeks for the effects of ISIS 147764 on liver enzyme (AST and ALT) levels. Increased levels of the liver enzymes ALT and AST indicate toxicity and liver damage. Control animals received saline treatment (50 mg/kg). Mice were dosed intraperitoneally every three days (twice a week), after fasting overnight, with 5, 25, 50 mg/kg ISIS 147764 (SEQ ID No: 109) or saline (50 mg/kg) for six weeks. AST and ALT levels were measured at 6 weeks.
[00783] Mice treated with ISIS 147764 showed no significant change in AST levels over the duration of the study compared to saline controls (105, 70 and 80 IU/L for doses of 5, 25 and 50 mg/kg, respectively compared to 65 IU/L for saline control). Mice treated with atorvastatin at a daily oral dose of 20 mg/kg had AST levels of 85 IU/L.
[00784] ALT levels were increased by all treatments with ISIS 147764 over the duration of the study compared to saline controls (50, 70 and 100 IU/L for doses of 5, 25 and 50 mg/kg,
respectively compared to 25 IU/L for saline control). Mice treated with atorvastatin at a daily oral dose of 20 mg/kg had AST levels of 40 IU/L.
2.25 Example 26 - Effects of antisense inhibition of ApoB (ISIS 147764) on serum glucose levels
[00785] Male C57BL/6 mice (n=8) receiving a high fat diet (60% kcal fat) were evaluated over the course of 6 weeks for the effects of ISIS 147764 on serum glucose levels. Control animals received saline treatment (50 mg/kg). Mice were dosed intraperitoneally every three days (twice a week), after fasting overnight, with 5, 25, 50 mg/kg ISIS 147764 (SEQ ID No: 109) or saline (50 mg/kg) for six weeks.
[00786] At study termination, animals were sacrificed 48 hours after the final injections and evaluated for serum glucose levels. ISIS 147764 showed a dose-response effect, reducing serum glucose levels to 225, 190 and 180 mg/dL at doses of 5, 25 and 50 mg/kg, respectively compared to the saline control of 300 mg/dL. Mice treated with atorvastatin at a daily oral dose of 20 mg/kg had serum glucose levels of 215 mg/dL. These data demonstrate that ISIS 147764 is capable of reducing serum glucose levels in high fat fed mice.
2.26 Example 27 - Effects of antisense inhibition of ApoB (ISIS 147764) on body, spleen, liver and fat pad weight
[00787] Male C57BL/6 mice (n=8) receiving a high fat diet (60% kcal fat) were evaluated over the course of 6 weeks for the effects of ISIS 147764 on body, spleen, liver and fat pad weight. Control animals received saline treatment (50 mg/kg). Mice were dosed intraperitoneally every three days (twice a week), after fasting overnight, with 5, 25, 50 mg/kg ISIS 147764 (SEQ ID No: 109) or saline (50 mg/kg) for six weeks.
[00788] At study termination, animals were sacrificed 48 hours after the final injections and body, spleen, liver and fat pad weights were measured. These data can be shown in Table 8. Values can be expressed as percent change in body weight or ogan weight compared to the saline -treated control animals. Data from mice treated with atorvastatin at a daily dose of 20 mg/kg can also be shown in the table. Negative values indicated a decrease in weight.
Table 8
Effects of antisense inhibition of mouse ApoB on body and organ weight

[00789] These data show a decrease in fat over the dosage range of ISIS 147764
counterbalanced by an increase in both spleen and liver weight with increased dose to give an overall decrease in total body weight.
2.27 Example 28 - Effects of antisense inhibition of ApoB (ISIS 147764) in B6. 129P-ApoetmlUnc knockout mice: Lean animals vs. High Fat Fed animals.
[00790] B6. 129P-ApoEtmlUnc knockout mice (herein referred to as ApoE knockout mice) obtained from The Jackson Laboratory (Bar Harbor, ME), can be homozygous for the
ApoetmlUnc mutation and show a marked increase in total plasma cholesterol levels that can be unaffected by age or sex. These animals present with fatty streaks in the proximal aorta at 3 months of age. These lesions increase with age and progress to lesions with less lipid but more elongated cells, typical of a more advanced stage of pre-atherosclerotic lesion.
[00791] The mutation in these mice resides in the apolipoprotein E (ApoE) gene. The primary role of the ApoE protein is to transport cholesterol and triglycerides throughout the body. It stabilizes lipoprotein structure, binds to the low density lipoprotein receptor (LDLR) and related proteins, and is present in a subclass of HDLs, providing them the ability to bind to LDLR. ApoE is expressed most abundantly in the liver and brain. Female B6. 129P- ApoetmlUnc knockout mice (ApoE knockout mice) were used in the following studies to evaluate antisense
oligonucleotides as potential lipid lowering compounds.
[00792] Female ApoE knockout mice ranged in age from 5 to 7 weeks and were placed on a normal diet for 2 weeks before study initiation. ApoE knockout mice were then fed ad libitum a 60% fat diet, with 0. 15% added cholesterol to induce dyslipidemia and obesity. Control animals
were maintained on a high-fat diet with no added cholesterol. After overnight fasting, mice from each group were dosed intraperitoneally every three days with saline, 50 mg/kg of a control antisense oligonucleotide (ISIS 29837; TCGATCTCCTTTTATGCCCG; SEQ ID NO. 124) or 5, 25 or 50 mg/kg ISIS 147764 (SEQ ID No: 109) for six weeks.
[00793] The control oligonucleotide is a chimeric oligonucleotides ("gapmers") 20 nucleotides in length, composed of a central "gap" region consisting of ten 2'-deoxynucleotides, which is flanked on both sides (5' and 3' directions) by five -nucleotide "wings". The wings can be composed of 2'-methoxyethyl (2'-MOE)nucleotides. The internucleoside (backbone) linkages can be phosphorothioate (P=S) throughout the oligonucleotide. All cytidine residues can be 5- methy lcytidines .
[00794] At study termination and forty eight hours after the final injections, animals were sacrificed and evaluated for target m NA levels in liver by RT-PCR methods verified by Northern Blot analysis, glucose levels, cholesterol and lipid levels by HPLC separation methods and triglyceride and liver enzyme levels (perfomed by LabCorp Preclinical Services; San Diego, CA). Data from ApoE knockout mice treated with atorvastatin at a daily dose of 20 mg/kg can also be shown in the table for comparison.
[00795] The results of the comparative studies can be shown in Table 9. Data can be normalized to saline controls.
Table 9
Effects of ISIS 147764 treatment on ApoB mRNA, cholesterol, glucose, lipid, triglyceride and liver enzyme levels in ApoE knockout mice.

[00796] It is evident from these data that treatment with ISIS 147764 lowered glucose and cholesterol as well as all lipoproteins investigated (HDL, LDL and VLDL) in ApoE knockout mice. Further, these decreases correlated with a decrease in both protein and RNA levels of ApoB, demonstrating an antisense mechanism of action. No significant changes in liver enzyme levels were observed, indicating that the antisense oligonucleotide was not toxic to either treatment group.
2.28 Example 29 - Antisense inhibition of human ApoB expression by chimeric phosphorothioate oligonucleotides having 2'-MOE wings and a deoxy gap:
Additional Oligonucleotides
[00797] In accordance with provided herein, another series of oligonucleotides was designed to target different regions of the human ApoB RNA, using published sequence (GenBank accession number NM 000384. 1, incorporated herein as SEQ ID NO: 3). The oligonucleotides can be shown in Table 10. "Target site" indicates the first (5 '-most) nucleotide number on the
particular target sequence to which the oligonucleotide binds. All compounds in Table 10 can be chimeric oligonucleotides ("gapmers") 20 nucleotides in length, composed of a central "gap" region consisting of ten 2'-deoxynucleotides, which is flanked on both sides (5' and 3' directions) by five -nucleotide "wings". The wings can be composed of 2'-methoxyethyl (2'- MOE)nucleotides. The internucleoside (backbone) linkages can be phosphorothioate (P=S) throughout the oligonucleotide. All cytidine residues can be 5-methylcytidines. The compounds were analyzed for their effect on human ApoB mRNA levels in HepG2 cells by quantitative realtime PCR as described in other examples hereiN.D.ata can be averages from two experiments in which HepG2 cells were treated with 150 nM of the compounds in Table 10. If present, "N.D." indicates "no data".
Table 10
Inhibition of human ApoB mRNA levels by chimeric phosphorothioate oligonucleotides having
2'-MOE wings and a deoxy gap

271003 coding 3 2299 GACACCATCAGGAACTTGAC 46 142
271004 coding 3 2459 GCTCCTCTCCCAAGATGCGG 10 143
271005 coding 3 2518 GGCACCCATCAGAAGCAGCT 32 144
271006 coding 3 2789 AGTCCGGAATGATGATGCCC 42 145
271007 coding 3 2919 CTGAGCAGCTTGACTGGTCT 26 146
271008 coding 3 3100 CCCGGTCAGCGGATAGTAGG 37 147
271010 exon : 3 3449 TGTCACAACTTAGGTGGCCC 57 148 exon
junction
271011 coding 3 3919 GTCTGGCAATCCCATGTTCT 51 149
271012 coding 3 4089 CCCACAGACTTGAAGTGGAG 55 150
271013 coding 3 4579 GAACTGCCCATCAATCTTGA 19 151
271014 coding 3 5146 CCCAGAGAGGCCAAGCTCTG 54 152
271015 coding 3 5189 TGTGTTCCCTGAAGCGGCCA 43 153
271016 coding 3 5269 ACCCAGAATCATGGCCTGAT 19 154
271017 coding 3 6049 GGTGCCTGTCTGCTCAGCTG 30 155
271018 coding 3 6520 ATGTGAAACTTGTCTCTCCC 44 156
271019 coding 3 6639 TATGTCTGCAGTTGAGATAG 15 157
271020 coding 3 6859 TTGAATCCAGGATGCAGTAC 35 158
271021 coding 3 7459 GAGTCTCTGAGTCACCTCAC 38 159
271022 coding 3 7819 GATAGAATATTGCTCTGCAA 100 160
271023 coding 3 7861 CCCTTGCTCTACCAATGCTT 44 161
271025 coding 3 8449 TCCATTCCCTATGTCAGCAT 16 162
271026 coding 3 8589 GACTCCTTCAGAGCCAGCGG 39 163
271027 coding 3 8629 CCCATGCTCCGTTCTCAGGT 26 164
271028 coding 3 8829 CGCAGGTCAGCCTGACTAGA 98 165
271030 coding 3 9119 CAGTTAGAACACTGTGGCCC 52 166
271031 coding 3 10159 CAGTGTGATGACACTTGATT 49 167
271032 coding 3 10301 CTGTGGCTAACTTCAATCCC 22 168
271033 coding 3 10349 CAGTACTGTTATGACTACCC 34 169
271034 coding 3 10699 CACTGAAGACCGTGTGCTCT 35 170
271035 coding 3 10811 TCGTACTGTGCTCCCAGAGG 23 171
271036 coding 3 10839 AAGAGGCCCTCTAGCTGTAA 95 172
271037 coding 3 11039 AAGACCCAGAATGAATCCGG 23 173
271038 coding 3 11779 GTCTACCTCAAAGCGTGCAG 29 174
271039 coding 3 11939 TAGAGGCTAACGTACCATCT 4 175
271041 coding 3 12149 CCATATCCATGCCCACGGTG 37 176
271042 coding 3 12265 AGTTTCCTCATCAGATTCCC 57 177
271043 coding 3 12380 CCCAGTGGTACTTGTTGACA 68 178
271044 coding 3 12526 CCCAGTGGTGCCACTGGCTG 22 179
271045 coding 3 12579 GTCAACAGTTCCTGGTACAG 19 180
271046 coding 3 12749 CCCTAGTGTATATCCCAGGT 61 181
271048 coding 3 13009 CTGAAGATTACGTAGCACCT 7 182
271049 coding 3 13299 GTCCAGCCAACTATACTTGG 54 183
271050 coding 3 13779 CCTGGAGCAAGCTTCATGTA 42 184
281586 exon : 3 229 TGGACAGACCAGGCTGACAT 80 185 exon
junction
ISIS # REGION TARGET TARGET SEQUENCE % INHIB SEQ ID SEQ ID SITE NO NO
281587 coding 3 269 ATGTGTACTTCCGGAGGTGC 77 186
281588 coding 3 389 TCTTCAGGATGAAGCTGCAG 80 187
281589 coding 3 449 TCAGCAAGGCTTTGCCCTCA 90 188
281590 coding 3 529 CTGCTTCCCTTCTGGAATGG 84 189
281591 coding 3 709 TGCCACATTGCCCTTCCTCG 90 190
281592 coding 3 829 GCTGATCAGAGTTGACAAGG 56 191
281593 coding 3 849 TACTGACAGGACTGGCTGCT 93 192
281594 coding 3 889 GATGGCTTCTGCCACATGCT 74 193
281595 coding 3 1059 GATGTGGATTTGGTGCTCTC 76 194
281596 coding 3 1199 TGACTGCTTCATCACTGAGG 77 195
281597 coding 3 1349 GGTAGGTGACCACATCTATC 36 196
281598 coding 3 1390 TCGCAGCTGCTGTGCTGAGG 70 197
281599 exon : 3 1589 TTCCAATGACCCGCAGAATC 74 198 exon
junction
281600 coding 3 1678 GATCATCAGTGATGGCTTTG 52 199
281601 coding 3 1699 AGCCTGGATGGCAGCTTTCT 83 200
281602 coding 3 1749 GTCTGAAGAAGAACCTCCTG 84 201
281603 coding 3 1829 TATCTGCCTGTGAAGGACTC 82 202
281604 coding 3 1919 CTGAGTTCAAGATATTGGCA 78 203
281605 exon : 3 2189 CTTCCAAGCCAATCTCGATG 82 204 exon
junction
281606 coding 3 2649 TGCAACTGTAATCCAGCTCC 86 205
281607 exon : 3 2729 CCAGTTCAGCCTGCATGTTG 84 206 exon
junction
281608 coding 3 2949 GTAGAGACCAAATGTAATGT 62 207
281609 coding 3 3059 CGTTGGAGTAAGCGCCTGAG 70 208
281610 exon : 3 3118 CAGCTCTAATCTGGTGTCCC 69 209 exon
junction
281611 coding 3 3189 CTGTCCTCTCTCTGGAGCTC 93 210
281612 coding 3 3289 CAAGGTCATACTCTGCCGAT 83 211
281613 coding 3 3488 GTATGGAAATAACACCCTTG 70 212
281614 coding 3 3579 TAAGCTGTAGCAGATGAGTC 63 213
281615 coding 3 4039 TAGATCTCTGGAGGATTTGC 81 214
281616 coding 3 4180 GTCTAGAACACCCAGGAGAG 66 215
281617 coding 3 4299 ACCACAGAGTCAGCCTTCAT 89 216
281618 coding 3 4511 AAGCAGACATCTGTGGTCCC 90 217
281619 coding 3 4660 CTCTCCATTGAGCCGGCCAG 96 218
281620 coding 3 4919 CCTGATATTCAGAACGCAGC 89 219
281621 coding 3 5009 CAGTGCCTAAGATGTCAGCA 53 220
281622 coding 3 5109 AGCACCAGGAGACTACACTT 88 221
281623 coding 3 5212 CCCATCCAGACTGAATTTTG 59 222
281624 coding 3 5562 GGTTCTAGCCGTAGTTTCCC 75 223
281625 coding 3 5589 AGGTTACCAGCCACATGCAG 94 224
281626 coding 3 5839 ATGTGCATCGATGGTCATGG 88 225
ISIS # REGION TARGET TARGET SEQUENCE % INHIB SEQ ID SEQ ID SITE NO NO
281627 coding 3 5869 CCAGAGAGCGAGTTTCCCAT 82 226
281628 coding 3 5979 CTAGACACGAGATGATGACT 81 227
281629 coding 3 6099 TCCAAGTCCTGGCTGTATTC 83 228
281630 coding 3 6144 CGTCCAGTAAGCTCCACGCC 82 229
281631 coding 3 6249 TCAACGGCATCTCTCATCTC 88 230
281632 coding 3 6759 TGATAGTGCTCATCAAGACT 75 231
281633 coding 3 6889 GATTCTGATTTGGTACTTAG 73 232
281634 coding 3 7149 CTCTCGATTAACTCATGGAC 81 233
281635 coding 3 7549 ATACACTGCAACTGTGGCCT 89 234
281636 coding 3 7779 GCAAGAGTCCACCAATCAGA 68 235
281637 coding 3 7929 AGAGCCTGAAGACTGACTTC 74 236
281638 coding 3 8929 TCCCTCATCTGAGAATCTGG 66 237
281640 coding 3 10240 CAGTGCATCAATGACAGATG 87 238
281641 coding 3 10619 CCGAACCCTTGACATCTCCT 72 239
281642 coding 3 10659 GCCTCACTAGCAATAGTTCC 59 240
281643 coding 3 10899 GACATTTGCCATGGAGAGAG 61 241
281644 coding 3 11209 CTGTCTCCTACCAATGCTGG 26 242
281645 exon : 3 11979 TCTGCACTGAAGTCACGGTG 78 243 exon
junction
281646 coding 3 12249 TCCCGGACCCTCAACTCAGT 76 244
281648 3'UTR 3 13958 GCAGGTCCAGTTCATATGTG 81 245
281649 3'UTR 3 14008 GCCATCCTTCTGAGTTCAGA 76 246
301012 exon : 3 3249 GCCTCAGTCTGCTTCGCACC 87 247 exon
junction
301013 5'UTR 3 3 CCCCGCAGGTCCCGGTGGGA 82 248
301014 5'UTR 3 6 CAGCCCCGCAGGTCCCGGTG 88 249
301015 5'UTR 3 23 CAACCGAGAAGGGCACTCAG 53 250
301016 5'UTR 3 35 CCTCAGCGGCAGCAACCGAG 62 251
301017 5'UTR 3 36 TCCTCAGCGGCAGCAACCGA 47 252
301018 5'UTR 3 37 CTCCTCAGCGGCAGCAACCG 45 253
301019 5'UTR 3 39 GGCTCCTCAGCGGCAGCAAC 70 254
301020 5'UTR 3 43 GGCGGGCTCCTCAGCGGCAG 85 255
301021 5'UTR 3 116 GGTCCATCGCCAGCTGCGGT 89 256
301022 Start 3 120 GGCGGGTCCATCGCCAGCTG 69 257
Codon
301023 Stop 3 13800 TAGAGGATGATAGTAAGTTC 69 258
Codon
301024 3'UTR 3 13824 AAATGAAGATTTCTTTTAAA 5 259
301025 3'UTR 3 13854 TATGTGAAAGTTCAATTGGA 76 260
301026 3'UTR 3 13882 ATATAGGCAGTTTGAATTTT 57 261
301027 3'UTR 3 13903 GCTCACTGTATGGTTTTATC 89 262
301028 3'UTR 3 13904 GGCTCACTGTATGGTTTTAT 93 263
301029 3'UTR 3 13908 GGCTGGCTCACTGTATGGTT 90 264
301030 3'UTR 3 13909 AGGCTGGCTCACTGTATGGT 90 265
301031 3'UTR 3 13910 AAGGCTGGCTCACTGTATGG 90 266
301032 3'UTR 3 13917 CTACTGCAAGGCTGGCTCAC 63 267
ISIS # REGION TARGET TARGET SEQUENCE % INHIB SEQ ID
SEQ ID SITE NO NO
301033 3'UTR 3 13922 ACTGCCTACTGCAAGGCTGG 77 268
301034 3'UTR 3 13934 TGCTTATAGTCTACTGCCTA 88 269
301035 3'UTR 3 13937 TTCTGCTTATAGTCTACTGC 82 270
301036 3'UTR 3 13964 TTTGGTGCAGGTCCAGTTCA 88 271
301037 3'UTR 3 13968 CAGCTTTGGTGCAGGTCCAG 90 272
301038 3'UTR 3 13970 GCCAGCTTTGGTGCAGGTCC 86 273
301039 3'UTR 3 13974 TGGTGCCAGCTTTGGTGCAG 73 274
301040 3'UTR 3 13978 GCCCTGGTGCCAGCTTTGGT 74 275
301041 3'UTR 3 13997 GAGTTCAGAGACCTTCCGAG 85 276
301042 3'UTR 3 14012 AAATGCCATCCTTCTGAGTT 81 277
301043 3'UTR 3 14014 AAAAATGCCATCCTTCTGAG 81 278
301044 3'UTR 3 14049 AAAATAACTCAGATCCTGAT 76 279
301045 3'UTR 3 14052 AGCAAAATAACTCAGATCCT 90 280
301046 3'UTR 3 14057 AGTTTAGCAAAATAACTCAG 80 281
301047 3'UTR 3 14064 TCCCCCAAGTTTAGCAAAAT 56 282
301048 3'UTR 3 14071 TTCCTCCTCCCCCAAGTTTA 67 283
301217 3'UTR 3 14087 AGACTCCATTTATTTGTTCC 81 284
2.29 Example 30 - Antisense inhibition of ApoB - Gene walk
[00798] In accordance with provided herein, a "gene walk" was conducted in which another series of oligonucleotides was designed to target the regions of the human ApoB RNA (GenBank accession number NM 000384. 1, incorporated herein as SEQ ID NO: 3) which can be near the target site of SEQ ID Nos 224 or 247. The oligonucleotides can be shown in Table 11. "Target site" indicates the first (5 '-most) nucleotide number on the particular target sequence to which the oligonucleotide binds. All compounds in Table 11 can be chimeric oligonucleotides
("gapmers") 20 nucleotides in length, composed of a central "gap" region consisting of ten 2'- deoxynucleotides, which is flanked on both sides (5 ' and 3 ' directions) by five-nucleotide "wings". The wings can be composed of 2'-methoxyethyl (2'-MOE)nucleotides. The
internucleoside (backbone) linkages can be phosphorothioate (P=S) throughout the
oligonucleotide. All cytidine residues can be 5-methylcytidines. The compounds were analyzed for their effect on human ApoB mRNA levels in HepG2 cells by quantitative real-time PCR as described in other examples herein. Treatment doses were 50 nM and 150 nM and can be indicated in Table 11. Data can be averages from two experiments. If present, "N.D." indicates "no data".
Table 11
Inhibition of human ApoB mRNA levels by chimeric phosphorothioate oligonucleotides having
2'-MOE wings and a deoxy gap - Gene walk

308611 coding 3 5586 TTACCAGCCACATGCAGCTT 95 59 307
308612 coding 3 5588 GGTTACCAGCCACATGCAGC 90 75 308
308613 coding 3 5590 TAGGTTACCAGCCACATGCA 87 43 309
308614 coding 3 5592 TTTAGGTTACCAGCCACATG 92 74 310
308615 coding 3 5594 CTTTTAGGTTACCAGCCACA 85 45 311
308616 coding 3 5596 TCCTTTTAGGTTACCAGCCA 81 39 312
308617 coding 3 5598 GCTCCTTTTAGGTTACCAGC 87 77 313
308618 coding 3 5600 AGGCTCCTTTTAGGTTACCA 77 61 314
308619 coding 3 5602 GTAGGCTCCTTTTAGGTTAC 74 69 315
308620 coding 3 5604 TGGTAGGCTCCTTTTAGGTT 88 69 316
308621 coding 3 5606 TTTGGTAGGCTCCTTTTAGG 91 56 317
[00799] As shown in Tables 10 and 11, SEQ ID Nos 124, 128, 129, 132, 133, 134, 138, 140,
141, 142, 144, 145, 147, 148, 149, 150, 152, 153, 155, 156, 158, 159, 160, 161, 163, 165, 166,
167, 169, 170, 172, 176, 177, 178, 181, 183, 184, 185, 186, 187, 188, 189, 190, 191, 192, 193,
194, 195, 196, 197, 198, 199, 200, 201, 202, 203, 204, 205, 206, 207, 208, 209, 210, 211, 212,
213, 214, 215, 216, 217, 218, 219, 220, 221, 222, 223, 224, 225, 226, 227, 228, 229, 230, 231,
232, 233, 234, 235, 236, 237, 238, 239, 240, 241, 243, 244, 245, 246, 247, 248, 249, 250, 251,
252, 253, 254, 255, 256, 257, 258, 260, 261, 262, 263, 264, 265, 266, 267, 268, 269, 270, 271,
272, 273, 274, 275, 276, 277, 278, 279, 280, 281, 282, 283, 284, 285, 286, 287, 288, 289, 290,
291, 292, 293, 294, 295, 296, 297, 298, 299, 300, 301, 302, 303, 304, 305, 306, 307, 308, 309,
310, 311, 312, 313, 314, 315, 316, and 317 demonstrated at least 30% inhibition of human ApoB expression in this assay and can be therefore . More can be SEQ ID Nos 224, 247, and 262. The target regions to which these sequences can be complementary can be herein referred to as " target segments" and can be therefore for targeting by compounds of provided herein,. These target segments can be shown in Table 18. The sequences represent the reverse complement of the antisense compounds shown in Tables 10 and 11. "Target site" indicates the first (5 '-most) nucleotide number on the particular target nucleic acid to which the oligonucleotide binds. Also shown in Table 18 is the species in which each of the target segments was found.
2.30 Example 31 - Antisense inhibition of human ApoB expression by chimeric phosphorothioate oligonucleotides having 2'-MOE wings and a deoxy gap:
Targeting GenBank Accession number M14162. 1
[00800] In accordance with provided herein, another series of oligonucleotides was designed to target different regions of the human ApoB RNA, using published sequence (GenBank accession number M 14162. 1, incorporated herein as SEQ ID NO: 318). The oligonucleotides can be shown in Table 12. "Target site" indicates the first (5 '-most) nucleotide number on the particular target sequence to which the oligonucleotide binds. All compounds in Table 12 can be chimeric oligonucleotides ("gapmers") 20 nucleotides in length, composed of a central "gap" region consisting of ten 2'-deoxynucleotides, which is flanked on both sides (5' and 3' directions) by five -nucleotide "wings". The wings can be composed of 2'-methoxyethyl (2'- MOE)nucleotides. The internucleoside (backbone) linkages can be phosphorothioate (P=S) throughout the oligonucleotide. All cytidine residues can be 5-methylcytidines. The compounds were analyzed for their effect on human ApoB mRNA levels in HepG2 cells by quantitative realtime PCR as described in other examples hereiN.D.ata can be averages from two experiments in which HepG2 cells were treated with 150 nM of the compounds in Table 12. If present, "N.D." indicates "no data".
Table 12
Inhibition of human ApoB mRNA levels by chimeric phosphorothioate oligonucleotides having
2'-MOE wings and a deoxy gap

2.31 Example 32 - Antisense Inhibition of human ApoB - Gene walk targeting GenBank Accession number M14162. 1
[00801] In accordance with provided herein, a "gene walk" was conducted in which another series of oligonucleotides was designed to target the regions of the human ApoB RNA (GenBank accession number M 14162. 1, incorporated herein as SEQ ID NO: 318) which can be near the target site of SEQ ID NO: 319. The oligonucleotides can be shown in Table 13. "Target site" indicates the first (5 '-most) nucleotide number on the particular target sequence to which the
oligonucleotide binds. All compounds in Table 13 can be chimeric oligonucleotides ("gapmers") 20 nucleotides in length, composed of a central "gap" region consisting of ten 2'- deoxynucleotides, which is flanked on both sides (5 ' and 3 ' directions) by five-nucleotide "wings". The wings can be composed of 2'-methoxyethyl (2'-MOE)nucleotides. The
internucleoside (backbone) linkages can be phosphorothioate (P=S) throughout the
oligonucleotide. All cytidine residues can be 5-methylcytidines. The compounds were analyzed for their effect on human ApoB mRNA levels in HepG2 cells by quantitative real-time PCR as described in other examples herein. Treatment doses were 50 nM and 150 nM and can be indicated in Table 13. Data can be averages from two experiments. If present, "N.D." indicates "no data".
Table 13
Inhibition of human ApoB mRNA levels by chimeric phosphorothioate oligonucleotides having
2'-MOE wings and a deoxy gap

[00802] As shown in Tables 12 and 13, SEQ ID Nos 319, 323, 324, 325, 326, 327, 328, 329, 330, 331, 332, and 333 demonstrated at least 30% inhibition of human ApoB expression in this assay and can be therefore . More is SEQ ID NO: 319. The target regions to which these sequences can be complementary can be herein referred to as " target segments" and can be therefore for targeting by compounds of provided herein,. These target segments can be shown in Table 18. The sequences represent the reverse complement of the antisense compounds shown in Tables 12 and 13. "Target site" indicates the first (5 '-most) nucleotide number on the particular target nucleic acid to which the oligonucleotide binds. Also shown in Table 18 is the species in which each of the target segments was found.
2.32 Example 33 - Antisense inhibition of human ApoB expression by chimeric phosphorothioate oligonucleotides having 2'-MOE wings and a deoxy gap - Targeting the Genomic sequence
[00803] In accordance with provided herein, another series of oligonucleotides was designed to target different regions of the human ApoB RNA, using published sequence (the complement of nucleotides 39835 to 83279 of the sequence with GenBank accession number NT 022227. 9, representing a genomic sequence, incorporated herein as SEQ ID NO: 334). The
oligonucleotides can be shown in Table 14. "Target site" indicates the first (5 '-most) nucleotide number on the particular target sequence to which the oligonucleotide binds. All compounds in Table 14 can be chimeric oligonucleotides ("gapmers") 20 nucleotides in length, composed of a central "gap" region consisting of ten 2'-deoxynucleotides, which is flanked on both sides (5' and 3' directions) by five-nucleotide "wings". The wings can be composed of 2'-methoxyethyl (2'-MOE)nucleotides. The internucleoside (backbone) linkages can be phosphorothioate (P=S) throughout the oligonucleotide. All cytidine residues can be 5-methylcytidines. The compounds were analyzed for their effect on human ApoB mRNA levels in HepG2 cells by quantitative realtime PCR as described in other examples hereiN.D.ata can be averages from two experiments in which HepG2 cells were treated with 150 nM of the oligonucleotides in Table 14. If present, "N.D." indicates "no data".
Table 14
Inhibition of human ApoB mRNA levels by chimeric phosphorothioate oligonucleotides having
2'-MOE wings and a deoxy gap

j unction
301055 intron 334 2722 TGGCTCATGTCTACCATATT 49 341
301056 intron 334 2791 CAGTTGAAATGCAGCTAATG 35 342
301057 intron 334 3045 TGCAGACTAGGAGTGAAAGT 30 343
301058 intron 334 3117 AGGAGGATGTCCTTTTATTG 27 344
301059 intron 334 3290 ATCAGAGCACCAAAGGGAAT 12 345
301060 intron : 334 3381 CCAGCTCAACCTGAGAATTC 17 346 exon
j unction
301061 exon : 334 3527 CATGACTTACCTGGACATGG 52 347 intron
j unction
301062 intron 334 3566 CCTCAGCGGACACACACACA 21 348
301063 intron 334 3603 GTCACATCCGTGCCTGGTGC 41 349
301064 intron 334 3864 CAGTGCCTCTGGGACCCCAC 60 350
301065 intron 334 3990 AGCTGCAGTGGCCGATCAGC 50 351
301066 intron 334 4251 GACCTCCCCAGCCACGTGGA 61 352
301067 intron 334 4853 TCTGATCACCATACATTACA 45 353
301068 intron 334 5023 ATTTCCCACTGGGTACTCTC 44 354
301069 intron 334 5055 GGCTGAAGCCCATGCTGACT 44 355
301070 intron 334 5091 GTTGGACAGTCATTCTTTTG 38 356
301071 intron 334 5096 CACTTGTTGGACAGTCATTC 48 357
301072 intron 334 5301 ATTTTAAATTACAGTAGATA 43 358
301073 intron 334 5780 CTGTTCTCCACCCATATCAG 37 359
301074 intron : 334 6353 GAGCTCATACCTGTCCCAGA 75 360 exon
j unction
301075 intron 334 6534 TTCAAGGGCCACTGCTATCA 52 361
301076 intron 334 6641 CCAGTATTTCACGCCAATCC 36 362
301077 intron 334 6661 GGCAGGAGGAACCTCGGGCA 55 363
301078 intron 334 6721 TTTTAAAATTAGACCCAACC 22 364
301079 intron 334 6727 TGACTGTTTTAAAATTAGAC 20 365
301080 intron 334 6788 CCCAGCAAACACAGGTGAAG 25 366
301081 intron 334 7059 GAGTGTGGTCTTGCTAGTGC 46 367
301082 intron 334 7066 CTATGCAGAGTGTGGTCTTG 41 368
301083 intron 334 7189 AGAAGATGCAACCACATGTA 29 369
301084 intron : 334 7209 ACACGGTATCCTATGGAGGA 49 370 exon
j unction
301085 exon : 334 7365 TGGGACTTACCATGCCTTTG 11 371 intron
j unction
301086 intron 334 7702 GGTTTTGCTGCCCTACATCC 30 372
301087 intron 334 7736 ACAAGGAGTCCTTGTGCAGA 40 373
301088 intron 334 8006 ATGTTCACTGAGACAGGCTG 41 374
301089 intron 334 8215 GAAGGTCCATGGTTCATCTG 0 375
301090 intron 334 8239 ATTAGACTGGAAGCATCCTG 39 376
ISIS # REGION TARGET TARGET SEQUENCE % SEQ ID SEQ ID SITE INHIB NO NO
301091 intron 334 8738 GAGATTGGAGACGAGCATTT 35 377
301092 exon : 334 8881 CATGACC ACTTG AGGAGA 22 378 intron
j unction
301093 intron 334 9208 TGGATTTGGATACACAAGTT 42 379
301094 intron 334 9244 ACTCAATATATATTCATTGA 22 380
301095 intron 334 9545 CAAGGAAGCACACCATGTCA 38 381
301096 intron : 334 9563 ATACTTATTCCTGGTAACCA 24 382 exon
j unction
301097 intron 334 9770 GGTAGCCAGAACACCAGTGT 50 383
301098 intron 334 9776 ACTAGAGGTAGCCAGAACAC 34 384
301099 intron 334 10149 ACCACCTGACATCACAGGTT 24 385
301100 intron 334 10341 TACTGTGACCTATGCCAGGA 55 386
301101 intron 334 10467 GGAGGTGCTACTGTTGACAT 42 387
301102 intron 334 10522 TCCAGACTTGTCTGAGTCTA 47 388
301103 intron 334 10547 TCTAAGAGGTAGAGCTAAAG 7 389
301104 intron 334 10587 CCAGAGATGAGCAACTTAGG 38 390
301105 intron 334 10675 GGCCATGTAAATTGCTCATC 7 391
301106 intron 334 10831 AAAGAAACTATCCTGTATTC 12 392
301107 intron : 334 10946 TTCTTAGTACCTGGAAGATG 23 393 exon
j unction
301108 exon : 334 11166 CATTAGATACCTGGACACCT 29 394 intron
j unction
301109 intron 334 11337 GTTTCATGGAACTCAGCGCA 44 395
301110 intron 334 11457 CTGGAGAGCACCTGCAATAG 35 396
301111 intron 334 11521 TGAAGGGTAGAGAAATCATA 9 397
301112 exon : 334 12111 GGAAACTCACTTGTTGACCG 25 398 intron
j unction
301113 intron 334 12155 AGGTGCAAGATGTTCCTCTG 46 399
301114 intron 334 12162 TGCACAGAGGTGCAAGATGT 16 400
301115 intron 334 12221 CACAAGAGTAAGGAGCAGAG 39 401
301116 intron 334 12987 GATGGATGGTGAGAAATTAC 33 402
301117 intron 334 13025 TAGACAATTGAGACTCAGAA 39 403
301118 intron 334 13057 ATGTGCACACAAGGACATAG 33 404
301119 intron 334 13634 ACATACAAATGGCAATAGGC 33 405
301120 intron 334 13673 TAGGCAAAGGACATGAATAG 30 406
301121 coding 334 14448 TTATGATAGCTACAGAATAA 29 407
301122 exon : 334 14567 CTGAGATTACCCGCAGAATC 32 408 intron
j unction
301123 intron 334 14587 GATGTATGTCATATAAAAGA 26 409
301124 intron : 334 14680 TTTCCAATGACCTGCATTGA 48 410 exon
j unction
ISIS # REGION TARGET TARGET SEQUENCE % SEQ ID SEQ ID SITE INHIB NO NO
301125 intron 334 15444 AGGGATGGTCAATCTGGTAG 57 411
301126 intron 334 15562 GGCTAATAAATAGGGTAGTT 22 412
301127 intron 334 15757 TCCTAGAGCACTATCAAGTA 41 413
301128 intron : 334 15926 CCTCCTGGTCCTGCAGTCAA 56 414 exon
j unction
301129 intron 334 16245 CATTTGCACAAGTGTTTGTT 35 415
301130 intron 334 16363 CTGACACACCATGTTATTAT 10 416
301131 intron : 334 16399 CTTTTTCAGAC AGA AAGA 0 417 exon
j unction
301132 exon : 334 16637 TCACACT ACCTCGATGAGG 29 418 intron
j unction
301133 intron 334 17471 AAGAAAATGGCATCAGGTTT 13 419
301134 intron : 334 17500 CCAAGCCAATCTGAGAAAGA 25 420 exon
j unction
301135 exon : 334 17677 AAATACACACCTGCTCATGT 20 421 intron
j unction
301136 exon : 334 17683 CTTCACAAATACACACCTGC 20 422 intron
j unction
301137 intron 334 18519 AGTGGAAGTTTGGTCTCATT 41 423
301138 intron 334 18532 TTGCTAGCTTCAAAGTGGAA 44 424
301139 intron 334 18586 TCAAGAATAAGCTCCAGATC 41 425
301140 intron 334 18697 GCATACAAGTCACATGAGGT 34 426
301141 intron 334 18969 TACAAGGTGTTTCTTAAGAA 38 427
301142 intron 334 19250 ATGCAGCCAGGATGGGCCTA 54 428
301143 intron : 334 19340 TTACCATATCCTGAGAGTTT 55 429 exon
j unction
301144 intron 334 19802 GCAAAGGTAGAGGAAGGTAT 32 430
301145 intron 334 19813 AAGGACCTTCAGCAAAGGTA 36 431
301146 intron 334 20253 CATAGGAGTACATTTATATA 23 432
301147 intron 334 20398 ATTATGATAAAATCAATTTT 19 433
301148 intron 334 20567 AGAAATTTCACTAGATAGAT 31 434
301149 intron 334 20647 AGCATATTTTGATGAGCTGA 44 435
301150 intron 334 20660 GAAAGGAAGGACTAGCATAT 39 436
301151 intron : 334 20772 CCTCTCCAATCTGTAGACCC 28 437 exon
j unction
301152 intron 334 21316 CTGGATAACTCAGACCTTTG 40 438
301153 intron 334 21407 AGTCAGAAAACAACCTATTC 11 439
301154 intron : 334 21422 CAGCCTGCATCTATAAGTCA 31 440 exon
j unction
301155 exon : 334 21634 AAAGAATTACCCTCCACTGA 33 441
ISIS # REGION TARGET TARGET SEQUENCE % SEQ ID SEQ ID SITE INHIB NO NO
intron
j unction
301156 intron 334 21664 TCTTTCAAACTGGCTAGGCA 39 442
301157 intron 334 21700 GCCTGGCAAAATTCTGCAGG 37 443
301158 intron 334 22032 CTACCTCAAATCAATATGTT 28 444
301159 intron 334 22048 TGCTTTACCTACCTAGCTAC 36 445
301160 intron 334 22551 ACCTTGTGTGTCTCACTCAA 49 446
301161 intron 334 22694 ATGCATTCCCTGACTAGCAC 34 447
301162 intron 334 22866 CATCTCTGAGCCCCTTACCA 24 448
301163 intron 334 22903 GCTGGGCATGCTCTCTCCCC 51 449
301164 intron 334 22912 GCTTTCGCAGCTGGGCATGC 55 450
301165 intron 334 23137 ACTCCTTTCTATACCTGGCT 47 451
301166 intron 334 23170 ATTCTGCCTCTTAGAAAGTT 38 452
301167 intron 334 23402 CCAAGCCTCTTTACTGGGCT 29 453
301168 intron 334 23882 CACTCATGACCAGACTAAGA 35 454
301169 intron 334 23911 ACCTCCCAGAAGCCTTCCAT 22 455
301170 intron 334 24184 TTCATATGAAATCTCCTACT 40 456
301171 intron 334 24425 TATTTAATTTACTGAGAAAC 7 457
301172 intron : 334 24559 TAATGTGTTGCTGGTGAAGA 35 458 exon
j unction
301173 exon : 334 24742 CATCTCTAACCTGGTGTCCC 21 459 intron
j unction
301174 intron 334 24800 GTGCCATGCTAGGTGGCCAT 37 460
301175 intron 334 24957 AGCAAATTGGGATCTGTGCT 29 461
301176 intron 334 24991 TCTGGAGGCTCAGAAACATG 57 462
301177 intron 334 25067 TGAAGACAGGGAGCCACCTA 40 463
301178 intron 334 25152 AGGATTCCCAAGACTTTGGA 38 464
301179 intron : 334 25351 CAGCTCTAATCTAAAGACAT 22 465 exon
j unction
301180 exon : 334 25473 GAATACTCACCTTCTGCTTG 6 466 intron
j unction
301181 intron 334 26047 ATCTCTCTGTCCTCATCTTC 28 467
301182 intron 334 26749 CCAACTCCCCCTTTCTTTGT 37 468
301183 intron 334 26841 TCTGGGCCAGGAAGACACGA 68 469
301184 intron 334 27210 TATTGTGTGCTGGGCACTGC 52 470
301185 intron : 334 27815 TGCTTCGCACCTGGACGAGT 51 471 exon
j unction
301186 exon : 334 28026 CCTTCTTTACCTTAGGTGGC 37 472 intron
j unction
301187 intron 334 28145 GCTCTCTCTGCCACTCTGAT 47 473
301188 intron 334 28769 AACTTCTAAAGCCAACATTC 27 474
301189 intron : 334 28919 TGTGTCACAACTATGGTAAA 63 475 exon
ISIS # REGION TARGET TARGET SEQUENCE % SEQ ID SEQ ID SITE INHIB NO NO
j unction
301190 exon : 334 29095 AGACACATACCATAATGCCA 22 476 intron
j unction
301191 intron : 334 29204 TTCTCTTCATCTGAAAATAC 21 477 exon
j unction
301192 intron 334 29440 TGAGGATGTAATTAGCACTT 27 478
301193 intron : 334 29871 AGCTCATTGCCTACAAAATG 31 479 exon
j unction
301194 intron 334 30181 GTTCTCATGTTTACTAATGC 40 480
301195 intron 334 30465 GAATTGAGACAACTTGATTT 26 481
301196 intron : 334 30931 CCGGCCATCGCTGAAATGAA 54 482 exon
j unction
301197 exon : 334 31305 CATAGCTCACCTTGCACATT 28 483 intron
j unction
301198 intron 334 31325 CGGTGCACCCTTTACCTGAG 28 484
301199 intron : 334 31813 TCTCCAGATCCTAACATAAA 19 485 exon
j unction
301200 intron 334 39562 TTGAATGACACTAGATTTTC 37 486
301201 intron 334 39591 AAAATCCATTTTCTTTAAAG 12 487
301202 intron 334 39654 CAGCTCACACTTATTTTAAA 7 488
301203 intron : 334 39789 GTTCCCAAAACTGTATAGGA 36 489 exon
j unction
301204 exon : 334 39904 AGCTCCATACTGAAGTCCTT 37 490 intron
j unction
301205 intron 334 39916 CAATTCAATAAAAGCTCCAT 31 491
301206 intron 334 39938 GTTTTCAAAAGGTATAAGGT 28 492
301207 intron : 334 40012 TTCCCATTCCCTGAAAGCAG 13 493 exon
j unction
301208 exon : 334 40196 TGGTATTTACCTGAGGGCTG 21 494 intron
j unction
301209 intron 334 40412 ATAAATAATAGTGCTGATGG 39 495
301210 intron 334 40483 CTATGGCTGAGCTTGCCTAT 33 496
301211 intron 334 40505 CTCTCTGAAAAATATACCCT 17 497
301212 intron 334 40576 TTGATGTATCTCATCTAGCA 41 498
301213 intron 334 40658 TAGAACCATGTTTGGTCTTC 35 499
301214 intron 334 40935 TTTCTCTTTATCACATGCCC 29 500
ISIS # REGION TARGET TARGET SEQUENCE % SEQ ID
SEQ ID SITE INHIB NO NO
301215 intron 334 41066 A AG ACAC AAAACTTCA 1 501
301216 intron : 334 41130 CTGGAGAGGAC AAACAGAG 49 502
exon
j unction
[00804] As shown in Table 14, SEQ ID Nos 335, 339, 341, 342, 343, 347, 349, 350, 351, 352,
353, 354, 355, 356, 357, 358, 359, 360, 361, 362, 363, 367, 368, 370, 372, 373, 374, 376, 377,
379, 381, 383, 384, 386, 387, 388, 390, 395, 396, 399, 401, 402, 403, 404, 405, 406, 408, 410,
411, 413, 414, 415, 423, 424, 425, 426, 427, 428, 429, 430, 431, 434, 435, 436, 438, 440, 441,
442, 443, 445, 446, 447, 449, 450, 451, 452, 454, 456, 458, 460, 462, 463, 464, 468, 469, 470,
471, 472, 473, 475, 479, 480, 482, 486, 489, 490, 491, 495, 496, 498, 499, and 502 demonstrated at least 30% inhibition of human ApoB expression in this assay and can be therefore . The target regions to which these sequences can be complementary can be herein referred to as " target segments" and can be therefore for targeting by compounds of provided herein,. These target segments can be shown in Table 18. The sequences represent the reverse complement of the antisense compounds shown in Table 14. "Target site" indicates the first (5 '-most) nucleotide number on the particular target nucleic acid to which the oligonucleotide binds. Also shown in
Table 18 is the species in which each of the target segments was found.
2.33 Example 34 - Antisense inhibition of human ApoB expression by chimeric phosphorothioate oligonucleotides having 2'-MOE wings and a deoxy gap - Targeting GenBank accession number AI249040. 1
[00805] In accordance with provided herein, another series of oligonucleotides was designed to target different regions of the human ApoB RNA, using published sequence (the complement of the sequence with GenBank accession number AI249040. 1, incorporated herein as SEQ ID NO: 503). The oligonucleotides can be shown in Table 15. "Target site" indicates the first (5'- most) nucleotide number on the particular target sequence to which the oligonucleotide binds. All compounds in Table 15 can be chimeric oligonucleotides ("gapmers") 20 nucleotides in length, composed of a central "gap" region consisting of ten 2'-deoxynucleotides, which is flanked on both sides (5' and 3' directions) by five -nucleotide "wings". The wings can be composed of 2'-methoxyethyl (2'-MOE)nucleotides. The internucleoside (backbone) linkages can be phosphorothioate (P=S) throughout the oligonucleotide. All cytidine residues can be 5-
methylcytidines. The compounds were analyzed for their effect on human ApoB mRNA levels in HepG2 cells by quantitative real-time PCR as described in other examples hereiN.D.ata can be averages from two experiments in which HepG2 cells were treated with 150 nM of the oligonucleotides in Table 15. If present, "N.D." indicates "no data".
Table 15
Inhibition of human ApoB mRNA levels by chimeric phosphorothioate oligonucleotides having
2'-MOE wings and a deoxy gap

[00806] As shown in Table 15, SEQ ID Nos 505, 506, 507, 510, and 512 demonstrated at least
30% inhibition of human ApoB expression in this assay and can be therefore . The target regions to which these sequences can be complementary can be herein referred to as " target segments" and can be therefore for targeting by compounds of provided herein,. These target segments can be shown in Table 18. The sequences represent the reverse complement of the antisense compounds shown in Table 15. "Target site" indicates the first (5 '-most) nucleotide number on the particular target nucleic acid to which the oligonucleotide binds. Also shown in Table 18 is the species in which each of the target segments was found.
2.34 Example 35 - Antisense inhibition of human ApoB expression by chimeric phosphorothioate oligonucleotides having 2'-MOE wings and a deoxy gap - Variation in position of the gap
[00807] In accordance with provided herein, a series of antisense compounds was designed to target different regions of the human ApoB RNA, using published sequences (GenBank accession number NM 000384. 1, incorporated herein as SEQ ID NO: 3). The compounds can be shown in Table 16. "Target site" indicates the first (5 '-most) nucleotide number on the particular target sequence to which the compound binds. All compounds in Table 16 can be
chimeric oligonucleotides ("gapmers") 20 nucleotides in length. The "gap" region consists of 2'- deoxynucleotides, which is flanked on one or both sides (5' and 3' directions) by "wings" composed of 2'-methoxyethyl (2'-MOE)nucleotides. The number of 2'-MOE nucleotides on either side of the gap varies such that the total number of 2'-MOE nucleotides always equals 10 and the total length of the chimeric oligonucleotide is 20 nucleotides. The exact structure of each oligonucleotide is designated in Table 16 as the "gap structure" and the 2'-deoxynucleotides can be in bold type. A designation of 8-10-2, for instance, indicates that the first (5'-most) 8 nucleotides and the last (3'-most) 2 nucleotides can be 2'-MOE nucleotides and the 10 nucleotides in the gap can be 2'-deoxynucleotides. The internucleoside (backbone) linkages can be phosphorothioate (P=S) throughout the oligonucleotide. All cytidine residues can be 5- methylcytidines. The compounds were analyzed for their effect on human ApoB mRNA levels by quantitative real-time PCR as described in other examples hereiN.D.ata, shown in Table 16, can be averages from three experiments in which HepG2 cells were treated with the antisense oligonucleotides of provided herein, at doses of 50 nM and 150 nM. If present, "N.D." indicates "no data".
Table 16
Inhibition of human ApoB mRNA levels by chimeric phosphorothioate oligonucleotides having
2'-MOE wings and a variable deoxy gap

308656 3 3249 GCCTCAGTCTGCTTCGCACC 97 37 8 - 10 -2 247
308658 3 5589 AGGTTACCAGCCACATGCAG 78 86 9 - 10 - 1 224
308659 3 3249 GCCTCAGTCTGCTTCGCACC 93 70 9 - 10 - 1 247
308660 3 3254 TGGTAGCCTCAGTCTGCTTC 92 72 2 - 10 - 8 514
308662 3 3254 TGGTAGCCTCAGTCTGCTTC 83 76 8 - 10 -2 514
[00808] As shown in Table 16, SEQ ID Nos 224, 247, and 514 demonstrated at least 30% inhibition of human ApoB expression in this assay at both doses. These data suggest that the oligonucleotides can be effective with a number of variations in the gap placement. The target regions to which these sequences can be complementary can be herein referred to as " target segments" and can be therefore for targeting by compounds of provided herein,. These target segments can be shown in Table 18. The sequences represent the reverse complement of the antisense compounds shown in Table 16. "Target site" indicates the first (5 '-most) nucleotide number on the particular target nucleic acid to which the oligonucleotide binds. Also shown in
Table 18 is the species in which each of the target segments was found.
2.35 Example 36 - Antisense inhibition of human ApoB expression by chimeric phosphorothioate oligonucleotides having 2'-MOE wings and a deoxy gap - Variation in position of the gap of SEQ ID Nos: 319 and 515
[00809] In accordance with provided herein, a series of antisense compounds was designed based on SEQ ID Nos 319 and 515, with variations in the gap structure. The compounds can be shown in Table 17. "Target site" indicates the first (5'-most) nucleotide number on the particular target sequence to which the compound binds. All compounds in Table 17 can be chimeric oligonucleotides ("gapmers") 20 nucleotides in length. The "gap" region consists of 2'- deoxynucleotides, which is flanked on one or both sides (5' and 3' directions) by "wings" composed of 2'-methoxyethyl (2'-MOE)nucleotides. The number of 2'-MOE nucleotides on either side of the gap varies such that the total number of 2'-MOE nucleotides always equals 10 and the total length of the chimeric oligonucleotide is 20 nucleotides. The exact structure of each oligonucleotide is designated in Table 17 as the "gap structure" and the 2'-deoxynucleotides can be in bold type. A designation of 8—10—2, for instance, indicates that the first (5'-most) 8 nucleotides and the last (3'-most) 2 nucleotides can be 2'-MOE nucleotides and the 10 nucleotides in the gap can be 2'-deoxynucleotides. The internucleoside (backbone) linkages can
be phosphorothioate (P=S) throughout the oligonucleotide. All cytidine residues can be 5- methylcytidines. The compounds were analyzed for their effect on human ApoB mRNA levels by quantitative real-time PCR as described in other examples herein. Data, shown in Table 17, can be averages from three experiments in which HepG2 cells were treated with the antisense oligonucleotides of provided herein, at doses of 50 nM and 150 nM. If present, "N.D." indicates "no data".
Table 17
Inhibition of human ApoB mRNA levels by chimeric phosphorothioate oligonucleotides having
2'-MOE wings and a variable deoxy gap

[00810] As shown in Table 17, SEQ ID Nos 319 and 515 demonstrated at least 44% inhibition of human ApoB expression in this assay for either dose. These data suggest that the compounds can be effective with a number of variations in gap placement. The target regions to which these sequences can be complementary can be herein referred to as " target segments" and can be therefore for targeting by compounds of provided herein,. These target segments can be shown in Table 18. The sequences represent the reverse complement of the antisense compounds shown in Table 17. "Target site" indicates the first (5 '-most) nucleotide number on the particular target nucleic acid to which the oligonucleotide binds. Also shown in Table 18 is the species in which each of the target segments was found.
Table 18
Sequence and position of target segments identified in ApoB.
 SITE ID TARGET TARGET SEQUENCE REV COMP ACTIVE IN SEQ ID SEQ ID SITE OF SEQ NO NO ID NO
SITE ID TARGET TARGET SEQUENCE REV COMP ACTIVE IN SEQ ID SEQ ID SITE OF SEQ NO NO ID NO
197729 3 709 CGAGGAAGGGCAATGTGGCA 190 H. sapiens 558
197730 3 829 CCTTGTCAACTCTGATCAGC 191 H. sapiens 559
197731 3 849 AGCAGCCAGTCCTGTCAGTA 192 H. sapiens 560
197732 3 889 AGCATGTGGCAGAAGCCATC 193 H. sapiens 561
197733 3 1059 GAGAGCACCAAATCCACATC 194 H. sapiens 562
197734 3 1199 CCTCAGTGATGAAGCAGTCA 195 H. sapiens 563
197735 3 1349 GATAGATGTGGTCACCTACC 196 H. sapiens 564
197736 3 1390 CCTCAGCACAGCAGCTGCGA 197 H. sapiens 565
197737 3 1589 GATTCTGCGGGTCATTGGAA 198 H. sapiens 566
197738 3 1678 CAAAGCCATCACTGATGATC 199 H. sapiens 567
197739 3 1699 AGAAAGCTGCCATCCAGGCT 200 H. sapiens 568
197740 3 1749 CAGGAGGTTCTTCTTCAGAC 201 H. sapiens 569
197741 3 1829 GAGTCCTTCACAGGCAGATA 202 H. sapiens 570
197742 3 1919 TGCCAATATCTTGAACTCAG 203 H. sapiens 571
197743 3 2189 CATCGAGATTGGCTTGGAAG 204 H. sapiens 572
197744 3 2649 GGAGCTGGATTACAGTTGCA 205 H. sapiens 573
197745 3 2729 CAACATGCAGGCTGAACTGG 206 H. sapiens 574
197746 3 2949 ACATTACATTTGGTCTCTAC 207 H. sapiens 575
197747 3 3059 CTCAGGCGCTTACTCCAACG 208 H. sapiens 576
197748 3 3118 GGGACACCAGATTAGAGCTG 209 H. sapiens 577
197749 3 3189 GAGCTCCAGAGAGAGGACAG 210 H. sapiens 578
197750 3 3289 ATCGGCAGAGTATGACCTTG 211 H. sapiens 579
197751 3 3488 CAAGGGTGTTATTTCCATAC 212 H. sapiens 580
197752 3 3579 GACTCATCTGCTACAGCTTA 213 H. sapiens 581
197753 3 4039 GCAAATCCTCCAGAGATCTA 214 H. sapiens 582
197754 3 4180 CTCTCCTGGGTGTTCTAGAC 215 H. sapiens 583
197755 3 4299 ATGAAGGCTGACTCTGTGGT 216 H. sapiens 584
197756 3 4511 GGGACCACAGATGTCTGCTT 217 H. sapiens 585
197757 3 4660 CTGGCCGGCTCAATGGAGAG 218 H. sapiens 586
197758 3 4919 GCTGCGTTCTGAATATCAGG 219 H. sapiens 587
197759 3 5009 TGCTGACATCTTAGGCACTG 220 H. sapiens 588
197760 3 5109 AAGTGTAGTCTCCTGGTGCT 221 H. sapiens 589
197761 3 5212 CAAAATTCAGTCTGGATGGG 222 H. sapiens 590
197762 3 5562 GGGAAACTACGGCTAGAACC 223 H. sapiens 591
197763 3 5589 CTGCATGTGGCTGGTAACCT 224 H. sapiens 592
197764 3 5839 CCATGACCATCGATGCACAT 225 H. sapiens 593
197765 3 5869 ATGGGAAACTCGCTCTCTGG 226 H. sapiens 594
197766 3 5979 AGTCATCATCTCGTGTCTAG 227 H. sapiens 595
197767 3 6099 GAATACAGCCAGGACTTGGA 228 H. sapiens 596
197768 3 6144 GGCGTGGAGCTTACTGGACG 229 H. sapiens 597
197769 3 6249 GAGATGAGAGATGCCGTTGA 230 H. sapiens 598
197770 3 6759 AGTCTTGATGAGCACTATCA 231 H. sapiens 599
197771 3 6889 CTAAGTACCAAATCAGAATC 232 H. sapiens 600
197772 3 7149 GTCCATGAGTTAATCGAGAG 233 H. sapiens 601
197773 3 7549 AGGCCACAGTTGCAGTGTAT 234 H. sapiens 602
SITE ID TARGET TARGET SEQUENCE REV COMP ACTIVE IN SEQ ID SEQ ID SITE OF SEQ NO NO ID NO
197774 3 7779 TCTGATTGGTGGACTCTTGC 235 H. sapiens 603
197775 3 7929 GAAGTCAGTCTTCAGGCTCT 236 H. sapiens 604
197776 3 8929 CCAGATTCTCAGATGAGGGA 237 H. sapiens 605
197778 3 10240 CATCTGTCATTGATGCACTG 238 H. sapiens 606
197779 3 10619 AGGAGATGTCAAGGGTTCGG 239 H. sapiens 607
197780 3 10659 GGAACTATTGCTAGTGAGGC 240 H. sapiens 608
197781 3 10899 CTCTCTCCATGGCAAATGTC 241 H. sapiens 609
197783 3 11979 CACCGTGACTTCAGTGCAGA 243 H. sapiens 610
197784 3 12249 ACTGAGTTGAGGGTCCGGGA 244 H. sapiens 611
197786 3 13958 CACATATGAACTGGACCTGC 245 H. sapiens 612
197787 3 14008 TCTGAACTCAGAAGGATGGC 246 H. sapiens 613
216825 3 3249 GGTGCGAAGCAGACTGAGGC 247 H. sapiens 614
216826 3 3 TCCCACCGGGACCTGCGGGG 248 H. sapiens 615
216827 3 6 CACCGGGACCTGCGGGGCTG 249 H. sapiens 616
216828 3 23 CTGAGTGCCCTTCTCGGTTG 250 H. sapiens 617
216829 3 35 CTCGGTTGCTGCCGCTGAGG 251 H. sapiens 618
216830 3 36 TCGGTTGCTGCCGCTGAGGA 252 H. sapiens 619
216831 3 37 CGGTTGCTGCCGCTGAGGAG 253 H. sapiens 620
216832 3 39 GTTGCTGCCGCTGAGGAGCC 254 H. sapiens 621
216833 3 43 CTGCCGCTGAGGAGCCCGCC 255 H. sapiens 622
216834 3 116 ACCGCAGCTGGCGATGGACC 256 H. sapiens 623
216835 3 120 CAGCTGGCGATGGACCCGCC 257 H. sapiens 624
216836 3 13800 GAACTTACTATCATCCTCTA 258 H. sapiens 625
216838 3 13854 TCCAATTGAACTTTCACATA 260 H. sapiens 626
216839 3 13882 AAAATTCAAACTGCCTATAT 261 H. sapiens 627
216840 3 13903 GATAAAACCATACAGTGAGC 262 H. sapiens 628
216841 3 13904 ATAAAACCATACAGTGAGCC 263 H. sapiens 629
216842 3 13908 AACCATACAGTGAGCCAGCC 264 H. sapiens 630
216843 3 13909 ACCATACAGTGAGCCAGCCT 265 H. sapiens 631
216844 3 13910 CCATACAGTGAGCCAGCCTT 266 H. sapiens 632
216845 3 13917 GTGAGCCAGCCTTGCAGTAG 267 H. sapiens 633
216846 3 13922 CCAGCCTTGCAGTAGGCAGT 268 H. sapiens 634
216847 3 13934 TAGGCAGTAGACTATAAGCA 269 H. sapiens 635
216848 3 13937 GCAGTAGACTATAAGCAGAA 270 H. sapiens 636
216849 3 13964 TGAACTGGACCTGCACCAAA 271 H. sapiens 637
216850 3 13968 CTGGACCTGCACCAAAGCTG 272 H. sapiens 638
216851 3 13970 GGACCTGCACCAAAGCTGGC 273 H. sapiens 639
216852 3 13974 CTGCACCAAAGCTGGCACCA 274 H. sapiens 640
216853 3 13978 ACCAAAGCTGGCACCAGGGC 275 H. sapiens 641
216854 3 13997 CTCGGAAGGTCTCTGAACTC 276 H. sapiens 642
216855 3 14012 AACTCAGAAGGATGGCATTT 277 H. sapiens 643
216856 3 14014 CTCAGAAGGATGGCATTTTT 278 H. sapiens 644
216857 3 14049 ATCAGGATCTGAGTTATTTT 279 H. sapiens 645
216858 3 14052 AGGATCTGAGTTATTTTGCT 280 H. sapiens 646
216859 3 14057 CTGAGTTATTTTGCTAAACT 281 H. sapiens 647
SITE ID TARGET TARGET SEQUENCE REV COMP ACTIVE IN SEQ ID SEQ ID SITE OF SEQ NO NO ID NO
216860 3 14064 ATTTTGCTAAACTTGGGGGA 282 H. sapiens 648
216861 3 14071 TAAACTTGGGGGAGGAGGAA 283 H. sapiens 649
217030 3 14087 GGAACAAATAAATGGAGTCT 284 H. sapiens 650
224316 3 3230 GTTTG AACTCAAGCAGAAG 285 H. sapiens 651
224317 3 3232 TTGTAACTCAAGCAGAAGGT 286 H. sapiens 652
224318 3 3234 GTAACTCAAGCAGAAGGTGC 287 H. sapiens 653
224319 3 3236 AACTCAAGCAGAAGGTGCGA 288 H. sapiens 654
224320 3 3238 CTCAAGCAGAAGGTGCGAAG 289 H. sapiens 655
224321 3 3240 CAAGCAGAAGGTGCGAAGCA 290 H. sapiens 656
224322 3 3242 AGCAGAAGGTGCGAAGCAGA 291 H. sapiens 657
224323 3 3244 CAGAAGGTGCGAAGCAGACT 292 H. sapiens 658
224324 3 3246 GAAGGTGCGAAGCAGACTGA 293 H. sapiens 659
224325 3 3248 AGGTGCGAAGCAGACTGAGG 294 H. sapiens 660
224326 3 3250 GTGCGAAGCAGACTGAGGCT 295 H. sapiens 661
224327 3 3252 GCGAAGCAGACTGAGGCTAC 296 H. sapiens 662
224328 3 3254 GAAGCAGACTGAGGCTACCA 297 H. sapiens 663
224329 3 3256 AGCAGACTGAGGCTACCATG 298 H. sapiens 664
224330 3 3258 CAGACTGAGGCTACCATGAC 299 H. sapiens 665
224331 3 3260 GACTGAGGCTACCATGACAT 300 H. sapiens 666
224332 3 3262 CTGAGGCTACCATGACATTC 301 H. sapiens 667
224333 3 3264 GAGGCTACCATGACATTCAA 302 H. sapiens 668
224334 3 3266 GGCTACCATGACATTCAAAT 303 H. sapiens 669
224335 3 3268 CTACCATGACATTCAAATAT 304 H. sapiens 670
224336 3 5582 CCTGAAGCTGCATGTGGCTG 305 H. sapiens 671
224337 3 5584 TGAAGCTGCATGTGGCTGGT 306 H. sapiens 672
224338 3 5586 AAGCTGCATGTGGCTGGTAA 307 H. sapiens 673
224339 3 5588 GCTGCATGTGGCTGGTAACC 308 H. sapiens 674
224340 3 5590 TGCATGTGGCTGGTAACCTA 309 H. sapiens 675
224341 3 5592 CATGTGGCTGGTAACCTAAA 310 H. sapiens 676
224342 3 5594 TGTGGCTGGTAACCTAAAAG 311 H. sapiens 677
224343 3 5596 TGGCTGGTAACCTAAAAGGA 312 H. sapiens 678
224344 3 5598 GCTGGTAACCTAAAAGGAGC 313 H. sapiens 679
224345 3 5600 TGGTAACCTAAAAGGAGCCT 314 H. sapiens 680
224346 3 5602 GTAACCTAAAAGGAGCCTAC 315 H. sapiens 681
224347 3 5604 AACCTAAAAGGAGCCTACCA 316 H. sapiens 682
224348 3 5606 CCTAAAAGGAGCCTACCAAA 317 H. sapiens 683
187366 318 3121 GGCGCGAAGCAGACTGAGGC 319 H. sapiens 684
187404 318 12651 CACTATGTTCATGAGGGAGG 323 H. sapiens 685
197777 318 9851 CCATCATAGGTTCTGACGTC 324 H. sapiens 686
197785 318 12561 GAAGCTGATTGACTCACTCA 325 H. sapiens 687
224349 318 3104 TTGTAACTCAAGCAGAAGGC 326 H. sapiens 688
224350 318 3106 GTAACTCAAGCAGAAGGCGC 327 H. sapiens 689
224351 318 3108 AACTCAAGCAGAAGGCGCGA 328 H. sapiens 690
224352 318 3110 CTCAAGCAGAAGGCGCGAAG 329 H. sapiens 691
224353 318 3116 CAGAAGGCGCGAAGCAGACT 330 H. sapiens 692
SITE ID TARGET TARGET SEQUENCE REV COMP ACTIVE IN SEQ ID SEQ ID SITE OF SEQ NO NO ID NO
224354 318 3118 GAAGGCGCGAAGCAGACTGA 331 H. sapiens 693
224355 318 3120 AGGCGCGAAGCAGACTGAGG 332 H. sapiens 694
224356 318 3122 GCGCGAAGCAGACTGAGGCT 333 H. sapiens 695
224328 3 3254 GAAGCAGACTGAGGCTACCA 514 H. sapiens 696
224353 318 3116 CAGAAGGCGCGAAGCAGACT 515 H. sapiens 697
216862 334 904 TCTTTCTCCTGTCTTACAGA 335 H. sapiens 698
216866 334 1988 CCCACGT AGAAGATGCGAC 339 H. sapiens 699
216868 334 2722 AATATGGTAGACATGAGCCA 341 H. sapiens 700
216869 334 2791 CATTAGCTGCATTTCAACTG 342 H. sapiens 701
216870 334 3045 ACTTTCACTCCTAGTCTGCA 343 H. sapiens 702
216874 334 3527 CCATGTCCAGGTAAGTCATG 347 H. sapiens 703
216876 334 3603 GCACCAGGCACGGATGTGAC 349 H. sapiens 704
216877 334 3864 GTGGGGTCCCAGAGGCACTG 350 H. sapiens 705
216878 334 3990 GCTGATCGGCCACTGCAGCT 351 H. sapiens 706
216879 334 4251 TCCACGTGGCTGGGGAGGTC 352 H. sapiens 707
216880 334 4853 TGTAATGTATGGTGATCAGA 353 H. sapiens 708
216881 334 5023 GAGAGTACCCAGTGGGAAAT 354 H. sapiens 709
216882 334 5055 AGTCAGCATGGGCTTCAGCC 355 H. sapiens 710
216883 334 5091 CAAAAGAATGACTGTCCAAC 356 H. sapiens 711
216884 334 5096 GAATGACTGTCCAACAAGTG 357 H. sapiens 712
216885 334 5301 TATCTACTGTAATTTAAAAT 358 H. sapiens 713
216886 334 5780 CTGATATGGGTGGAGAACAG 359 H. sapiens 714
216887 334 6353 TCTGGGACAGGTATGAGCTC 360 H. sapiens 715
216888 334 6534 TGATAGCAGTGGCCCTTGAA 361 H. sapiens 716
216889 334 6641 GGATTGGCGTGAAATACTGG 362 H. sapiens 717
216890 334 6661 TGCCCGAGGTTCCTCCTGCC 363 H. sapiens 718
216894 334 7059 GCACTAGCAAGACCACACTC 367 H. sapiens 719
216895 334 7066 CAAGACCACACTCTGCATAG 368 H. sapiens 720
216897 334 7209 TCCTCCATAGGATACCGTGT 370 H. sapiens 721
216899 334 7702 GGATGTAGGGCAGCAAAACC 372 H. sapiens 722
216900 334 7736 TCTGCACAAGGACTCCTTGT 373 H. sapiens 723
216901 334 8006 CAGCCTGTCTCAGTGAACAT 374 H. sapiens 724
216903 334 8239 CAGGATGCTTCCAGTCTAAT 376 H. sapiens 725
216904 334 8738 AAATGCTCGTCTCCAATCTC 377 H. sapiens 726
216906 334 9208 AACTTGTGTATCCAAATCCA 379 H. sapiens 727
216908 334 9545 TGACATGGTGTGCTTCCTTG 381 H. sapiens 728
216910 334 9770 ACACTGGTGTTCTGGCTACC 383 H. sapiens 729
216911 334 9776 GTGTTCTGGCTACCTCTAGT 384 H. sapiens 730
216913 334 10341 TCCTGGCATAGGTCACAGTA 386 H. sapiens 731
216914 334 10467 ATGTCAACAGTAGCACCTCC 387 H. sapiens 732
216915 334 10522 TAGACTCAGACAAGTCTGGA 388 H. sapiens 733
216917 334 10587 CCTAAGTTGCTCATCTCTGG 390 H. sapiens 734
216922 334 11337 TGCGCTGAGTTCCATGAAAC 395 H. sapiens 735
216923 334 11457 CTATTGCAGGTGCTCTCCAG 396 H. sapiens 736
216926 334 12155 CAGAGGAACATCTTGCACCT 399 H. sapiens 737
SITE ID TARGET TARGET SEQUENCE REV COMP ACTIVE IN SEQ ID SEQ ID SITE OF SEQ NO NO ID NO
216928 334 12221 CTCTGCTCCTTACTCTTGTG 401 H. sapiens 738
216929 334 12987 GTAATTTCTCACCATCCATC 402 H. sapiens 739
216930 334 13025 TTCTGAGTCTCAATTGTCTA 403 H. sapiens 740
216931 334 13057 CTATGTCCTTGTGTGCACAT 404 H. sapiens 741
216932 334 13634 GCCTATTGCCATTTGTATGT 405 H. sapiens 742
216933 334 13673 CTATTCATGTCCTTTGCCTA 406 H. sapiens 743
216935 334 14567 GATTCTGCGGGTAATCTCAG 408 H. sapiens 744
216937 334 14680 TCAATGCAGGTCATTGGAAA 410 H. sapiens 745
216938 334 15444 C ACCAGATTGACCATCCCT 411 H. sapiens 746
216940 334 15757 TACTTGATAGTGCTCTAGGA 413 H. sapiens 747
216941 334 15926 TTGACTGCAGGACCAGGAGG 414 H. sapiens 748
216942 334 16245 AACAAACACTTGTGCAAATG 415 H. sapiens 749
216950 334 18519 AATGAGACCAAACTTCCACT 423 H. sapiens 750
216951 334 18532 TTCCACTTTGAAGCTAGCAA 424 H. sapiens 751
216952 334 18586 GATCTGGAGCTTATTCTTGA 425 H. sapiens 752
216953 334 18697 ACCTCATGTGACTTGTATGC 426 H. sapiens 753
216954 334 18969 TTCTTAAGAAACACCTTGTA 427 H. sapiens 754
216955 334 19250 TAGGCCCATCCTGGCTGCAT 428 H. sapiens 755
216956 334 19340 AAACTCTCAGGATATGGTAA 429 H. sapiens 756
216957 334 19802 ATACCTTCCTCTACCTTTGC 430 H. sapiens 757
216958 334 19813 TACCTTTGCTGAAGGTCCTT 431 H. sapiens 758
216961 334 20567 ATCTATCTAGTGAAATTTCT 434 H. sapiens 759
216962 334 20647 TCAGCTCATCAAAATATGCT 435 H. sapiens 760
216963 334 20660 ATATGCTAGTCCTTCCTTTC 436 H. sapiens 761
216965 334 21316 CAAAGGTCTGAGTTATCCAG 438 H. sapiens 762
216967 334 21422 TGACTTATAGATGCAGGCTG 440 H. sapiens 763
216968 334 21634 TCAGTGGAGGGTAATTCTTT 441 H. sapiens 764
216969 334 21664 TGCCTAGCCAGTTTGAAAGA 442 H. sapiens 765
216970 334 21700 CCTGCAGAATTTTGCCAGGC 443 H. sapiens 766
216972 334 22048 GTAGCTAGGTAGGTAAAGCA 445 H. sapiens 767
216973 334 22551 TTGAGTGAGACACACAAGGT 446 H. sapiens 768
216974 334 22694 GTGCTAGTCAGGGAATGCAT 447 H. sapiens 769
216976 334 22903 GGGGAGAGAGCATGCCCAGC 449 H. sapiens 770
216977 334 22912 GCATGCCCAGCTGCGAAAGC 450 H. sapiens 771
216978 334 23137 AGCCAGGTATAGAAAGGAGT 451 H. sapiens 772
216979 334 23170 AACTTTCTAAGAGGCAGAAT 452 H. sapiens 773
216981 334 23882 TCTTAGTCTGGTCATGAGTG 454 H. sapiens 774
216983 334 24184 AGTAGGAGATTTCATATGAA 456 H. sapiens 775
216985 334 24559 TCTTCACCAGCAACACATTA 458 H. sapiens 776
216987 334 24800 ATGGCCACCTAGCATGGCAC 460 H. sapiens 777
216989 334 24991 CATGTTTCTGAGCCTCCAGA 462 H. sapiens 778
216990 334 25067 TAGGTGGCTCCCTGTCTTCA 463 H. sapiens 779
216991 334 25152 TCCAAAGTCTTGGGAATCCT 464 H. sapiens 780
216995 334 26749 ACAAAGAAAGGGGGAGTTGG 468 H. sapiens 781
216996 334 26841 TCGTGTCTTCCTGGCCCAGA 469 H. sapiens 782
SITE ID TARGET TARGET SEQUENCE REV COMP ACTIVE IN SEQ ID SEQ ID SITE OF SEQ NO NO ID NO
216997 334 27210 GCAGTGCCCAGCACACAATA 470 H. sapi ens 783
216998 334 27815 ACTCGTCCAGGTGCGAAGCA 471 H. sapi ens 784
216999 334 28026 GCCACCTAAGGTAAAGAAGG 472 H. sapi ens 785
217000 334 28145 ATCAGAGTGGCAGAGAGAGC 473 H. sapi ens 786
217002 334 28919 TTTACCATAGTTGTGACACA 475 H. sapi ens 787
217006 334 29871 CATTTTGTAGGCAATGAGCT 479 H. sapi ens 788
217007 334 30181 GCATTAGTAAACATGAGAAC 480 H. sapi ens 789
217009 334 30931 TTCATTTCAGCGATGGCCGG 482 H. sapi ens 790
217013 334 39562 GAAAATCTAGTGTCATTCAA 486 H. sapi ens 791
217016 334 39789 TCCTATACAGTTTTGGGAAC 489 H. sapi ens 792
217017 334 39904 AAGGACTTCAGTATGGAGCT 490 H. sapi ens 793
217018 334 39916 ATGGAGCTTTTATTGAATTG 491 H. sapi ens 794
217022 334 40412 CCATCAGCACTATTATTTAT 495 H. sapi ens 795
217023 334 40483 ATAGGCAAGCTCAGCCATAG 496 H. sapi ens 796
217025 334 40576 TGCTAGATGAGATACATCAA 498 H. sapi ens 797
217026 334 40658 GAAGACCAAACATGGTTCTA 499 H. sapi ens 798
217029 334 41130 CTCTGTTTAGTCCTCTCCAG 502 H. sapi ens 799
217032 503 490 CATTGATAAAATGTTCTGGC 505 H. sapi ens 800
217033 503 504 TCTGGCACAGCAAAACCTCT 506 H. sapi ens 801
217034 503 506 TGGCACAGCAAAACCTCTAG 507 H. sapi ens 802
217037 503 523 TAGAACACATAGTGTGATTT 510 H. sapi ens 803
217039 503 526 AACACATAGTGTGATTTAAG 512 H. sapi ens 804
[00811] As these " target segments" have been found by experimentation to be open to, and accessible for, hybridization with the antisense compounds of provided herein, one of skill in the art will recognize or be able to ascertain, using no more than routine experimentation, further embodiments, that encompass other compounds that specifically hybridize to these target segments and consequently inhibit the expression of ApoB.
[00812] According to provided herein, antisense compounds include antisense oligomeric compounds, antisense oligonucleotides, ribozymes, external guide sequence (EGS)
oligonucleotides, alternate splicers, primers, probes, and other short oligomeric compounds which hybridize to at least a portion of the target nucleic acid.
2.36 Example 37 - Antisense inhibition of human ApoB expression - dose response of oligonucleotides
[00813] In accordance with provided herein, 12 oligonucleotides described in Examples 29 and 31 were further investigated in a dose response study. The control oligonucleotides used in this study were ISIS 18076 (SEQ ID NO: 805) and ISIS 13650 (SEQ ID NO: 806).
[00814] All compounds in this study, including the controls, were chimeric oligonucleotides ("gapmers") 20 nucleotides in length, composed of a central "gap" region consisting of ten 2'- deoxynucleotides, which is flanked on both sides (5 ' and 3 ' directions) by five-nucleotide "wings". The wings can be composed of 2'-methoxyethyl (2'-MOE) nucleotides. The internucleoside (backbone) linkages can be phosphorothioate (P=S) throughout the
oligonucleotides. All cytidine residues can be 5-methylcytidines.
[00815] In the dose-response experiment, with mRNA levels as the endpoint, HepG2 cells were treated with the antisense oligonucleotides or the control oligonucleotides at doses of 37, 75, 150, and 300 nM oligonucleotide. Data were obtained by real-time quantitative PCR as described in other examples herein and can be averaged from two experiments with mRNA levels in the treatment groups being normalized to an untreated control group. The data can be shown in Table 19.
Table 19
Inhibition of ApoB mRNA levels by chimeric phosphorothioate oligonucleotides having 2'-
MOE wings and a deoxy gap - Dose Response

2.37 Example 38 - Antisense inhibition of human ApoB expression - dose response - Lower dose range
[00816] In accordance with provided herein, seven oligonucleotides described in Examples 29, 31, 35, and 36 were further investigated in a dose response study. The control
oligonucleotides used in this study were ISIS 18076 (SEQ ID NO: 805), ISIS 13650 (SEQ ID NO: 806), and ISIS 129695 (SEQ ID NO: 807).
[00817] All compounds in this study, including the controls, were chimeric oligonucleotides ("gapmers") 20 nucleotides in length, composed of a central "gap" region consisting of ten 2'- deoxynucleotides, which is flanked on both sides (5 ' and 3 ' directions) by five-nucleotide "wings". The wings can be composed of 2'-methoxyethyl (2'-MOE) nucleotides. The internucleoside (backbone) linkages can be phosphorothioate (P=S) throughout the
oligonucleotides. All cytidine residues can be 5-methylcytidines.
[00818] In the dose-response experiment, with mRNA levels as the endpoint, HepG2 cells were treated with the antisense oligonucleotides or the control oligonucleotides at doses of 12. 5, 37, 75, 150, and 300 nM oligonucleotide. Data were obtained by real-time quantitative PCR as described in other examples herein and can be averaged from two experiments with mRNA levels in the treatment groups being normalized to an untreated control group. The data can be shown in Table 20.
Table 20
Inhibition of ApoB mRNA levels by chimeric phosphorothioate oligonucleotides having 2'-
MOE wings and a deoxy gap - Dose Response

2.38 Example 39 - RNA Synthesis
[00819] In general, RNA synthesis chemistry is based on the selective incorporation of various protecting groups at strategic intermediary reactions. Although one of ordinary skill in the art will understand the use of protecting groups in organic synthesis, a useful class of protecting groups includes silyl ethers. In particular bulky silyl ethers can be used to protect the
5 '-hydroxyl in combination with an acid-labile orthoester protecting group on the 2'-hydroxyl. This set of protecting groups is then used with standard solid-phase synthesis technology. It is important to lastly remove the acid labile orthoester protecting group after all other synthetic steps. Moreover, the early use of the silyl protecting groups during synthesis ensures facile removal when desired, without undesired deprotection of 2' hydroxyl.
[00820] Following this procedure for the sequential protection of the 5 '-hydroxyl in combination with protection of the 2 '-hydroxyl by protecting groups that can be differentially removed and can be differentially chemically labile, RNA oligonucleotides were synthesized.
[00821] RNA oligonucleotides can be synthesized in a stepwise fashion. Each nucleotide is added sequentially (3 '- to 5 '-direction) to a solid support-bound oligonucleotide. The first nucleoside at the 3 '-end of the chain is covalently attached to a solid support. The nucleotide precursor, a ribonucleoside phosphoramidite, and activator can be added, coupling the second base onto the 5 '-end of the first nucleoside. The support is washed and any unreacted 5 '- hydroxyl groups can be capped with acetic anhydride to yield 5 '-acetyl moieties. The linkage is then oxidized to the more stable and ultimately desired P(V) linkage. At the end of the nucleotide addition cycle, the 5 '-silyl group is cleaved with fluoride. The cycle is repeated for each subsequent nucleotide.
[00822] Following synthesis, the methyl protecting groups on the phosphates can be cleaved in 30 minutes utilizing 1 M disodium-2-carbamoyl-2-cyanoethylene-l,l-dithiolate trihydrate (S2Na2) in DMF. The deprotection solution is washed from the solid support-bound
oligonucleotide using water. The support is then treated with 40% methylamine in water for 10 minutes at 55 °C. This releases the RNA oligonucleotides into solution, deprotects the exocyclic amines, and modifies the 2'- groups. The oligonucleotides can be analyzed by anion exchange HPLC at this stage.
[00823] The 2 '-orthoester groups can be the last protecting groups to be removed. The ethylene glycol monoacetate orthoester protecting group developed by Dharmacon Research, Inc. (Lafayette, CO), is one example of a useful orthoester protecting group which, has the following important properties. It is stable to the conditions of nucleoside phosphoramidite synthesis and oligonucleotide synthesis. However, after oligonucleotide synthesis the
oligonucleotide is treated with methylamine which not only cleaves the oligonucleotide from the solid support but also removes the acetyl groups from the orthoesters. The resulting 2-ethyl-
hydroxyl substituents on the orthoester can be less electron withdrawing than the acetylated precursor. As a result, the modified orthoester becomes more labile to acid-catalyzed hydrolysis. Specifically, the rate of cleavage is approximately 10 times faster after the acetyl groups can be removed. Therefore, this orthoester possesses sufficient stability in order to be compatible with oligonucleotide synthesis and yet, when subsequently modified, permits deprotection to be carried out under relatively mild aqueous conditions compatible with the final RNA
oligonucleotide product.
[00824] Additionally, methods of RNA synthesis can be well known in the art (Scaringe, S. A. Ph. D. Thesis, University of Colorado, 1996; Scaringe, S. A., et al., J. Am. Chem. Soc. , 1998, 120, 11820-11821; Matteucci, M. D. and Caruthers, M. H. J. Am. Chem. Soc. , 1981, 103, 3185- 3191; Beaucage, S. L. and Caruthers, M. H. Tetrahedron Lett. , 1981, 22, 1859-1862; Dahl, B. J., et al., Acta Chem. Scand,. 1990, 44, 639-641; Reddy, M. P., et al, Tetrahedrom Lett. , 1994, 25, 4311-4314; Wincott, F. et al, Nucleic Acids Res. , 1995, 23, 2677-2684; Griffin, B. E., et al, Tetrahedron, 1967, 23, 2301-2313; Griffin, B. E., et al, Tetrahedron, 1967, 23, 2315-2331).
[00825] RNA antisense compounds (RNA oligonucleotides) of provided herein, can be synthesized by the methods herein or purchased from Dharmacon Research, Inc (Lafayette, CO). Once synthesized, complementary RNA antisense compounds can then be stably annealed by methods known in the art to form double stranded (duplexed) antisense compounds. For example, duplexes can be formed by combining 30 μΐ of each of the complementary strands of RNA oligonucleotides (50 uM RNA oligonucleotide solution) and 15 μΐ of 5X annealing buffer (100 mM potassium acetate, 30 mM HEPES-KOH pH 7. 4, 2 mM magnesium acetate) followed by heating for 1 minute at 90°C, then 1 hour at 37°C. The resulting duplexed antisense compounds can be used in kits, assays, screens, or other methods to investigate the role of a target nucleic acid.
2.39 Example 40 - Design and screening of duplexed antisense compounds targeting ApoB
[00826] In accordance with provided herein, a series of nucleic acid duplexes comprising the antisense compounds of provided herein, and their complements can be designed to target ApoB. The nucleobase sequence of the antisense strand of the duplex comprises at least a portion of an oligonucleotide described herein. The ends of the strands can be modified by the addition of one or more natural or modified nucleobases to form an overhang. The sense strand of the dsRNA is
then designed and synthesized as the complement of the antisense strand and can also contain modifications or additions to either terminus. For example, in one embodiment, both strands of the dsR A duplex would be complementary over the central nucleobases, each having overhangs at one or both termini. The antisense and sense strands of the duplex comprise from about 17 to 25 nucleotides, or from about 19 to 23 nucleotides. Alternatively, the antisense and sense strands comprise 20, 21 or 22 nucleotides.
[00827] For example, a duplex comprising an antisense strand having the sequence
CGAGAGGCGGACGGGACCG and having a two-nucleobase overhang of deoxythymidine(dT) would have the following structure:
cgagaggcggacgggaccgTT Antisense Strand
I I I I I I I I I I I I I I I I I I I TTgctctccgcctgccctggc Complement
[00828] In another embodiment, a duplex comprising an antisense strand having the same sequence CGAGAGGCGGACGGGACCG can be prepared with blunt ends (no single stranded overhang) as shown:
cgagaggcggacgggaccg Antisense Strand
I I I I I I I I I I I I I I I I
gctctccgcctgccctggc Complement
[00829] R A strands of the duplex can be synthesized by methods disclosed herein or purchased from Dharmacon Research Inc., (Lafayette, CO). Once synthesized, the
complementary strands can be stably annealed. The single strands can be aliquoted and diluted to a concentration of 50 uM. Once diluted, 30 uL of each strand is combined with 15uL of a 5X solution of annealing buffer. The final concentration of said buffer is 100 mM potassium acetate, 30 mM HEPES-KOH pH 7. 4, and 2mM magnesium acetate. The final volume is 75 uL. This solution is incubated for 1 minute at 90°C and then centrifuged for 15 seconds. The tube is allowed to sit for 1 hour at 37°C at which time the dsRNA duplexes can be used in
experimentation. The final concentration of the dsRNA duplex is 20 uM. This solution can be stored frozen (-20°C) and freeze-thawed up to 5 times.
[00830] Once prepared, the duplexed antisense compounds can be evaluated for their ability to modulate ApoB expression.
[00831 ] When cells reached 80% confluency, they can be treated with duplexed antisense compounds . For cells grown in 96-well plates, wells can be washed once with 200 μΐ, OPTI
CS -
MEM-1 reduced-serum medium (Gibco BRL) and then treated with 130 μΐ. of OPTI-MEM-1 containing 12 μg/mL LIPOFECTIN (Gibco BRL) and the desired duplex antisense compound at a final concentration of 200 nM. After 5 hours of treatment, the medium is replaced with fresh medium. Cells can be harvested 16 hours after treatment, at which time RNA is isolated and target reduction measured by RT-PCR.
2.40 Example 41 - Design of phenotypic assays and in vivo studies for the use of ApoB inhibitors
2.40.1. Phenotypic assays
[00832] Once ApoB inhibitors have been identified by the methods disclosed herein, the compounds can be further investigated in one or more phenotypic assays, each having measurable endpoints predictive of efficacy in the treatment of a particular disease state or condition. Phenotypic assays, kits and reagents for their use can be well known to those skilled in the art and can be herein used to investigate the role and/or association of ApoB in health and disease. Representative phenotypic assays, which can be purchased from any one of several commercial vendors, include those for determining cell viability, cytotoxicity, proliferation or cell survival (Molecular Probes, Eugene, OR; PerkinElmer, Boston, MA), protein-based assays including enzymatic assays (Panvera, LLC, Madison, WI; BD Biosciences, Franklin Lakes, NJ; Oncogene Research Products, San Diego, CA), cell regulation, signal transduction,
inflammation, oxidative processes and apoptosis (Assay Designs Inc., Ann Arbor, MI), triglyceride accumulation (Sigma-Aldrich, St. Louis, MO), angiogenesis assays, tube formation assays, cytokine and hormone assays and metabolic assays (Chemicon International Inc., Temecula, CA; Amersham Biosciences, Piscataway, NJ).
[00833] In one non-limiting example, cells determined to be appropriate for a particular phenotypic assay {i.e., MCF-7 cells selected for breast cancer studies; adipocytes for obesity studies) can be treated with ApoB inhibitors identified from the in vitro studies as well as control compounds at optimal concentrations which can be determined by the methods described above. At the end of the treatment period, treated and untreated cells can be analyzed by one or more methods specific for the assay to determine phenotypic outcomes and endpoints.
[00834] Phenotypic endpoints include changes in cell morphology over time or treatment dose as well as changes in levels of cellular components such as proteins, lipids, nucleic acids, hormones, saccharides or metals. Measurements of cellular status which include pH, stage of the
cell cycle, intake or excretion of biological indicators by the cell, can also be endpoints of interest.
[00835] Analysis of the genotype of the cell (measurement of the expression of one or more of the genes of the cell) after treatment is also used as an indicator of the efficacy or potency of the ApoB inhibitors. Hallmark genes, or those genes suspected to be associated with a specific disease state, condition, or phenotype, can be measured in both treated and untreated cells.
2.40.2. In vivo studies
[00836] The subject subjects of the in vivo studies described herein can be warm-blooded vertebrate animals, which includes humans.
[00837] The clinical trial is subjected to rigorous controls to ensure that s are not
unnecessarily put at risk and that they can be fully informed about their role in the study.
[00838] To account for the psychological effects of receiving treatments, volunteers can be randomly given placebo or ApoB inhibitor. Furthermore, to prevent the doctors from being biased in treatments, they are not informed as to whether the medication they can be
administering is a ApoB inhibitor or a placebo. Using this randomization approach, each volunteer has the same chance of being given either the new treatment or the placebo.
[00839] Volunteers receive either the ApoB inhibitor or placebo for eight week period with biological parameters associated with the indicated disease state or condition being measured at the beginning (baseline measurements before any treatment), end (after the final treatment), and at regular intervals during the study period. Such measurements include the levels of nucleic acid molecules encoding ApoB or ApoB protein levels in body fluids, tissues or organs compared to pre-treatment levels. Other measurements include, but are not limited to, indices of the disease state or condition being treated, body weight, blood pressure, serum titers of pharmacologic indicators of disease or toxicity as well as ADME (absorption, distribution, metabolism and excretion) measurements.
[00840] Information recorded for each patient includes age (years), gender, height (cm), family history of disease state or condition (yes/no), motivation rating (some/moderate/great) and number and type of previous treatment regimens for the indicated disease or condition.
[00841] Volunteers taking part in this study can be healthy adults (age 18 to 65 years) and roughly an equal number of males and females participate in the study. Volunteers with certain characteristics can be equally distributed for placebo and ApoB inhibitor treatment. In general,
the volunteers treated with placebo have little or no response to treatment, whereas the volunteers treated with the ApoB inhibitor show positive trends in their disease state or condition index at the conclusion of the study.
2.41 Example 42 - Antisense inhibition of rabbit ApoB expression by chimeric phosphorothioate oligonucleotides having 2'-MOE wings and a deoxy gap
[00842] In accordance with provided herein, a series of oligonucleotides was designed to target different regions of rabbit ApoB, using published sequences (GenBank accession number X07480. 1, incorporated herein as SEQ ID NO: 808, GenBank accession number M17780. 1, incorporated herein as SEQ ID NO: 809, and a sequence was derived using previously described primers (Tanaka, Journ. Biol. Chem. , 1993,268, 12713-12718) representing an mRNA of the rabbit ApoB, incorporated herein as SEQ ID NO: 810). The oligonucleotides can be shown in Table 21. "Target site" indicates the first (5 '-most) nucleotide number on the particular target sequence to which the oligonucleotide binds. All compounds in Table 21 can be chimeric oligonucleotides ("gapmers") 20 nucleotides in length, composed of a central "gap" region consisting of ten 2'-deoxynucleotides, which is flanked on both sides (5' and 3' directions) by five-nucleotide "wings". The wings can be composed of 2'-methoxyethyl (2'-MOE)nucleotides. The internucleoside (backbone) linkages can be phosphorothioate (P=S) throughout the oligonucleotide. All cytidine residues can be 5-methylcytidines. The compounds were analyzed for their effect on rabbit ApoB mRNA levels in primary rabbit hepatocytes by quantitative realtime PCR as described in other examples herein. Primary rabbit hepatocytes were treated with 150 nM of the compounds in Table 21. For rabbit ApoB the PCR primers were:
[00843] forward primer: AAGCACCCCCAATGTCACC (SEQ ID NO: 811)
[00844] reverse primer: GGGATGGCAGAGCCAATGTA (SEQ ID NO: 812) and the PCR probe was: FAM- TCCTGGATTCAAGCTTCTATGTGCCTTCA -TAMRA (SEQ ID NO: 813) where FAM (PE-Applied Biosystems, Foster City, CA) is the fluorescent reporter dye) and TAMRA (PE-Applied Biosystems, Foster City, CA) is the quencher dye. Data can be averages from two experiments. If present, "N.D." indicates "no data".
Table 21
Inhibition of rabbit ApoB mRNA levels by chimeric phosphorothioate oligonucleotides having
2'-MOE wings and a deoxy gap

[00845] In accordance with provided herein, a subset of the antisense oligonuclotides in Example 42 was further investigated in dose-response studies. Treatment doses were 10, 50, 150 and 300 nM. ISIS 233160 (SEQ ID NO: 824), ISIS 233166 (SEQ ID NO: 830), ISIS 233172 (SEQ ID NO: 835), ISIS 233175 (SEQ ID NO: 838), and ISIS 233183 (SEQ ID NO: 846) were analyzed for their effect on rabbit ApoB mRNA levels in primary rabbit hepatocytes by quantitative real-time PCR as described in other examples hereiN.D.ata can be averages from two experiments and can be shown in Table 22.
Table 22
Inhibition of rabbit ApoB mRNA levels by chimeric phosphorothioate oligonucleotides having
2'-MOE wings and a deoxy gap

2.43 Example 44 - Effects of antisense inhibition of ApoB in LDLr-/- mice - Dose Response
[00846] LDL receptor-deficient mice (LDLr(-/-)mice), a strain that cannot edit the ApoB mRNA and therefore synthesize exclusively ApoB- 100, have markedly elevated LDL cholesterol and ApoB- 100 levels and develop extensive atherosclerosis.
[00847] LDLr(-/-) mice, purchased from Taconic (Germantown, NY) were used to evaluate antisense oligonucleotides for their potential to lower ApoB mRNA or protein levels, as well as phenotypic endpoints associated with ApoB. LDLr(-/-) mice were separated into groups of males and females. LDLr(-/-) mice were dosed intraperitoneally twice a week for six weeks with either 10, 25, or 50 mg/kg of ISIS 147764 (SEQ ID NO: 109) or ISIS 270906 (SEQ ID NO: 856) which is a 4 base mismatch of ISIS 147764, or with saline, or 20 mg/kg of Atorvastatin. At study termination animals were sacrificed and evaluated for several phenotypic markers.
[00848] ISIS 147764 was able to lower cholesterol, triglycerides, and mR A levels in a dose- dependent manner in both male and female mice while the 4-base mismatch ISIS 270906 was not able to do this. The results of the study can be summarized in Table 23.
Table 23
Effects of ISIS 147764 treatment in male and female LDLr-/- mice on ApoB mRNA, liver enzyme, cholesterol, and triglyceride levels.
Dose Liver Enzymes Lipoproteins mRNA
IU/L mg/dL
ISIS No. mg/kg AST ALT CHOL HDL LDL TRIG control
Males
Saline 68. 4 26. 6 279. 2 125. 4 134. 7 170. 6 100. 0
10 57. 6 29. 8 314. 2 150. 0 134. 7 198. 6 61. 7
147764 25 112. 6 78. 8 185. 0 110. 6 66. 2 104. 2 30. 7
50 163. 6 156. 8 165. 6 107. 8 51. 2 113. 4 16. 6
270906 50 167. 4 348. 0 941. 0 244. 2 541. 9 844. 8 N.D.
Atorvastatin 20 N.D. N.D. N.D. N.D. N.D. N.D. 110. 9
Females
Saline 65. 0 23. 4 265. 8 105. 8 154. 9 121. 4 100. 0
10 82. 0 27. 2 269. 6 121. 0 127. 8 140. 8 64. 2
147764 25 61. 4 32. 2 175. 8 99. 5 68. 9 100. 4 41. 3
50 134. 6 120. 4 138. 2 92. 2 45. 9 98. 0 18. 5
270906 50 96. 0 88. 6 564. 6 200. 0 310. 0 240. 4 N.D.
Atorvastatin 20 N.D. N.D. N.D. N.D. N.D. N.D. 109. 0
2.44 Example 45 - Effects of antisense inhibition of ApoB in Cynomolgus monkeys
[00849] Cynomolgus monkeys fed an atherogenic diet develop atherosclerosis with many similarities to atherosclerosis of human beings. Female Cynomolgus macaques shcan be several similarities in lipoproteins and the cardiovascular system with humans. In addition to these characteristics, there can be similarities in reproductive biology. The Cynomolgus female has a 28-day menstrual cycle like that of women. Plasma hormone concentrations have been measured throughout the Cynomolgus menstrual cycle, and the duration of the follicular and luteal phases, as well as plasma estradiol and progesterone concentrations across the cycle, can also be remarkably similar to those in women.
[00850] Cynomolgus monkeys (male or female) can be used to evaluate antisense
oligonucleotides for their potential to lower ApoB mRNA or protein levels, as well as phenotypic endpoints associated with ApoB including, but not limited to cardiovascular indicators, atherosclerosis, lipid diseases, obesity, and plaque formation. One study could include
normal and induced hypercholesterolemic monkeys fed diets that can be normal or high in lipid and cholesterol. Cynomolgus monkeys can be dosed in a variety of regimens, one being subcutaneously with 10-20 mg/kg of the oligomeric compound for 1-2 months. Parameters that can observed during the test period could include: total plasma cholesterol, LDL-cholesterol, HDL-cholesterol, triglyceride, arterial wall cholesterol content, and coronary intimal thickening.
2.45 Example 46 - Sequencing of Cynomolgus monkey (Macaca fascicularis) ApoB target segment
[00851] In accordance with provided herein, a portion of the cynomolgus monkey ApoB mRNA not available in the art, was amplified. Positions 2920 to 3420 of the human ApoB mRNA sequence (GenBank accession number NM_000384. 1 , incorporated herein as SEQ ID NO: 3) contain the target segment to which ISIS 301012 hybridizes and the corresponding segment of cynomolgus monkey ApoB mRNA was amplified and sequenced. The site to which ISIS 301012 hybridizes in the human ApoB was amplified by placing primers at 5' position 2920 and 3' position 3420. The cynomolgus monkey hepatocytes were purchased from //? vitro Technologies (Gaithersburg, MD). The 500 bp fragments were produced using human and cynomolgus monkey 1° tiepatocyte cDNA and were produced by reverse transcription of purified total RNA followed by 40 rounds of PCR amplification. Following gel purification of the human and cynomolgus amplicons, the forward and reverse sequencing reactions of each product were performed by Retrogen (Invitrogen kit was used to create the single-stranded cDNA and provided reagents for Amplitaq PCR reaction). This cynomolgus monkey sequence is incorporated herein as SEQ ID NO: 855 and is 96% identical to positions 2920 to 3420 of the human ApoB mRNA.
2.46 Example 47 - Effects of antisense inhibition of human ApoB gene (ISIS 281625 and 301012) in C57BL/6NTac-TgN(APOB 100) transgenic mice
[00852] C57BL/6NTac-TgN(APOB 100) transgenic mice have the human ApoB gene "knocked-in". These mice express high levels of human ApoB 100 resulting in mice with elevated serum levels of LDL cholesterol. These mice can be useful in identifying and evaluating compounds to reduce elevated levels of LDL cholesterol and the risk of atherosclerosis. When fed a high fat cholesterol diet, these mice develop significant foam cell accumulation underlying the endothelium and within the media, and have significantly more complex atherosclerotic lesions than control animals.
[00853] C57BL/6NTac-TgN(APOB 100) mice were divided into two groups - one group receiving oligonucleotide treatment and control animals receiving saline treatment. After overnight fasting, mice were dosed intraperitoneally twice a week with saline or 25 mg/kg ISIS 281625 (SEQ ID No: 224) or ISIS 301012 (SEQ ID No: 247) for eight weeks. At study termination and forty eight hours after the final injections, animals were sacrificed and evaluated for target mRNA levels in liver, cholesterol and triglyceride levels, and liver enzyme levels. In addition, the endogenous mouse ApoB levels in liver were measured to evaluate any effects of these antisense oligonucletides targeted to the human ApoB.
[00854] Upon treatment with either ISIS 281625 or ISIS 301012, the AST and ALT levels were increased, yet did not exceed normal levels (-300 IU/L). Cholesterol levels were slightly increased relative to saline treatment, while triglyceride levels were slightly decreased.
Treatment with either of these oligonucleotides targeted to the human ApoB which is expressed in these mice markedly decreased the mRNA levels of the human apolipoprotein, while the levels of the endogenous mouse ApoB were unaffected, indicating that these oligonucleotides exhibit specificity for the human ApoB. The results of the comparative studies can be shown in Table 24.
Table 24
Effects of ISIS 281625 and 301012 treatment in mice on ApoB mRNA, liver enzyme, cholesterol, and triglyceride levels.

[00855] Following 2 and 4 weeks of ISIS 301012 treatment, LDL-cholesterol levels were significantly reduced to 22 mg/dL and 17 mg/dL, respectively.
[00856] ApoB protein levels in liver were also evaluated at the end of the 8 week treatment period. Liver protein was isolated and subjected to immunoblot analysis using antibodies specific for human or mouse ApoB protein (US Biologicals, Swampscott, MA and Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, respectively). Immunoblot analysis of liver protein samples reveals a reduction in the expression of both forms of human ApoB , ApoB- 100 and ApoB-48. Mouse ApoB levels in liver were not significantly changed, as judged by immunoblot analysis.
[00857] Serum samples were also collected at 2, 4, 6 and 8 weeks and were evaluated for human ApoB expression by using a human ApoB specific ELISA kit (ALerCHEK Inc., Portland, ME). Quantitation of serum human ApoB protein by ELISA revealed that treatment with ISIS 281625 reduced serum human ApoB protein by 31, 26, 11 and 26% at 2, 4, 6 and 8 weeks, respectively, relative to saline-treated animals. Treatment with ISIS 301012 reduced serum human ApoB protein by 70, 87, 81 and 41% at 2, 4, 6 and 8 weeks, respectively, relative to saline -treated control animals. Serum from transgenic mice was also subjected to immunoblot analysis using both human and mouse specific ApoB antibodies (US Biologicals, Swampscott, MA and Santa Cruz Biotechnology, Inc., Santa Cruz, CA, respectively). Immunoblot analysis of serum samples taken from animals shows a similar pattern of human ApoB expression, with a significant reduction in serum ApoB protein after 2, 4 and 6 weeks of treatment and a slight reduction at 8 weeks. Mouse ApoB in serum was not significantly changed, as judged by immunoblot analysis.
2.47 Example 48 - Effects of antisense inhibition of ApoB (ISIS 233172, 233175, 281625, 301012, and 301027) in C57BL/6 mice
[00858] C57BL/6 mice, a strain reported to be susceptible to hyperlipidemia-induced atherosclerotic plaque formation were used in the following studies to evaluate the toxicity in mice of several antisense oligonucleotides targeted to human or rabbit ApoB.
[00859] C57BL/6 mice were divided into two groups - one group receiving oligonucleotide treatment and control animals receiving saline treatment. After overnight fasting, mice were dosed intraperitoneally twice a week with saline or 25 mg/kg of one of several oligonucleotides for two weeks. The antisense oligonucleotides used in the present study were ISIS 233172 (SEQ ID NO: 835) and ISIS 233175 (SEQ ID NO: 838), both targeted to rabbit ApoB, and ISIS 281625 (SEQ ID NO: 224), ISIS 301012 (SEQ ID NO: 247), and ISIS 301027 (SEQ ID NO:
262), targeted to human ApoB. At study termination and forty eight hours after the final injections, animals were sacrificed and evaluated for liver enzyme levels, body weight, liver weight, and spleen weight.
[00860] The levels of liver enzymes in mice were decreased relative to saline treatment for three of the antisense oligonucleotide. However, the rabbit oligonucleotide ISIS 233175 and the human oligonucleotide ISIS 301027 both elicited drastically increased levels of these liver enzymes, indicating toxicity. For all of the oligonucleotides tested, the change in weight of body, liver, and spleen were minor. The results of the comparative studies can be shown in Table 25.
Table 25
Effects of antisense oligonucleotides targeted to human or rabbit ApoB on mouse ApoB mRNA, liver enzyme, cholesterol, and triglyceride levels.

2.48 Example 49 - Time course evaluation of oligonucleotide at two different doses
[00861] C57BL/6 mice, a strain reported to be susceptible to hyperlipidemia-induced atherosclerotic plaque formation were used in the following studies to evaluate the toxicity in mice of several antisense oligonucleotides targeted to human ApoB.
[00862] Female C57BL/6 mice were divided into two groups - one group receiving oligonucleotide treatment and control animals receiving saline treatment. After overnight fasting, mice were dosed intraperitoneally twice a week with saline or 25 mg/kg or 50 mg/kg of ISIS 281625 (SEQ ID NO: 224), ISIS 301012 (SEQ ID NO: 247), or ISIS 301027 (SEQ ID NO: 262). After 2 weeks, a blood sample was taken from the tail of the mice and evaluated for liver enzyme. After 4 weeks, and study termination, animals were sacrificed and evaluated for liver enzyme levels.
[00863] For ISIS 281625 and ISIS 301012, AST and ALT levels remained close to those of saline at either dose after 2 weeks. After 4 weeks, AST and ALT levels showed a moderate increase over saline treated animals for the lower dose, but a large increase at the higher dose. ISIS 301027, administered at either dose, showed a small increase in AST and ALT levels after 2 weeks and a huge increase in AST and ALT levels after 4 weeks. The results of the studies can be summarized in Table 26.
Table 26
AST and ALT levels in mice treated with ISIS 281625, 301012, or 301027 after 2 and 4 weeks

2.49 Example 50 - Effects of antisense inhibition of ApoB (ISIS 147483 and 147764) in ob/ob mice
[00864] Leptin is a hormone produced by fat that regulates appetite. Deficiencies in this hormone in both humans and non-human animals leads to obesity, ob/ob mice have a mutation in the leptin gene which results in obesity and hyperglycemia. As such, these mice can be a useful model for the investigation of obesity and diabetes and treatments designed to treat these conditions.
[00865] Ob/ob mice receiving a high fat, high cholesterol diet (60% kcal fat supplemented with 0. 15% cholesterol) were treated with one of several oligonucleotides to evaluate their effect on ApoB-related phenotypic endpoints in ob/ob mice. After overnight fasting, mice from each group were dosed intraperitoneally twice a week with 50 mg/kg of ISIS 147483 (SEQ ID NO: 79), or 147764 (SEQ ID NO: 109), or the controls ISIS 116847 (SEQ ID NO: 857), or 141923 (SEQ ID NO: 858), or saline for six weeks. At study termination and forty eight hours after the
final injections, animals were sacrificed and evaluated for target mRNA levels in liver, cholesterol and triglyceride levels, liver enzyme levels, serum glucose levels, and PTEN levels.
[00866] ISIS 147483 and 147764 were both able to lower ApoB mRNA levels, as well as glucose, cholesterol, and triglyceride levels. The results of the comparative studies can be shown in Table 27.
Table 27
Effects of ISIS 147483 and 147764 treatment in ob/ob mice on ApoB mRNA, cholesterol, lipid, triglyceride, liver enzyme, glucose, and PTEN levels.
ISIS No .
SALINE 116847 141923 147483 147764
Glucose mg/dL 269. 6 135. 5 328. 5 213. 2 209. 2
Liver Enzymes
AST 422. 3 343. 2 329. 3 790. 2 406. 5
IU/L
ALT 884. 3 607. 5 701. 7 941. 7 835. 0
Lipoproteins
CHOL 431. 9 287. 5 646. 3 250. 0 286. 3
mg/dL
TRIG 128. 6 196. 5 196. 5 99. 8 101. 2
mRNA % control
ApoB 100. 0 77. 0 100. 0 25. 2 43. 1
PTEN 100. 0 20. 0 113. 6 143. 2 115. 3
2.50 Example 51 - Antisense inhibition of ApoB in high fat fed mice: time- dependent effects
[00867] In a further embodiment , the inhibition of ApoB mRNA in mice was compared to liver oligonucleotide concentration, total cholesterol, LDL-cholesterol and HDL-cholesterol. Male C57B1/6 mice receiving a high fat diet (60% fat) were evaluated over the course of 6 weeks for the effects of treatment with twice weekly intraperitoneal injections of 50 mg/kg ISIS 147764 (SEQ ID NO: 109) or 50 mg/kg of the control oligonucleotide ISIS 141923 (SEQ ID NO: 858). Control animals received saline treatment. Animals were sacrificed after 2 days, 1, 2, 4 and 6 weeks of treatment. Each treatment group at each time point consisted of 8 mice.
[00868] Target expression in liver was measured by real-time PCR as described by other examples herein and is expressed as percent inhibition relative to saline treated mice. Total, LDL- and HDL-cholesterol levels were measured by routine clinical analysis using an Olympus Clinical Analyzer (Olympus America Inc., Melville, NY) and can be presented in mg/dL. Results from saline-treated animals can be shown for comparison. Intact oligonucleotide in liver tissue
was measured by capillary gel electrophoresis and is presented as micrograms of oligonucleotide per gram of tissue. All results can be the average of 8 animals and can be shown in Table 28.
Table 28
Correlation between liver drug concentration, ApoB mRNA expression and serum lipids during ISIS 147764 treatment

[00869] These results illustrate that inhibition of ApoB mRNA by ISIS 147764 occurred within 2 days of treatment, increased with successive treatments and persisted for 6 weeks of treatment. Quantitation of liver oligonucleotide levels reveals a strong correlation between the extent of target inhibition and liver drug concentration. Furthermore, at 1, 2, 3 and 4 weeks of treatment, a inverse correlation between inhibition of target mRNA and cholesterol levels (total, HDL and LDL) is observed, with cholesterol levels lowering as percent inhibition of ApoB mRNA becomes greater. Serum samples were subjected to immunoblot analysis using an antibody to detect mouse ApoB protein (Gladstone Institute, San Francisco, CA). The expression of protein follows the same pattern as that of the mRNA, with ApoB protein in serum markedly reduced within 48 hours and lowered throughout the 6 week treatment period.
[00870] The oligonucleotide treatments described in this example were duplicated to investigate the extent to which effects of ISIS 147764 persist following cessation of treatment. Mice were treated as described, and sacrificed 1, 2, 4, 6 and 8 weeks following the cessation of
oligonucleotide treatment. The same parameters were analyzed and the results can be shown in Table 29.
Table 29
Correlation between liver drug concentration, ApoB mR A expression, and serum lipids after cessation of dosing

[00871] These data demonstrate that after termination of oligonucleotide treatment, the effects of ISIS 147764, including ApoB mRNA inhibition, and cholesterol lowering, persist for up to 8 weeks, Immimoblot analysis demonstrates that ApoB protein levels follow a pattern similar that observed for mRNA expression levels.
2.51 Example 52 - Effects of antisense inhibition of human ApoB gene by 301012 in C57BL/6NTac-TgN(APOB 100) transgenic mice: dosing study
[00872] C57BL/6NTac-TgN(APOB 100) transgenic mice have the human ApoB gene "knocked-in". These mice express high levels of human ApoB resulting in mice with elevated serum levels of LDL cholesterol. These mice can be useful in identifying and evaluating compounds to reduce elevated levels of LDL cholesterol and the risk of atherosclerosis. When fed a high fat cholesterol diet, these mice develop significant foam cell accumulation underlying the endothelium and within the media, and have significantly more complex atherosclerotic plaque lesions than control animals.
[00873] A long-term study of inhibition of human ApoB by ISIS 301012 in C57BL/6NTac- TgN(APOB 100) mice (Taconic, Germantown, NY) was conducted for a 3 month period. Mice were dosed intraperitoneally twice a week with 10 or 25 mg/kg ISIS 301012 (SEQ ID No: 247) for 12 weeks. Saline-injected animals served as controls. Each treatment group comprised 4 animals.
[00874] After 2, 4, 6, 8 and 12 weeks of treatment, serum samples were collected for the purpose of measuring human ApoB protein. Serum protein was quantitated using an ELISA kit specific for human ApoB (ALerCHEK Inc., Portland, ME). The data can be shown in Table 30 and each result represents the average of 4 animals. Data can be normalized to saline-treated control animals.
Table 30
Reduction of human ApoB protein in transgenic mouse serum following ISIS 301012 treatment

[00875] These data illustrate that following 2, 4, 6 or 12 weeks of treatment with ISIS 301012, the level of human ApoB protein in serum from transgenic mice is lowered by approximately 80%, demonstrating that in addition to inhibiting mRNA expression, ISIS 301012 effectively inhibits human ApoB protein expression in mice carrying the human ApoB transgene. ApoB protein in serum was also assessed by immunoblot analysis using an antibody directed to human ApoB protein (US Biologicals, Swampscott, MA). This analysis shows that the levels human ApoB protein, both the ApoB- 100 and ApoB-48 forms, can be lowered at 2, 4, 6 and 12 weeks of treatment. Immunoblot analysis using a mouse ApoB specific antibody (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA) reveals no significant change in the expression of the mouse protein in serum.
[00876] At the beginning of the treatment (start) and after 2, 4, 6 and 8 weeks of treatment, serum samples were collected and total, LDL- and HDL-cholesterol levels were measured by routine clinical analysis using an Olympus Clinical Analyzer (Olympus America Inc., Melville, NY), and these data can be presented in Table 31. Results can be presented as mg/dL in serum
and represent the average of 4 animals. Results from the saline control animals can also be shown.
Table 31
Effects of ISIS 301012 on serum lipids in human ApoB transgenic mice

[00877] These data demonstrate that LDL-cholesterol is lowered by treatment with 10 or 25 mg/kg of ISIS 147764 during the first 4 weeks of treatment.
[00878] The study was terminated forty eight hours after the final injections in the eighth week of treatment, when animals were sacrificed and evaluated for target mRNA levels in liver, ApoB protein levels in liver and serum cholesterol and liver enzyme levels. In addition, the expression of endogenous mouse ApoB levels in liver was measured to evaluate any effects of ISIS 301012 on mouse ApoB mRNA expression.
[00879] Human and mouse ApoB mRNA levels in livers of animals treated for 12 weeks were measured by real-time PCR as described herein. Each result represents the average of data from 4 animals. The data were normalized to saline controls and can be shown in Table 32.
Table 32
Effects of ISIS 301012 on human and mouse ApoB mRNA levels in transgenic mice

[00880] These data demonstrate that following 12 weeks of treatment with ISIS 301012, human ApoB mRNA is reduced by as much as 75% in the livers of transgenic mice, whereas mouse liver ApoB mRNA was unaffected. Furthermore, ELISA analysis of ApoB protein in livers of transgenic mice reveals an 80% and 82% reduction in the human protein following 10 and 20 mg/kg ISIS 301012, respectively. Immunoblot analysis using an antibody directed to human ApoB also demonstrates a reduction in the expression of human ApoB, both the ApoB-
100 and ApoB-48 forms, in the livers of transgenic mice. Immunoblot analysis using an antibody directed to mouse ApoB protein (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) reveals that expression of the mouse protein in liver does not change significantly.
[00881] ALT and AST levels in serum were also measured using the Olympus Clinical
Analyzer (Olympus America Inc., Melville, NY) and showed that following treatment with ISIS
301012, the AST and ALT levels were increased, yet did not exceed normal levels (-300 IU/L), indicating a lack of toxicity due to ISIS 301012 treatment.
2.52 Example 53 - Assessment of in vitro immunostimulatory effects of ISIS 301012
[00882] Immunostimulatory activity is defined by the production of cytokines upon exposure to a proinflammatory agent. In a further embodiment , ISIS 301012 was tested for
immunostimulatory, or proinflammatory, activity. These studies were performed by MDS Pharma Services (Saint Germain sur l'Arbresle, France). Whole blood was collected from naive B6C3F1 mice, which had not been knowingly exposed to viral, chemical or radiation treatment. Cultured blood cells were exposed to 0. 5, 5 or 50 μΜ of ISIS 301012 for a period of 14 to 16
hours. Antisense oligonucleotides known to possess proinflammatory activity served as positive controls. Each treatment was performed in triplicate. At the end of the treatment period, supematants were collected and cytokine analysis was performed using a flow cytometry method with the mouse Inflammation CBA kit (Becton Dickinson, Franklin Lakes, NJ). The results revealed that ISIS 301012 does not stimulate the release of any of the tested cytokines, which were interleukin-12p70 (IL-12p70), tumor necrosis factor-alpha (TNF-alpha), interferon-gamma (IFN-gamma), interleukin-6 (IL-6), macrophage chemoattractant protein- 1 (MCP-1) and interleukin-10 (IL-10). Thus, ISIS 301012 does not possess immunostimulatory activity, as determined by the in vitro immunostimulatory assay.
2.53 Example 54 - Comparative genomic analysis of ApoB
[00883] In accordance with provided herein, a comparative genomic analysis of ApoB sequences from human, mouse and monkey was performed and illustrated that ApoB sequences can be conserved across species. The organization of human and mouse ApoB genes is also highly conserved. The human and mouse genes can be comprised of 29 and 26 exons, respectively. The mouse mRNA is approximately 81% homologous to the human sequence. The complete sequence and gene structure of the ApoB gene in non-human primates have not been identified. However, as illustrated in Example 46, a 500 base pair fragment which contains the ISIS 301012 target sequence exhibits approximately 96% identity to the human sequence.
[00884] The binding site for ISIS 301012 lies within the coding region, within exon 22 of the human ApoB mRNA. When the ISIS 301012 binding sites from human, mouse and monkey were compared, significant sequence diversity was observed. Although the overall sequence conservation between human and monkey over a 500 nucleotide region was approximately 96%, the ISIS 301012 binding site of the monkey sequence contains 2 mismatches relative to the human sequence. Likewise, though the mouse ApoB mRNA sequence is approximately 81% homologous to human, within the ISIS 301012 binding site, 5 nucleotides can be divergent. The sequence comparisons for the ISIS 301012 binding site for human, mouse and monkey ApoB sequences can be shown in Table 33. Mismatched nucleotides relative to the ISIS 301012 target sequence can be underlined.
Table 33
Comparison of ISIS 301012 binding site among human, monkey and mouse ApoB sequences

[00885] The target sequence to which the mouse antisense oligonucleotide ISIS 147764 hybridizes lies within exon 24 of the mouse ApoB gene. The sequence comparisons for the ISIS 147764 binding site in mouse and human ApoB sequences can be shown in Table 34.
Mismatched nucleotides relative to the ISIS 147764 target sequence can be underlined.
Table 34
Comparison of ISIS 147764 binding site between mouse and human ApoB sequences

2.54 Example 55 - BLAST analysis of ISIS 301012
[00886] In accordance with provided herein, the number of regions in the human genome to which ISIS 301012 will hybridize with perfect complementarity was determined. Percent complementarity of an antisense compound with a region of a target nucleic acid was determined using BLAST programs (basic local alignment search tools) and PowerBLAST programs known in the art (Altschul et al., J. Mol. Biol, 1990, 215, 403-410; Zhang and Madden, Genome Res., 1997, 7, 649-656). This analysis assessed sequence complementarity in genomic or pre-mRNA regions and in coding sequences.
[00887] In genomic regions, ISIS 301012 shows perfect sequence complementarity to the ApoB gene only. No target sequences with one mismatch relative to ISIS 301012 were found. Two mismatches can be found between the ISIS 301012 target sequence and the heparanase gene, and 3 mismatches can be found between the ISIS 301012 target sequence and 28 unique genomic sites.
[00888] In RNA sequences, perfect sequence complementarity is found between ISIS 301012 and the ApoB mRNA and three expressed sequence tags that bear moderate similarity to a human ApoB precursor. A single mismatch is found between ISIS 301012 and an expressed sequence tag similar to the smooth muscle form of myosin light chain.
2.55 Example 56 - Antisense inhibition of ApoB in primary human hepatocytes: dose response studies
[00889] In accordance with provided herein, antisense oligonucleotides targeted to human ApoB were tested in dose response studies in primary human hepatocytes. Pre -plated primary human hepatocytes were purchased from In Vitro Technologies (Baltimore, MD). Cells were cultured in high-glucose DMEM (Invitrogen Corporation, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen Corporation, Carlsbad, CA), 100 units/mL and 100 μg/mL streptomycin (Invitrogen Corporation, Carlsbad, CA).
[00890] Human primary hepatocytes were treated with ISIS 301012 (SEQ ID NO: 247) at 10, 50, 150 or 300 nM. Untreated cells and cells treated with the scrambled control oligonucleotide ISIS 113529 (CTCTTACTGTGCTGTGGACA, SEQ ID NO: 859) served as two groups of control cells. ISIS 113529 is a chimeric oligonucleotide ("gapmer") 20 nucleotides in length, composed of a central "gap" region consisting of ten 2'-deoxynucleotides, which is flanked on both sides (5' and 3' directions) by five-nucleotide "wings". The wings can be composed of 2'- methoxyethyl (2'-MOE)nucleotides. The internucleoside (backbone) linkages can be
phosphorothioate (P=S) throughout the oligonucleotide. All cytidines can be 5-methylcytidines.
[00891] Oligonucleotides were introduced into cells through LIPOFECTIN-mediated transfection as described by other examples herein. Cells were harvested both 24 and 48 hours after treatment with oligonucleotide, and both RNA and protein were isolated. Additionally, the culture media from treated cells was collected for ELISA analysis of ApoB protein secretion.
[00892] ApoB mRNA expression was determined by real-time PCR of RNA samples as described by other examples herein. Each result represents 6 experiments. The data can be normalized to untreated control cells and can be shown in Table 35.
Table 35
Inhibition of ApoB mRNA by antisense oligonucleotides in human primary hepatocytes

[00893] These data demonstrate that ISIS 301012 inhibits ApoB expression in a dose- dependent manner in human primary hepatocytes.
[00894] ApoB protein secreted from into the cultured cell media was measured in the samples treated with 50 and 150 nM of oligonucleotide, using a target protein specific ELISA kit (ALerCHEK Inc., Portland, ME). Each result represents 3 experiments. The data can be normalized to untreated control cells and can be shown in Table 36.
Table 36
Inhibition of ApoB protein secretion from human primary hepatocytes by ISIS 301012

(Swampscott, MA). Immunoblot analysis further demonstrates that ApoB protein in human hepatocytes is reduced in a dose-dependent manner following antisense oligonucleotide treatment with ISIS 301012.
[00896] An additional experiment was performed to test the effects of ISIS 271009 (SEQ ID NO: 319), ISIS 281625 (SEQ ID NO: 224) and ISIS 301027 (SEQ ID NO: 262) on human ApoB mRNA in human primary hepatocytes. Cells were cultured as described herein and treated with 5, 10, 50 or 150 nM of ISIS 271009, ISIS 281625 or ISIS 301027 for a period of 24 hours. The control oligonucleotides ISIS 13650 (SEQ ID NO: 806) and ISIS 113529 (SEQ ID NO: 859) were used at 50 or 150 nM. Human ApoB mRNA expression was evaluated by real-time PCR as described by other examples herein. ApoB protein secreted into the cultured cell media was measured in the samples treated with 50 and 150 nM of oligonucleotide, using a target protein specific ELISA kit (ALerCHEK Inc., Portland, ME).
[00897] The data, shown in Table 37, represent the average 2 experiments and can be normalized to untreated control cells. Where present, a "+" indicates that gene expression was increased.
Table 37
Antisense inhibition of human ApoB mRNA by
ISIS 271009, ISIS 281625 and ISIS 301027

ISIS 301027 inhibit the secretion of ApoB protein from cells in a dose-dependent manner.
2.56 Example 57 - Effects of apolipoproteinB-100 antisense oligonucleotides on apolipoprotein(a) expression
[00899] Lipoprotein(a) [Lp(a)] contains two disulfide-linked distinct proteins,
apolipoprotein(a) and ApoB (Rainwater and Kammerer, J. Exp. Zool., 1998, 282, 54-61). In accordance with provided herein, antisense oligonucleotides targeted to ApoB were tested for effects on the expression of the apolipoprotein(a) component of the lipoprotein(a) particle in primary human hepatocytes.
[00900] Primary human hepatocytes (In Vitro Technologies, Baltimore, MD), cultured and transfected as described herein, were treated with 5, 10, 50 or 150 nM of ISIS 271009 (SEQ ID NO: 319), 281625 (SEQ ID NO: 224), 301012 (SEQ ID NO: 247) or 301027 (SEQ ID NO: 262). Cells were also treated with 50 or 150 nM of the control oligonucleotides ISIS 113529 (SEQ ID NO: 859) or ISIS 13650 (SEQ ID NO: 806). Untreated cells served as a control. Following 24 hours of oligonucleotide treatment, apolipoprotein(a) mRNA expression was measured by quantitative real-time PCR as described in other examples herein.
[00901] Probes and primers to human apolipoprotein(a) were designed to hybridize to a human apolipoprotein(a) sequence, using published sequence information (GenBank accession number NM 005577. 1, incorporated herein as SEQ ID NO: 860). For human apolipoprotein(a) the PCR primers were:
[00902] forward primer: CAGCTCCTTATTGTTATACGAGGGA (SEQ ID NO: 861)
[00903] reverse primer: TGCGTCTGAGCATTGCGT (SEQ ID NO: 862) and the PCR probe was: FAM-CCCGGTGTCAGGTGGGAGTACTGC-TAMRA
[00904] (SEQ ID NO: 863) where FAM is the fluorescent dye and TAMRA is the quencher dye.
[00905] Data can be the average of three experiments and can be expressed as percent inhibition relative to untreated controls. The results can be shown in Table 38. A "+" or "-" preceding the number indicates that apolipoprotein(a) expression was increased or decreased, respectively, following treatment with antisense oligonucleotides.
Table 38
Effects of ApoB antisense oligonucleotides on apolipoprotein(a) expression

[00906] These results illustrate that ISIS 301012 did not inhibit the expression of
apolipoprotein(a) in human primary hepatocytes. ISIS 271009 inhibited apolipoprotein(a) expression at the highest dose. ISIS 281625 and ISIS 301027 decreased the levels of
apolipoprotein(a) mRNA.
2.57 Example 58 - Inhibition of lipoprotein(a) particle secretion with antisense oligonucleotides targeted to apolipoproteinB-100
[00907] In accordance with provided herein, the secretion of lipoprotein(a) particles, which can be comprised of one apolipoprotein(a) molecule covalently linked to one ApoB molecule, was evaluated in primary human hepatocytes treated with antisense oligonucleotides targeted to the ApoB component of lipoprotein(a).
[00908] Primary human hepatocytes (In Vitro Technologies, Baltimore, MD), cultured and transfected as described herein, were treated for 24 hours with 50 or 150 nM of ISIS 271009 (SEQ ID NO: 319), 281625 (SEQ ID NO: 224), 301012 (SEQ ID NO: 247) or 301027 (SEQ ID NO: 262). Cells were also treated with 150 nM of the control oligonucleotides ISIS 113529 (SEQ ID NO: 859) or ISIS 13650 (SEQ ID NO: 806). Untreated cells served as a control.
Following 24 hours of oligonucleotide treatment, the amount of lipoprotein (a) in the culture medium collected from the treated cells was measured using a commercially available ELISA kit (ALerCHEK Inc., Portland, ME). The results can be the average of three experiments and can be expressed as percent change in lipoprotein(a) secretion relative to untreated controls. The data can be shown in Table 39. A "+" or "-" preceding the number indicates that lipoprotein(a) particle secretion was increased or decreased, respectively, following treatment with antisense oligonucleotides targeted to ApoB.
Table 39
Inhibition of lipoprotein(a) particle secretion with antisense oligonucleotides targeted to ApoB

[00909] These data demonstrate that antisense inhibition of ApoB, a component of the lipoprotein(a) particle, can reduce the secretion of lipoprotein(a) from human primary hepatocytes. In addition, this reduction in lipoprotein(a) secretion is not necessarily concomitant with a decrease in apolipoprotein(a) mRNA expression, as shown in Example 57.
2.58 Example 59 - Mismatched and trunctated derivatives of ISIS 301012
[00910] As demonstrated herein, ISIS 301012 (SEQ ID NO: 247) reduces ApoB mRNA levels in cultured human cell lines as well as in human primary hepatocytes. In a further embodiment , a study was performed using nucleotide sequence derivatives of ISIS 301012. A series of oligonucleotides containing from 1 to 7 base mismatches, starting in the center of the ISIS 301012 sequence, was designed. This series was designed to introduce the consecutive loss of Watson-Crick base pairing between ISIS 301012 and its target mRNA sequence. These compounds can be shown in Table 40. The antisense compounds with mismatched nucleotides relative to ISIS 301012 can be chimeric oligonucleotides ("gapmers") 20 nucleotides in length, composed of a central "gap" region consisting of ten 2'-deoxynucleotides, which is flanked on both sides (5' and 3' directions) by five-nucleotide "wings". The wings can be composed of 2'- methoxyethyl (2'-MOE) nucleotides. The internucleoside (backbone) linkages can be phosphorothioate (P=S) throughout the oligonucleotide.
[00911] An additional derivative of ISIS 301012 was designed, comprising the ISIS 301012 sequence with 2'MOE nucleotides throughout the oligonucleotide (uniform 2'-MOE). This compound is 20 nucleotides in length, with phosphorothioate linkages throughout the oligonucleotide. This compound is also shown in Table 40.
[00912] HepG2 cells were treated with 50 or 150 nM of the compounds in Table 40 for a 24 hour period, after which RNA was isolated and target expression was measured by real-time PCR as described herein. Untreated cells served as controls. The results can be shown in Tables 40 and can be normalized to untreated control samples.
Table 40
Effects of ISIS 301012 mismatched oligonucleotides and a uniform 2'MOE oligonucleotide on
ApoB expression in HepG2 cells

[00913] The results of treatment of HepG2 cells with the compounds in Table 40 reveals that none of the compounds displays the dose-dependent inhibition observed following treatment with the parent ISIS 301012 sequence. ISIS 332770, which has only a single thymidine to cytosine substitution in the center of the oligonucleotide, was 3-fold less potent than ISIS 301012. Further nucleotide substitutions abrogated antisense inhibition of ApoB expression.
[00914] Phosphorothioate chimeric oligonucleotides can be metabolized in vivo
predominantly by endonucleolytic cleavage. In accordance with provided herein, a series of oligonucleotides was designed by truncating the ISIS 301012 sequence in 1 or 2 base increments from the 5' and/or 3' end. The truncated oligonucleotides represent the possible products that result from endonucleotlytic cleavage. These compounds can be shown in Table 41. The compounds in Table 41 can be chimeric oligonucleotides ("gapmers") of varying lengths, composed of a central "gap" region consisting of 2'-deoxynucleotides, which is flanked on both ends by 2'-methoxyethyl (2'-MOE)nucleotides. The exact structure of each chimeric
oligonucleotide is designated in Table 41 as the "chimera structure". For example, a designation
of 4-10-4 indicates that the first 4 (5' most) and last 4 (3' most) nucleotides can be 2'-MOE nucleotides, and the 10 nucleotides in the gap can be 2'-deoxynucleotides. 2'-MOE nucleotides can be indicated by bold type. The internucleoside (backbone) linkages can be phosphodiester (P=0) between underscored nucleotides; all other internucleoside linkages can be
phosphorothioate (P=S).
[00915] These compounds were tested for their ability to reduce the expression of ApoB mRNA. HepG2 cells were treated with 10, 50 or 150 nM of each antisense compound in Table 41 for a 24 hour period, after which RNA was isolated and target expression was measured by real-time PCR as described herein. Untreated cells served as controls. The results can be shown in Tables 41 and can be normalized to untreated control samples.
Table 41
Effect of ISIS 301012 truncation mutants on ApoB expression in HepG2 cells

[00916] The results in Table 41 illustrate that inhibition of ApoB is dependent upon sequence length, as well as upon sequence complementarity and dose, as demonstrated in Table 41, but truncated versions of ISIS 301012 can be to a certain degree capable of inhibiting ApoB mRNA expression.
2.59 Example 60 - Design and screening of dsRNAs targeting human ApoB
[00917] In accordance with provided herein, a series of nucleic acid duplexes comprising the antisense compounds of provided herein, and their complements were designed to target ApoB and can be shown in Table 42. All compounds in Table 42 can be oligoribonucleotides 20 nucleotides in length with phosphodiester internucleoside linkages (backbones) throughout the compound. The compounds were prepared with blunt ends. Table 41 shows the antisense strand of the dsRNA, and the sense strand is synthesized as the complement of the antisense strand. These sequences can be shown to contain uracil (U) but one of skill in the art will appreciate that uracil (U) is generally replaced by thymine (T) in DNA sequences. "Target site" indicates the first (5 '-most) nucleotide number on the particular target sequence to which the compound binds. A subset of the compounds in Table 42 can be the RNA equivalents of DNA antisense oligonucleotides described herein, and, where applicable, this is noted by the ISIS # of the DNA oligonucleotide in the column "RNA equivalent of ISIS #".
Table 42
dsRNAs targeted to human ApoB

[00918] The dsRNA compounds in Table 42 were tested for their effects on human
apolipoprotein mRNA in HepG2 cells. HepG2 cells were treated with 100 nM of dsRNA
compounds mixed with 5 μ§/ι Ι, LIPOFECTIN (Invitrogen Corporation, Carlsbad, CA) for a period of 16 hours. In the same experiment, HepG2 cells were also treated with 150 nM of subset of the antisense oligonucleotides described herein mixed with 3. 75 μg/mL LIPOFECTIN; these compounds can be listed in Table 43. Control oligonucleotides included ISIS 18078
(GTGCGCGCGAGCCCGAAATC, SEQ ID NO: 888). ISIS 18078 is a chimeric oligonucleotide ("gapmer") 20 nucleotides in length, composed of a central "gap" region consisting of 9 2'- deoxynucleotides, which is flanked on the 5' and 3' ends by a five-nucleotide "wing" and a six- nucleotide "wing", respectively. The wings can be composed of 2'-methoxyethyl (2'- MOE)nucleotides. The internucleoside (backbone) linkages can be phosphorothioate (P=S) throughout the oligonucleotide. All cytidines can be 5-methylcytidines.
[00919] The duplex of ISIS 263188 (CUUCUGGCAUCCGGUUUAGTT, SEQ ID NO: 889) and its complement was also used as a control. ISIS 263188 is an oligoribonucleotide 21 nucleotides in length with the 2 nucleotides on the 3 ' end being oligodeoxyribonucleotides (TT) and with phosphodiester internucleoside linkages (backbones) throughout the compound.
[00920] Cells were treated for 4 hours, after which human ApoB mRNA expression was measured as described by examples herein. Results were normalized to untreated control cells, which were not treated with LIPOFECTIN or oligonucleotide. Data can be the average of 4 experiments and can be presented in Table 43.
Table 43
Inhibition of ApoB mRNA by dsRNAs in
HepG2 cells

Dose SEQ ID #
# Inhibition
342868 100 nM 36 881
342869 100 nM 78 882
342870 100 nM 71 883
342871 100 nM 9 883
342872 100 nM 2 885
342873 100 nM 53 886
342874 100 nM 73 887
281625 150 nM 79 224
301012 150 nM 77 247
301014 150 nM 88 249
301021 150 nM 67 256
301027 150 nM 79 262
301028 150 nM 85 263
301029 150 nM 77 264
301030 150 nM 70 265
301031 150 nM 73 266
301037 150 nM 80 272
301038 150 nM 84 273
301045 150 nM 77 280
263188 150 nM 26 888
18078 150 nM 13 889
2.60 Example 61 - Antisense inhibition of ApoB in Cynomolgous monkey primary hepatocytes
[00921] As demonstrated in Example 46, the region containing the target site to which ISIS 301012 hybridizes to 96% identity with the corresponding region of Cynomolgus monkey ApoB mRNA sequence. ISIS 301012 contains two mismatched nucleotides relative to the
Cynomolgous monkey ApoB mRNA sequence to which it hybridizes. In a further embodiment , oligonucleotides were designed to target regions of the monkey ApoB mRNA, using the partial Cynomologous monkey ApoB sequence described herein (SEQ ID NO: 855) and an additional portion of Cynomolgous monkey ApoB RNA sequence, incorporated herein as SEQ ID NO: 890. The target site indicates the first (5 '-most) nucleotide number on the particular target sequence to which the oligonucleotide binds. For ISIS 326358
(GCCTCAGTCTGCTTTACACC, SEQ ID NO: 891) the target site is nucleotide 168 of SEQ ID NO: 855 and for ISIS 315089 (AG ATT AC C AGC C AT ATGC AG, SEQ ID NO: 892) the target site is nucleotide 19 of SEQ ID NO: 890. ISIS 326358 and ISIS 315089 can be chimeric oligonucleotides ("gapmers") 20 nucleotides in length, composed of a central "gap" region consisting of ten 2'-deoxynucleotides, which is flanked on both sides (5' and 3' directions) by
five-nucleotide "wings". The wings can be composed of 2'-methoxyethyl (2'-MOE)nucleotides. The internucleoside (backbone) linkages can be phosphorothioate (P=S) throughout the oligonucleotide. All cytidine residues can be 5-methylcytidines. ISIS 326358 and ISIS 315089 can be the Cynomolgous monkey equivalents of the human ApoB antisense oligonucleotides ISIS 301012 (SEQ ID NO: 247) and ISIS 281625 (SEQ ID NO: 224), respectively.
[00922] Antisense inhibition by ISIS 301012 was compared to that of ISIS 326358, which is a perfect match to the Cynomolgous monkey ApoB sequence to which ISIS 301012 hybridizes. The compounds were analyzed for their effect on Cynomolgous monkey ApoB mRNA levels in primary Cynomolgous monkey hepatocytes purchased from In vitro Technologies (Gaithersburg, MD). Pre-plated primary Cynonomolgous monkey hepatocytes were purchased from In Vitro Technologies (Baltimore, MD). Cells were cultured in high-glucose DMEM (Invitrogen
Corporation, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen Corporation, Carlsbad, CA), 100 units/mL and 100 μg/mL streptomycin (Invitrogen Corporation, Carlsbad, CA).
[00923] Primary Cynomolgous monkey hepatocytes were treated with 10, 50, 150 or 300 nM of antisense oligonucleotides for 48 hours. ISIS 113529 (SEQ ID NO: 859) was used as a control oligonucleotide. Untreated cells also served as a control. Cynomolgous monkey ApoB mRNA levels were quantitated by real-time PCR using the human ApoB and GAPDH primers and probes described by other examples herein. The results, shown in Table 44, can be the average of 6 experiments and can be expressed as percent inhibition of ApoB mRNA normalized to untreated control cells.
Table 44
Inhibition of Cynomolgous monkey ApoB mRNA by ISIS 301012 and ISIS 326358
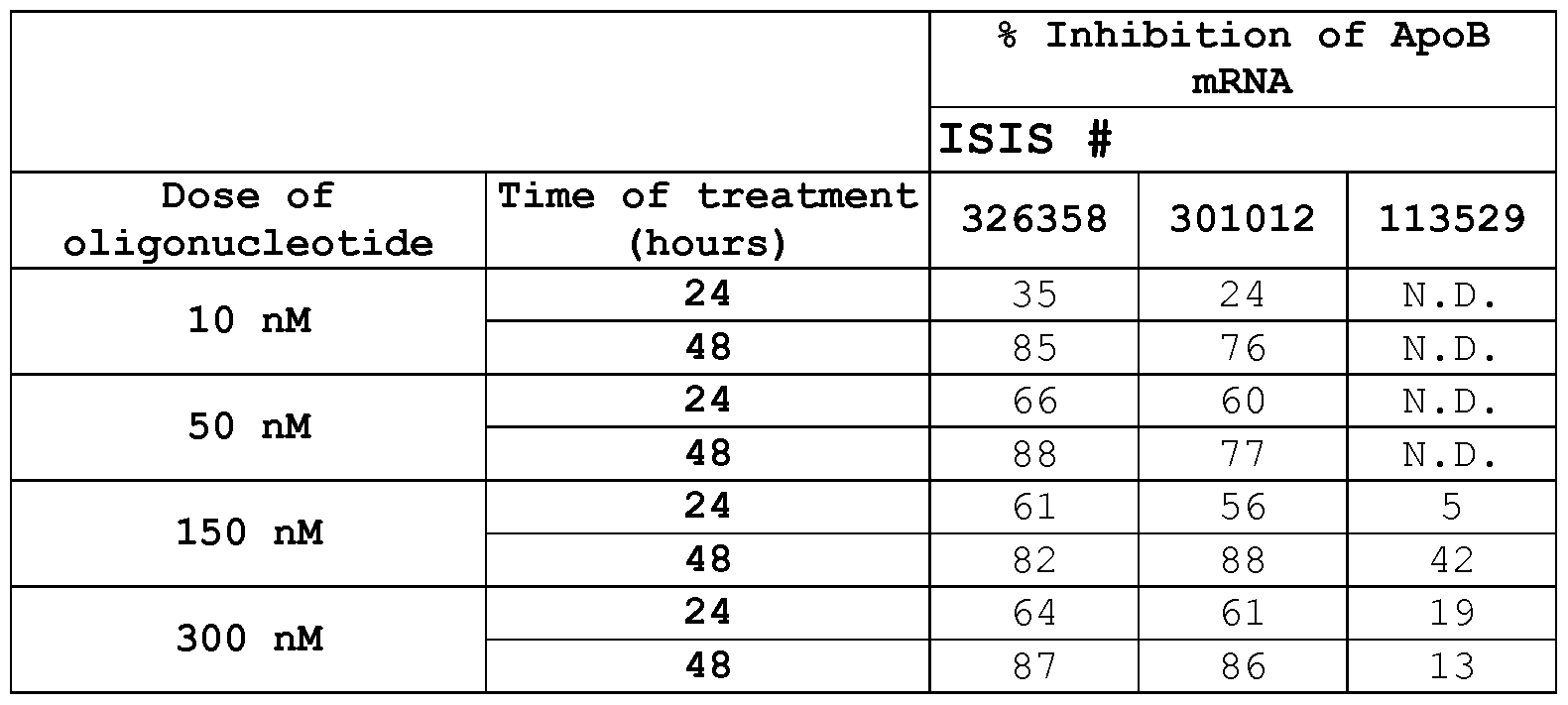
[00925] ApoB protein secreted from primary Cynomolgous hepatocytes treated with 150 and 300 nM of oligonucleotide was measured by ELISA using an ApoB protein specific kit
(ALerCHEK Inc., Portland, ME). Each result represents the average of 3 experiments. The data can be normalized to untreated control cells and can be shown in Table 45.
Table 45
Reduction in ApoB protein secreted from Cynomolgous monkey hepatocytes following antisense oligonucleotide treatment

[00926] These results demonstrate that antisense inhibition by ISIS 301012 and ISIS 326358 leads to a decrease in the secretion of ApoB protein from cultured primary Cynomolgous hepatocytes.
[00927] Additionally, protein was isolated from oligonucleotide-treated primary
Cynomolgous monkey hepatocytes and subjected to immunoblot analysis to further assess ApoB protein expression. Immunoblotting was performed as described herein, using an antibody to human ApoB protein (US Biologicals, Swampscott, MA). Immunoblot analysis of ApoB expression following antisense oligonucleotide treatment with ISIS 326358 and ISIS 301012 reveals a substantial reduction in ApoB expression.
[00928] In a further embodiment, antisense inhibition by ISIS 281625 was compared to that by ISIS 315089, which is a perfect match to the Cynomolgous monkey ApoB sequence to which
ISIS 281625 hybridizes. Primary Cynomolgous monkey hepatocytes, cultured as described herein, were treated with 10, 50, 150 or 300 nM of ISIS 315089 or ISIS 281625 for 24 hours. Cells were treated with the control oligonucleotide ISIS 13650 (SEQ ID NO: 806) at 150 and 300 nM or ISIS 113529 (SEQ ID NO: 859) at 300 nM. Untreated cells also served as a control. Cynomolgous monkey ApoB mRNA levels in primary Cynomolgous monkey hepatocytes was quantitated using real-time PCR with human primers and probe as described by other examples herein. The results, shown in Table 46, can be the average of 3 experiments and can be expressed as percent inhibition of ApoB mRNA normalized to untreated control cells. Where present, a "+" preceding the value indicates that mRNA expression was increased.
Table 46
Antisense inhibition of ApoB mRNA expression in Cynomolgous monkey hepatocytes

[00929] These data demonstrate that both ISIS 315089 and ISIS 281625 can inhibit the expression of ApoB mRNA in Cynomolgous monkey primary hepatocytes, in a dose-dependent manner.
[00930] ApoB protein secreted primary Cynomolgous hepatocytes treated with 50 and 150 nM of ISIS 315089 and ISIS 281625 was measured by ELISA using an ApoB protein specific kit (ALerCHEK Inc., Portland, ME). Each result represents the average of 3 experiments. The data can be normalized to untreated control cells and can be shown in Table 47.
Table 47
Reduction in ApoB protein secreted from Cynomolgous monkey hepatocytes following antisense oligonucleotide treatment
% Reduction of monkey ApoB
protein secretion
ISIS #
Dose of
315089 281625 13650 113529
oligonucleotide
50 nM 1 1 6 1 6 N . D .
150 nM 25 13 13 12
[00931] These results demonstrate that antisense inhibition by 150 nM of ISIS 315089 leads to a decrease in the secretion of ApoB protein from cultured primary Cynomolgous hepatocytes.
[00932] ISIS 271009 (SEQ ID NO: 319) and ISIS 301027 (SEQ ID NO: 262) were also tested for their effects on ApoB mRNA and protein expression in Cynomolgous primary hepatoctyes. Cells, cultured as described herein, were treated with 10, 50 and 150 nM of ISIS 271009 or ISIS 301027 for 24 hours. Cells were treated with the control oligonucleotide ISIS 113529 (SEQ ID NO: 859) at 150 nM. Untreated cells also served as a control. Cynomolgous monkey ApoB mRNA levels in primary Cynomolgous monkey hepatocytes was quantitated using real-time PCR with human primers and probe as described by other examples herein. The results, shown in Table 48, can be the average of 2 experiments and can be expressed as percent inhibition of ApoB mRNA normalized to untreated control cells.
Table 48
Antisense inhibition of ApoB mRNA expression in Cynomolgous monkey hepatocytes

[00933] These data demonstrate that both ISIS 271009 and ISIS 301027 can inhibit the expression of ApoB mRNA in Cynomolgous monkey primary hepatocytes, in a dose-dependent manner.
[00934] ApoB protein secreted from primary Cynomolgous hepatocytes treated with 50 and 150 nM of ISIS 271009 and ISIS 301027 was measured by ELISA using an ApoB protein
specific kit (ALerCHEK Inc., Portland, ME). Each result represents the average of 3 experiments. The data can be shown as percent reduction in secreted protein, normalized to untreated control cells, and can be shown in Table 49. Where present, a "+" indicates that protein secretion was increased.
Table 49
Reduction in ApoB protein secreted from Cynomolgous monkey hepatocytes following antisense oligonucleotide treatment

[00935] These results demonstrate that antisense inhibition by ISIS 315089 and ISIS 281625 leads to a decrease in the secretion of ApoB protein from cultured primary Cynomolgous hepatocytes.
2.61 Example 62 - Methods for evaluating hepatic steatosis
[00936] Hepatic steatosis refers to the accumulation of lipids in the liver, or "fatty liver", which is frequently caused by alcohol consumption, diabetes and hyperlipidemia. Livers of animals treated with antisense oligonucleotides targeted to ApoB were evaluated for the presence of steatosis. Steatosis is assessed by histological analysis of liver tissue and measurement of liver triglyceride levels.
[00937] Tissue resected from liver is immediately immersed in Tissue Tek OCT embedding compound (Ted Pella, Inc., Redding, CA) and frozen in a 2-methyl-butane dry ice slurry. Tissue sections can be cut at a thickness of 4-5 μιη and then fixed in 5% neutral-buffered formalin. Tissue sections can be stained with hematoxylin and eosin following standard histological procedures to visualize nuclei and cytoplasm, respectively, and oil red O according to the manufacturer's instructions (Newcomers Supply, Middleton, WI) to visualize lipids.
[00938] Alternatively, tissues can be fixed in 10% neutral-buffered formalin, embedded in paraffin, sectioned at a thickness of 4-5 μιη, deparaffinized and stained with hematoxylin and eosin, all according to standard histological procedures.
[00939] Quantitation of liver triglyceride content is also used to assess steatosis. Tissue triglyceride levels can be measured using a Triglyceride GPO Assay (Sigma- Aldrich, St. Louis, MO).
2.62 Example 63 - Effects of antisense inhibition by ISIS 301012 in lean mice: long-term study
[00940] In accordance with provided herein, the toxicity of ISIS 301012 (SEQ ID NO: 247) is investigated in a long-term, 3 month study in mice. Two-month old male and female CD-I mice (Charles River Laboratories, Wilmington, MA) can be dosed with 2, 5, 12. 5, 25 or 50 mg/kg of ISIS 301012 twice per week for first week, and every 4 days thereafter. The mice can be maintained on a standard rodent diet. Saline and control oligonucleotide animals serve as controls and can be injected on the same schedule. Each treatment group contains 6 to 10 mice of each sex, and each treatment group is duplicated, one group for a 1 month study termination, the other for a 3 month study termination. After the 1 or 3 month treatment periods, the mice can be sacrificed and evaluated for target expression in liver, lipid levels in serum and indicators of toxicity. Liver samples can be procured, RNA is isolated and ApoB mRNA expression is measured by real-time PCR as described in other examples herein. Serum lipids, including total cholesterol, LDL-cholesterol, HDL-cholesterol and triglycerides, can be evaluated by routine clinical analysis using an Olympus Clinical Analyzer (Olympus America Inc., Melville, NY). Ratios of LDL-cholesterol to HDL-cholesterol and total cholesterol to HDL-cholesterol can also be calculated. Analyses of serum ALT and AST, inflammatory infiltrates in tissue and basophilic granules in tissue provide an assessment of toxicities related to the treatment. Hepatic steatosis, or accumulation of lipids in the liver, is assessed by routine histological analysis with oil red O stain and measurement of liver tissue triglycerides using a Triglyceride GPO Assay (Sigma- Aldrich, St. Louis, MO).
[00941] The toxicity study also includes groups of animals allowed to recover following cessation of oligonucleotide treatment. Both male and female CD-I mice (Charles River
Laboratories, Wilmington, MA) can be treated with 5, 10, 50 mg/kg of ISIS 301012 twice per week for the first week and every 4 days thereafter. Saline and control oligonucleotide injected
animals serve as controls. Each treatment group includes 6 animals per sex. After 3 months of treatment, animals remain untreated for an additional 3 months, after which they can be sacrificed. The same parameters can be evaluated as in the mice sacrificed immediately after 3 months of treatment.
[00942] After one month of treatment, real-time PCR quantitation reveals that mouse ApoB mRNA levels in liver can be reduced by 53%. Additionally, the expected dose-response toxicities were observed. ALT and AST levels, measured by routine clinical procedures on an
Olympus Clinical Analyzer (Olympus America Inc., Melville, NY), can be increased in mice treated with 25 or 50 mg/kg of ISIS 301012. Tissues were prepared for analysis by routine histological procedures. Basophilic granules in liver and kidney tissue were observed at doses of
ISIS 301012 above 12. 5 mg/kg. Mild lymphohistiocytic infiltrates were observed in various tissues at doses greater than 12. 5 mg/kg of ISIS 301012. Staining of tissue sections with oil red
O reveals no steatosis present following the oligonucleotide treatments.
2.63 Example 64 - Effects of antisense inhibition by ISIS 301012 in lean
Cynomolgous monkeys: long-term study
[00943] As discussed in Example 45, Cynomolgus monkeys (male or female) can be used to evaluate antisense oligonucleotides for their potential to lower ApoB mRNA or protein levels, as well as phenotypic endpoints associated with ApoB including, but not limited to cardiovascular indicators, atherosclerosis, lipid diseases, obesity, and plaque formation. Accordingly, in a further embodiment , ISIS 301012 (SEQ ID NO: 247) is investigated in a long-term study for its effects on ApoB expression and serum lipids in Cynomolgous monkeys. Such a long-term study is also used to evaluate the toxicity of antisense compounds.
[00944] Male and female Cynomologous monkeys can be treated with 2, 4 or 12 mg/kg of ISIS 301012 intravenously or 2 or 20 mg/kg subcutaneously at a frequency of every two days for the first week, and every 4 days thereafter, for 1 and 3 month treatment periods. Saline-treated animals serve as controls. Each treatment group includes 2 to 3 animals of each sex.
[00945] At a one month interval and at the 3 month study termination, the animals can be sacrificed and evaluated for target expression in liver, lipid levels in serum and indicators of toxicity. Liver samples can be procured, RNA is isolated and ApoB mRNA expression is measured by real-time PCR as described in other examples herein. Serum lipids, including total cholesterol, LDL-cholesterol, HDL-cholesterol and triglycerides, can be evaluated by routine
clinical analysis using an Olympus Clinical Analyzer (Olympus America Inc., Melville, NY). Ratios of LDL-cholesterol to HDL-cholesterol and total cholesterol to HDL-cholesterol can also be calculated. Analyses of serum ALT and AST, inflammatory infiltrates in tissue and basophilic granules in tissue provide an assessment of toxicities related to the treatment. Hepatic steatosis, or accumulation of lipids in the liver, is assessed by routine histological analysis with oil red O stain and measurement of liver tissue triglycerides using a Triglyceride GPO Assay (Sigma- Aldrich, St. Louis, MO).
[00946] Additional treatment groups consisting of 2 animals per sex can be treated with saline (0 mg/kg), 12 or 20 mg/kg ISIS 301012 at a frequency of every two days for the first week, and every 4 days thereafter, for a 3 month period. Following the treatment period, the animals receive no treatment for an additional three months. These treatment groups can be for the purpose of studying the effects of ApoB inhibition 3 months after cessation of treatment. At the end of the 3 month recovery period, animals can be sacrificed and evaluated for the same parameters as the animals sacrificed immediately after 1 and 3 months of treatment.
[00947] The results from the one month interval of the long term treatment can be shown in Table 50 and can be normalized to saline-treated animals for mRNA and to untreated baseline values for lipid levels. Total cholesterol, LDL-cholesterol, HDL-cholesterol, LDL particle concentration and triglyceride levels in serum were measured by nuclear magnetic resonance spectroscopy by Liposcience (Raleigh, NC). Additionally, the concentration of intact
oligonucleotide in liver was measured by capillary gel electrophoresis and is presented as micrograms of oligonucleotide per gram of liver tissue. Each result represents the average of data from 4 animals (2 males and 2 females).
Table 50
Effects of antisense inhibition by ISIS 301012 in lean Cynomolgous monkeys

[00948] These data show that ISIS 301012 inhibits ApoB expression in a dose-dependent manner in a primate species and concomitantly lowers lipid levels at higher doses of ISIS 301012. Furthermore, these results demonstrate that antisense oligonucleotide accumulates in the liver in a dose-dependent manner.
[00949] Hepatic steatosis, or accumulation of lipids in the liver, was not observed following 4 weeks of treatment with the doses indicated. Expected dose-related toxicities were observed at the higher doses of 12 and 20 mg/kg, including a transient 1. 2-1. 3 fold increase in activated partial thromboplastin time (APTT) during the first 4 hours and basophilic granules in the liver and kidney (as assessed by routine histological examination of tissue samples). No functional changes in kidney were observed.
[00950] In a similar experiment, male and female Cynomolgous monkeys received an intravenous dose of ISIS 301012 at 4 mg/kg, every two days for the first week and every 4 days thereafter. Groups of animals were sacrificed after the first dose and the fourth dose, as well as 11, 15 and 23 days following the fourth and final dose. Liver RNA was isolated and ApoB mRNA levels were evaluated by real-time PCR as described herein. The results of this experiment demonstrate a 40% reduction in ApoB mRNA expression after a single intravenous
dose of 4 mg/kg ISIS 301012. Furthermore, after 4 doses of ISIS 301012 at 4 mg/kg, target mRNA was reduced by approximately 85% and a 50% reduction in target mRNA was sustained for up to 16 days following the cessation of antisense oligonucleotide treatment.
2.64 Example 65 - Microarray analysis: gene expression patterns in lean versus high-fat fed mice
[00951] Male C57B1/6 mice were divided into the following groups, consisting of 5 animals each: (1) mice on a lean diet, injected with saline (lean control); (2) mice on a high fat diet; (3) mice on a high fat diet injected with 50 mg/kg of the control oligonucleotide 141923 (SEQ ID NO: 858); (4) mice on a high fat diet given 20 mg/kg atorvastatin calcium (Lipitor®, Pfizer Inc. ); (5) mice on a high fat diet injected with 10, 25 or 50 mg/kg ISIS 147764 (SEQ ID NO: 109). Saline and oligonucleotide treatments were administered intraperitoneally twice weekly for 6 weeks. Atorvastatin was administered daily for 6 weeks. At study termination, liver samples were isolated from each animal and RNA was isolated for Northern blot qualitative assessment, DNA microarray and quantitative real-time PCR. Northern blot assessment and quantitative realtime PCR were performed as described by other examples herein.
[00952] For DNA microarray analysis, hybridization samples were prepared from 10 μg of total RNA isolated from each mouse liver according to the Affymetrix Expression Analysis Technical Manual (Affymetrix, Inc., Santa Clara, CA). Samples were hybridized to a mouse gene chip containing approximately 22,000 genes, which was subsequently washed and double- stained using the Fluidics Station 400 (Affymetrix, Inc., Santa Clara, CA) as defined by the manufacturer's protocol. Stained gene chips were scanned for probe cell intensity with the GeneArray scanner (Affymetrix, Inc., Santa Clara, CA). Signal values for each probe set were calculated using the Affymetrix Microarray Suite v5. 0 softwcan be (Affymetrix, Inc., Santa Clara, CA). Each condition was profiled from 5 biological samples per group, one chip per sample. Fold change in expression was computed using the geometric means of signal values as generated by Microarray Suite v5. 0. Statistical analysis utilized one-way ANOVA followed by 9 pair-wise comparisons. All groups were compared to the high fat group to determine gene expression changes resulting from ISIS 147764 treatment. Microarray data was interpreted using hierarchical clustering to visualize global gene expression patterns.
[00953] The results of the microarray analysis reveal that treatment with ISIS 147764 drives the gene expression profile in high fat fed mice to the profile observed in lean mice. Real-time
PCR analysis confirmed the reduction in mRNA expression for the following genes involved in the lipid metabolism: hepatic lipase, fatty acid synthase ATP -binding cassette, sub-family D (ALD) member 2, intestinal fatty acid binding protein 2, stearol CoA desaturase-1 and HMG Co A reductase.
[00954] Mouse ApoB mRNA and serum cholesterol levels, measured as described herein, were evaluated to confirm antisense inhibition by ISIS 147764 and ISIS 147483. Both mRNA and cholesterol levels were lowered in a dose-dependent manner following treatment with ISIS 147764 or ISIS 147483, as demonstrated in other examples herein. The 50 mg/kg dose of ISIS 147483 increased ALT and AST levels. The 10, 25 and 50 mg/kg doses of ISIS 147764 and the 10 and 25 mg/kg doses of ISIS 147483 did not significantly elevate ALT or AST levels.
2.65 Example 66
2.65.1. Evaluation of hepatic steatosis in animals treated with ApoB antisense oligonucleotides
[00955] Livers of animals treated with antisense oligonucleotides targeted to ApoB were evaluated for the presence of steatosis. Steatosis is assessed by histological analysis of liver tissue and measurement of liver triglyceride levels.
2.65.2. Evaluation of steatosis in high fat fed animals treated with ISIS
147764 for 6 weeks
[00956] Liver tissue from ISIS 147764 (SEQ ID NO: 109) and control-treated animals described in Example 21 was evaluated for steatosis at study termination following 6 weeks of treatment. Tissue sections were stained with oil red O and hematoxylin to visualize lipids and nuclei, respectively. Tissue sections were also stained with hematoxylin and eosin to visualize nuclei and cytoplasm, respectively. Histological analysis of tissue sections stained by either method reveal no difference in steatosis between saline treated and ISIS 147764 treated animals, demonstrating that a 6 week treatment with ISIS 147764 does not lead to accumulation of lipids in the liver.
2.65.3. Evaluation of steatosis following long-term treatment with ApoB
inhibitor in high-fat fed animals
[00957] Male C57B1/6 mice were treated with twice weekly intraperitoneal injections of 25 mg/kg ISIS 147764 (SEQ ID NO: 109) or 25 mg/kg ISIS 141923 (SEQ ID NO: 858) for 6, 12 and 20 weeks. Saline treated animals served as controls. Each treatment group contained 4
animals. Animals were sacrificed at 6, 12 and 20 weeks and liver tissue was procured for histological analysis and measurement of tissue triglyeride content. The results reveal no significant differences in liver tissue triglyceride content when ISIS 147764 treated animals can be compared to saline treated animals. Furthermore, histological analysis of liver tissue section demonstrates that steatosis is reduced at 12 and 20 weeks following treatment of high fat fed mice with ISIS 147764, in comparison to saline control animals that received a high fat diet.
2.65.4. Evaluation of steatosis in lean mice
[00958] The accumulation of lipids in liver tissue was also evaluated in lean mice. Male C67B1/6 mice (Charles River Laboratories (Wilmington, MA) at 6 to 7 weeks of age were maintained on a standard rodent diet and were treated twice weekly with intraperitoneal injections of 25 or 50 mg/kg 147764 (SEQ ID NO: 109) or 147483 (SEQ ID NO: 79) for 6 weeks. Saline treated animals served as controls. Each treatment group was comprised of 4 animals. Animals were sacrificed after the 6 week treatment period, at which point liver tissue and serum were collected.
[00959] ApoB mRNA levels were measured by real-time PCR as described by other examples herein. The data, shown in Table 51 , represent the average of 4 animals and can be presented as inhibition relative to saline treated controls. The results demonstrate that both ISIS 147483 and ISIS 147764 inhibit ApoB mRNA expression in lean mice in a dose-dependent manner.
Table 51
Antisense inhibition of ApoB mRNA in lean mice

[00960] Total cholesterol, LDL-cholesterol, HDL-cholesterol and triglycerides in serum were measured by routine clinical analysis using an Olympus Clinical Analyzer (Olympus America Inc., Melville, NY). The liver enzymes ALT and ALT in serum were also measured using the Olympus Clinical Analyzer. These results demonstrate that ISIS 147764 lowers serum lipids relative to saline-treated control animals. ALT and AST levels do not exceed the normal range
for mice (300 IU/L), indicating a lack of treatment-associated toxicity. The results can be the average of data from 4 animals and can be shown in Table 52.
Table 52
Serum lipids and liver enzyme levels in lean mice treated with ISIS 147764 and ISIS 147483

[00961] Liver tissue was prepared by routine histological methods to evaluate steatosis, as described herein. Examination of tissue samples stained with oil red O or hematoxylin and eosin reveals that treatment of lean mice with ApoB antisense oligonucleotides does not result in steatosis.
2.65.5. Six month study to further evaluate steatosis in mice treated with ApoB antisense oligonucleotides
[00962] A long-term treatment of mice with antisense oligonucleotides targeted to ApoB is used to evaluate the toxicological and pharmacological effects of extended treatment with antisense compounds. Both male and female C57B1/6 mice at 2 months of age can be treated with 2, 5, 25 or 50 mg/kg of ApoB antisense oligonucleotide. Treatments can be administered intraperitoneally every 2 days for the first week and every 4 days thereafter. Mice treated with saline alone or control oligonucleotide serve as control groups. Each treatment group contains 25 to 30 mice. After 6 months of treatment, a subset of the mice in each treatment group is
sacrificed. The remaining mice can be allowed a 3 month recovery period without treatment, after which they can be sacrificed. ApoB m NA expression in liver is measured by real-time
PCR as described by other methods herein. Liver tissue is also prepared for measurement of triglyceride content using a Triglyceride GPO Assay (Sigma-Aldrich, St. Louis, MO). Serum is collected and evaluated for lipid content, including total cholesterol, LDL-cholesterol, HDL- cholesterol and triglyceride, using an Olympus Clinical Analyzer (Olympus America Inc.,
Melville, NY). The liver enzymes ALT and AST can also be measured in serum, also using the clinical analyzer. Serum samples can be subjected to immunoblot analysis using an antibody directed to ApoB (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Liver, kidney and other tissues can be prepared by routine procedures for histological analyses. Tissues can be evaluated for the presence of basophilic granules and inflammatory infiltrates. Steatosis is evaluated by oil red O stain of liver tissue sections.
2.66 Example 67 - A mouse model for atherosclerotic plaque formation: human ApoB transgenic mice lacking the LDL receptor gene
[00963] The LDL receptor is responsible for clearing ApoB-containing LDL particles.
Without the LDL receptor, animals cannot effectively clear ApoB-containing LDL particles from the plasma. Thus the serum levels of ApoB and LDL cholesterol can be markedly elevated. Mice expressing the human ApoB transgene (TgN-hApoB +/+) and mice deficient for the LDL receptor (LDLr -/-) can be both used as animal models of atherosclerotic plaque development. When the LDL receptor deficiency genotype is combined with a human ApoB transgenic genotype (TgN-hApoB +/+; LDLr -/-), atherosclerotic plaques develop rapidly. In accordance with provided herein, mice of this genetic background can be used to investigate the ability of compounds to prevent atherosclerosis and plaque formation.
[00964] Male TgN-hApoB +/+;LDLr -/- mice can be treated twice weekly with 10 or 20 mg/kg of human ApoB antisense oligonucleotides for 12 weeks. Control groups can be treated with saline or control oligonucleotide. Serum total cholesterol, HDL-cholesterol, LDL- cholesterol and triglycerides can be measured at 2, 4, 6, 8 and 12 weeks by routine clinical analysis using an Olympus Clinical Analyzer (Olympus America Inc., Melville, NY). Serum human ApoB protein is measured at 2, 4, 6, 8 and 12 weeks using an ELISA kit (ALerCHEK Inc., Portland, ME). Human and mouse apolipoprotein mRNA in liver is measured at 12 weeks.
The results of the 12 week study serve to evaluate the pharmacological behavior of ISIS 301012 in a doubly transgenic model.
[00965] Additionally, a four month study is performed in TgN-hApoB +/+;LDLr -/- mice, with treatment conditions used in the 12 week study. Mice can be treated for 4 months with antisense oligonucleotides targeted to human ApoB to evaluate the ability of such compounds to prevent atherosclerotic plaque formation. At the end of the 4 month treatment period, mice can be anesthetized and perfused with 10% formalin. The perfused arterial tree is isolated and examined for the presence of atherosclerotic plaques. Sections of the arterial tree can be embedded in paraffin and prepared for histological analysis using routine methods. Serum total cholesterol, HDL-cholesterol, LDL-cholesterol and triglycerides can be measured at 2, 4, 6, 8, 12 and 16 weeks by routine clinical analysis using an Olympus Clinical Analyzer (Olympus America Inc., Melville, NY). Serum human ApoB protein is measured at 2, 4, 6, 8, 12 and 16 weeks using an ELISA kit (ALerCHEK Inc., Portland, ME). Human and mouse apolipoprotein mRNA in liver at 16 weeks is measured by real-time PCR.
2.67 Example 68 - Rabbit models for study of atherosclerotic plaque formation
[00966] The Watanabe heritable hyperlipidemic (WHHL) strain of rabbit is used as a model for atherosclerotic plaque formation. New Zealand white rabbits on a high-fat diet can also be used as a model of atherosclerotic plaque formation. Treatment of WHHL or high fat fed New Zealand white rabbits with ApoB antisense compounds is used to test their potential as therapeutic or prophylactic treatments for atherosclerotic plaque disease. Rabbits can be injected with 5, 10, 25 or 50 mg/kg of antisense oligonucleotides targeted to ApoB. Animals treated with saline alone or a control oligonucleotide serve as controls. Throughout the treatment, serum samples can be collected and evaluated for ApoB protein levels by ELISA (kit from ALerCHEK Inc., Portland, ME) and serum lipids (cholesterol, LDL-cholesterol, VLDL-cholesterol, HDL- cholesterol, triglycerides) by routine clinical analysis. Liver tissue triglyceride content is measured using a Triglyceride GPO Assay (Sigma-Aldrich, St. Louis, MO). Liver, kidney, heart, aorta and other tissues can be procured and processed for histological analysis using routine procedures. Liver and kidney tissues can be examined for evidence of basophilic granules and inflammatory infiltrates. Liver tissue is evaluated for steatosis using oil red O stain. Additionally, aortic sections stained with oil red O stain and hematoxylin can be examined to evaluate the formation of atherosclerotic lesions.
2.68 Example 69
2.68.1. Subjects
[00967] Three subjects were identified as having homozygous familial hypercholesterolemia using the following criteria: (1) documented history of LDL-C above 500 mg/dL in the absence of lipid- lowering therapy; and (2) at least one of (a) genetic testing confirming 2 mutated LDL- receptor genes; (b) tendinous and/or cutaneous xanthoma prior to age 10 years; or (c)
documentation of elevated LDL-C prior to lipid-lowering therapy consistent with heterozygous familial hypercholesterolemia in both parents (if a parent's medical history was not available, a history of coronary heart disease in a first degree male relative younger than 55 years old or first degree female relative younger than 60 years old was used as a criterion in place of a parent's medical history).
2.68.2. Dose Regimen
[00968] The three subjects received a 300 mg dose of ISIS 301012 on Days 1, 4, 8, 11, 15, 22, 29, 36, 43, 50, 57, 64, 71, 78, and 85, as summarized in the table, below. ISIS 301012 was administered subcutaneously. The four doses on Days 1, 4, 8, and 11 were administered to achieve estimated ISIS 301012 levels in liver tissue that are approximately 60-90% of steady- state concentration. The subjects were monitored for concentrations of ApoB, LDL-C, VLDL-C, non-HDL-C, HDL-C, ApoAl, total cholesterol, triglycerides, and Lp(a). Subjects were also monitored to ensure acceptable safety profiles. The subjects also received one or more lipid- lowering therapies.
Table 53
Co-administration of ISIS 301012 with one or more additional lipid-lowering therapies in subjects with homozygous familial hypercholesterolemia.

[00969] At Day 43, after 9 doses of 301012, both ApoB and LDL-C were reduced by approximately 30% (n=3) compared to the subject's concentration at baseline, prior to ISIS 301012 administration.
[00970] At Day 78, after 14 doses of ISIS 301012, (n=2; one subject had not yet received reached the 14th dose at time of this writing) both ApoB and LDL-C were reduced by
approximately 50%. VLDL-C concentration was reduced by approximately 30%. Non-HDL-C concentration reductions ranged from 32 to 50%. Total cholesterol concentration reductions ranged from 30% to 46%. Triglyceride concentration reductions ranged from 29% to 33%. Lp(a) concentration reductions of 19%>, 20%>, and 54% were observed. HDL-C concentration increases of 5%), 7%), and 40%> were observed.
[00971] These data demonstrate that ISIS 301012 lowered lipid concentrations in subjects having familial hypercholesterolemia. LDL-C reductions in subjects having familial
hypercholesterolemia were observed later during the administration period relative to LDL-C reductions in subjects having non-familial hypercholesterolemia, suggesting that a longer induction period in familial hypercholesterolemic subjects may provide a therapeutic benefit such subjects. For example, an induction period of at least 8 weeks may provide a therapeutic benefit to subjects having familial hypercholesterolemia, whereas shorter induction periods or induction periods with lower doses may be sufficient for subjects with non-familial
hypercholesterolemia.
2.69 Example 70 - Rate and Magnitude of ApoB reduction with ISIS 301012
[00972] Without being bound to a particular theory, it is believed that mild increases in ALT levels seen during initial treatment with ISIS 301012 in dose regimens having a high multi-dose loading period, reflect extreme lipid lowering activity. Particularly, the rise in ALT can be attributed to the rate and magnitude of lipid lowering. An ALT rise can be lessened or prevented by a dosing regimen that limits the rate and magnitude of ApoB reduction during the induction or initial dosing period, approximately the first 15 to 90 days of treatment. Thus, for the first time, the rate and magnitude of ApoB reduction has been correlated with ALT levels.
2.69.1. Subjects
[00973] Hypercholesterolemics on stable statin therapy. ApoB levels and ALT levels were determined for the subjects in the trial. Table 54 provides the ApoB levels for individual subjects and resulting ALT levels.
Table 54
ApoB and ALT Levels for Individual Subjects

2.69.2. Results
[00974] The data provided indicate that increases in ALT levels can occur when subjects fall below two thresholds, a minimum ApoB concentration of about 60 mg/dL or less during the first 15 to 60 days and an average rate of reduction of 1.5 mg/dL/day or greater within the first 15 to 60 days of treatment can result in an elevated ALT of about 100 U/L or greater. This may also be considered in terms of approximately a 15% drop per day or greater or a change of more than 30mg/dL over the first 30 days. An ALT of 100 U/L is considered greater than three times the
upper limit of normal. The upper limit of normal for ALT concentration is approximately 30 U/L. A value three times above such limit would be considered unfavorable.
[00975] Two hundred and forty four subjects from 6 different clinical trials including healthy volunteers, familial hypercholesterolemic subject, polygenic hypercholesterolemic subject and subject on statin therapy have been evaluated and threshold levels of ApoB have been associated with ALT levels. Table 54 shows the number of subjects having a positive slope (rate of reduction of ApoB greater than Img/dL/day) and who violated the threshold for magnitude of reduction (ApoB concentration of less than 60mg/dL). 80% of such subjects (32 of 41) experienced and ALT rise of 3XULN or above.
Table 55
Slope and Threshold Analysis

N = 244
18(0) - uninterpretable due to borderline threshold levels
2.70 Example 71 - Reduction in ApoB with ISIS 301012 in Subjects with
Polygenic Hypersholeterolemia
2.70.1. Subjects
[00976] Subjects were identified as having polygenic hypercholesterolemia. Subjects typically had LDL-C levels greater than about 130 mg/dL in the absence of lipid-lowering therapy.
2.70.2. Dosing
[00977] Three groups of 8 patients each were dosed. The groups were dosed with 200, 300 or 400 mg of ISIS 301012 once per week for 13 weeks with no initial loading. ISIS 301012 was administered by subcutaneous injection.
Table 56
ApoB levels and and ALT Elevations

*ALT elevations > 3XULN on two consecutive measurements at least 7 days apart
**4 of 8 patients reached the lower limit of detection for ApoB
2.70.3. Results
[00978] The drop in ApoB from baseline at 2 weeks for the 200 and 300 mg/kg dose groups resulted, on average in a rate and/or magnitude drop that did not meet or exceed the threshold requirements identified in example 2. There were no ALT elevations in the three treatment groups during the dosing period. The drop in ApoB from baseline at 2 weeks for the 400 mg/kg dose group resulted on average in a rate and/or magnitude drop that did exceed the threshold requirements identified in example 2 and resulted in ALT increases 3XULN.
2.71 Example 72 - Predictive Effect of Long Induction at High Dose
2.71.1. Subjects
[00979] Subjects are identified as having familial or polygenic hypercholesterolemia.
Subjects typically have LDL-C levels greater than about 120 mg/dL in the absence of lipid- lowering therapy.
2.71.2. Dose Regimen
[00980] Subjects are initially dosed using a long induction at higher dose then the
maintenance dose. Group A receives a 200 mg dose of ISIS 301012 once a week for 13 weeks. Group B receives a 300 mg dose of ISIS 301012 once a week for 13 weeks. After 13 weeks, subjects are placed on a reduced maintenance dose regimen. Group A receives a 100 mg dose of ISIS 301012 once weekly and Group B receives a 200 mg dose of ISIS 301012 once weekly. ISIS 301012 can be administered subcutaneously or by any other method provided herein. The 13 week induction doses are administered to achieve estimated ISIS 301012 levels in liver tissue that are approximately 60-90% of steady- state concentration. The subjects are monitored for concentrations of ApoB, LDL-C, VLDL-C, non-HDL-C, HDL-C, ApoAl, total cholesterol, triglycerides, and Lp(a). Subjects are also monitored to ensure acceptable safety profiles including ALT, AST and bilirubin.
Table 57
Administration of ISIS 301012

[00981] Although 100 and 200 mg/wk are exemplified above, maintenance doses can be higher or lower. Table 55, 56, 57 and 58 provide predicted values based on modeled dosing regimens. The models are based on the clinical trial data obtained to date and particularly the polygenic monotherapy trials in Example 71. Based on an induction of either 200 or 300 mg/wk, ApoB and LDL levels as well as plasma trough and liver concentrations are predicted for the induction and maintenance phases,
Table 58
Predicted Effect (%change from baseline) at end of 13-week 200 mg/wk induction and 13-week maintenance

Table 59
Predicted PK at end of 13 -week 200 mg/wk induction and 13 -week maintenance

2.71.3. Group A Results
[00982] At the end of the 13 week induction phase, the mean liver concentration of 301012 in Group A is predicted to be approximately 123 ug/g. This concentration is maintained at a maintenance dose of about 100 mg/wk. At the end of the 13 week induction, the mean percent change in ApoB levels from baseline is approximately 46.9%. At week 13 of maintenance, the mean percent change in ApoB levels from baseline is maintained. At the end of the 13 week induction, the mean percent change in LDL levels from baseline is approximately 43.3%. At week 13 of maintenance, the mean percent change in LDL levels from baseline is maintained. At the end of the 13 week induction phase, plasma trough concentration is approximately 24.5 ng/mL. At week 13 of maintenance, the plasma trough concentration is maintained.
Table 60
Predicted Effect (%change from baseline) at end of 13-week 300 mg induction and 13-week maintenance

Table 61
Predicted PK at end of 13 -week 300 mg induction (based on polygenic monotherapy trials in
Example 71) and 13 -week maintenance

2.71.4. Group B Results
[00983] At the end of the 13 week induction phase, the mean liver concentration of 301012 in Group B is predicted to be approximately 184 ug/g. This concentration is maintained at a maintenance dose of about 200 mg/wk. At the end of the 13 week induction, the mean percent change in ApoB levels from baseline is approximately 56%. At week 13 of maintenance, the mean percent change in ApoB levels from baseline is maintained. At the end of the 13 week induction, the mean percent change in LDL levels from baseline is approximately 52.5%. At week 13 of maintenance, the mean percent change in LDL levels from baseline is maintained. At the end of the 13 week induction phase, plasma trough concentration is approximately 36.8 ng/mL. At week 13 of maintenance, the plasma trough concentration is maintained.
2.72 Example 73 - Predictive Effect of Long Induction at Low Dose
2.72.1. Subjects
[00984] Subjects are identified as having polygenic or familial hypercholesterolemia. Subjects typically have LDL-C levels greater than about 130 mg/dL in the absence of lipid-lowering therapy.
2.72.2. Dose Regimen
[00985] Subjects are initially dosed using a long induction at lower dose then the maintenance dose. Group A receives a 100 mg dose of ISIS 301012 once a week for 13 weeks. Group B receives a 200 mg dose of ISIS 301012 once a week for 13 weeks. After 13 weeks, subjects are evaluated based on tolerability and effectiveness with respect to treatment goals. If dosing is well tolerated and treatment goals are not being met, patients are placed on an elevated maintenance dose regimen. Group A receives a 200 mg dose of ISIS 301012 once weekly and Group B receives a 300 mg dose of ISIS 301012 once weekly. ISIS 301012 is administered subcutaneously. The 13 week induction doses are administered to achieve estimated ISIS 301012 levels in liver tissue that are approximately 60-90% of steady- state concentration. The subjects are monitored for concentrations of ApoB, LDL-C, VLDL-C, non-HDL-C, HDL-C, ApoAl, total cholesterol, triglycerides, and Lp(a). Subjects were also monitored to ensure acceptable safety profiles (including ALT, AST and bilirubin.
Table 62
Administration of ISIS 301012

[00986] Table 62 and 63 provide predicted values based on a modeled dosing regimen. The model is based on the clinical trial data obtained to date and particularly the polygenic monotherapy trials in Example 71. Based on an induction of either 100 mg/wk, ApoB and LDL levels as well as plasma trough and liver concentrations are predicted for the induction and maintenance phases,
Table 63
Predicted Effect (%change from baseline) for a 6-month trial with a 13 week priming regimen of
100 mg/wk and a 26 week maintenance of 200 mg/wk

Table 64
Predicted PK for a 6-month trial with a 13 week priming regimen and a 26 week maintenance

2.72.3. Group A Results
[00987] At the end of the 13 week induction phase, the mean liver concentration of 301012 in Group A is predicted to be approximately 61.3 ug/g. This concentration is increased to 141 after 13 weeks of the 200 mg/wk maintenance dose. At the end of the 13 week induction, the mean percent change in ApoB levels from baseline is approximately 28.4%. At week 26 of dosing, the mean percent change in ApoB levels from baseline is 53.1%. At the end of the 13 week induction, the mean percent change in LDL levels from baseline is approximately 24.5%. After 13 weeks of maintenance, the mean percent change in LDL levels from baseline is increased to 49.6%. At the end of the 13 week induction phase, plasma trough concentration is
approximately 12.3 ng/mL. At week 13 of maintenance, the plasma trough concentration is predicted to increased to 28.2 ng/mL.
2.73 Example 74 - Once Every Other Week Interval Dosing
2.73.1. Subjects
[00988] Subjects were identified as having polygenic hypercholesterolemia. Subjects typically had LDL-C levels greater than about 120 mg/dL in the absence of lipid-lowering therapy.
2.73.2. Dose Regimen
[00989] ISIS 301012 was administered by s.c. injection at 200 mg twice weekly for two weeks followed by a dose of 200 mg every other week for 11 weeks.
Table 65
ApoB levels at Two and Fourteen Weeks of Treatment

*ALT elevations > 3XULN
**(% change from baseline)
2.73.3. Results
[00990] The results show the effectiveness of delivering ISIS 301012 in a once every other week regimen with no associated ALT elevations.
2.74 Example 75 - Predicted Effect of Once Every Other Week Interval Dosing With Induction Phase
2.74.1. Subjects
[00991] Subjects are identified as having familial hypercholesterolemia and represent a lower tolerance population such as diabetics and mix-hyperlipidemics. Subjects typically have LDL-C levels greater than about 130 mg/dL in the absence of lipid- lowering therapy.
2.74.2. Dose regimen
[00992] ISIS 301012 is administered by s.c. injection at a dose of 200 mg every week for 13 weeks and then 200 mg every other week for 52 weeks.
Table 66
Predicted Effect (%change from baseline) for a 6-month trial with a 13 week induction of 200 mg/wk and a predicted 13 week maintenance of 200 mg every other week

Table 67
Predicted PK for a 6-month trial with a 13 week induction of 200 mg/wk and a predicted 13 week maintenance of 200 mg every other week

2.74.3. Results
[00993] The results show the delivering ISIS 301012 in a once every other week regimen can be and effective regimen and is not likely to result in elevated ALT levels.
2.75 Example 76 - Predicted Effect of Once Monthly Interval Dosing With Induction Phase
2.75.1. Subjects
[00994] Subjects are identified as having familial hypercholesterolemia and represent a lower tolerance population such as diabetics and mix-hyperlipidemics. Subjects typically have LDL-C levels greater than about 130 mg/dL in the absence of lipid- lowering therapy.
2.75.2. Dose Regimen
[00995] ISIS 301012 is administered by s.c. injection or by any method provided herein. In certain embodiments, subjects can be dosed at 200, 400 or 800 mg every other month for 12 months. In other embodiments, subjects are dosed at 200 mg/wk for 1 month and then 200, 400
or 600 mg once monthly for 11 months. In other embodiments, subjects are dosed at 200 mg/wk for 3 month and then 200, 400 or 600 mg once monthly for 9 months.
2.75.3. Results
[00996] The results predict that delivering ISIS 301012 in a once monthly regimen with or without an induction period can be an effective regimen and is not likely to result in elevated ALT levels.
2.76 Example 77 - Decreased Cardiovascular Events in Familial
Hypercholesterolemic Patients Treated with Mipomersen, an Antisense Inhibitor of Apolipoprotein B Translation
2.76.1. Background
[00997] Familial hypercholesterolemia (FH) is associated with a 10-20 fold increase in cardiovascular (CV) events. Mipomersen significantly lowers levels of atherogenic lipoproteins in plasma. Previous safety analysis of all patients in phase 3 trials found no imbalance in CV events between placebo and mipomersen arms.
[00998] In this study, it was investigated whether taking mipomersen for at least 1 year would reduce CV events in FH patients taking maximally tolerated lipid-lowering therapy.
2.76.2. Study Design
[00999] Randomized, double blinded, placebo controlled study was conducted. All patients provided written, informed consent before the start of the study.
[001000] Patients, investigator staff, persons performing the assessment, and data analysts remained blinded to the identity of the treatment from the time of randomization until data base lock. Treatments were all identical in packaging, labeling, schedule of administration and appearance.
2.76.3. Patients and Study Procedure
[001001] Rates of MACE during 2 years prior to mipomersen treatment were compared to MACE after 1 year of mipomersen treatment in 104 FH patient who participated in one of three phase 3 blinded randomized placebo-controlled 6-month trials and an open-label extension study (NCT00607373, NCT00706849, NCT00794664, NCT00694109). In each study, patients received 200 mg (1 ml) (or 160 mg (0.8 ml) in patients weighing less than 50 kg) mipomersen (ISIS 301012) subcutaneous injection per week. Negative control patients received the same amount/volume of a placebo s.c. per week. Patient subpopulations analyzed: homozygous FH
((NCT00607373); heterozygous FH with CAD (NCT00706849); severe hypercholesterolemia (NCT00794664); hypercholesterolemia (NCT00694109).
[001002] Retrospective, explanatory analyses were performed using datasets from phase 3 clinical trials involving 4 distinct populations, as described above, to determine if any differences exist between mipomersen-treated patients and placebo-treated patients with respect to MACE.
[001003] One third of patients (n=34) received placebo for the initial 6 months followed by mipomersen for at least 1 year. Two thirds (n=70) received blinded mipomersen for 6 months followed by at least 6 months in open label treatment. In this study, MACE were defined as nonfatal myocardial infarction, stroke, unstable angina, and revascularization procedures
(percutaneous coronary intervention (PCI), coronary artery bypass graft (CABG) surgery).
[001004] MACE occurring before randomization were identified in medical history records.
[001005] On- study MACE, including those for placebo-treated patients, were adjudicated by an independent committee.
2.76.4. Results
[001006] MACE were identified in 62% of patients (64 patients with 146 events [39 MI, 99 PCI/CABG, 5 UA, 3 stroke]) during 24 months prior to mipomersen treatment, and 9% of patients (9 patients with 12 events [2 UA+MI, 6 PCI/CABG, 4 UA]) during a mean of 24. 4 months after initiation of mipomersen treatment (MACE rate 25. 7/1000 patient-months vs 3. 6/1000 patient-months, OR = 0. 035 [95% CI 0. 009 - 0. 144], p<0. 0001 by exact McNemar's test). The marked reduction in MACE coincided with the absolute refers to reductions in LDL cholesterol levels (-49 to -113 mg/dL) reported for the phase 3 FH clinical trials.
[001007] MACE rates were significantly lower in 104 FH patients treated with mipomersen for 1 year versus the rate 24 months prior to treatment. Indeed, MACE rates were remarkably lower during mipomersen treatment compared with 24 months prior to mipomersen (p<0. 0001).
Table 68
MACE rate reduced in FH patients after 1 year of treatment with mipomersen

[001008] Thus, results from this analysis suggest that treatment with mipomersen reduces cardiovascular events in patients with FH. ALT levels.
2.77 Example 78 - Decreased Cardiovascular Events in Familial
Hypercholesterolemic Patients Treated with Mipomersen, an Antisense Inhibitor of Apolipoprotein B Translation
2.77.1. Patients and Study Procedure
[001009] FIG. 1 depicts the general study design of Phase 3 randomized, placebo-controlled studies.
[001010] Rates of MACE during 2 years prior to mipomersen treatment were compared to MACE after 1 year of mipomersen treatment in 104 FH patient who participated in one of three phase 3 blinded randomized placebo-controlled 6-month trials and an open-label extension study (NCT00607373, NCT00706849, NCT00794664, NCT00694109). In each study, patients received 200 mg (1 ml) (or 160 mg (0.8 ml) in patients weighing less than 50 kg) mipomersen (ISIS 301012) subcutaneous injection per week. Negative control patients received the same amount/volume of a placebo s.c. per week. Patient subpopulations analyzed: homozygous FH ((NCT00607373); heterozygous FH with CAD (NCT00706849); severe hypercholesterolemia (NCT00794664); hypercholesterolemia (NCT00694109).
[001011] Retrospective, explanatory analyses were performed using datasets from phase 3 clinical trials involving 4 distinct populations, as described above, to determine if any differences exist between mipomersen-treated patients and placebo-treated patients with respect to MACE.
[001012] One third of patients (n=34) received placebo for the initial 6 months followed by mipomersen for at least 1 year. Two thirds (n=70) received blinded mipomersen for 6 months followed by at least 6 months in open label treatment. In this study, MACE wee defined as nonfatal myocardial infarction, stroke, unstable angina, and revascularization procedures
(percutaneous coronary intervention (PCI), coronary artery bypass graft (CABG) surgery).
[001013] MACE occurring before randomization were identified in medical history records.
[001014] On-study MACE, including those for placebo-treated patients, were adjudicated by an independent committee.
2.77.2. Results
[001015] As shown in Table 69, MACE rate was significantly reduced in FH patients treatd with mipomersen for at least one year.
Table 69
MACE rate reduced in FH patients after at least 1 year of treatment with mipomersen

[001017] In summary, these results show that patients treated with mipomersen for at least 1 year (mean 2 years) had a significantly reduced incidence of MACE compared to 2 years prior to therapy
[001018] Moreover, LDL-C reduction with mipomersen appears to be associated with improved cardiovascular outcomes in patients with familial hypercholesterolemia.
2.78 Example 79: A Study of the Safety and Efficacy of Two Different Regimens of Mipomersen in Patients With Familial Hypercholesterolemia and Inadequately Controlled Low-Density Lipoprotein Cholesterol
2.78.1. Patients and Study Procedure
[001019] The study consisted of a Screening period of up to 4 weeks, blinded treatment phase of 60 weeks, open-label continuation period of 26 weeks, and post-treatment phase of 24 weeks.
[001020] Study Design, masking - Study treatment was blinded (double-blinded) through the primary efficacy assessment visit in the blinded treatment period. Study treatment is open-label in the Open-Label continuation period. The two experimental consisted of a blinded 200 mg (lml) once weekly mipomersen sodium, subcutantous injections administered once every other week for the first 8 weeks followed by once every week for 52 weeks. Placebo comparator of subcutaneous injection (lml) administered once every other week for the first 8 weeks followed by once every week for 52 weeks. Second experimental arm of mipomersen sodium 70 mg thrice weekly, 70 mg (0.5ml), subcutaneous injections administered three times a week every other week for the first 8 weeks followed by three times a week every week for 52 weeks.
Placebo Comparator: Placebo thrice weekly.
[001021] Placebo subcutaneous injection (0.5 mL) three times a week every other week for the first 8 weeks followed by three times every week for 52 weeks.
[001022] Primary objective:: Determine whether mipomersen (ISIS 301012) significantly reduces atherogenic lipid levels in patients with severe heterozygous familial
hypercholesterolemia (severe HeFH), defined as low-density lipoprotein cholesterol (LDL-C) levels ^200 mg/dL plus the presence of coronary heart disease (CHD)/risk equivalents or LDL-
C levels ^300 mg/dL regardless of the presence of CHD/risk equivalents (referred to as Cohort 1) compared to placebo. Two different mipomersen dosing regimens were studied: subcutaneous (SC) mipomersen 200 mg once weekly versus placebo, and SC mipomersen 70 mg thrice weekly versus placebo.
[001023] Secondary Objectives: Determine whether there are qualitative differences between the safety profiles of the 2 dosing regimens and placebo in Cohort 1 , patients with HeFH with LDL-C levels ^ 160 mg/dL and <200 mg/dL plus the presence of CHD/risk equivalents (referred to as Cohort 2), and the overall study population.
[001024] Determine whether there are qualitative differences between the tolerability of the 2 dosing regimens and placebo in Cohort 1, Cohort 2, and the overall study population
[001025] Further characterize the pharmacokinetics (PK) of the 2 dosing regimens in Cohort 1, Cohort 2, and the overall study population.
[001026] Determine whether the 2 mipomersen dosing regimens significantly reduce atherogenic lipid levels in Cohort 2 compared to placebo.
[001027] Additional data regarding ongoing safety and efficacy of mipomersen in patients with FH and inadequately controlled LDL-C who complete the primary efficacy assessment visit (PET) in the Blinded Treatment Period and continue treatment in Open-Label Continuation Period was obtained.
[001028] Results: It is expected that those pateints who received mipomersen in conjuction with or in the absence of statin therapy in both dosing regimes will have the prolonged benefit of a reduction in MACE events.
SEQUENCE LISTING
[001029] The present specification is being filed with a computer readable form (CRF) copy of the Sequence Listing. The CRF entitled 10103-069-888_SEQLIST.txt, which was created on December 4, 2014 and is 343,213 bytes in size, is identical to the paper copy of the Sequence Listing and is incorporated herein by reference in its entirety.
Claims
1. A method of treating, preventing, or managing a major adverse cardiovascular event (MACE) in a hypercholesterolemia patient in need thereof, wherein the method comprises administering to the patient a therapeutically effect amount of an antisense olionucleotide complementary to a nucleic acid encoding human apolipoprotein B.
2. The method of claim 1, wherein the MACE is a myocardial infarction, reinfarction, stroke, unstable angina, cardiogenic shock, pulmonary edema, cardiac arrest, coronary revascularization, investigational angioplasty, interventional angioplasty, a percutaneous transluminal coronary angioplasty, percutaneous coronary intervention, a coronary artery bypass graft, or any combination thereof.
3. The method of claim 2, wherein the MACE is myocardial infarction.
4. The method of any one of claims 1 to 3, wherein the patient is statin-resistant or statin intolerant.
5. The method of any one of claims lto 4, wherein the oligonucleotide is administered for at least 12 months.
6. The method of any one of claims 1 to 5, wherein the oligonucleotide is administered for 12 to 24 months.
7. The method of any one of claims 1 to 6, wherein the patient has an established cardiovascular disease.
8. The method of any one of claims 1 to 7, wherein the administration rsults in reversed cardiac injury.
9. The method any one of claims 1 to 8, wherein the patient is homozygous for familial hypercholestrolemia.
10. The method any one of claims 1 to 8, wherein the patient is heterozygous for familial hypercholestrolemia.
11. The method of claim 10, wherein the patient is heterozygous for familial hypercholestrolemia with coronary artery disease.
12. The method of any one of claims 1 to 8, wherein the patient has severe
hypercholesterolemia.
13. The method of any one of claims 1 to 12, wherein the patient has not previously been treated for MACE.
14. The method of any one of claims 1 to 12, wherein the patient has been previously been treated for MACE.
15. The method of any one of claims 1 to 12, wherein the method reduces the occurrence of or prevents MACE in a patient having an established CVD.
16. The method of any claims 1 to 12, wherein the method reduces the occurrence of MACE in a patient at risk of CVD.
17. The method of any one of claims 1 to Error! Reference source not found., wherein the antisense oligonucleotide is 20 nucleobases in length.
18. The method of any one of claims 1 to Error! Reference source not found., wherein the antisense oligonucleotide has a nucleobase sequence consisting of the nucleobase sequence of GCCTCAGTCTGCTTCGCACC (SEQ ID NO: 247).
19. The method of any one of claims Error! Reference source not found, to 18, wherein the antisense oligonucleotide comprises a modified internucleoside linkage, a modified sugar moiety, a modified nucleobase, or a combination thereof.
20. The method of claim 19, wherein the modified sugar moiety is a 2'-0-methoxyethyl sugar moiety or a bicyclic sugar moiety.
21. The method of claim 19 or 20, wherein the modified internucleoside linkage is a phosphorothioate linkage.
22. The method of any one of claims 20 to 21, wherein the modified nucleobase is a 5- methylcytosine.
23. The method of any one of claims Error! Reference source not found, to Error! Reference source not found., wherein the antisense oligonucleotide is a chimeric
oligonucleotide.
24. The method of claim 23, wherein the chimeric oligonucleotide comprises a gap segment often linked 2'-deoxynucleotides, wherein the gap segment is positioned between wing segments, wherein each nucleoside of each wing segment comprises a modified sugar moiety.
25. The method of claim 24, wherein the modified sugar moiety is a 2 '-O-methoxy ethyl sugar moiety.
26. The method of claim Error! Reference source not found, or Error! Reference source not found., wherein the gap segment is 10 2'-deoxynucleosides in length, and each wing segment comprises from 1 to 8 2'-0-methoxyethyl.
27. The method of claim Error! Reference source not found., wherein each wing segment comprises 2'-methoxyethoxyl nucleotides.
28. The method of any one of claims 1 to 27, wherein the antisense oligonucleotide is an antisense oligonucleotide 20 nucleotides in length having the nucleobase sequence of
GCCTCAGTCTGCTTCGCACC (SEQ ID NO: 247), and comprising a 5-methylcytosine at nucleobases 2, 3, 5, 9, 12, 15, 17, 19, and 20, wherein every internucleoside linkage is a phosphorothioate linkage, nucleotides 1-5 and 16-20 can be 2 '-O-methoxy ethyl nucleotides, and nucleotides 6-15 can be 2'-deoxynucleotides, or wherein said antisense oligonucleotide is a pharmaceutically acceptable salt form thereof.
29. The method any one of claims 1 to 28, wherein the antisense oligonucleotide is administered in a dosage to achieve at least a 60% reduction in MACE.
30. The method of any one of claims 1 to 29, wherein the antisense oligonucleotide is administered in a dosage to achieve at least a 65%, at least a 70%, at least a 75%, at least a 80%, at least a 85%, or at least a 90% reduction in MACE.
31. The method of any one of claims 1 to 30, wherein the antisense oligonucleotide is administered at 200 mg per week.
32. The method of any one of claims 1-31, wherein the administering comprises an induction phase, wherein a 210 mg dose of the antisense oligonucleotide per week is administered in two or more administrations for at least 13 weeks, followed by a maintenance phase, wherein a 210 mg dose of the antisense oligonucleotide per week is administered in two or more administrations.
33. The method of any one of claims lto 32, wherein the patient has a reduction of serum cholesterol, ApoB, serum low density lipoprotein (LDL), serum very low density lipoprotein (VLDL), serum triglycerides, serum apolipoprotein (a) and/or free fatty acids after
administration of the antisence oligonucleotide.
34. A method of treating, preventing, or managing a major adverse cardiovascular event (MACE) in a patient comprising; selecting a patient having a disease or condition that increases the risk of MACE, and administering to the patient a therapeutically effect amount of an antisense olionucleotide complementary to a nucleic acid encoding human apolipoprotein B.
35. The method of claim 34, wherein the patient has a history of MACE.
36. The method of any one of claims 34 to 35, wherein the MACE is a myocardial infarction, reinfarction, stroke, unstable angina, cardiogenic shock, pulmonary edema, cardiac arrest, coronary revascularization, investigational angioplasty, interventional angioplasty, a percutaneous transluminal coronary angioplasty, percutaneous coronary intervention, a coronary artery bypass graft, or any combination thereof.
37. The method any one of claims 34 to 36, wherein the MACE is myocardial infarction.
38. The method of any one of claims 34 to 37, wherein the patient is statin-resistant or statin intolerant.
39. The method of any one of claims 34 to 38, wherein the oligonucleotide is administered for at least 12 months.
40. The method of any one of claims 34 to 39, wherein the oligonucleotide is administerd for 12 to 24 months.
41. The method of any one of claims 34 to 40, wherein the patient has an established cardiovascular disease.
42. The method of any one of claims 34 to 41, wherein the administration rsults in reversed cardiac injury.
43. The method of any one of claims 34 to 42, wherein the patient is homozygous for FH.
44. The method of any one of claims 34 to 43, wherein the patient is heterozygous for FH.
45. The method of claim 44, wherein the patient is heterozygous for FH with coronary artery disease.
46. The method of any one of claims 34 to 45, wherein the patient has severe
hypercholesterolemia.
47. The method of any one of claims 34 to 46, wherein the patient has not previously been treated for MACE.
48. The method of any one of claims 34 to 46, wherein the patient has been previously been treated for MACE.
49. The method of of any one of claims 34 to 46, wherein the method reduces the occurrence of or prevents MACE in a patient having an established CVD.
50. The method of any one of claims 34 to 49, wherein the method reduces the occurrence of MACE in a patient at risk of CVD.
51. The method of any one of claims 34 to 50, wherein the antisense oligonucleotide is 20 nucleobases in length.
52. The method of any one of claims 34 to 51 , wherein the antisense oligonucleotide is less than 20 nucleobases in length.
53. The method of any one of claims 34 to 52, wherein the antisense oligonucleotide has a nucleobase sequence consisting of the nucleobase sequence of SEQ ID NO: 247.
54. The method of any one of claims 34 to 53, wherein the antisense oligonucleotide comprises a modified internucleoside linkage, a modified sugar moiety, a modified nucleobase, or a combination thereof.
55. The method of claim 54, wherein the modified sugar moiety is a 2'-0-methoxyethyl sugar moiety or a bicyclic sugar moiety.
56. The method of claim 54 or 55, wherein the modified internucleoside linkage is a phosphorothioate linkage.
57. The method of any one of claims 54 to 56, wherein the modified nucleobase is a 5- methylcytosine.
58. The method of any one of claims 34 to 57, wherein the antisense oligonucleotide is a chimeric oligonucleotide.
59. The method of claim 58, wherein the chimeric oligonucleotide comprises a gap segment often linked 2'-deoxynucleotides, wherein the gap segment is positioned between wing segments, wherein each nucleoside of each wing segment comprises a modified sugar moiety.
60. The method of claim 59, wherein the modified sugar moiety is a 2'-0-methoxyethyl sugar moiety.
61. The method of claim 59 or 60, wherein the gap segment is 10 2'-deoxynucleosides in length, and each wing segment comprises from 1 to 8 2'-0-methoxyethyl.
62. The method of claim 61, wherein each wing segment comprises 2'-methoxyethoxyl nucleotides.
63. The method of any one of claims 34 to 62, wherein the antisense oligonucleotide is an antisense oligonucleotide 20 nucleotides in length having the nucleobase sequence of SEQ ID NO: 247, and comprising a 5-methylcytosine at nucleobases 2, 3, 5, 9, 12, 15, 17, 19, and 20,
wherein every internucleoside linkage is a phosphorothioate linkage, nucleotides 1-5 and 16-20 can be 2'-0-methoxyethyl nucleotides, and nucleotides 6-15 can be 2'-deoxynucleotides, or wherein said antisense oligonucleotide is a pharmaceutically acceptable salt form thereof.
64. The method any one of claims 34 to 63, wherein the antisense oligonucleotide is administered in a dosage to achieve at least a 60% reduction in MACE.
65. The method of any one of claims 34 to 64, wherein the antisense oligonucleotide is administered in a dosage to achieve at least a 65%, at least a 70%, at least a 75%, at least a 80%, at least a 85%, or at least a 90% reduction in MACE.
66. The method of any one of claims 34 to 65, wherein the antisense oligonucleotide is administered at 200 mg per week.
67. The method of any one of claims 34 to 66, wherein the administering comprises an induction phase, wherein a 210 mg dose of the antisense oligonucleotide per week is
administered in two or more administrations for at least 13 weeks, followed by a maintenance phase, wherein a 210 mg dose of the antisense oligonucleotide per week is administered in two or more administrations.
68. A method of treating, preventing, or managing a major adverse cardiovascular event (MACE) in a patient with familial hypercholesterolemia (FH) in need thereof, wherein the method comprises administering to the patient a therapeutically effective amount of an antisense compound having a nucleobase sequence of SEQ ID NO: 247.
69. The method of claim 68, wherein the MACE is a myocardial infarction, reinfarction, stroke, unstable angina, cardiogenic shock, pulmonary edema, cardiac arrest, atrial dysrhythmia, coronary revascularization, investigational angioplasty, interventional angioplasty, a
percutaneous transluminal coronary angioplasty, percutaneous coronary intervention, a coronary artery bypass graft, or any combination thereof.
70. The method any one of claims 68 to 69, wherein the patient is homozygous for FH.
71. The method any one of claims 68 to 70, wherein the patient is heterozygous for FH.
72. The method of claim 71, wherein the patient is heterozygous for FH with coronary artery disease.
73. The method of any one of claims 68 to 71, wherein the patient has severe
hypercholesterolemia.
74. The method of claim 73, wherein the patient has a high risk of CVD.
75. The method of any one of claims 68 to 74, wherein the patient has not previously been treated for MACE.
76. The method of any one of claims 68 to 75, wherein the patient has been previously been treated for MACE.
77. The method of any one of claims 68 to 76, wherein the method reduces the occurrence of or prevents MACE in a patient having an established CVD.
78. The method of any claims 68 to 77, wherein the method reduces the occurrence of MACE in a patient at risk of CVD.
79. The method of anyone of claims 68 to Error! Reference source not found., wherein the antisense compound is an antisense oligonucleotide.
80. The method of any one of claims 68 to 79, wherein the antisense compound is 20 nucleobases in length.
81. The method of claim Error! Reference source not found., wherein the antisense compound is an antisense oligonucleotide 20 nucleobases in length.
82. The method of any one of claims 68 to Error! Reference source not found., wherein the antisense compound has a nucleobase sequence consisting of the nucleobase sequence of SEQ ID NO: 247.
83. The method of any one of claims 79 to 82, wherein the antisense compound comprises a modified internucleoside linkage, a modified sugar moiety, a modified nucleobase, or a combination thereof.
84. The method of claim 83, wherein the modified sugar moiety is a 2'-0-methoxyethyl sugar moiety or a bicyclic sugar moiety.
85. The method of claim Error! Reference source not found, or Error! Reference source not found., wherein the modified internucleoside linkage is a phosphorothioate linkage.
86. The method of any one of claims Error! Reference source not found, to 85, wherein the modified nucleobase is a 5-methylcytosine.
87. The method of any one of claims 83 to 86, wherein the antisense oligonucleotide is a chimeric oligonucleotide.
88. The method of claim 87, wherein the chimeric oligonucleotide comprises a gap segment often linked 2'-deoxynucleotides, wherein the gap segment is positioned between wing segments, wherein each nucleoside of each wing segment comprises a modified sugar moiety.
89. The method of claim Error! Reference source not found., wherein the modified sugar moiety is a 2'-0-methoxyethyl sugar moiety.
90. The method of claim Error! Reference source not found, or Error! Reference source not found., wherein the gap segment is 10 2'-deoxynucleosides in length, and each wing segment comprises from 1 to 8 2'-0-methoxyethyl.
91. The method of claim Error! Reference source not found., wherein each wing segment comprises 2'-methoxyethoxyl nucleotides.
92. The method of any one of claims 68 to Error! Reference source not found., wherein the antisense compound is an antisense oligonucleotide 20 nucleotides in length having the nucleobase sequence of SEQ ID NO: 247, and comprising a 5-methylcytosine at nucleobases 2, 3, 5, 9, 12, 15, 17, 19, and 20, wherein every internucleoside linkage is a phosphorothioate linkage, nucleotides 1-5 and 16-20 can be 2'-0-methoxyethyl nucleotides, and nucleotides 6-15 can be 2'-deoxynucleotides, or wherein said antisense oligonucleotide is a pharmaceutically acceptable salt form thereof.
93. The method any one of claims 68 to Error! Reference source not found., wherein the antisense compound is administered in a dosage to achieve at least a 60% reduction in MACE.
94. The method of claim 93, wherein the antisense compound is administered in a dosage to achieve at least a 65%, at least a 70%, at least a 75%, at least a 80%, at least a 85%, or at least a 90% reduction in MACE.
95. The method of claim Error! Reference source not found, or 94, wherein the antisense compound is administered at 200 mg per day.
96. The method of claim 93 or 94, wherein the antisense compound is administered at 200 mg per week.
97. The method any one of claims 68 to 96, wherein the antisense compound is
administered for at least 12 months.
98. The method of any one of claims 1 to 97, wherein administration of the antisense compound decreases total serum cholesterol, ApoB , serum LDL, serum VLDL, serum triglycerides, serum apolipoprotein(a) and/or free fatty acids in the animal.
99. The method of claim 98, wherein administration of the antisense compound decreases LDL cholesterol.
100. The method of claim 98, wherein the LDL level is reduced to about 100 mg/dl or lower, about 70 mg/dl or lower, or about 50 mg/dl or lower.
101. The method of any one of claims 1-100, wherein administration of the antisense oligonucleotide cause reductions in atherogenic lipoproteins in plasma.
102. The method of any one of claims 1-101, wherein the antisense oligonucleotide is administered with one or more additional compounds selected from the group consisting of angiotensin-converting-enzyme inhibitors, microsomal triglyceride transfer protein inhibitor (MTP inhibitor), angiotensin receptor blockers, renin inhibitors, HMG CoA reductase inhibitors, dihydropyridine calcium channel blockers, antiarrhythmic agents, azetidinone-based cholesterol absorption inhibitors, PCSK9 inhibitor, niacin, niacin derivatives, PPAR agonists, PPAR
antagonists, bile acid sequestrants; and antiplatelet drugs; or any pharmaceutically acceptable esters, derivatives, conjugates, precursors or salts thereof.
103. The method of any one of claims 1 to 102, wherein the method reduces the occurrence of MACE as compared to a patient that has not been administered the antisense oligonucleotide.
104. The method of claim 103, wherein the patient that has not received the antisense oligonucleotide is a patient that has been administered a placebo.
105. The method of any one of claims 1 to 104, wherein the method reduces the occurrence of MACE as compared to the occurrence of the MACE prior to administration of the antisense oligonucleotide.
106. The method of claim 105, wherein the reduction in the occurrence of the MACE is as compared to the occurrence of the MACE in the 24 months prior to administration of the antisense oligonucleotide.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462043989P | 2014-08-29 | 2014-08-29 | |
| US62/043,989 | 2014-08-29 | ||
| US201462087712P | 2014-12-04 | 2014-12-04 | |
| US62/087,712 | 2014-12-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016033424A1 true WO2016033424A1 (en) | 2016-03-03 |
Family
ID=54066237
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2015/047372 WO2016033424A1 (en) | 2014-08-29 | 2015-08-28 | Methods for the prevention and treatment of major adverse cardiovascular events using compounds that modulate apolipoprotein b |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2016033424A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4035659A1 (en) | 2016-11-29 | 2022-08-03 | PureTech LYT, Inc. | Exosomes for delivery of therapeutic agents |
Citations (225)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US568194A (en) | 1896-09-22 | Skylight-support | ||
| US3687808A (en) | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| US4426330A (en) | 1981-07-20 | 1984-01-17 | Lipid Specialties, Inc. | Synthetic phospholipid compounds |
| US4469863A (en) | 1980-11-12 | 1984-09-04 | Ts O Paul O P | Nonionic nucleic acid alkyl and aryl phosphonates and processes for manufacture and use thereof |
| US4476301A (en) | 1982-04-29 | 1984-10-09 | Centre National De La Recherche Scientifique | Oligonucleotides, a process for preparing the same and their application as mediators of the action of interferon |
| US4501729A (en) | 1982-12-13 | 1985-02-26 | Research Corporation | Aerosolized amiloride treatment of retained pulmonary secretions |
| US4534899A (en) | 1981-07-20 | 1985-08-13 | Lipid Specialties, Inc. | Synthetic phospholipid compounds |
| US4587044A (en) | 1983-09-01 | 1986-05-06 | The Johns Hopkins University | Linkage of proteins to nucleic acids |
| US4605735A (en) | 1983-02-14 | 1986-08-12 | Wakunaga Seiyaku Kabushiki Kaisha | Oligonucleotide derivatives |
| US4667025A (en) | 1982-08-09 | 1987-05-19 | Wakunaga Seiyaku Kabushiki Kaisha | Oligonucleotide derivatives |
| WO1988004924A1 (en) | 1986-12-24 | 1988-07-14 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US4762779A (en) | 1985-06-13 | 1988-08-09 | Amgen Inc. | Compositions and methods for functionalizing nucleic acids |
| US4818542A (en) | 1983-11-14 | 1989-04-04 | The University Of Kentucky Research Foundation | Porous microspheres for drug delivery and methods for making same |
| US4824941A (en) | 1983-03-10 | 1989-04-25 | Julian Gordon | Specific antibody to the native form of 2'5'-oligonucleotides, the method of preparation and the use as reagents in immunoassays or for binding 2'5'-oligonucleotides in biological systems |
| US4828979A (en) | 1984-11-08 | 1989-05-09 | Life Technologies, Inc. | Nucleotide analogs for nucleic acid labeling and detection |
| US4835263A (en) | 1983-01-27 | 1989-05-30 | Centre National De La Recherche Scientifique | Novel compounds containing an oligonucleotide sequence bonded to an intercalating agent, a process for their synthesis and their use |
| US4837028A (en) | 1986-12-24 | 1989-06-06 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US4845205A (en) | 1985-01-08 | 1989-07-04 | Institut Pasteur | 2,N6 -disubstituted and 2,N6 -trisubstituted adenosine-3'-phosphoramidites |
| US4876335A (en) | 1986-06-30 | 1989-10-24 | Wakunaga Seiyaku Kabushiki Kaisha | Poly-labelled oligonucleotide derivative |
| US4885172A (en) | 1985-06-26 | 1989-12-05 | The Liposome Company, Inc. | Composition for targeting, storing and loading of liposomes |
| US4904582A (en) | 1987-06-11 | 1990-02-27 | Synthetic Genetics | Novel amphiphilic nucleic acid conjugates |
| WO1990004384A1 (en) | 1988-10-20 | 1990-05-03 | Royal Free Hospital School Of Medicine | Liposomes |
| US4948882A (en) | 1983-02-22 | 1990-08-14 | Syngene, Inc. | Single-stranded labelled oligonucleotides, reactive monomers and methods of synthesis |
| US4958013A (en) | 1989-06-06 | 1990-09-18 | Northwestern University | Cholesteryl modified oligonucleotides |
| US4981957A (en) | 1984-07-19 | 1991-01-01 | Centre National De La Recherche Scientifique | Oligonucleotides with modified phosphate and modified carbohydrate moieties at the respective chain termini |
| WO1991005545A1 (en) | 1989-10-20 | 1991-05-02 | Liposome Technology, Inc. | Liposome microreservoir composition and method |
| US5013830A (en) | 1986-09-08 | 1991-05-07 | Ajinomoto Co., Inc. | Compounds for the cleavage at a specific position of RNA, oligomers employed for the formation of said compounds, and starting materials for the synthesis of said oligomers |
| US5023243A (en) | 1981-10-23 | 1991-06-11 | Molecular Biosystems, Inc. | Oligonucleotide therapeutic agent and method of making same |
| US5034506A (en) | 1985-03-15 | 1991-07-23 | Anti-Gene Development Group | Uncharged morpholino-based polymers having achiral intersubunit linkages |
| US5059421A (en) | 1985-07-26 | 1991-10-22 | The Liposome Company, Inc. | Preparation of targeted liposome systems of a defined size distribution |
| US5082830A (en) | 1988-02-26 | 1992-01-21 | Enzo Biochem, Inc. | End labeled nucleotide probe |
| US5109124A (en) | 1988-06-01 | 1992-04-28 | Biogen, Inc. | Nucleic acid probe linked to a label having a terminal cysteine |
| US5108921A (en) | 1989-04-03 | 1992-04-28 | Purdue Research Foundation | Method for enhanced transmembrane transport of exogenous molecules |
| US5112963A (en) | 1987-11-12 | 1992-05-12 | Max-Planck-Gesellschaft Zur Foerderung Der Wissenschaften E.V. | Modified oligonucleotides |
| US5118800A (en) | 1983-12-20 | 1992-06-02 | California Institute Of Technology | Oligonucleotides possessing a primary amino group in the terminal nucleotide |
| US5118802A (en) | 1983-12-20 | 1992-06-02 | California Institute Of Technology | DNA-reporter conjugates linked via the 2' or 5'-primary amino group of the 5'-terminal nucleoside |
| US5130302A (en) | 1989-12-20 | 1992-07-14 | Boron Bilogicals, Inc. | Boronated nucleoside, nucleotide and oligonucleotide compounds, compositions and methods for using same |
| US5134066A (en) | 1989-08-29 | 1992-07-28 | Monsanto Company | Improved probes using nucleosides containing 3-dezauracil analogs |
| US5138045A (en) | 1990-07-27 | 1992-08-11 | Isis Pharmaceuticals | Polyamine conjugated oligonucleotides |
| US5149797A (en) | 1990-02-15 | 1992-09-22 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of rna and production of encoded polypeptides |
| US5166315A (en) | 1989-12-20 | 1992-11-24 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5171578A (en) | 1985-06-26 | 1992-12-15 | The Liposome Company, Inc. | Composition for targeting, storing and loading of liposomes |
| US5175273A (en) | 1988-07-01 | 1992-12-29 | Genentech, Inc. | Nucleic acid intercalating agents |
| US5177198A (en) | 1989-11-30 | 1993-01-05 | University Of N.C. At Chapel Hill | Process for preparing oligoribonucleoside and oligodeoxyribonucleoside boranophosphates |
| US5177196A (en) | 1990-08-16 | 1993-01-05 | Microprobe Corporation | Oligo (α-arabinofuranosyl nucleotides) and α-arabinofuranosyl precursors thereof |
| US5185444A (en) | 1985-03-15 | 1993-02-09 | Anti-Gene Deveopment Group | Uncharged morpolino-based polymers having phosphorous containing chiral intersubunit linkages |
| US5188897A (en) | 1987-10-22 | 1993-02-23 | Temple University Of The Commonwealth System Of Higher Education | Encapsulated 2',5'-phosphorothioate oligoadenylates |
| US5194599A (en) | 1988-09-23 | 1993-03-16 | Gilead Sciences, Inc. | Hydrogen phosphonodithioate compositions |
| US5214134A (en) | 1990-09-12 | 1993-05-25 | Sterling Winthrop Inc. | Process of linking nucleosides with a siloxane bridge |
| US5214136A (en) | 1990-02-20 | 1993-05-25 | Gilead Sciences, Inc. | Anthraquinone-derivatives oligonucleotides |
| US5216141A (en) | 1988-06-06 | 1993-06-01 | Benner Steven A | Oligonucleotide analogs containing sulfur linkages |
| US5218105A (en) | 1990-07-27 | 1993-06-08 | Isis Pharmaceuticals | Polyamine conjugated oligonucleotides |
| US5220007A (en) | 1990-02-15 | 1993-06-15 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of RNA and production of encoded polypeptides |
| US5223618A (en) | 1990-08-13 | 1993-06-29 | Isis Pharmaceuticals, Inc. | 4'-desmethyl nucleoside analog compounds |
| US5225212A (en) | 1989-10-20 | 1993-07-06 | Liposome Technology, Inc. | Microreservoir liposome composition and method |
| US5227170A (en) | 1989-06-22 | 1993-07-13 | Vestar, Inc. | Encapsulation process |
| US5235033A (en) | 1985-03-15 | 1993-08-10 | Anti-Gene Development Group | Alpha-morpholino ribonucleoside derivatives and polymers thereof |
| US5245022A (en) | 1990-08-03 | 1993-09-14 | Sterling Drug, Inc. | Exonuclease resistant terminally substituted oligonucleotides |
| US5254469A (en) | 1989-09-12 | 1993-10-19 | Eastman Kodak Company | Oligonucleotide-enzyme conjugate that can be used as a probe in hybridization assays and polymerase chain reaction procedures |
| US5256775A (en) | 1989-06-05 | 1993-10-26 | Gilead Sciences, Inc. | Exonuclease-resistant oligonucleotides |
| US5258506A (en) | 1984-10-16 | 1993-11-02 | Chiron Corporation | Photolabile reagents for incorporation into oligonucleotide chains |
| US5262536A (en) | 1988-09-15 | 1993-11-16 | E. I. Du Pont De Nemours And Company | Reagents for the preparation of 5'-tagged oligonucleotides |
| US5264221A (en) | 1991-05-23 | 1993-11-23 | Mitsubishi Kasei Corporation | Drug-containing protein-bonded liposome |
| US5264564A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences | Oligonucleotide analogs with novel linkages |
| US5264562A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences, Inc. | Oligonucleotide analogs with novel linkages |
| US5264423A (en) | 1987-03-25 | 1993-11-23 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| WO1993024510A1 (en) | 1992-05-25 | 1993-12-09 | Centre National De La Recherche Scientifique (Cnrs) | Phosphotriester-type biologically active compounds |
| US5272250A (en) | 1992-07-10 | 1993-12-21 | Spielvogel Bernard F | Boronated phosphoramidate compounds |
| US5276019A (en) | 1987-03-25 | 1994-01-04 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| WO1994000203A1 (en) | 1992-06-30 | 1994-01-06 | Velke, Willi, H. | Tennis racquet having a surface structure adapted for the reduction of air resistance |
| US5278302A (en) | 1988-05-26 | 1994-01-11 | University Patents, Inc. | Polynucleotide phosphorodithioates |
| WO1994002501A1 (en) | 1992-07-23 | 1994-02-03 | Isis Pharmaceuticals, Inc. | Novel 2'-o-alkyl nucleosides and phosphoramidites processes for the preparation and uses thereof |
| WO1994002499A1 (en) | 1992-07-27 | 1994-02-03 | Hybridon, Inc. | Oligonucleotide alkylphosphonothioates |
| US5292873A (en) | 1989-11-29 | 1994-03-08 | The Research Foundation Of State University Of New York | Nucleic acids labeled with naphthoquinone probe |
| US5317098A (en) | 1986-03-17 | 1994-05-31 | Hiroaki Shizuya | Non-radioisotope tagging of fragments |
| US5319080A (en) | 1991-10-17 | 1994-06-07 | Ciba-Geigy Corporation | Bicyclic nucleosides, oligonucleotides, process for their preparation and intermediates |
| US5321131A (en) | 1990-03-08 | 1994-06-14 | Hybridon, Inc. | Site-specific functionalization of oligodeoxynucleotides for non-radioactive labelling |
| WO1994017093A1 (en) | 1993-01-25 | 1994-08-04 | Hybridon, Inc. | Oligonucleotide alkylphosphonates and alkylphosphonothioates |
| WO1994020073A1 (en) | 1993-03-03 | 1994-09-15 | Liposome Technology, Inc. | Lipid-polymer conjugates and liposomes |
| US5354844A (en) | 1989-03-16 | 1994-10-11 | Boehringer Ingelheim International Gmbh | Protein-polycation conjugates |
| US5356633A (en) | 1989-10-20 | 1994-10-18 | Liposome Technology, Inc. | Method of treatment of inflamed tissues |
| US5359044A (en) | 1991-12-13 | 1994-10-25 | Isis Pharmaceuticals | Cyclobutyl oligonucleotide surrogates |
| US5367066A (en) | 1984-10-16 | 1994-11-22 | Chiron Corporation | Oligonucleotides with selectably cleavable and/or abasic sites |
| WO1994026764A1 (en) | 1993-05-12 | 1994-11-24 | Centre National De La Recherche Scientifique (Cnrs) | Triester phosphorothioate oligonucleotides and method of preparation |
| US5371241A (en) | 1991-07-19 | 1994-12-06 | Pharmacia P-L Biochemicals Inc. | Fluorescein labelled phosphoramidites |
| US5378825A (en) | 1990-07-27 | 1995-01-03 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs |
| US5386023A (en) | 1990-07-27 | 1995-01-31 | Isis Pharmaceuticals | Backbone modified oligonucleotide analogs and preparation thereof through reductive coupling |
| US5391723A (en) | 1989-05-31 | 1995-02-21 | Neorx Corporation | Oligonucleotide conjugates |
| US5399331A (en) | 1985-06-26 | 1995-03-21 | The Liposome Company, Inc. | Method for protein-liposome coupling |
| US5399676A (en) | 1989-10-23 | 1995-03-21 | Gilead Sciences | Oligonucleotides with inverted polarity |
| US5403711A (en) | 1987-11-30 | 1995-04-04 | University Of Iowa Research Foundation | Nucleic acid hybridization and amplification method for detection of specific sequences in which a complementary labeled nucleic acid probe is cleaved |
| US5405939A (en) | 1987-10-22 | 1995-04-11 | Temple University Of The Commonwealth System Of Higher Education | 2',5'-phosphorothioate oligoadenylates and their covalent conjugates with polylysine |
| US5405938A (en) | 1989-12-20 | 1995-04-11 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5414077A (en) | 1990-02-20 | 1995-05-09 | Gilead Sciences | Non-nucleoside linkers for convenient attachment of labels to oligonucleotides using standard synthetic methods |
| US5417978A (en) | 1993-07-29 | 1995-05-23 | Board Of Regents, The University Of Texas System | Liposomal antisense methyl phosphonate oligonucleotides and methods for their preparation and use |
| US5432272A (en) | 1990-10-09 | 1995-07-11 | Benner; Steven A. | Method for incorporating into a DNA or RNA oligonucleotide using nucleotides bearing heterocyclic bases |
| US5434257A (en) | 1992-06-01 | 1995-07-18 | Gilead Sciences, Inc. | Binding compentent oligomers containing unsaturated 3',5' and 2',5' linkages |
| US5446137A (en) | 1993-12-09 | 1995-08-29 | Syntex (U.S.A.) Inc. | Oligonucleotides containing 4'-substituted nucleotides |
| US5451463A (en) | 1989-08-28 | 1995-09-19 | Clontech Laboratories, Inc. | Non-nucleoside 1,3-diol reagents for labeling synthetic oligonucleotides |
| US5457187A (en) | 1993-12-08 | 1995-10-10 | Board Of Regents University Of Nebraska | Oligonucleotides containing 5-fluorouracil |
| US5459255A (en) | 1990-01-11 | 1995-10-17 | Isis Pharmaceuticals, Inc. | N-2 substituted purines |
| US5459127A (en) | 1990-04-19 | 1995-10-17 | Vical, Inc. | Cationic lipids for intracellular delivery of biologically active molecules |
| US5462854A (en) | 1993-04-19 | 1995-10-31 | Beckman Instruments, Inc. | Inverse linkage oligonucleotides for chemical and enzymatic processes |
| US5466786A (en) | 1989-10-24 | 1995-11-14 | Gilead Sciences | 2'modified nucleoside and nucleotide compounds |
| US5466677A (en) | 1993-03-06 | 1995-11-14 | Ciba-Geigy Corporation | Dinucleoside phosphinates and their pharmaceutical compositions |
| US5470307A (en) | 1994-03-16 | 1995-11-28 | Lindall; Arnold W. | Catheter system for controllably releasing a therapeutic agent at a remote tissue site |
| US5470967A (en) | 1990-04-10 | 1995-11-28 | The Dupont Merck Pharmaceutical Company | Oligonucleotide analogs with sulfamate linkages |
| US5469854A (en) | 1989-12-22 | 1995-11-28 | Imarx Pharmaceutical Corp. | Methods of preparing gas-filled liposomes |
| US5476925A (en) | 1993-02-01 | 1995-12-19 | Northwestern University | Oligodeoxyribonucleotides including 3'-aminonucleoside-phosphoramidate linkages and terminal 3'-amino groups |
| US5484908A (en) | 1991-11-26 | 1996-01-16 | Gilead Sciences, Inc. | Oligonucleotides containing 5-propynyl pyrimidines |
| US5486603A (en) | 1990-01-08 | 1996-01-23 | Gilead Sciences, Inc. | Oligonucleotide having enhanced binding affinity |
| US5489677A (en) | 1990-07-27 | 1996-02-06 | Isis Pharmaceuticals, Inc. | Oligonucleoside linkages containing adjacent oxygen and nitrogen atoms |
| US5491133A (en) | 1987-11-30 | 1996-02-13 | University Of Iowa Research Foundation | Methods for blocking the expression of specifically targeted genes |
| US5502177A (en) | 1993-09-17 | 1996-03-26 | Gilead Sciences, Inc. | Pyrimidine derivatives for labeled binding partners |
| US5506351A (en) | 1992-07-23 | 1996-04-09 | Isis Pharmaceuticals | Process for the preparation of 2'-O-alkyl guanosine and related compounds |
| WO1996010391A1 (en) | 1994-09-30 | 1996-04-11 | The University Of British Columbia | Polyethylene glycol modified ceramide lipids and liposome uses thereof |
| US5508270A (en) | 1993-03-06 | 1996-04-16 | Ciba-Geigy Corporation | Nucleoside phosphinate compounds and compositions |
| US5510475A (en) | 1990-11-08 | 1996-04-23 | Hybridon, Inc. | Oligonucleotide multiple reporter precursors |
| US5512295A (en) | 1994-11-10 | 1996-04-30 | The Board Of Trustees Of The Leland Stanford Junior University | Synthetic liposomes for enhanced uptake and delivery |
| US5512667A (en) | 1990-08-28 | 1996-04-30 | Reed; Michael W. | Trifunctional intermediates for preparing 3'-tailed oligonucleotides |
| US5512439A (en) | 1988-11-21 | 1996-04-30 | Dynal As | Oligonucleotide-linked magnetic particles and uses thereof |
| US5514785A (en) | 1990-05-11 | 1996-05-07 | Becton Dickinson And Company | Solid supports for nucleic acid hybridization assays |
| US5519126A (en) | 1988-03-25 | 1996-05-21 | University Of Virginia Alumni Patents Foundation | Oligonucleotide N-alkylphosphoramidates |
| US5519134A (en) | 1994-01-11 | 1996-05-21 | Isis Pharmaceuticals, Inc. | Pyrrolidine-containing monomers and oligomers |
| US5521291A (en) | 1991-09-30 | 1996-05-28 | Boehringer Ingelheim International, Gmbh | Conjugates for introducing nucleic acid into higher eucaryotic cells |
| US5525711A (en) | 1994-05-18 | 1996-06-11 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Pteridine nucleotide analogs as fluorescent DNA probes |
| US5525465A (en) | 1987-10-28 | 1996-06-11 | Howard Florey Institute Of Experimental Physiology And Medicine | Oligonucleotide-polyamide conjugates and methods of production and applications of the same |
| US5527528A (en) | 1989-10-20 | 1996-06-18 | Sequus Pharmaceuticals, Inc. | Solid-tumor treatment method |
| US5534259A (en) | 1993-07-08 | 1996-07-09 | Liposome Technology, Inc. | Polymer compound and coated particle composition |
| US5539082A (en) | 1993-04-26 | 1996-07-23 | Nielsen; Peter E. | Peptide nucleic acids |
| US5541307A (en) | 1990-07-27 | 1996-07-30 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs and solid phase synthesis thereof |
| US5540935A (en) | 1993-12-06 | 1996-07-30 | Nof Corporation | Reactive vesicle and functional substance-fixed vesicle |
| US5543152A (en) | 1994-06-20 | 1996-08-06 | Inex Pharmaceuticals Corporation | Sphingosomes for enhanced drug delivery |
| US5543158A (en) | 1993-07-23 | 1996-08-06 | Massachusetts Institute Of Technology | Biodegradable injectable nanoparticles |
| US5545730A (en) | 1984-10-16 | 1996-08-13 | Chiron Corporation | Multifunctional nucleic acid monomer |
| US5547932A (en) | 1991-09-30 | 1996-08-20 | Boehringer Ingelheim International Gmbh | Composition for introducing nucleic acid complexes into higher eucaryotic cells |
| US5550111A (en) | 1984-07-11 | 1996-08-27 | Temple University-Of The Commonwealth System Of Higher Education | Dual action 2',5'-oligoadenylate antiviral derivatives and uses thereof |
| US5552540A (en) | 1987-06-24 | 1996-09-03 | Howard Florey Institute Of Experimental Physiology And Medicine | Nucleoside derivatives |
| US5556948A (en) | 1993-01-22 | 1996-09-17 | Mitsubishi Chemical Corporation | Phospholipid derivatized with PEG bifunctional linker and liposome containing it |
| US5561225A (en) | 1990-09-19 | 1996-10-01 | Southern Research Institute | Polynucleotide analogs containing sulfonate and sulfonamide internucleoside linkages |
| US5565552A (en) | 1992-01-21 | 1996-10-15 | Pharmacyclics, Inc. | Method of expanded porphyrin-oligonucleotide conjugate synthesis |
| US5565350A (en) | 1993-12-09 | 1996-10-15 | Thomas Jefferson University | Compounds and methods for site directed mutations in eukaryotic cells |
| US5567811A (en) | 1990-05-03 | 1996-10-22 | Amersham International Plc | Phosphoramidite derivatives, their preparation and the use thereof in the incorporation of reporter groups on synthetic oligonucleotides |
| US5571799A (en) | 1991-08-12 | 1996-11-05 | Basco, Ltd. | (2'-5') oligoadenylate analogues useful as inhibitors of host-v5.-graft response |
| US5574142A (en) | 1992-12-15 | 1996-11-12 | Microprobe Corporation | Peptide linkers for improved oligonucleotide delivery |
| US5576427A (en) | 1993-03-30 | 1996-11-19 | Sterling Winthrop, Inc. | Acyclic nucleoside analogs and oligonucleotide sequences containing them |
| US5578718A (en) | 1990-01-11 | 1996-11-26 | Isis Pharmaceuticals, Inc. | Thiol-derivatized nucleosides |
| US5580575A (en) | 1989-12-22 | 1996-12-03 | Imarx Pharmaceutical Corp. | Therapeutic drug delivery systems |
| US5580731A (en) | 1994-08-25 | 1996-12-03 | Chiron Corporation | N-4 modified pyrimidine deoxynucleotides and oligonucleotide probes synthesized therewith |
| US5583020A (en) | 1992-11-24 | 1996-12-10 | Ribozyme Pharmaceuticals, Inc. | Permeability enhancers for negatively charged polynucleotides |
| US5585481A (en) | 1987-09-21 | 1996-12-17 | Gen-Probe Incorporated | Linking reagents for nucleotide probes |
| WO1996040062A1 (en) | 1995-06-07 | 1996-12-19 | Georgetown University | A method of transfection of cells using liposomally encapsulated nucleic acids |
| US5587361A (en) | 1991-10-15 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides having phosphorothioate linkages of high chiral purity |
| US5587371A (en) | 1992-01-21 | 1996-12-24 | Pharmacyclics, Inc. | Texaphyrin-oligonucleotide conjugates |
| US5591722A (en) | 1989-09-15 | 1997-01-07 | Southern Research Institute | 2'-deoxy-4'-thioribonucleosides and their antiviral activity |
| US5591721A (en) | 1994-10-25 | 1997-01-07 | Hybridon, Inc. | Method of down-regulating gene expression |
| US5593974A (en) | 1991-06-28 | 1997-01-14 | Massachusetts Institute Of Technology | Localized oligonucleotide therapy |
| US5594121A (en) | 1991-11-07 | 1997-01-14 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified purines |
| US5595756A (en) | 1993-12-22 | 1997-01-21 | Inex Pharmaceuticals Corporation | Liposomal compositions for enhanced retention of bioactive agents |
| US5595726A (en) | 1992-01-21 | 1997-01-21 | Pharmacyclics, Inc. | Chromophore probe for detection of nucleic acid |
| US5596091A (en) | 1994-03-18 | 1997-01-21 | The Regents Of The University Of California | Antisense oligonucleotides comprising 5-aminoalkyl pyrimidine nucleotides |
| US5596086A (en) | 1990-09-20 | 1997-01-21 | Gilead Sciences, Inc. | Modified internucleoside linkages having one nitrogen and two carbon atoms |
| US5597909A (en) | 1994-08-25 | 1997-01-28 | Chiron Corporation | Polynucleotide reagents containing modified deoxyribose moieties, and associated methods of synthesis and use |
| US5597696A (en) | 1994-07-18 | 1997-01-28 | Becton Dickinson And Company | Covalent cyanine dye oligonucleotide conjugates |
| US5599928A (en) | 1994-02-15 | 1997-02-04 | Pharmacyclics, Inc. | Texaphyrin compounds having improved functionalization |
| US5602240A (en) | 1990-07-27 | 1997-02-11 | Ciba Geigy Ag. | Backbone modified oligonucleotide analogs |
| WO1997004787A1 (en) | 1995-08-01 | 1997-02-13 | Novartis Ag | Liposomal oligonucleotide compositions |
| US5608046A (en) | 1990-07-27 | 1997-03-04 | Isis Pharmaceuticals, Inc. | Conjugated 4'-desmethyl nucleoside analog compounds |
| US5610300A (en) | 1992-07-01 | 1997-03-11 | Ciba-Geigy Corporation | Carbocyclic nucleosides containing bicyclic rings, oligonucleotides therefrom, process for their preparation, their use and intermediates |
| US5610289A (en) | 1990-07-27 | 1997-03-11 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogues |
| US5609629A (en) | 1995-06-07 | 1997-03-11 | Med Institute, Inc. | Coated implantable medical device |
| US5614617A (en) | 1990-07-27 | 1997-03-25 | Isis Pharmaceuticals, Inc. | Nuclease resistant, pyrimidine modified oligonucleotides that detect and modulate gene expression |
| US5618704A (en) | 1990-07-27 | 1997-04-08 | Isis Pharmacueticals, Inc. | Backbone-modified oligonucleotide analogs and preparation thereof through radical coupling |
| WO1997013499A1 (en) | 1995-10-11 | 1997-04-17 | The University Of British Columbia | Liposomal formulations of mitoxantrone |
| US5623070A (en) | 1990-07-27 | 1997-04-22 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5623065A (en) | 1990-08-13 | 1997-04-22 | Isis Pharmaceuticals, Inc. | Gapped 2' modified oligonucleotides |
| US5625050A (en) | 1994-03-31 | 1997-04-29 | Amgen Inc. | Modified oligonucleotides and intermediates useful in nucleic acid therapeutics |
| US5627053A (en) | 1994-03-29 | 1997-05-06 | Ribozyme Pharmaceuticals, Inc. | 2'deoxy-2'-alkylnucleotide containing nucleic acid |
| US5633360A (en) | 1992-04-14 | 1997-05-27 | Gilead Sciences, Inc. | Oligonucleotide analogs capable of passive cell membrane permeation |
| US5639873A (en) | 1992-02-05 | 1997-06-17 | Centre National De La Recherche Scientifique (Cnrs) | Oligothionucleotides |
| US5645985A (en) | 1991-11-26 | 1997-07-08 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified pyrimidines |
| US5646269A (en) | 1994-04-28 | 1997-07-08 | Gilead Sciences, Inc. | Method for oligonucleotide analog synthesis |
| US5646265A (en) | 1990-01-11 | 1997-07-08 | Isis Pharmceuticals, Inc. | Process for the preparation of 2'-O-alkyl purine phosphoramidites |
| US5652356A (en) | 1995-08-17 | 1997-07-29 | Hybridon, Inc. | Inverted chimeric and hybrid oligonucleotides |
| US5652355A (en) | 1992-07-23 | 1997-07-29 | Worcester Foundation For Experimental Biology | Hybrid oligonucleotide phosphorothioates |
| US5658873A (en) | 1993-04-10 | 1997-08-19 | Degussa Aktiengesellschaft | Coated sodium percarbonate particles, a process for their production and detergent, cleaning and bleaching compositions containing them |
| WO1997030731A2 (en) | 1996-02-21 | 1997-08-28 | The Immune Response Corporation | Method of preparing polynucleotide-carrier complexes for delivery to cells |
| US5663312A (en) | 1993-03-31 | 1997-09-02 | Sanofi | Oligonucleotide dimers with amide linkages replacing phosphodiester linkages |
| US5665710A (en) | 1990-04-30 | 1997-09-09 | Georgetown University | Method of making liposomal oligodeoxynucleotide compositions |
| US5670161A (en) | 1996-05-28 | 1997-09-23 | Healy; Kevin E. | Biodegradable stent |
| US5670633A (en) | 1990-01-11 | 1997-09-23 | Isis Pharmaceuticals, Inc. | Sugar modified oligonucleotides that detect and modulate gene expression |
| US5672697A (en) | 1991-02-08 | 1997-09-30 | Gilead Sciences, Inc. | Nucleoside 5'-methylene phosphonates |
| US5674242A (en) | 1995-06-06 | 1997-10-07 | Quanam Medical Corporation | Endoprosthetic device with therapeutic compound |
| US5677437A (en) | 1990-07-27 | 1997-10-14 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5677439A (en) | 1990-08-03 | 1997-10-14 | Sanofi | Oligonucleotide analogues containing phosphate diester linkage substitutes, compositions thereof, and precursor dinucleotide analogues |
| US5688941A (en) | 1990-07-27 | 1997-11-18 | Isis Pharmaceuticals, Inc. | Methods of making conjugated 4' desmethyl nucleoside analog compounds |
| US5700922A (en) | 1991-12-24 | 1997-12-23 | Isis Pharmaceuticals, Inc. | PNA-DNA-PNA chimeric macromolecules |
| US5705188A (en) | 1993-02-19 | 1998-01-06 | Nippon Shinyaku Company, Ltd. | Drug composition containing nucleic acid copolymer |
| US5714331A (en) | 1991-05-24 | 1998-02-03 | Buchardt, Deceased; Ole | Peptide nucleic acids having enhanced binding affinity, sequence specificity and solubility |
| US5719262A (en) | 1993-11-22 | 1998-02-17 | Buchardt, Deceased; Ole | Peptide nucleic acids having amino acid side chains |
| US5721218A (en) | 1989-10-23 | 1998-02-24 | Gilead Sciences, Inc. | Oligonucleotides with inverted polarity |
| US5749915A (en) | 1988-08-24 | 1998-05-12 | Focal, Inc. | Polymeric endoluminal paving process |
| US5792608A (en) | 1991-12-12 | 1998-08-11 | Gilead Sciences, Inc. | Nuclease stable and binding competent oligomers and methods for their use |
| US5792747A (en) | 1995-01-24 | 1998-08-11 | The Administrators Of The Tulane Educational Fund | Highly potent agonists of growth hormone releasing hormone |
| WO1998039352A1 (en) | 1997-03-07 | 1998-09-11 | Takeshi Imanishi | Novel bicyclonucleoside and oligonucleotide analogues |
| US5824049A (en) | 1995-06-07 | 1998-10-20 | Med Institute, Inc. | Coated implantable medical device |
| US5830653A (en) | 1991-11-26 | 1998-11-03 | Gilead Sciences, Inc. | Methods of using oligomers containing modified pyrimidines |
| WO1999014226A2 (en) | 1997-09-12 | 1999-03-25 | Exiqon A/S | Bi- and tri-cyclic nucleoside, nucleotide and oligonucleotide analogues |
| US6159946A (en) | 1993-01-07 | 2000-12-12 | Thomas Jefferson University | Antisense inhibition of c-myc to modulate the proliferation of smooth muscle cells |
| US6200304B1 (en) | 1997-07-11 | 2001-03-13 | Cardiogene Gentherapeutische Systeme Ag | Transfection system, its preparation and use in somatic gene therapy |
| US6217886B1 (en) | 1997-07-14 | 2001-04-17 | The Board Of Trustees Of The University Of Illinois | Materials and methods for making improved micelle compositions |
| US6270790B1 (en) | 1998-08-18 | 2001-08-07 | Mxneil-Ppc, Inc. | Soft, convex shaped chewable tablets having reduced friability |
| US6287285B1 (en) | 1998-01-30 | 2001-09-11 | Advanced Cardiovascular Systems, Inc. | Therapeutic, diagnostic, or hydrophilic coating for an intracorporeal medical device |
| US6344035B1 (en) | 1998-04-27 | 2002-02-05 | Surmodics, Inc. | Bioactive agent release coating |
| US6344028B1 (en) | 1994-06-30 | 2002-02-05 | Boston Scientific Corporation | Replenishable stent and delivery system |
| US6368356B1 (en) | 1996-07-11 | 2002-04-09 | Scimed Life Systems, Inc. | Medical devices comprising hydrogel polymers having improved mechanical properties |
| US6433012B1 (en) | 1998-03-25 | 2002-08-13 | Large Scale Biology Corp. | Method for inhibiting inflammatory disease |
| US6495532B1 (en) | 1997-03-19 | 2002-12-17 | Sky High, Llc | Compositions containing lysophosphotidic acids which inhibit apoptosis and uses thereof |
| US6547787B1 (en) | 1997-03-13 | 2003-04-15 | Biocardia, Inc. | Drug delivery catheters that attach to tissue and methods for their use |
| US6562864B1 (en) | 1999-09-02 | 2003-05-13 | Drake Larson | Catechin multimers as therapeutic drug delivery agents |
| US20030215503A1 (en) | 2002-04-04 | 2003-11-20 | Tanya Havlir | Palatable chewable tablet |
| US20040265372A1 (en) | 2003-06-27 | 2004-12-30 | David Wynn | Soft tablet containing high molecular weight cellulosics |
| US20060009410A1 (en) * | 2002-11-13 | 2006-01-12 | Crooke Rosanne M | Effects of apolipoprotein B inhibition on gene expression profiles in animals |
| WO2010076248A1 (en) * | 2008-12-31 | 2010-07-08 | Santaris Pharma A/S | Use of lna apob antisense oligomers for the treatment of acute coronary syndromes |
| US20100297105A1 (en) | 2007-03-24 | 2010-11-25 | Genzyme Corporation | Administering antisense oligonucleotides complementary to human apolipoprotein b |
-
2015
- 2015-08-28 WO PCT/US2015/047372 patent/WO2016033424A1/en active Application Filing
Patent Citations (256)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US568194A (en) | 1896-09-22 | Skylight-support | ||
| US3687808A (en) | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| US4469863A (en) | 1980-11-12 | 1984-09-04 | Ts O Paul O P | Nonionic nucleic acid alkyl and aryl phosphonates and processes for manufacture and use thereof |
| US4426330A (en) | 1981-07-20 | 1984-01-17 | Lipid Specialties, Inc. | Synthetic phospholipid compounds |
| US4534899A (en) | 1981-07-20 | 1985-08-13 | Lipid Specialties, Inc. | Synthetic phospholipid compounds |
| US5023243A (en) | 1981-10-23 | 1991-06-11 | Molecular Biosystems, Inc. | Oligonucleotide therapeutic agent and method of making same |
| US4476301A (en) | 1982-04-29 | 1984-10-09 | Centre National De La Recherche Scientifique | Oligonucleotides, a process for preparing the same and their application as mediators of the action of interferon |
| US4789737A (en) | 1982-08-09 | 1988-12-06 | Wakunaga Seiyaku Kabushiki Kaisha | Oligonucleotide derivatives and production thereof |
| US4667025A (en) | 1982-08-09 | 1987-05-19 | Wakunaga Seiyaku Kabushiki Kaisha | Oligonucleotide derivatives |
| US4501729A (en) | 1982-12-13 | 1985-02-26 | Research Corporation | Aerosolized amiloride treatment of retained pulmonary secretions |
| US4835263A (en) | 1983-01-27 | 1989-05-30 | Centre National De La Recherche Scientifique | Novel compounds containing an oligonucleotide sequence bonded to an intercalating agent, a process for their synthesis and their use |
| US4605735A (en) | 1983-02-14 | 1986-08-12 | Wakunaga Seiyaku Kabushiki Kaisha | Oligonucleotide derivatives |
| US4948882A (en) | 1983-02-22 | 1990-08-14 | Syngene, Inc. | Single-stranded labelled oligonucleotides, reactive monomers and methods of synthesis |
| US5541313A (en) | 1983-02-22 | 1996-07-30 | Molecular Biosystems, Inc. | Single-stranded labelled oligonucleotides of preselected sequence |
| US4824941A (en) | 1983-03-10 | 1989-04-25 | Julian Gordon | Specific antibody to the native form of 2'5'-oligonucleotides, the method of preparation and the use as reagents in immunoassays or for binding 2'5'-oligonucleotides in biological systems |
| US4587044A (en) | 1983-09-01 | 1986-05-06 | The Johns Hopkins University | Linkage of proteins to nucleic acids |
| US4818542A (en) | 1983-11-14 | 1989-04-04 | The University Of Kentucky Research Foundation | Porous microspheres for drug delivery and methods for making same |
| US5118800A (en) | 1983-12-20 | 1992-06-02 | California Institute Of Technology | Oligonucleotides possessing a primary amino group in the terminal nucleotide |
| US5118802A (en) | 1983-12-20 | 1992-06-02 | California Institute Of Technology | DNA-reporter conjugates linked via the 2' or 5'-primary amino group of the 5'-terminal nucleoside |
| US5550111A (en) | 1984-07-11 | 1996-08-27 | Temple University-Of The Commonwealth System Of Higher Education | Dual action 2',5'-oligoadenylate antiviral derivatives and uses thereof |
| US4981957A (en) | 1984-07-19 | 1991-01-01 | Centre National De La Recherche Scientifique | Oligonucleotides with modified phosphate and modified carbohydrate moieties at the respective chain termini |
| US5552538A (en) | 1984-10-16 | 1996-09-03 | Chiron Corporation | Oligonucleotides with cleavable sites |
| US5545730A (en) | 1984-10-16 | 1996-08-13 | Chiron Corporation | Multifunctional nucleic acid monomer |
| US5578717A (en) | 1984-10-16 | 1996-11-26 | Chiron Corporation | Nucleotides for introducing selectably cleavable and/or abasic sites into oligonucleotides |
| US5258506A (en) | 1984-10-16 | 1993-11-02 | Chiron Corporation | Photolabile reagents for incorporation into oligonucleotide chains |
| US5367066A (en) | 1984-10-16 | 1994-11-22 | Chiron Corporation | Oligonucleotides with selectably cleavable and/or abasic sites |
| US4828979A (en) | 1984-11-08 | 1989-05-09 | Life Technologies, Inc. | Nucleotide analogs for nucleic acid labeling and detection |
| US4845205A (en) | 1985-01-08 | 1989-07-04 | Institut Pasteur | 2,N6 -disubstituted and 2,N6 -trisubstituted adenosine-3'-phosphoramidites |
| US5235033A (en) | 1985-03-15 | 1993-08-10 | Anti-Gene Development Group | Alpha-morpholino ribonucleoside derivatives and polymers thereof |
| US5034506A (en) | 1985-03-15 | 1991-07-23 | Anti-Gene Development Group | Uncharged morpholino-based polymers having achiral intersubunit linkages |
| US5185444A (en) | 1985-03-15 | 1993-02-09 | Anti-Gene Deveopment Group | Uncharged morpolino-based polymers having phosphorous containing chiral intersubunit linkages |
| US4762779A (en) | 1985-06-13 | 1988-08-09 | Amgen Inc. | Compositions and methods for functionalizing nucleic acids |
| US4885172A (en) | 1985-06-26 | 1989-12-05 | The Liposome Company, Inc. | Composition for targeting, storing and loading of liposomes |
| US5399331A (en) | 1985-06-26 | 1995-03-21 | The Liposome Company, Inc. | Method for protein-liposome coupling |
| US5171578A (en) | 1985-06-26 | 1992-12-15 | The Liposome Company, Inc. | Composition for targeting, storing and loading of liposomes |
| US5059421A (en) | 1985-07-26 | 1991-10-22 | The Liposome Company, Inc. | Preparation of targeted liposome systems of a defined size distribution |
| US5317098A (en) | 1986-03-17 | 1994-05-31 | Hiroaki Shizuya | Non-radioisotope tagging of fragments |
| US4876335A (en) | 1986-06-30 | 1989-10-24 | Wakunaga Seiyaku Kabushiki Kaisha | Poly-labelled oligonucleotide derivative |
| US5013830A (en) | 1986-09-08 | 1991-05-07 | Ajinomoto Co., Inc. | Compounds for the cleavage at a specific position of RNA, oligomers employed for the formation of said compounds, and starting materials for the synthesis of said oligomers |
| WO1988004924A1 (en) | 1986-12-24 | 1988-07-14 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US4837028A (en) | 1986-12-24 | 1989-06-06 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5286717A (en) | 1987-03-25 | 1994-02-15 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5276019A (en) | 1987-03-25 | 1994-01-04 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5264423A (en) | 1987-03-25 | 1993-11-23 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US4904582A (en) | 1987-06-11 | 1990-02-27 | Synthetic Genetics | Novel amphiphilic nucleic acid conjugates |
| US5552540A (en) | 1987-06-24 | 1996-09-03 | Howard Florey Institute Of Experimental Physiology And Medicine | Nucleoside derivatives |
| US5585481A (en) | 1987-09-21 | 1996-12-17 | Gen-Probe Incorporated | Linking reagents for nucleotide probes |
| US5188897A (en) | 1987-10-22 | 1993-02-23 | Temple University Of The Commonwealth System Of Higher Education | Encapsulated 2',5'-phosphorothioate oligoadenylates |
| US5405939A (en) | 1987-10-22 | 1995-04-11 | Temple University Of The Commonwealth System Of Higher Education | 2',5'-phosphorothioate oligoadenylates and their covalent conjugates with polylysine |
| US5525465A (en) | 1987-10-28 | 1996-06-11 | Howard Florey Institute Of Experimental Physiology And Medicine | Oligonucleotide-polyamide conjugates and methods of production and applications of the same |
| US5112963A (en) | 1987-11-12 | 1992-05-12 | Max-Planck-Gesellschaft Zur Foerderung Der Wissenschaften E.V. | Modified oligonucleotides |
| US5491133A (en) | 1987-11-30 | 1996-02-13 | University Of Iowa Research Foundation | Methods for blocking the expression of specifically targeted genes |
| US5403711A (en) | 1987-11-30 | 1995-04-04 | University Of Iowa Research Foundation | Nucleic acid hybridization and amplification method for detection of specific sequences in which a complementary labeled nucleic acid probe is cleaved |
| US5082830A (en) | 1988-02-26 | 1992-01-21 | Enzo Biochem, Inc. | End labeled nucleotide probe |
| US5519126A (en) | 1988-03-25 | 1996-05-21 | University Of Virginia Alumni Patents Foundation | Oligonucleotide N-alkylphosphoramidates |
| US5278302A (en) | 1988-05-26 | 1994-01-11 | University Patents, Inc. | Polynucleotide phosphorodithioates |
| US5453496A (en) | 1988-05-26 | 1995-09-26 | University Patents, Inc. | Polynucleotide phosphorodithioate |
| US5109124A (en) | 1988-06-01 | 1992-04-28 | Biogen, Inc. | Nucleic acid probe linked to a label having a terminal cysteine |
| US5216141A (en) | 1988-06-06 | 1993-06-01 | Benner Steven A | Oligonucleotide analogs containing sulfur linkages |
| US5175273A (en) | 1988-07-01 | 1992-12-29 | Genentech, Inc. | Nucleic acid intercalating agents |
| US5749915A (en) | 1988-08-24 | 1998-05-12 | Focal, Inc. | Polymeric endoluminal paving process |
| US5262536A (en) | 1988-09-15 | 1993-11-16 | E. I. Du Pont De Nemours And Company | Reagents for the preparation of 5'-tagged oligonucleotides |
| US5194599A (en) | 1988-09-23 | 1993-03-16 | Gilead Sciences, Inc. | Hydrogen phosphonodithioate compositions |
| US5565555A (en) | 1988-09-23 | 1996-10-15 | Gilead Sciences, Inc. | Nucleoside hydrogen phosphonodithioate diesters and activated phosphonodithioate analogues |
| EP0445131B1 (en) | 1988-10-20 | 1994-04-27 | PolyMASC Pharmaceuticals plc | Liposomes |
| WO1990004384A1 (en) | 1988-10-20 | 1990-05-03 | Royal Free Hospital School Of Medicine | Liposomes |
| US5512439A (en) | 1988-11-21 | 1996-04-30 | Dynal As | Oligonucleotide-linked magnetic particles and uses thereof |
| US5599923A (en) | 1989-03-06 | 1997-02-04 | Board Of Regents, University Of Tx | Texaphyrin metal complexes having improved functionalization |
| US5354844A (en) | 1989-03-16 | 1994-10-11 | Boehringer Ingelheim International Gmbh | Protein-polycation conjugates |
| US5108921A (en) | 1989-04-03 | 1992-04-28 | Purdue Research Foundation | Method for enhanced transmembrane transport of exogenous molecules |
| US5416016A (en) | 1989-04-03 | 1995-05-16 | Purdue Research Foundation | Method for enhancing transmembrane transport of exogenous molecules |
| US5391723A (en) | 1989-05-31 | 1995-02-21 | Neorx Corporation | Oligonucleotide conjugates |
| US5256775A (en) | 1989-06-05 | 1993-10-26 | Gilead Sciences, Inc. | Exonuclease-resistant oligonucleotides |
| US5416203A (en) | 1989-06-06 | 1995-05-16 | Northwestern University | Steroid modified oligonucleotides |
| US4958013A (en) | 1989-06-06 | 1990-09-18 | Northwestern University | Cholesteryl modified oligonucleotides |
| US5227170A (en) | 1989-06-22 | 1993-07-13 | Vestar, Inc. | Encapsulation process |
| US5451463A (en) | 1989-08-28 | 1995-09-19 | Clontech Laboratories, Inc. | Non-nucleoside 1,3-diol reagents for labeling synthetic oligonucleotides |
| US5134066A (en) | 1989-08-29 | 1992-07-28 | Monsanto Company | Improved probes using nucleosides containing 3-dezauracil analogs |
| US5254469A (en) | 1989-09-12 | 1993-10-19 | Eastman Kodak Company | Oligonucleotide-enzyme conjugate that can be used as a probe in hybridization assays and polymerase chain reaction procedures |
| US5591722A (en) | 1989-09-15 | 1997-01-07 | Southern Research Institute | 2'-deoxy-4'-thioribonucleosides and their antiviral activity |
| WO1991005545A1 (en) | 1989-10-20 | 1991-05-02 | Liposome Technology, Inc. | Liposome microreservoir composition and method |
| EP0496813B1 (en) | 1989-10-20 | 1994-12-14 | SEQUUS PHARMACEUTICALS, INC. (a Delaware Corporation) | Liposome microreservoir composition and method |
| US5213804A (en) | 1989-10-20 | 1993-05-25 | Liposome Technology, Inc. | Solid tumor treatment method and composition |
| US5013556A (en) | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5225212A (en) | 1989-10-20 | 1993-07-06 | Liposome Technology, Inc. | Microreservoir liposome composition and method |
| US5356633A (en) | 1989-10-20 | 1994-10-18 | Liposome Technology, Inc. | Method of treatment of inflamed tissues |
| US5527528A (en) | 1989-10-20 | 1996-06-18 | Sequus Pharmaceuticals, Inc. | Solid-tumor treatment method |
| US5399676A (en) | 1989-10-23 | 1995-03-21 | Gilead Sciences | Oligonucleotides with inverted polarity |
| US5527899A (en) | 1989-10-23 | 1996-06-18 | Gilead Sciences, Inc. | Oligonucleotides with inverted polarity |
| US5721218A (en) | 1989-10-23 | 1998-02-24 | Gilead Sciences, Inc. | Oligonucleotides with inverted polarity |
| US5466786B1 (en) | 1989-10-24 | 1998-04-07 | Gilead Sciences | 2' Modified nucleoside and nucleotide compounds |
| US5264562A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences, Inc. | Oligonucleotide analogs with novel linkages |
| US5264564A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences | Oligonucleotide analogs with novel linkages |
| US5466786A (en) | 1989-10-24 | 1995-11-14 | Gilead Sciences | 2'modified nucleoside and nucleotide compounds |
| US5292873A (en) | 1989-11-29 | 1994-03-08 | The Research Foundation Of State University Of New York | Nucleic acids labeled with naphthoquinone probe |
| US5455233A (en) | 1989-11-30 | 1995-10-03 | University Of North Carolina | Oligoribonucleoside and oligodeoxyribonucleoside boranophosphates |
| US5177198A (en) | 1989-11-30 | 1993-01-05 | University Of N.C. At Chapel Hill | Process for preparing oligoribonucleoside and oligodeoxyribonucleoside boranophosphates |
| US5166315A (en) | 1989-12-20 | 1992-11-24 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5130302A (en) | 1989-12-20 | 1992-07-14 | Boron Bilogicals, Inc. | Boronated nucleoside, nucleotide and oligonucleotide compounds, compositions and methods for using same |
| US5405938A (en) | 1989-12-20 | 1995-04-11 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5469854A (en) | 1989-12-22 | 1995-11-28 | Imarx Pharmaceutical Corp. | Methods of preparing gas-filled liposomes |
| US5580575A (en) | 1989-12-22 | 1996-12-03 | Imarx Pharmaceutical Corp. | Therapeutic drug delivery systems |
| US5486603A (en) | 1990-01-08 | 1996-01-23 | Gilead Sciences, Inc. | Oligonucleotide having enhanced binding affinity |
| US5587469A (en) | 1990-01-11 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides containing N-2 substituted purines |
| US5578718A (en) | 1990-01-11 | 1996-11-26 | Isis Pharmaceuticals, Inc. | Thiol-derivatized nucleosides |
| US5459255A (en) | 1990-01-11 | 1995-10-17 | Isis Pharmaceuticals, Inc. | N-2 substituted purines |
| US5670633A (en) | 1990-01-11 | 1997-09-23 | Isis Pharmaceuticals, Inc. | Sugar modified oligonucleotides that detect and modulate gene expression |
| US5646265A (en) | 1990-01-11 | 1997-07-08 | Isis Pharmceuticals, Inc. | Process for the preparation of 2'-O-alkyl purine phosphoramidites |
| US5366878A (en) | 1990-02-15 | 1994-11-22 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of RNA and production of encoded polypeptides |
| US5149797A (en) | 1990-02-15 | 1992-09-22 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of rna and production of encoded polypeptides |
| US5220007A (en) | 1990-02-15 | 1993-06-15 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of RNA and production of encoded polypeptides |
| US5214136A (en) | 1990-02-20 | 1993-05-25 | Gilead Sciences, Inc. | Anthraquinone-derivatives oligonucleotides |
| US5414077A (en) | 1990-02-20 | 1995-05-09 | Gilead Sciences | Non-nucleoside linkers for convenient attachment of labels to oligonucleotides using standard synthetic methods |
| US5541306A (en) | 1990-03-08 | 1996-07-30 | Worcester Foundation For Biomedical Research | Aminoalkylphosphotriester oligonucleotide derivatives |
| US5536821A (en) | 1990-03-08 | 1996-07-16 | Worcester Foundation For Biomedical Research | Aminoalkylphosphorothioamidate oligonucleotide deratives |
| US5563253A (en) | 1990-03-08 | 1996-10-08 | Worcester Foundation For Biomedical Research | Linear aminoalkylphosphoramidate oligonucleotide derivatives |
| US5321131A (en) | 1990-03-08 | 1994-06-14 | Hybridon, Inc. | Site-specific functionalization of oligodeoxynucleotides for non-radioactive labelling |
| US5470967A (en) | 1990-04-10 | 1995-11-28 | The Dupont Merck Pharmaceutical Company | Oligonucleotide analogs with sulfamate linkages |
| US5459127A (en) | 1990-04-19 | 1995-10-17 | Vical, Inc. | Cationic lipids for intracellular delivery of biologically active molecules |
| US5665710A (en) | 1990-04-30 | 1997-09-09 | Georgetown University | Method of making liposomal oligodeoxynucleotide compositions |
| US5567811A (en) | 1990-05-03 | 1996-10-22 | Amersham International Plc | Phosphoramidite derivatives, their preparation and the use thereof in the incorporation of reporter groups on synthetic oligonucleotides |
| US5514785A (en) | 1990-05-11 | 1996-05-07 | Becton Dickinson And Company | Solid supports for nucleic acid hybridization assays |
| US5610289A (en) | 1990-07-27 | 1997-03-11 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogues |
| US5614617A (en) | 1990-07-27 | 1997-03-25 | Isis Pharmaceuticals, Inc. | Nuclease resistant, pyrimidine modified oligonucleotides that detect and modulate gene expression |
| US5489677A (en) | 1990-07-27 | 1996-02-06 | Isis Pharmaceuticals, Inc. | Oligonucleoside linkages containing adjacent oxygen and nitrogen atoms |
| US5378825A (en) | 1990-07-27 | 1995-01-03 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs |
| US5677437A (en) | 1990-07-27 | 1997-10-14 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5602240A (en) | 1990-07-27 | 1997-02-11 | Ciba Geigy Ag. | Backbone modified oligonucleotide analogs |
| US5688941A (en) | 1990-07-27 | 1997-11-18 | Isis Pharmaceuticals, Inc. | Methods of making conjugated 4' desmethyl nucleoside analog compounds |
| US5541307A (en) | 1990-07-27 | 1996-07-30 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs and solid phase synthesis thereof |
| US5608046A (en) | 1990-07-27 | 1997-03-04 | Isis Pharmaceuticals, Inc. | Conjugated 4'-desmethyl nucleoside analog compounds |
| US5623070A (en) | 1990-07-27 | 1997-04-22 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5218105A (en) | 1990-07-27 | 1993-06-08 | Isis Pharmaceuticals | Polyamine conjugated oligonucleotides |
| US5138045A (en) | 1990-07-27 | 1992-08-11 | Isis Pharmaceuticals | Polyamine conjugated oligonucleotides |
| US5386023A (en) | 1990-07-27 | 1995-01-31 | Isis Pharmaceuticals | Backbone modified oligonucleotide analogs and preparation thereof through reductive coupling |
| US5618704A (en) | 1990-07-27 | 1997-04-08 | Isis Pharmacueticals, Inc. | Backbone-modified oligonucleotide analogs and preparation thereof through radical coupling |
| US5567810A (en) | 1990-08-03 | 1996-10-22 | Sterling Drug, Inc. | Nuclease resistant compounds |
| US5245022A (en) | 1990-08-03 | 1993-09-14 | Sterling Drug, Inc. | Exonuclease resistant terminally substituted oligonucleotides |
| US5677439A (en) | 1990-08-03 | 1997-10-14 | Sanofi | Oligonucleotide analogues containing phosphate diester linkage substitutes, compositions thereof, and precursor dinucleotide analogues |
| US5623065A (en) | 1990-08-13 | 1997-04-22 | Isis Pharmaceuticals, Inc. | Gapped 2' modified oligonucleotides |
| US5223618A (en) | 1990-08-13 | 1993-06-29 | Isis Pharmaceuticals, Inc. | 4'-desmethyl nucleoside analog compounds |
| US5177196A (en) | 1990-08-16 | 1993-01-05 | Microprobe Corporation | Oligo (α-arabinofuranosyl nucleotides) and α-arabinofuranosyl precursors thereof |
| US5512667A (en) | 1990-08-28 | 1996-04-30 | Reed; Michael W. | Trifunctional intermediates for preparing 3'-tailed oligonucleotides |
| US5214134A (en) | 1990-09-12 | 1993-05-25 | Sterling Winthrop Inc. | Process of linking nucleosides with a siloxane bridge |
| US5561225A (en) | 1990-09-19 | 1996-10-01 | Southern Research Institute | Polynucleotide analogs containing sulfonate and sulfonamide internucleoside linkages |
| US5596086A (en) | 1990-09-20 | 1997-01-21 | Gilead Sciences, Inc. | Modified internucleoside linkages having one nitrogen and two carbon atoms |
| US5432272A (en) | 1990-10-09 | 1995-07-11 | Benner; Steven A. | Method for incorporating into a DNA or RNA oligonucleotide using nucleotides bearing heterocyclic bases |
| US5510475A (en) | 1990-11-08 | 1996-04-23 | Hybridon, Inc. | Oligonucleotide multiple reporter precursors |
| US5672697A (en) | 1991-02-08 | 1997-09-30 | Gilead Sciences, Inc. | Nucleoside 5'-methylene phosphonates |
| US5264221A (en) | 1991-05-23 | 1993-11-23 | Mitsubishi Kasei Corporation | Drug-containing protein-bonded liposome |
| US5714331A (en) | 1991-05-24 | 1998-02-03 | Buchardt, Deceased; Ole | Peptide nucleic acids having enhanced binding affinity, sequence specificity and solubility |
| US5593974A (en) | 1991-06-28 | 1997-01-14 | Massachusetts Institute Of Technology | Localized oligonucleotide therapy |
| US5371241A (en) | 1991-07-19 | 1994-12-06 | Pharmacia P-L Biochemicals Inc. | Fluorescein labelled phosphoramidites |
| US5571799A (en) | 1991-08-12 | 1996-11-05 | Basco, Ltd. | (2'-5') oligoadenylate analogues useful as inhibitors of host-v5.-graft response |
| US5521291A (en) | 1991-09-30 | 1996-05-28 | Boehringer Ingelheim International, Gmbh | Conjugates for introducing nucleic acid into higher eucaryotic cells |
| US5547932A (en) | 1991-09-30 | 1996-08-20 | Boehringer Ingelheim International Gmbh | Composition for introducing nucleic acid complexes into higher eucaryotic cells |
| US5587361A (en) | 1991-10-15 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides having phosphorothioate linkages of high chiral purity |
| US5393878A (en) | 1991-10-17 | 1995-02-28 | Ciba-Geigy Corporation | Bicyclic nucleosides, oligonucleotides, process for their preparation and intermediates |
| US5319080A (en) | 1991-10-17 | 1994-06-07 | Ciba-Geigy Corporation | Bicyclic nucleosides, oligonucleotides, process for their preparation and intermediates |
| US5594121A (en) | 1991-11-07 | 1997-01-14 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified purines |
| US5484908A (en) | 1991-11-26 | 1996-01-16 | Gilead Sciences, Inc. | Oligonucleotides containing 5-propynyl pyrimidines |
| US5830653A (en) | 1991-11-26 | 1998-11-03 | Gilead Sciences, Inc. | Methods of using oligomers containing modified pyrimidines |
| US5645985A (en) | 1991-11-26 | 1997-07-08 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified pyrimidines |
| US5792608A (en) | 1991-12-12 | 1998-08-11 | Gilead Sciences, Inc. | Nuclease stable and binding competent oligomers and methods for their use |
| US5359044A (en) | 1991-12-13 | 1994-10-25 | Isis Pharmaceuticals | Cyclobutyl oligonucleotide surrogates |
| US5700922A (en) | 1991-12-24 | 1997-12-23 | Isis Pharmaceuticals, Inc. | PNA-DNA-PNA chimeric macromolecules |
| US5565552A (en) | 1992-01-21 | 1996-10-15 | Pharmacyclics, Inc. | Method of expanded porphyrin-oligonucleotide conjugate synthesis |
| US5595726A (en) | 1992-01-21 | 1997-01-21 | Pharmacyclics, Inc. | Chromophore probe for detection of nucleic acid |
| US5587371A (en) | 1992-01-21 | 1996-12-24 | Pharmacyclics, Inc. | Texaphyrin-oligonucleotide conjugates |
| US5639873A (en) | 1992-02-05 | 1997-06-17 | Centre National De La Recherche Scientifique (Cnrs) | Oligothionucleotides |
| US5633360A (en) | 1992-04-14 | 1997-05-27 | Gilead Sciences, Inc. | Oligonucleotide analogs capable of passive cell membrane permeation |
| WO1993024510A1 (en) | 1992-05-25 | 1993-12-09 | Centre National De La Recherche Scientifique (Cnrs) | Phosphotriester-type biologically active compounds |
| US5434257A (en) | 1992-06-01 | 1995-07-18 | Gilead Sciences, Inc. | Binding compentent oligomers containing unsaturated 3',5' and 2',5' linkages |
| WO1994000203A1 (en) | 1992-06-30 | 1994-01-06 | Velke, Willi, H. | Tennis racquet having a surface structure adapted for the reduction of air resistance |
| US5610300A (en) | 1992-07-01 | 1997-03-11 | Ciba-Geigy Corporation | Carbocyclic nucleosides containing bicyclic rings, oligonucleotides therefrom, process for their preparation, their use and intermediates |
| US5700920A (en) | 1992-07-01 | 1997-12-23 | Novartis Corporation | Carbocyclic nucleosides containing bicyclic rings, oligonucleotides therefrom, process for their preparation, their use and intermediates |
| US5272250A (en) | 1992-07-10 | 1993-12-21 | Spielvogel Bernard F | Boronated phosphoramidate compounds |
| WO1994002501A1 (en) | 1992-07-23 | 1994-02-03 | Isis Pharmaceuticals, Inc. | Novel 2'-o-alkyl nucleosides and phosphoramidites processes for the preparation and uses thereof |
| US5652355A (en) | 1992-07-23 | 1997-07-29 | Worcester Foundation For Experimental Biology | Hybrid oligonucleotide phosphorothioates |
| US5506351A (en) | 1992-07-23 | 1996-04-09 | Isis Pharmaceuticals | Process for the preparation of 2'-O-alkyl guanosine and related compounds |
| WO1994002499A1 (en) | 1992-07-27 | 1994-02-03 | Hybridon, Inc. | Oligonucleotide alkylphosphonothioates |
| US5583020A (en) | 1992-11-24 | 1996-12-10 | Ribozyme Pharmaceuticals, Inc. | Permeability enhancers for negatively charged polynucleotides |
| US5574142A (en) | 1992-12-15 | 1996-11-12 | Microprobe Corporation | Peptide linkers for improved oligonucleotide delivery |
| US6159946A (en) | 1993-01-07 | 2000-12-12 | Thomas Jefferson University | Antisense inhibition of c-myc to modulate the proliferation of smooth muscle cells |
| US5556948A (en) | 1993-01-22 | 1996-09-17 | Mitsubishi Chemical Corporation | Phospholipid derivatized with PEG bifunctional linker and liposome containing it |
| WO1994017093A1 (en) | 1993-01-25 | 1994-08-04 | Hybridon, Inc. | Oligonucleotide alkylphosphonates and alkylphosphonothioates |
| US5476925A (en) | 1993-02-01 | 1995-12-19 | Northwestern University | Oligodeoxyribonucleotides including 3'-aminonucleoside-phosphoramidate linkages and terminal 3'-amino groups |
| US5705188A (en) | 1993-02-19 | 1998-01-06 | Nippon Shinyaku Company, Ltd. | Drug composition containing nucleic acid copolymer |
| US5395619A (en) | 1993-03-03 | 1995-03-07 | Liposome Technology, Inc. | Lipid-polymer conjugates and liposomes |
| WO1994020073A1 (en) | 1993-03-03 | 1994-09-15 | Liposome Technology, Inc. | Lipid-polymer conjugates and liposomes |
| US5466677A (en) | 1993-03-06 | 1995-11-14 | Ciba-Geigy Corporation | Dinucleoside phosphinates and their pharmaceutical compositions |
| US5508270A (en) | 1993-03-06 | 1996-04-16 | Ciba-Geigy Corporation | Nucleoside phosphinate compounds and compositions |
| US5576427A (en) | 1993-03-30 | 1996-11-19 | Sterling Winthrop, Inc. | Acyclic nucleoside analogs and oligonucleotide sequences containing them |
| US5663312A (en) | 1993-03-31 | 1997-09-02 | Sanofi | Oligonucleotide dimers with amide linkages replacing phosphodiester linkages |
| US5658873A (en) | 1993-04-10 | 1997-08-19 | Degussa Aktiengesellschaft | Coated sodium percarbonate particles, a process for their production and detergent, cleaning and bleaching compositions containing them |
| US5462854A (en) | 1993-04-19 | 1995-10-31 | Beckman Instruments, Inc. | Inverse linkage oligonucleotides for chemical and enzymatic processes |
| US5539082A (en) | 1993-04-26 | 1996-07-23 | Nielsen; Peter E. | Peptide nucleic acids |
| WO1994026764A1 (en) | 1993-05-12 | 1994-11-24 | Centre National De La Recherche Scientifique (Cnrs) | Triester phosphorothioate oligonucleotides and method of preparation |
| US5770713A (en) | 1993-05-12 | 1998-06-23 | Centre National De La Recherche Scientifique | Phosphorothioate triester oligonucleotides and method of preparation |
| US5534259A (en) | 1993-07-08 | 1996-07-09 | Liposome Technology, Inc. | Polymer compound and coated particle composition |
| US5543158A (en) | 1993-07-23 | 1996-08-06 | Massachusetts Institute Of Technology | Biodegradable injectable nanoparticles |
| US5417978A (en) | 1993-07-29 | 1995-05-23 | Board Of Regents, The University Of Texas System | Liposomal antisense methyl phosphonate oligonucleotides and methods for their preparation and use |
| US5763588A (en) | 1993-09-17 | 1998-06-09 | Gilead Sciences, Inc. | Pyrimidine derivatives for labeled binding partners |
| US5502177A (en) | 1993-09-17 | 1996-03-26 | Gilead Sciences, Inc. | Pyrimidine derivatives for labeled binding partners |
| US6005096A (en) | 1993-09-17 | 1999-12-21 | Gilead Sciences, Inc. | Pyrimidine derivatives |
| US5719262A (en) | 1993-11-22 | 1998-02-17 | Buchardt, Deceased; Ole | Peptide nucleic acids having amino acid side chains |
| US5540935A (en) | 1993-12-06 | 1996-07-30 | Nof Corporation | Reactive vesicle and functional substance-fixed vesicle |
| US5457187A (en) | 1993-12-08 | 1995-10-10 | Board Of Regents University Of Nebraska | Oligonucleotides containing 5-fluorouracil |
| US5446137B1 (en) | 1993-12-09 | 1998-10-06 | Behringwerke Ag | Oligonucleotides containing 4'-substituted nucleotides |
| US5446137A (en) | 1993-12-09 | 1995-08-29 | Syntex (U.S.A.) Inc. | Oligonucleotides containing 4'-substituted nucleotides |
| US5565350A (en) | 1993-12-09 | 1996-10-15 | Thomas Jefferson University | Compounds and methods for site directed mutations in eukaryotic cells |
| US5595756A (en) | 1993-12-22 | 1997-01-21 | Inex Pharmaceuticals Corporation | Liposomal compositions for enhanced retention of bioactive agents |
| US5519134A (en) | 1994-01-11 | 1996-05-21 | Isis Pharmaceuticals, Inc. | Pyrrolidine-containing monomers and oligomers |
| US5599928A (en) | 1994-02-15 | 1997-02-04 | Pharmacyclics, Inc. | Texaphyrin compounds having improved functionalization |
| US5470307A (en) | 1994-03-16 | 1995-11-28 | Lindall; Arnold W. | Catheter system for controllably releasing a therapeutic agent at a remote tissue site |
| US5596091A (en) | 1994-03-18 | 1997-01-21 | The Regents Of The University Of California | Antisense oligonucleotides comprising 5-aminoalkyl pyrimidine nucleotides |
| US5627053A (en) | 1994-03-29 | 1997-05-06 | Ribozyme Pharmaceuticals, Inc. | 2'deoxy-2'-alkylnucleotide containing nucleic acid |
| US5625050A (en) | 1994-03-31 | 1997-04-29 | Amgen Inc. | Modified oligonucleotides and intermediates useful in nucleic acid therapeutics |
| US5646269A (en) | 1994-04-28 | 1997-07-08 | Gilead Sciences, Inc. | Method for oligonucleotide analog synthesis |
| US5525711A (en) | 1994-05-18 | 1996-06-11 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Pteridine nucleotide analogs as fluorescent DNA probes |
| US5543152A (en) | 1994-06-20 | 1996-08-06 | Inex Pharmaceuticals Corporation | Sphingosomes for enhanced drug delivery |
| US6344028B1 (en) | 1994-06-30 | 2002-02-05 | Boston Scientific Corporation | Replenishable stent and delivery system |
| US5597696A (en) | 1994-07-18 | 1997-01-28 | Becton Dickinson And Company | Covalent cyanine dye oligonucleotide conjugates |
| US5580731A (en) | 1994-08-25 | 1996-12-03 | Chiron Corporation | N-4 modified pyrimidine deoxynucleotides and oligonucleotide probes synthesized therewith |
| US5591584A (en) | 1994-08-25 | 1997-01-07 | Chiron Corporation | N-4 modified pyrimidine deoxynucleotides and oligonucleotide probes synthesized therewith |
| US5597909A (en) | 1994-08-25 | 1997-01-28 | Chiron Corporation | Polynucleotide reagents containing modified deoxyribose moieties, and associated methods of synthesis and use |
| WO1996010391A1 (en) | 1994-09-30 | 1996-04-11 | The University Of British Columbia | Polyethylene glycol modified ceramide lipids and liposome uses thereof |
| US5591721A (en) | 1994-10-25 | 1997-01-07 | Hybridon, Inc. | Method of down-regulating gene expression |
| US5512295A (en) | 1994-11-10 | 1996-04-30 | The Board Of Trustees Of The Leland Stanford Junior University | Synthetic liposomes for enhanced uptake and delivery |
| US5792747A (en) | 1995-01-24 | 1998-08-11 | The Administrators Of The Tulane Educational Fund | Highly potent agonists of growth hormone releasing hormone |
| US5674242A (en) | 1995-06-06 | 1997-10-07 | Quanam Medical Corporation | Endoprosthetic device with therapeutic compound |
| US5609629A (en) | 1995-06-07 | 1997-03-11 | Med Institute, Inc. | Coated implantable medical device |
| WO1996040062A1 (en) | 1995-06-07 | 1996-12-19 | Georgetown University | A method of transfection of cells using liposomally encapsulated nucleic acids |
| US5824049A (en) | 1995-06-07 | 1998-10-20 | Med Institute, Inc. | Coated implantable medical device |
| WO1997004787A1 (en) | 1995-08-01 | 1997-02-13 | Novartis Ag | Liposomal oligonucleotide compositions |
| US5652356A (en) | 1995-08-17 | 1997-07-29 | Hybridon, Inc. | Inverted chimeric and hybrid oligonucleotides |
| WO1997013499A1 (en) | 1995-10-11 | 1997-04-17 | The University Of British Columbia | Liposomal formulations of mitoxantrone |
| WO1997030731A2 (en) | 1996-02-21 | 1997-08-28 | The Immune Response Corporation | Method of preparing polynucleotide-carrier complexes for delivery to cells |
| US5670161A (en) | 1996-05-28 | 1997-09-23 | Healy; Kevin E. | Biodegradable stent |
| US6368356B1 (en) | 1996-07-11 | 2002-04-09 | Scimed Life Systems, Inc. | Medical devices comprising hydrogel polymers having improved mechanical properties |
| WO1998039352A1 (en) | 1997-03-07 | 1998-09-11 | Takeshi Imanishi | Novel bicyclonucleoside and oligonucleotide analogues |
| US6547787B1 (en) | 1997-03-13 | 2003-04-15 | Biocardia, Inc. | Drug delivery catheters that attach to tissue and methods for their use |
| US6495532B1 (en) | 1997-03-19 | 2002-12-17 | Sky High, Llc | Compositions containing lysophosphotidic acids which inhibit apoptosis and uses thereof |
| US6200304B1 (en) | 1997-07-11 | 2001-03-13 | Cardiogene Gentherapeutische Systeme Ag | Transfection system, its preparation and use in somatic gene therapy |
| US6217886B1 (en) | 1997-07-14 | 2001-04-17 | The Board Of Trustees Of The University Of Illinois | Materials and methods for making improved micelle compositions |
| WO1999014226A2 (en) | 1997-09-12 | 1999-03-25 | Exiqon A/S | Bi- and tri-cyclic nucleoside, nucleotide and oligonucleotide analogues |
| US6287285B1 (en) | 1998-01-30 | 2001-09-11 | Advanced Cardiovascular Systems, Inc. | Therapeutic, diagnostic, or hydrophilic coating for an intracorporeal medical device |
| US6433012B1 (en) | 1998-03-25 | 2002-08-13 | Large Scale Biology Corp. | Method for inhibiting inflammatory disease |
| US6344035B1 (en) | 1998-04-27 | 2002-02-05 | Surmodics, Inc. | Bioactive agent release coating |
| US6270790B1 (en) | 1998-08-18 | 2001-08-07 | Mxneil-Ppc, Inc. | Soft, convex shaped chewable tablets having reduced friability |
| US6562864B1 (en) | 1999-09-02 | 2003-05-13 | Drake Larson | Catechin multimers as therapeutic drug delivery agents |
| US20030215503A1 (en) | 2002-04-04 | 2003-11-20 | Tanya Havlir | Palatable chewable tablet |
| US20060009410A1 (en) * | 2002-11-13 | 2006-01-12 | Crooke Rosanne M | Effects of apolipoprotein B inhibition on gene expression profiles in animals |
| US20040265372A1 (en) | 2003-06-27 | 2004-12-30 | David Wynn | Soft tablet containing high molecular weight cellulosics |
| US20100297105A1 (en) | 2007-03-24 | 2010-11-25 | Genzyme Corporation | Administering antisense oligonucleotides complementary to human apolipoprotein b |
| WO2010076248A1 (en) * | 2008-12-31 | 2010-07-08 | Santaris Pharma A/S | Use of lna apob antisense oligomers for the treatment of acute coronary syndromes |
Non-Patent Citations (137)
| Title |
|---|
| "Antisense Research and Applications", 1993, CRC PRESS, pages: 276 - 278 |
| "Carbohydrate Modifications in Antisense Research", 1994, AMERICAN CHEMICAL SOCIETY |
| "Coordinating Committee", 2004, NCEP |
| "guidelines established by the Adult Treatment Panel III", 2001, NATIONAL CHOLESTEROL EDUCATION PROGRAM |
| "Peptide Nucleic Acids (PNA): Synthesis, Properties and Potential Applications", BIOORGANIC & MEDICINAL CHEMISTRY, vol. 4, 1996, pages 5 - 23 |
| "Pharmaceutical Dosage Forms", vol. 1, 1988, MARCEL DEKKER, INC., pages: 245 |
| "Physician's Desk Reference", 2006 |
| "Remington's Pharmaceutical Sciences", 1990, MACK PUBLISHING CO. |
| "The Concise Encyclopedia Of Polymer Science And Engineering", 1990, JOHN WILEY & SONS, pages: 858 - 859 |
| "The Merck Manual of Diagnosis and Therapy", 1987, pages: 1206 - 1228 |
| "The Merck Manual of Diagnosis and Therapy", 1987, pages: 2499 - 2506,46- |
| ALLEN ET AL., FEBS LETTERS, vol. 223, 1987, pages 42 |
| ALTSCHUL ET AL., J. MOL. BIOL., vol. 215, 1990, pages 403 - 410 |
| AUSUBEL, F. M. ET AL.: "Current Protocols in Molecular Biology", vol. 1, 1993, JOHN WILEY & SONS, INC., pages: 4. 5. 1 - 4. 5. 3 |
| AUSUBEL, F. M. ET AL.: "Current Protocols in Molecular Biology", vol. 1, 1993, JOHN WILEY & SONS, INC., pages: 4.1.1 - 4.2.9,4. |
| AUSUBEL, F. M. ET AL.: "Current Protocols in Molecular Biology", vol. 2, 1991, JOHN WILEY & SONS, INC., pages: 11. 2. 1 - 11. 2. 2 |
| AUSUBEL, F. M. ET AL.: "Current Protocols in Molecular Biology", vol. 2, 1997, JOHN WILEY & SONS, INC., pages: 10. 8. 1 - 10. 8. 2 |
| AUSUBEL, F. M. ET AL.: "Current Protocols in Molecular Biology", vol. 2, 1997, JOHN WILEY & SONS, INC., pages: 11. 12. - 11. 12. |
| AUSUBEL, F. M. ET AL.: "Current Protocols in Molecular Biology", vol. 2, 1997, JOHN WILEY & SONS, INC., pages: 11.4. 1 - 11. 11.5 |
| AUSUBEL, F. M. ET AL.: "Current Protocols in Molecular Biology", vol. 2, 1998, JOHN WILEY & SONS, INC., pages: 10. 16. - 10. 16. |
| AUSUBEL, F.M. ET AL.: "Current Protocols in Molecular Biology", vol. 1, 1996, JOHN WILEY & SONS, INC., pages: 4. 2. 1 - 4. 2. 9 |
| BASRA SUKHDEEP S ET AL: "Acute coronary syndromes: unstable angina and non-ST elevation myocardial infarction.", CARDIOLOGY CLINICS AUG 2014, vol. 32, no. 3, 2 August 2014 (2014-08-02), pages 353 - 370, XP009187386, ISSN: 1558-2264 * |
| BEAUCAGE, S. L.; CARUTHERS, M. H., TETRAHEDRON LETT., vol. 22, 1981, pages 1859 - 1862 |
| BERGE ET AL.: "Pharmaceutical Salts", J. OF PHARMA SCI., vol. 66, 1977, pages 1 - 19 |
| BLOCK: "Pharmaceutical Dosage Forms", vol. 1, 1988, MARCEL DEKKER, INC., pages: 335 |
| BLOCK: "Pharmaceutical Dosage Forms", vol. 2, 1988, MARCEL DEKKER, INC., pages: 335 |
| BLUME ET AL., BIOCHIMICA ET BIOPHYSICA ACTA, vol. 1029, 1990, pages 91 |
| BONOW, CIRCULATION, vol. 106, 2002, pages 3140 - 3141 |
| BRAZMA; VILO, FEBS LETT., vol. 480, 2000, pages 17 - 24 |
| BRUNTON ET AL.: "Goodman & Gilman's The Pharmacological Basis of Therapeutics", 1996, MCGRAW-HILL, pages: 934 - 935 |
| BUUR ET AL., J. CONTROL REL., vol. 14, 1990, pages 43 - 51 |
| CARULLI ET AL., J. CELL BIOCHEM., vol. 31, 1998, pages 286 - 96 |
| CELIS ET AL., FEBS LETT., vol. 480, 2000, pages 2 - 16 |
| CHAN ET AL., DIABETES, vol. 51, 2002, pages 2377 - 2386 |
| CHAN ET AL., METABOLISM, vol. 51, 2002, pages 1041 - 1046 |
| CHIANG ET AL., J. BIOL. CHEM., vol. 266, 1991, pages 18162 - 18171 |
| CLIN BIOCHEM., vol. 46, no. 15, October 2013 (2013-10-01), pages 1353 - 71 |
| CONSTANTINIDES ET AL., PHARMACEUTICAL RESEARCH, vol. 11, 1994, pages 1385 |
| CONSTANTINIDES ET AL., PHARMACEUTICAL RESEARCH, vol. 11, 1994, pages 1385 - 1390 |
| CROOKE ET AL., J. PHARMACOL. EXP. THER., vol. 277, 1996, pages 923 - 937 |
| DAHL, B. J. ET AL., ACTA CHEM. SCAND, vol. 44, 1990, pages 639 - 641 |
| DAVIDSON; SHELNESS, ANNU. REV. NUTR., vol. 20, 2000, pages 169 - 193 |
| DAVIS; HUI, ARTERIOSCLER. THROMB. VASE. BIOL., vol. 21, 2001, pages 887 - 898 |
| DU PLESSIS ET AL., ANTIVIRAL RESEARCH, vol. 18, 1992, pages 259 - 265 |
| EL HARIRI ET AL., J. PHARM. PHARMACOL., vol. 44, 1992, pages 651 - 654 |
| ENGLISCH ET AL., ANGEWANDTE CHEMIE, INTERNATIONAL EDITION, vol. 30, 1991, pages 613 |
| FARESE ET AL., PROC. NATL. ACAD. SCI. U.S.A., vol. 92, 1995, pages 1774 - 1778 |
| FATIMA AKDIM ET AL: "Efficacy and Safety of Mipomersen, an Antisense Inhibitor of Apolipoprotein B, in Hypercholesterolemic Subjects Receiving Stable Statin Therapy", JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY., vol. 55, no. 15, 1 April 2010 (2010-04-01), US, pages 1611 - 1618, XP055229790, ISSN: 0735-1097, DOI: 10.1016/j.jacc.2009.11.069 * |
| FLAIM ET AL., J AM HEART ASSOC., vol. 3, 2014 |
| FLAIM ET AL.: "A phase I study in Healthy Volunteers to Evaluate the Pharokinetics, Safety and th Tolerability of Mipomersen in Three dosing Regimens", POSTER PRESENTATION, 2011 |
| FUCHS ET AL., ANAL. BIOCHEM., vol. 286, 2000, pages 91 - 98 |
| GABIZON ET AL., PROC. NATL. ACAD. SCI. U.S. A., vol. 85, 1988, pages 6949 |
| GOING; GUSTERSON, EUR. J. CANCER, vol. 35, 1999, pages 1895 - 904 |
| GRIFFIN, B. E. ET AL., TETRAHEDRON, vol. 23, 1967, pages 2301 - 2313 |
| GRIFFIN, B. E. ET AL., TETRAHEDRON, vol. 23, 1967, pages 2315 - 2331 |
| GRUNDY ET AL., CIRCULATION, vol. 110, 2004, pages 227 - 239 |
| GRUNDY, AM. J. CARDIOL., vol. 81, 1998, pages 18B - 25B |
| HIGUCHI ET AL.: "Remington's Pharmaceutical Sciences", 1985, MACK PUBLISHING CO., pages: 301 |
| HO ET AL., J. PHARM. SCI., vol. 85, 1996, pages 138 - 143 |
| HU ET AL., S. T. P. PHARMA. SCI., vol. 4, 6, 1994, pages 466 |
| IDSON: "Pharmaceutical Dosage Forms", vol. 1, 1988, MARCEL DEKKER, INC., pages: 199 |
| ILIUM ET AL., FEBS LETT., vol. 167, 1984, pages 79 |
| JARRETT, J. CHROMATOGR., vol. 618, 1993, pages 315 - 339 |
| JONES, L. J. ET AL., ANALYTICAL BIOCHEMISTRY, vol. 265, 1998, pages 368 - 374 |
| JUNGBLUT ET AL., ELECTROPHORESIS, vol. 20, 1999, pages 2100 - 10 |
| JURECIC; BELMONT, CURR. OPIN. MICROBIOL., vol. 3, 2000, pages 316 - 21 |
| KABANOV ET AL., FEBS LETT., vol. 259, 1990, pages 327 - 330 |
| KANE; HAVEL: "The Metabolic and Molecular Bases of Inherited Diseases", vol. 8, 2001, pages: 2717 - 2751 |
| KASTELEIN JOHN J P ET AL: "Potent reduction of apolipoprotein B and low-density lipoprotein cholesterol by short-term administration of an antisense inhibitor of apolipoprotein B", CIRCULATION, LIPPINCOTT WILLIAMS & WILKINS, US, vol. 114, no. 16, 1 October 2006 (2006-10-01), pages 1729 - 1735, XP002487104, ISSN: 0009-7322, DOI: 10.1161/CIRCULATIONAHA.105.606442 * |
| KAWASAKI, J. MED. CHEM., vol. 36, 1993, pages 831 - 841 |
| KIM; YOUNG, J. LIPID RES., vol. 39, 1998, pages 703 - 723 |
| KLIBANOV ET AL., FEBS LETT., vol. 268, 1990, pages 235 |
| KNOPP, NEW ENGL. J. MEDICINE, vol. 341, 1999, pages 498 - 511 |
| LARSON ET AL., CYTOMETRY, vol. 41, 2000, pages 203 - 208 |
| LARSSON ET AL., J. BIOTECHNOL., vol. 80, 2000, pages 143 - 57 |
| LEE ET AL., CRITICAL REVIEWS IN THERAPEUTIC DRUG CARRIER SYSTEMS, 1991, pages 92 |
| LETSINGER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 86, 1989, pages 6553 - 6556 |
| LEUNG; SHAH: "Controlled Release of Drugs: Polymers and Aggregate Systems", 1989, VCH PUBLISHERS, pages: 185 - 215 |
| MADDEN ET AL., DRUG DISCOV. TODAY, vol. 5, 2000, pages 415 - 425 |
| MANOHARA ET AL., TETRAHEDRON LETT., vol. 36, 1995, pages 3651 - 3654 |
| MANOHARAN ET AL., ANN. N. Y. ACAD. SCI., vol. 660, 1992, pages 306 - 309 |
| MANOHARAN ET AL., BIOORG. MED. CHEM. LET., vol. 3, 1993, pages 2765 - 2770 |
| MANOHARAN ET AL., BIOORG. MED. CHEM. LET., vol. 4, 1994, pages 1053 - 1060 |
| MANOHARAN ET AL., NUCLEOSIDES & NUCLEOTIDES, vol. 14, 1995, pages 969 - 973 |
| MANOHARAN ET AL., TETRAHEDRON LETT., vol. 36, 1995, pages 3651 - 3654 |
| MARTIN ET AL., HELV. CHIM. ACTA, vol. 78, 1995, pages 486 - 504 |
| MARTIN, P., HELVETICA CHIMICA ACTA, vol. 78, 1995, pages 486 - 504 |
| MATTEUCCI, M. D.; CARUTHERS, M. H., J. AM. CHEM. SOC., vol. 103, 1981, pages 3185 - 3191 |
| MISHRA ET AL., BIOCHIM. BIOPHYS. ACTA, vol. 1264, 1995, pages 229 - 237 |
| MIURA ET AL., CLIN. CHEM., vol. 42, 1996, pages 1758 - 1764 |
| MIYAO ET AL., ANTISENSE RES. DEV., vol. 5, 1995, pages 115 - 121 |
| MURANISHI, CRITICAL REVIEWS IN THERAPEUTIC DRUG CARRIER SYSTEMS, vol. 7, 1990, pages 1 - 33 |
| NIELSEN ET AL., SCIENCE, vol. 254, 1991, pages 1497 - 1500 |
| NISHINA ET AL., J. LIPID RES., vol. 31, 1990, pages 859 - 869 |
| NOGRADY: "Medicinal Chemistry A Biochemical Approach", 1985, OXFORD UNIVERSITY PRESS, pages: 388 - 392 |
| NOWAK-GOTTL ET AL., PEDIATRICS, vol. 99, 1997, pages E11 |
| NUTR METAB CARDIOVASC DIS., vol. 24, no. 10, October 2014 (2014-10-01), pages 1057 - 66 |
| OBERHAUSER, NUCL. ACIDS RES., vol. 20, 1992, pages 533 - 538 |
| PAPAHADJOPOULOS ET AL., ANN. N. Y. ACAD. SCI., vol. 507, 1987, pages 64 |
| PRASHAR; WEISSMAN, METHODS ENZYMOL., vol. 303, 1999, pages 258 - 72 |
| RAAL F J ET AL: "Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial", THE LANCET, THE LANCET PUBLISHING GROUP, GB, vol. 375, no. 9719, 20 March 2010 (2010-03-20), pages 998 - 1006, XP026978121, ISSN: 0140-6736, [retrieved on 20100319], DOI: 10.1016/S0140-6736(10)60284-X * |
| RAINWATER; KAMMERER, J. EXP. ZOOL., vol. 282, 1998, pages 54 - 61 |
| REDDY, M. P. ET AL., TETRAHEDROM LETT., vol. 25, 1994, pages 4311 - 4314 |
| RIEGER: "Pharmaceutical Dosage Forms", 1988, MARCEL DEKKER, INC., pages: 285 |
| RIEGER: "Pharmaceutical Dosage Forms", vol. 1, 1988, MARCEL DEKKER, INC., pages: 285 |
| RITSCHEL, METH. FIND. EXP. CLIN. PHARMACOL., vol. 13, 1993, pages 205 |
| ROSOFF: "Pharmaceutical Dosage Forms", vol. 1, 1988, MARCEL DEKKER, INC., pages: 245 |
| SAHEBJAR; WATTS, CARDIOVASCULAR DRUGS THER., vol. 27, 2013, pages 559 - 567 |
| SAISON-BEHMOARAS ET AL., EMBO J., vol. 10, 1991, pages 1111 - 1118 |
| SANDKAMP ET AL., CLIN. CHEM., vol. 36, 1990, pages 20 - 23 |
| SANGHVI, NUCLEIC ACIDS RESEARCH, vol. 21, 1993, pages 3197 - 3203 |
| SANGHVI, Y. S.: "Antisense Research and Applications", 1993, CRC PRESS, pages: 289 - 302 |
| SCARINGE, S. A. ET AL., J. AM. CHEM. SOC., vol. 120, 1998, pages 11820 - 11821 |
| SCARINGE, S. A., PH. D. THESIS, 1996 |
| SCHONFELD ET AL., J. LIPID RES., vol. 44, 2003, pages 878 - 883 |
| SCHOTT: "Remington's Pharmaceutical Sciences", 1985, MACK PUBLISHING CO., pages: 271 |
| SEED ET AL., N. ENGL. J. MED., vol. 322, 1990, pages 1494 - 1499 |
| SHEA ET AL., NUCL. ACIDS RES., vol. 18, 1990, pages 3777 - 3783 |
| SIMONS ET AL., NATURE, vol. 359, 1992, pages 67 - 70 |
| STANLEY CROOKE: "Antisense Drug Technology: Principles, Strategies, and Applications", 2008, TAYLOR & FRANCIS GROUP |
| SUNAMOTO ET AL., BULL. CHEM. SOC. JPN., vol. 53, 1980, pages 2778 |
| SUTCLIFFE ET AL., PROC. NATL. ACAD. SCI. U.S. A., vol. 97, 2000, pages 1976 - 81 |
| SVINARCHUK ET AL., BIOCHIMIE, vol. 75, 1993, pages 49 - 54 |
| SWINYARD: "Remington's Pharmaceutical Sciences", 1990, MACK PUBLISHING CO., pages: 782 - 783 |
| TAKAHASHI ET AL., J. PHARM. PHARMACOL., vol. 40, 1988, pages 252 |
| TAKAKURA ET AL., ANTISENSE & NUCL. ACID DRUG DEV., vol. 6, 1996, pages 177 - 183 |
| TANAKA, JOURN. BIOL. CHEM., vol. 268, 1993, pages 12713 - 12718 |
| TO, COMB. CHEM. HIGH THROUGHPUT SCREEN, vol. 3, 2000, pages 235 - 41 |
| WANG ET AL., BIOCHEM. BIOPHYS. RES. COMMUN., vol. 147, 1987, pages 980 - 985 |
| WEINER ET AL., JOURNAL OF DRUG TARGETING, vol. 2, 1992, pages 405 - 410 |
| WINCOTT, F. ET AL., NUCLEIC ACIDS RES., vol. 23, 1995, pages 2677 - 2684 |
| WU ET AL., CANCER RESEARCH, vol. 53, 1993, pages 3765 |
| YAMAMOTO ET AL., J. PHARM. EXP. THER., vol. 263, 1992, pages 25 |
| YAMASHITA ET AL., J. PHARM. PHARMACOL., vol. 39, 1987, pages 621 - 626 |
| YAMASHITA ET AL., J. PHARM. SCI., vol. 79, 1990, pages 579 - 583 |
| ZHANG; MADDEN, GENOME RES., vol. 7, 1997, pages 649 - 656 |
| ZHOU ET AL., JOURNAL OF CONTROLLED RELEASE, vol. 19, 1992, pages 269 - 274 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4035659A1 (en) | 2016-11-29 | 2022-08-03 | PureTech LYT, Inc. | Exosomes for delivery of therapeutic agents |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1569695B1 (en) | Antisense modulation of apolipoprotein b expression | |
| EP2174945B1 (en) | Antisense modulation of apolipoprotein B expression | |
| USRE44760E1 (en) | Antisense modulation of apolipoprotein B-expression | |
| AU2002326481A1 (en) | Antisense modulation of apolipoprotein B expression | |
| WO2004005460A2 (en) | Antisense modulation of hmg-coa reductase expression | |
| WO2003097662A1 (en) | Antisense modulation of apolipoprotein b expression | |
| WO2003014307A2 (en) | Antisense modulation of apolipoprotein(a) expression | |
| AU2002355552A1 (en) | Antisense modulation of apolipoprotein(A) expression | |
| WO2003066805A2 (en) | Antisense modulation of complement component c3 expression | |
| WO2003011888A1 (en) | Antisense modulation of sap-1 expression | |
| WO2002070535A1 (en) | Antisense modulation of recql5 expression | |
| WO2002068690A1 (en) | Antisense modulation of wrn expression | |
| EP1436310A1 (en) | Antisense modulation of vascular endothelial growth factor receptor-2 expression | |
| EP1461463A2 (en) | Antisense modulation of hematopoietic cell protein tyrosine kinase expression | |
| WO2002088163A1 (en) | Antisense modulation of interferon gamma receptor 2 expression | |
| WO2016033424A1 (en) | Methods for the prevention and treatment of major adverse cardiovascular events using compounds that modulate apolipoprotein b | |
| WO2002068590A2 (en) | Antisense modulation of recql expression | |
| WO2002092855A1 (en) | Antisense modulation of helicase-moi expression | |
| EP1451353A2 (en) | Antisense modulation of phospholipase a2 group v expression | |
| ZA200503690B (en) | Antisense modulation of apolipoprotein B expression | |
| WO2003010139A2 (en) | Antisense modulation of hormone-sensitive lipase expression | |
| AU2002355257A1 (en) | Antisense modulation of hormone-sensitive lipase expression | |
| WO2004035676A2 (en) | Antisense modulation of notch (drosophila) homolog 4 expression | |
| WO2003023011A2 (en) | Antisense modulation of ribonuclease l expression |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15760593 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 15760593 Country of ref document: EP Kind code of ref document: A1 |